


Abstract
CRISPR–Cas (Clustered regularly interspaced short palindromic repeats-CRISPR-associated proteins) is a prokaryotic immune system that destroys foreign nucleic acids in a sequence-specific manner using Cas nucleases guided by short RNAs (crRNAs). Staphylococcus epidermidis harbours a Type III-A CRISPR–Cas system that encodes the Cas10–Csm interference complex and crRNAs that are subjected to multiple processing steps. The final step, called maturation, involves a concerted effort between Csm3, a ruler protein in Cas10–Csm that measures six-nucleotide increments, and the activity of a nuclease(s) that remains unknown. Here, we elucidate the contributions of the Cas10–Csm complex toward maturation and explore roles of non-Cas nucleases in this process. Using genetic and biochemical approaches, we show that charged residues in Csm3 facilitate its self-assembly and dictate the extent of maturation cleavage. Additionally, acidic residues in Csm5 are required for efficient maturation, but recombinant Csm5 fails to cleave crRNAs in vitro. However, we detected cellular nucleases that co-purify with Cas10–Csm, and show that Csm5 regulates their activities through distinct mechanisms. Altogether, our results support roles for non-Cas nuclease(s) during crRNA maturation and establish a link between Type III-A CRISPR–Cas immunity and central nucleic acid metabolism.
INTRODUCTION
Prokaryotes destroy viral predators and other nucleic acid invaders with an RNA-guided immune system called CRISPR–Cas (Clustered regularly interspaced short palindromic repeats- CRISPR-associated proteins) (1–4). In this system, CRISPR loci maintain a genetic memory of past invaders in the form of short (∼30–40 nucleotide) invader-derived sequences called ‘spacers’, which are integrated between repeat sequences of similar length (5–7). Nucleic acids complementary to spacers (called ‘protospacers’) are targeted for destruction by Cas nucleases in a mechanism that involves three stages: adaptation, crRNA biogenesis and interference (8,9). During adaptation, challenge with foreign nucleic acids of plasmid or viral origin stimulate the integration of spacers and repeats into the CRISPR locus, creating an archive of targets for future interference. During crRNA biogenesis, transcription of the repeat-spacer array produces a long precursor transcript (pre-crRNA), which is subsequently chopped to liberate mature crRNAs that each specify a single target. During interference, crRNAs assemble with Cas nucleases to form a ribonucleoprotein effector complex that recognizes and degrades cognate nucleic acid targets. While all CRISPR–Cas systems share this general pathway, they exhibit remarkable structural and mechanistic diversity. The current classification scheme groups CRISPR–Cas systems into two broad classes, six types (I–VI) and nineteen subtypes based on their architecture and cas gene content (10,11). Moreover, variants exist outside of this classification scheme that have yet to be characterized.
Essential for the function of all CRISPR–Cas systems is proper crRNA processing, which can involve multiple distinct steps carried out by both Cas and non-Cas nucleases. The first step, known as primary processing, entails the cleavage of pre-crRNAs within repeat sequences. In Types I and III systems, Cas6 or Cas5d serve as the primary processing endonuclease (12–15). In Type II systems, primary processing is carried out by host RNase III, which cleaves pre-crRNAs paired with antisense trans-activating crRNAs (16). While little is known about the putative Types IV, V and VI systems (10,11), primary processing in a Type V system was shown to be carried out by the effector protein Cpf1 (17). Primary processing generates intermediate crRNAs that contain a single spacer flanked by partial repeats on both ends. While in Type I systems these constitute the final crRNA product (2), other systems employ a less well characterized maturation step that further trims repeat and spacer sequences from one end (11,16–19).
Staphylococcus epidermidis RP62a possesses a Type III-A CRISPR–Cas system that encodes three spacers (spc1-3) and nine cas/csm genes (Figure 1A) (3). This system degrades both DNA and RNA targets with a ribonucleoprotein complex called Cas10–Csm, composed of Cas10, Csm2, Csm3, Csm4, Csm5 and a crRNA (15,20–23). Previous work in this system showed that primary processing is carried out by Cas6, which cleaves within direct repeats to liberate 71 nucleotide intermediates (Figure 1B) (15). The final maturation step involves further degradation of intermediate 3΄ ends in discrete 6-nucleotide increments, to generate a collection of mature crRNAs that are 31–67 nucleotides in length, with 31, 37 and 43 being the most abundant (Figure 1C) (23). These mature species possess uniform 5΄ ends with eight nucleotides of repeat sequence (called the 5΄-tag) and variable 3΄ ends composed of spacer-derived sequences. The extent of maturation cleavage on 3΄ ends is dictated by Csm3, the ‘backbone’ of Cas10–Csm (Figure 1D) (24), which acts as a ruler that polymerizes on crRNAs and measures 6-nucleotide segments (23). However, the nuclease(s) responsible for 3΄-end degradation remains unknown.
Figure 1.

CRISPR RNA maturation in a Type III-A CRISPR–Cas system. (A) Illustration of the CRISPR–Cas locus in S. epidermidis RP62a. This system contains four direct repeats (white squares), three spacers (coloured squares), and nine CRISPR-associated (cas and csm) genes. (B) Primary processing of precursor crRNAs is catalysed by Cas6, which cleaves within repeat sequences to yield 71 nucleotide intermediates. (C) The final maturation step involves Csm3, which acts as a ruler that protects 6-nucleotide segments, and the activity of an unknown nuclease that cleaves the unprotected portion of the 3΄ end. These combined activities yield a collection of mature crRNAs (mostly 31–43 nucleotide lengths) that share a uniform 5΄-tag derived from repeat sequences, and variable 3΄-ends that vary by 6-nucleotide increments. (D) A schematic of the Cas10–Csm complex, illustrating the predicted organization of the subunits relative to the crRNA. (E) Csm2, Csm3 and Csm5 are each essential for the maturation process. Csm2 is the small subunit specific to Type III-A systems with unknown function, while Csm3 and Csm5 belong to the Repeat-associated mysterious protein (RAMP) superfamily that possess conserved glycine-rich regions predicted to bind RNA. Conserved residues throughout these proteins that have been explored in previous studies (I) and this study (*) are indicated. See Supplementary Figure S1 for a more detailed depiction of this information. RRM, RNA Recognition Motif.
Previous work showed that in-frame deletions of csm2, csm3 or csm5 selectively eliminate maturation and cause an accumulation of intermediate species (19), therefore establishing essential roles for Csm2, Csm3 and Csm5 in 3΄-end cleavage (Figure 1E). While there are no predictions for Csm2 function, Csm3 and Csm5 belong to the repeat-associated mysterious protein (RAMP) superfamily and the Cas7 family of CRISPR-associated HD nucleases, which possess RNA recognition motifs and conserved acidic amino acids with predicted and/or demonstrated nucleolytic activity (10,25). Csm3 was shown to be one of the nucleases responsible for RNA degradation during the interference stage of CRISPR immunity (20,24,26); however, ablation of its active site had no impact on crRNA maturation (20). Low resolution structures of related Type III-A complexes place Csm5 proximal to crRNA 3΄ ends (24,26) (Figure 1D), leading to speculations that crRNA maturation is catalyzed by Csm5 (26,27). However, in previous work, alanine-scanning mutations introduced into predicted catalytic residues of Csm5 (summarized in Figure 1E and Supplementary Figure S1) failed to disrupt the efficiency of maturation (15,23). Additionally, purified Cas10–Csm complexes fail to cleave intermediate crRNAs, and when complexes are overexpressed and purified from Escherichia coli, a non-native background, mature crRNAs retain their six nucleotide periodicity, but final lengths are each two nucleotides shorter than crRNAs generated in the native S. epidermidis background (23). These observations lead to the hypothesis that the final maturation cleavage involves the activity of cellular nuclease(s) external to the CRISPR–Cas locus (23).
In this study, we sought to clarify the contributions of Csm2, Csm3 and Csm5 toward crRNA maturation and assess potential roles of non-Cas nucleases during this process. We introduced alanine-scanning mutations in conserved residues throughout Csm2, Csm3 and Csm5, and analysed mutations that specifically disrupt maturation without impacting Cas10–Csm complex formation. Through this analysis, we found charged residues in Csm3 that dictate the extent of crRNA maturation by facilitating Csm3 multimerization, therefore playing a role in the assembly of the maturation ruler. Further, acidic amino acids in Csm5 are essential for efficient maturation; however, Csm5 fails to exhibit ribonucleolytic activity in vitro. To explore the potential involvement of non-Cas nucleases, we looked for cellular nucleases that co-purify with Cas10–Csm using mass spectrometry, and found several, including all known 3΄-5΄ exoribonucleases in staphylococci: Cbf1, RNase R and polyribonucleotide phosphorylase (PNPase). Using a purified system, we show that Csm5 differentially modulates the activity of these nucleases–while blocking Cbf1 and RNase R activity, Csm5 causes a striking stimulation of PNPase. We further demonstrate a physical association between recombinant Csm5 and PNPase in vitro. Altogether, these results expand our understanding of the molecular requirements for crRNA maturation in the Cas10–Csm complex and support roles for non-Cas nuclease(s) in this process. Our results also establish a link between Type III-A CRISPR–Cas immunity and central nucleic acid metabolism in staphylococci.
MATERIALS AND METHODS
Strains and growth conditions
S. epidermidis LM1680 (23) was grown in Brain Heart Infusion (BHI) media (Difco). S. aureus RN4220 (28) was grown in Tryptic Soy Broth (Difco). E. coli DH5α and BL21 (DE3) codon plus cells (EMD Millipore) were grown in Luria Bertani broth (LB, Difco) or Terrific broth (Amresco) for protein purification. Media was supplemented with the following as needed: 10 μg/ml chloramphenicol (for selection of pcrispr-based plasmids), 15 μg/mL neomycin (for selection of S. epidermidis), 5 μg/ml mupirocin (for selection of pG0400), 30 μg/ml chloramphenicol (for selection of E. coli BL21 (DE3)) and 50 μg/ml kanamycin (for selection of pET28b-based plasmids).
Plasmid construction
Amino acid substitutions were introduced into pcrispr-cas (23) using either inverse PCR or Gibson assembly (29) with the primers listed in Supplementary Table S1. Following PCR, products were purified using the EZNA Cycle Pure Kit (Omega). For inverse PCR, the products were 5΄ phosphorylated by T4 polynucleotide kinase and then circularized by T4 DNA ligase (NEB). Ligated and Gibson assembled constructs were first transformed intro S. aureus RN4220 via electroporation. At least two transformants were characterized and at least one of each construct was purified and sequenced using the primers listed in Supplementary Table S1 to confirm the intended mutation was introduced in exclusion of any second-site mutations. Pcrispr-cas constructs that showed a phenotype were further transformed into S. epidermidis LM1680 using electroporation to confirm observed phenotypes.
pET28b-His10Smt3-cbf1 was constructed by amplifying cbf1 from the genome of S. epidermidis RP62a using primers A146 and A147. The PCR product and pET28b were digested with BamHI and XhoI (NEB) and purified. The fragments were ligated using T4 DNA ligase (NEB) and transformed into E. coli DH5α. The remaining pET28b-His10Smt3 constructs (encoding Csm3, Csm3K4A, Csm3D179A, Csm5, PNPase and RNase R) were created using Gibson assembly with primers listed in Supplementary Table S1. Constructs were introduced by heat-shock into E. coli DH5α, two transformants were selected, and their plasmids were purified and sequenced with primers T7T and T7P. At least one confirmed plasmid for each construct was transformed into BL21 (DE3) cells for protein purification.
CRISPR RNA capture
Spc1 crRNAs were captured from total RNA extracts using a biotinylated probe complementary to the spc1 (primer A17 in Supplementary Table S1) as described previously (19).
Conjugation
Conjugation was carried out using filter mating as described previously (3). Briefly, equal numbers of cells from mid-log cultures of S. aureus RN4220 carrying the conjugative plasmid pG0400 and S. epidermidis LM1680 carrying pcrispr-cas or its mutant derivatives were combined on a 0.45 μm filter and incubated overnight on non-selective BHI agar. The growth on the filter was resuspended, serially diluted, and plated on media selective for pcrispr-cas or pcrispr-cas and pG0400, to enumerate the number of recipients and transconjugants, respectively. Conjugation efficiency was determined by dividing the number of transconjugants by the number of recipients. Each pcrispr-cas mutant was assayed in triplicate.
Purification of Cas10–Csm from staphylococci and Csm3 quantification
Cas10–Csm complexes containing a 6-His tag on the N-terminus of Csm2 (Csm2H6N) were purified as described previously (23). Complexes were resolved using SDS PAGE and visualized with Coomassie G-250. Gels were imaged using the FluorChem™ system (Protein Simple) and AlphaView software was used to quantify band intensities. % Csm3 in each lane was calculated using the following formula: ((Csm3 band intensity) / (band intensities of Csm2 + Csm3 + Csm4 + Csm5 + Cas10)) × 100. Csm3 was quantified in three independent protein preparations.
Purification of recombinant Csm3, Csm3K4A, Csm3D179A, Csm5, Cbf1, RNase R and PNPase from E. coli
Recombinant proteins cloned into pET28b-His10Smt3 were overexpressed and purified from E. coli BL21 (DE3) as previously described (23) with slight modifications. Briefly, overnight cultures were diluted 1:100 in 1 L Terrific Broth containing 50 μg/ml kanamycin and 30 μg/ml chloramphenicol. Once A600 reached 0.5–0.6, protein expression was induced by the addition of 0.3 mM isopropyl-1-thio-β-d-galactopyranoside and 2% ethanol. Induction proceeded overnight at 17°C, (or 3 h at 37°C for Cbf1). Cells were harvested and washed with PBS Buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). All subsequent steps were performed at 4°C. For Csm5 purification, buffers with pH 7.0 were used; all other proteins were purified at pH 7.5 as indicated. Pellets were re-suspended in 30 ml of Buffer A (50 mM Tris–HCl, pH 7.5, 1.25 M NaCl, 200 mM Li2SO4, 10% sucrose, 15 mM Imidazole) containing one complete EDTA-free protease inhibitor tablet (Roche), 0.1 mg/ml lysozyme, and 0.1% Triton X-100. After 1hr incubation, lysed cells were sonicated, and insoluble material removed by centrifugation and filtration. Cleared lysates were mixed with 2 ml of Ni2+-NTA-agarose (Thermo, pre-equilibrated with Buffer A), and incubated for 1 h. with constant rotation. Resin was pelleted using centrifugation, and washed with 40 ml of Buffer A and 5 ml of 3 M KCl. The resin was re-suspended with 25 ml of Buffer A and applied to a column. Protein was eluted stepwise with 3 ml of IMAC buffer (50 mM Tris–HCl pH 7.5, 250 mM NaCl, 10% glycerol) containing 50, 100, 200 and 500 mM imidazole, respectively. The 200 mM Imidazole elutions were pooled and mixed with SUMO Protease (Mclab, 1000 U) and supplied buffer (salt-free), and dialyzed against Dialysis Buffer (25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 15 mM imidazole, 5% glycerol) for 3 h. The dialysate was incubated for 1 h. with 1 ml of Ni2+-NTA-agarose resin (pre-equilibrated with Dialysis Buffer), applied to a column, and the untagged protein was collected in the flow-through. Protein concentrations were determined using Bradford reagent (Bio-Rad).
Blue native PAGE
BN-PAGE (30) adapted from (31) was carried out using Mini-PROTEAN® Tetra Cell (Bio-Rad). Recombinant Csm3 and mutant variants (10 pmol each) were resolved in a 10% native polyacrylamide gel (29:1 acrylamide/bisacrylamide) using cathode buffer (25 mM Tris, 250 mM glycine, 0.025% Commasie blue G-250) in the inner chamber and the anode buffer (25 mM Tris, 250 mM Glycine) in the outer chamber. The system was submerged in an ice-water bath and run at 60 V for 2 h. Proteins were visualized with Coomassie G-250. NativeMark Protein Standard (Thermo Fisher Scientific) was used to estimate molecular weight.
Construction of Csm3 oligomer model
The S. epidermidis Csm3 amino acid sequence was submitted to both the Phyre2 and I-TASSER webservers for homology modeling (32,33). Both algorithms identified the coordinates contained in PDB ID 4N0L, an X-ray structure of the M. kandleri Csm3, as the best template for homology modeling. The Phyre2 derived model of S. epidermidis Csm3 was then aligned to each of the Cmr4 protomers in the X-ray structure of the high resolution Type III-B complex (PDB ID: 3x1L) (34). Alignment was performed in the PyMOL Molecular Graphics System (Version 1.8 Schrödinger, LLC) using the residues of ‘motif I’ and ‘motif II’ of the RAMP domain as these two regions of Csm3 and Cmr4 have high sequence identity and structural similarity (25).
Double filter-binding assay
The double-filter protein–nucleic acid binding assay described by Wong and Lohman (35), as adapted by Tanaka and Schwer (36), was used. 0.2 μm nitrocellulose membranes (BioTrace) were soaked for 10 min in 0.5 M NaOH and rinsed with dH2O until pH became neutral. 0.45 μm nylon membranes (PerkinElmer) were soaked for ten minutes in 0.1 M EDTA (pH 8.0), washed three times (10 min) in 1.0 M NaCl, rinsed in 0.5 M NaOH, and rinsed with dH2O until pH became neutral. After washing, the membranes were equilibrated in binding buffer (40 mM Tris–HCl pH 7.5, 2 mM DTT, 1 mM EDTA pH 8.0, 10% glycerol) at 4°C for at least an hour. The membranes were assembled with the nitrocellulose on top of the nylon in a 96-well dot-blot vacuum apparatus (Bio-Rad). Purified Csm5 and trace amounts of 5΄-end labeled RNA substrate (5΄-ACGAGAACACGUAUGCCGAAGUAUAUAAAUC) were combined in RNA-binding buffer (25 mM Tris–HCl pH 7.5, 2 mM DTT and 10 mM MgCl2) and incubated at room temperature for 10 min. Each well was washed with 200 ul of ice cold binding buffer, samples were applied, and wells were washed twice with 100 ul of ice cold binding buffer. Filters were air-dried, exposed to a storage phosphor screen, and imaged using a Typhoon FLA 7000 phosphorimager (GE). The fraction of RNA bound to Csm5 was calculated using ImageQuant software with the formula: RNA-bound fraction = (intensity of nitrocellulose signal)/(intensity of nitrocellulose signal + intensity of nylon signal) for each well. Each data point is the average of three technical replicates.
Mass spectrometry
Freshly purified Cas10–Csm(Csm2H6N) (∼100 μg) was submitted to the Comprehensive Cancer Center at the University of Alabama, Birmingham. Mass spectrometry and data analysis was conducted as described in (37) using an Orbitrap Velos Pro hybrid mass spectrometer, equipped with a nano-electrospray source (Thermo Fisher Scientific). Duplicate samples were run and data represents pooled results.
Nuclease assays
5΄ end-labeled substrates were combined with Csm5 (1–8 pmol as indicated) and/or 1 pmol of cellular nucleases in nuclease buffer (25 mM Tris–HCl pH 7.5, 2 mM DTT) containing the appropriate metals (10 mM MnCl2 for Cbf1, 10 mM MgCl2 for PNPase and 1 mM MgCl2 for RNase R). Nuclease reactions were allowed to proceed at 37°C for 10 min (unless otherwise indicated), and stopped by adding EDTA (10 mM) and placing on ice for 5 min, followed by a 20 min digestion with Proteinase K (1 μg) at room temperature. Samples were combined with an equal volume of 95% formamide loading buffer and resolved using denaturing PAGE.
Affinity pull-down assay
Csm5-Smt3 (1 nmol), Csm3–Smt3 (0.5 nmol), or dialysis buffer (input 1) were loaded onto a column containing 150 μl of packed Ni2+-NTA-agarose that was pre-equilibrated with IMAC buffer (50 mM Tris–HCl pH 7.0, 250 mM NaCl, 10% glycerol) containing 15 mM imidazole. The column was washed with 5 ml of IMAC buffer containing 50 mM imidazole. PNPase (0.7 nmol) was passed through each column. Columns were washed twice with 4 ml of IMAC buffer containing 50 mM imidazole. Proteins were eluted in 250 μl of IMAC buffer containing 500 mM imidazole. The assay was repeated at least three times for each input 1.
Two-dimensional native/SDS PAGE
Two-dimensional Native/SDS PAGE was carried out as described in (38) with some modifications. Briefly, vertical (Mini-PROTEAN® Tetra Cell, Bio-Rad) and horizontal formats (CBS Scientific) were used where indicated. Recombinant Csm5 and PNPase (100 pmols each) alone or combined were resolved in 5% native polyacrylamide gels (29:1 acrylamide/bisacrylamide), ∼3 mm thick in the horizontal format and 0.75 mm thick in the vertical format), and run in pre-chilled tris-glycine buffer (25 mM Tris, 250 mM glycine, pH 9.0) on an ice-water bath at 70 V for 140 min. Proteins were visualized with Coomassie G-250. NativeMark Protein Standard (Thermo Fisher Scientific) was used to estimate molecular weight. For the second dimension run, bands of interest were excised from native gels and soaked in 5× SDS sample buffer (0.5 M Tris, pH 6.8, 10% glycerol, 10% SDS, 1% bromophenol blue) for 1 h at room temperature. Soaked gel slices were inserted in between glass plates, and a 10% SDS-polyacrylamide resolving gel was cast ∼3 mm below the slices. Gel slices were then completely submerged in a 6% stacking gel. Gels were run at 100 V for ∼15 min (until the dye front passed the stacking gel), and then 130 V until the dye front reached the bottom of the gel. Proteins were visualized with Coomassie G-250 and molecular weight was estimated using a prestained protein standard (NEB).
RESULTS
Conserved residues in Csm3 and Csm5 impact crRNA maturation
To elucidate the specific contributions of the Cas10–Csm complex toward maturation, we conducted a thorough mutagenic analysis of the three subunits that are essential to this process: Csm2, Csm3 and Csm5 (19). Into the plasmid pcrispr-cas, which contains the entire functional CRISPR–Cas locus of S. epidermidis RP62a (23) (Figure 1A), we introduced alanine-scanning mutations in conserved residues (Figure 1E and Supplementary Figure S1). In addition to acidic amino acids, which might participate in metal-dependent nuclease activity, basic residues were mutated, as they might facilitate protein–protein or protein–RNA interactions. Pcrispr-cas mutants were assayed for maturation defects using a two-tiered approach: Mutant constructs were first expressed in S. aureus RN4220, a relative of S. epidermidis that lacks its own CRISPR–Cas system (15). Any mutants that exhibited maturation defects in this heterologous system were then confirmed in the native background of S. epidermidis LM1680, a derivative of S. epidermidis RP62a that harbours a deletion in the CRISPR–Cas locus (23). Defects in maturation were detected using a crRNA capture assay, in which spc1 crRNAs were purified from a total RNA extract using a biotinylated antisense oligonucleotide. We observed that while alanine-scanning mutations in Csm2 had no effect on maturation (Supplementary Figure S2A), mutations in Csm3 and Csm5 impacted maturation in two distinct fashions (Supplementary Figure S2B and C): (i) by shifting crRNA size distribution in a manner that favours shorter lengths and (ii) by abrogating or eliminating maturation, while causing a concomitant accumulation of intermediate crRNAs. We examine both classes of mutants further in the results described below.
Charged residues in Csm3 mediate self-interactions and assemble the maturation ruler
In previous work, mutation of a highly conserved acidic residue in Csm3 (D100) was found to promote maturation cleavage and shift the crRNA size distribution to shorter lengths (23). The precise mechanism that causes this phenotype remains unclear. Here, we discovered two additional charged residues, K4 and D179, that impact crRNA maturation in a similar fashion (Supplementary Figure S2B). We first assessed the functionality of these mutants by measuring the efficiency of conjugative transfer of the plasmid pG0400, which contains a protospacer targeted by spc1 (3). Consistent with previous observations of Csm3 D100A (23), both mutants promote efficient anti-plasmid immunity comparable to that of wild-type Cas10–Csm (Supplementary Figure S2D and E). To further characterize these, we purified mutant Cas10–Csm complexes in which Csm2 harbours an N-terminal 6-His tag (Csm26HN). Mutant pcrispr-cas constructs were expressed in S. epidermidis LM1680, and complexes were purified from whole cell lysates using Ni2+ affinity chromatography. Consistent with the functional data, all members of the Cas10–Csm complex assemble with the K4A and D179A Csm3 mutants (Figure 2A). We further purified and radiolabeled the crRNAs contained within these complexes to confirm the shift in crRNA size distribution (Figure 2B), which is the only discernible phenotype associated with these mutants.
Figure 2.

Charged residues mediate Csm3 self-interactions and assemble the maturation ruler. (A) Cas10–Csm complexes containing the indicated Csm3 mutations are shown. Mutations were introduced into pcrispr-cas encoding a 6-His tag on the N-terminus of Csm2. Constructs were expressed in S. epidermidis LM1680, and whole cell lysates were subjected to Ni2+ affinity chromatography and a second purification using a biotinylated oligonucleotide antisense to spc1 crRNAs. Complexes were resolved and visualized using SDS-PAGE and Coomassie G-250 staining. Shown is a representative of at least three independent trials. (B) RNA was extracted from each complex shown in panel A, radiolabeled on the 5΄-end, and resolved using denaturing PAGE. (C) Purified recombinant Csm3 and mutant variants resolved by SDS-PAGE and visualized using Coomassie G-250 staining are shown. (D) Csm3 and mutant variants (10 pmol each) were resolved alongside the NativeMark unstained protein standard (Thermo Fisher Scientific) by blue native PAGE in which the cathode buffer contained 0.025% Coomassie G-250. Proteins were visualized using Coomassie G-250 staining. Shown is a representative of at least three independent trials. (E) Homology model of a Csm3 oligomer. The Csm3 structure was derived from the high resolution crystal structure of M. kandleri Csm3 (39), and then docked into the A. fulgidus Cmr4 backbone of a Type III-B complex (34). K4 and D179 residues are shown in space-fill.
Csm3 is integral to the formation of the Cas10–Csm complex (23), and in related Type III-A systems, multiple copies of Csm3 form a backbone that spans the length of the crRNA (24,26), with shorter crRNAs being associated with complexes containing fewer copies of Csm3 (26). We hypothesized that the conserved charged residues that maintain crRNA length facilitate Csm3 self-association within the complex. Consistent with this hypothesis, complexes harbouring Csm3D179A contain a smaller fraction of Csm3 when compared to wild-type, as evidenced by Csm3D179A band intensity in Coomassie-stained gels, which is significantly less than that of wild-type Csm3 when compared with total band intensity of their respective complexes (12% ± 2.6 in D179A versus 18% ± 2.9 in wild-type; P = 0.02) (Figure 2A). To visualize multimerization of Csm3 directly, we overexpressed and purified recombinant Csm3 and its mutant variants in E. coli (Figure 2C) and resolved them using blue-native PAGE. Indeed, wild-type Csm3 exhibits a banding pattern consistent with the assembly of higher molecular-weight complexes (Figure 2D). When compared with the native ladder, the 24 kDa Csm3 appears to assemble into groups of six (i.e. hexamerize), consistent with the stoichiometry of Csm3 found in a related Cas10–Csm complex from Thermus thermophilus (24). Importantly, the K4A and D179A mutants harbor pronounced defects in multimerization (Figure 2D). Together, these observations suggest that K4 and D179 lie at the Csm3–Csm3 binding interface and facilitate its multimerization, thereby assembling the maturation ruler. To further explore this possibility, we constructed a homology model of a Csm3 oligomer based on the M. kandleri Csm3 structure (39) and docked into the Cmr4 backbone of the high resolution crystal structure of a Type III-B complex (34) (Figure 2E). Strikingly, this model predicts K4 and D179 in adjacent Csm3 protomers form a salt bridge, supporting a direct interaction between these residues during complex assembly.
Acidic residues in Csm5 are necessary but insufficient to conduct crRNA maturation
Alanine-scanning mutations in both Csm3 (K52A, K54A, R56A triple mutant) and Csm5 (D162A or E191A) abrogate crRNA maturation (Supplementary Figure S2B and C). Because the complete deletion of either subunit causes a similar phenotype (19), we first wanted to determine if Cas10–Csm complexes remain intact in the presence of these mutations. Using Ni2+-affinity chromatography, we purified mutant complexes containing Csm2H6N from S. epidermidis LM1680. Consistent with the effect of a Csm3 deletion (15), the Csm3 triple mutant (K52A, K54A, R56A) failed to promote complex assembly (data not shown), suggesting these mutations might disrupt Csm3 folding, or its ability to assemble into the complex, thereby causing an effective knockout. Since either scenario prevents our further investigation of complex-associated crRNAs, we did not consider this mutant further. In contrast, all members of Cas10–Csm were detected in the presence of Csm5D162A and Csm5E191A (Figure 3A), excluding a role for these residues in associating with the complex and/or maintaining complex stability. CrRNAs purified from these complexes confirm that both mutants cause severe maturation defects (Figure 3B), particularly Csm5E191A, which exhibits a phenotype almost identical to that of a Csm5 knockout. This mutation also significantly impairs anti-plasmid immunity (Supplementary Figure S2D and E), allowing for a 10-fold higher efficiency of pG0400 conjugation when compared to that of wild-type Cas10–Csm. Together these observations support a direct role for D162 and E191 in the process of maturation and interference, and suggest at least two possible functions for Csm5: (i) as a metal-dependent nuclease that uses acidic amino acids to catalyse crRNA cleavage or (ii) as a binding protein that recruits the activity of an external nuclease to the crRNA.
Figure 3.

Acidic amino acids in Csm5 are necessary but insufficient to conduct crRNA maturation. (A) Cas10–Csm complexes containing the indicated Csm5 mutations are shown. Mutations were introduced into pcrispr-cas encoding a 6-His tag on the N-terminus of Csm2. Constructs were expressed in S. epidermidis LM1680, and whole cell lysates were subjected to Ni2+ affinity chromatography and a second purification using a biotinylated oligonucleotide antisense to spc1 crRNAs. Complexes were resolved and visualized using SDS-PAGE followed by Coomassie G-250 staining. The dotted line separates non-contiguous lanes within the same gel. (B) RNA was extracted from each complex in panel A, radiolabeled on the 5΄-end, and resolved using denaturing PAGE. (C) Purified recombinant Csm5 resolved by SDS-PAGE and visualized using Coomassie G-250 staining is shown. (D) 71-nucleotide intermediate crRNAs were extracted from Cas10–Csm/ΔCsm5 complexes (shown in panel A) and used as substrates in a nuclease assay containing purified Csm5 (8 pmol) and EDTA or various metals as indicated. The reaction was allowed to proceed at 37°C for 10 min. RNAs were resolved by denaturing PAGE. Shown is a representative of at least five independent trials with three different Csm5 preparations. (E) The 31-nucleotide spc1 crRNA substrate used for Csm5 binding and nuclease assays is shown. The 5΄ tag sequence is boxed in white, and spacer 1-derived sequence is shaded. The asterisk indicates the radiolabel on the 5΄-end. (F) A double-filter binding assay is shown. Increasing amounts of Csm5 (0, 0.5, 1, 2, 8 and 32 pmol) and trace amounts of 5΄-end labeled RNA substrate (panel E) were combined and applied to wells in triplicate. Csm5-bound probe (upper membrane) and free RNA (lower membrane) are indicated. (G) Quantification of the double-filter assay showing the fraction of RNA bound as an average of triplicate measurements (±S.D.)
To test the first possibility, we overexpressed and purified recombinant Csm5 from E. coli (Figure 3C), and assayed for its ability to cleave a variety of RNA substrates, including intermediate crRNAs extracted from Cas10–Csm/ΔCsm5 complexes, and in the presence of a variety of metals (Figure 3D and data not shown). Despite our best efforts, we were unable to reconstitute ribonuclease activity. This result is consistent with previous work in which the entire Cas10–Csm complex failed to cleave crRNAs in a purified system (23). To begin to explore the second possibility, we assayed for Csm5-RNA binding, which could support a role in nuclease recruitment to the crRNA. To test this, we used a radiolabeled 31-nucleotide RNA substrate that corresponds to spc1 crRNAs (Figure 3E) in a double-filter binding assay (35). In this assay, a nitrocellulose membrane is layered on top of a nylon membrane, and a 96-well vacuum apparatus is used to pull Csm5–RNA mixtures through both filters; RNA bound to Csm5 is retained on the top membrane, while free RNA flows through and binds to the bottom (Figure 3F). Using this assay, we observed that Csm5 indeed binds RNA, with a disassociation constant in the nanomolar range (Kd = 100.8 nM ± 11.5) (Figure 3G).
Csm5 differentially modulates the activity of cellular nucleases
The absence of ribonuclease activity in Csm5, coupled with its crRNA binding activity, prompted an investigation of cellular nucleases that might be recruited during maturation. Using Mass Spectrometry, we looked for candidate nucleases that co-purify with Cas10–Csm. As expected, the most abundant proteins detected were members of the Cas10–Csm complex (Supplementary Table S2); however, a relatively low abundance of cellular nucleases were also detected (Table 1). While this analysis does not provide conclusive evidence of specific binding to the Cas10–Csm complex, these nucleases constitute candidates for further investigation. Of these, all known staphylococcal 3΄-5΄ exonucleases were present: Cbf1, RNase R and polynucleotide phosphorylase (PNPase) (40). Little is known about these enzymes in S. epidermidis, however, some information is available on the function of their homologs in S. aureus and other organisms. Cbf1 in S. aureus is an HD nuclease that cleaves RNA in a Mn2+-dependent manner (41). RNase R is a 3΄-5΄ processive exoribonuclease (42) that in E. coli is involved in bulk RNA turnover and the degradation of structured RNAs, including rRNAs and tRNAs (43,44). The RNase R homolog in S. aureus is essential (45); however, little is known about its contributions toward RNA processing and turnover in this organism. PNPase is a highly-conserved and multifunctional enzyme that both degrades and synthesizes RNA in a Mg2+-dependent manner—inorganic phosphate is used for RNA phosphorylysis, and ribonucleotide diphosphates are used as substrates for polymerization in a template-independent manner (46–50). PNPase in S. aureus is a subunit of the RNA degradosome (51), a multisubunit complex in bacteria that plays a central role in RNA processing and degradation. Importantly, all three nucleases exhibit non-specific 3΄-end RNA degradation that leaves behind a 3΄-OH group, consistent with the products of crRNA maturation. We therefore selected these for further characterization.
Table 1. Mass spectrometry analysis of cellular ribonucleases that co-purify with Cas10–Csm(Csm2H6N) in S. epidermidis and their putative functions.
| Ribonuclease | Theoretical mass (kDa) | Unique peptide count | Normalized spectral countse |
|---|---|---|---|
| PNPasea, b | 77 | 28 | 74.55 |
| Ribonuclease J1c | 62 | 10 | 21.3 |
| Cbf1a, b | 36 | 7 | 19.53 |
| Ribonuclease J2c | 63 | 10 | 16.86 |
| RNase IIId | 28 | 5 | 12.43 |
| RNase Ra | 91 | 5 | 7.1 |
a3΄-5΄ exoribonuclease on single-stranded RNA.
b3΄-5΄ exonuclease on single-stranded DNA.
c5΄-3΄ exonuclease on single-stranded RNA.
dendoribonuclease on double-stranded RNA.
eValues reflect relative protein abundance.
To examine their function more closely, we overexpressed and purified recombinant S. epidermidis RP62a Cbf1, RNase R and PNPase in E. coli (Figure 4A). We first wanted to assess their ability to cleave intermediate crRNAs, which share two 3΄ end features that might present a barrier to exonucleases in isolation (Supplementary Figure S3A)—a hairpin structure derived from the 3΄ end of the crRNA repeat, and a 2΄,3΄-cyclic phosphate generated by Cas6 during primary processing (19). To test this, we extracted and end-labeled crRNAs from Cas10–Csm complexes and used these as substrates in a nuclease assay. Importantly, all three nucleases are capable of degrading intermediate crRNAs in vitro (Supplementary Figure S3B and C). This observation is consistent with previous reports on PNPase and RNase R, in which both nucleases were shown to degrade RNA substrates containing a 3΄ phosphate (42,52).
Figure 4.

Csm5 differentially modulates the activity of cellular nucleases. (A) Purified recombinant Cbf1, PNPase and RNase R resolved by SDS-PAGE and visualized using Coomassie G-250 staining are shown. (B and C) Csm5 protection against Cbf1 (B) and RNase R (C) activity is shown. 5΄-end labelled 31-nucleotide spc1 crRNA substrate (Figure 3E) was pre-incubated with increasing amounts of Csm5 (0, 1, 2, 4 and 8 pmol) before nuclease addition (1 pmol each). The reaction mixture was incubated at 37°C for 10 min. RNAs were resolved using denaturing PAGE. Full-length substrate (arrow), degradation intermediates (asterisks, *), and fully degraded substrate (brackets) are indicated. (D) Quantification of the Csm5 protection assays shown in panels B and C. Percent substrate protected was calculated as follows: ((band intensities of full length substrate + degradation intermediates)/total substrate added)) × 100. Shown is the average (±S.D.) of three independent trials. (E) PNPase (1 pmol) was combined with a radiolabeled 31 nucleotide spc1 crRNA substrate and Csm5 (4 pmols) where indicated. The reaction mixture was incubated at 37°C for increasing time points (0, 2, 5, or 10 minutes.) RNAs were resolved using denaturing PAGE. (F) The 5΄-end labelled 22-nucleotide DNA substrate used in nuclease assays. (G) PNPase or Cbf1 (1 pmol each) were combined with the DNA substrate (panel F) in the presence of Csm5 (4 pmol) where indicated. The reaction mixture was incubated at 37°C for 10 min, and DNAs were resolved using denaturing PAGE. All gel images are a representative of at least three independent trials.
We next assessed the impact of Csm5 on nuclease activity. Given that Csm5 exhibits RNA binding activity in the nanomolar range (Figure 3E–G), we hypothesized that Csm5 would protect RNA against nucleolytic degradation. To test this, we pre-incubated a radiolabeled synthetic crRNA substrate (Figure 3E) with Csm5 before adding the nucleases. As expected, Csm5 affords a dose-dependent protection against activities of Cbf1 and RNase R, as evidenced by the persistence of full-length substrate and its degradation intermediates in the presence of increasing amounts of Csm5 (Figure 4B–D). In striking contrast, Csm5 had the opposite effect on PNPase, causing a stimulation of RNA degradation and simultaneous inhibition of its polymerization activity (Figure 4E). To determine if these effects are specific to Csm5, we conducted the same assays using Csm3, and show that Csm3 has no impact on nuclease activity (Supplementary Figure S4).
We next wanted to test the effect of Csm5 on alternate functions of these nucleases. Cbf1 is a homolog of the B. subtilis exonuclease YhaM, which also exhibits DNase activity in a Mn2+-dependent manner (41). Moreover, PNPase in this organism also degrades and synthesizes single-stranded DNA in the presence of Mn2+ (53). We wondered if the S. epidermidis homologs exhibit similar functions, and if so, how Csm5 might have an impact. To test this, we conducted nuclease assays using a 22-nucleotide DNA substrate (Figure 4F) and found that both nucleases act on single-stranded DNA similarly to their B. subtilis homologs (Figure 4G). Importantly, while Csm5 has no discernible effect on Cbf1 DNase activity, stimulation of PNPase-mediated DNA degradation was observed. Taken together, these results demonstrate that Csm5 differentially modulates the functions of cellular nucleases.
Csm5 physically interacts with PNPase
While the Csm5-mediated repression of Cbf1 and RNase R activity might be explained by a protection mechanism caused by Csm5's affinity to RNA, we hypothesized that stimulation of PNPase by Csm5 might result from their direct interaction. Supporting this possibility, PNPase was the most abundant non-Cas nuclease that co-purified with Cas10–Csm according to the mass spectrometry analysis (Table 1), with a relative protein abundance of about one-third that of Csm5 (Supplementary Table S2). To test for a physical association between Csm5 and PNPase in a purified system, we first used a pull-down assay coupled with Ni2+-affinity chromatography in which Csm5 harbours an N-terminal His10Smt3 tag (Figure 5A). In this assay, purified tagged Csm5 (Csm5–Smt3) is first loaded onto a column containing Ni2+-agarose beads. Any unbound Csm5 is washed, and purified untagged PNPase is added. The column is then washed thoroughly to remove unbound PNPase, and proteins retained in the column are finally eluted with imidazole. Indeed, we were able to detect an interaction between Csm5 and PNPase, as evidenced by their co-elution at the last step (Figure 5B and Supplementary Figure S5A and B). The interaction is likely very weak, with the PNPase signal intensity constituting 5.9% ± 2.4 of total signal (average of three independent replicates). Importantly, PNPase is not retained in the column in the absence of Csm5 (Figure 5B and Supplementary Figure S5C). To demonstrate that this interaction is specific to Csm5, we performed the same experiment with Csm3–Smt3, and show that there is no detectible interaction (Supplementary Figure S5D).
Figure 5.

Csm5 physically interacts with PNPase. (A) Abbreviated experimental flow of a Ni2+ affinity pulldown assay, in which His10Smt3 tagged Csm5 (Csm5-Smt3, ‘input 1’, 1 nmol) is loaded onto a column containing Ni2+-agarose beads and washed. Untagged PNPase (‘input 2’, 0.7 nmol) is then passed through the column, and anything unbound is thoroughly washed. Proteins retained in the column are eluted using 500 mM imidazole. Abbreviations are as follows: FT, flow-through; W, wash; imid., imidazole. (B) Samples from the Csm5–Smt3 (input 1), untagged PNPase (input 2), and final elution were resolved using SDS-PAGE. The final elution from a negative control is also shown in which dialysis buffer was used as input 1. See Supplementary Figure S5 for a detailed description of the experiment and all samples collected during the experiment. The dotted line separates non-contiguous lanes within the same gel. Shown is a representative of four independent trials. (C) Csm5 and PNPase (100 pmol each) were resolved on a 5% vertical native gel alone or in combination (1:1 ratio). The position of (+) and (-) electrodes are indicated. (D) Numbered bands shown in panel C were excised from the native gel and the proteins within were resolved using denaturing SDS-PAGE. Arrowheads mark the top of each excised band and indicate band orientation when run in second dimension. Shown is a representative of three independent trials. Proteins were visualized with Coomassie G-250.
To confirm the physical association between Csm5 and PNPase, we used two-dimensional Native/SDS PAGE (Figure 5C and D and Supplementary Figure S6). Since the isoelectric point of Csm5 is basic (theoretical pI ∼ 9.5), its charge at physiological pH is predicted to be positive, which would cause it to migrate toward the negative electrode in a native gel. To confirm this, we first examined the migration of Csm5 and PNPase using a horizontal native gel with wells cast in the center (Supplementary Figure S6). As expected, Csm5 migrates toward the negative electrode, while PNPase migrates in the opposite direction. PNPase migrates as a trimer, consistent with previous observations (54). When Csm5 and PNPase are combined, their migration is shifted to an intermediate position, suggesting an interaction. To obtain a higher resolution of band separation, Csm5 and PNPase were resolved on a thinner (0.75 mm) vertical gel (Figure 5C). Csm5 alone does not travel into the gel in this format since wells are cast at the top; however, when combined with PNPase, Csm5 causes a prominent shift in PNPase mobility. To determine the protein content of each band, indicated bands were excised from the gel and subjected to a second separation under denaturing conditions (Figure 5D). Importantly, the shifted band (labelled ‘1’) contains both Csm5 and PNPase. Altogether these results provide evidence that Csm5 recruits PNPase to the Cas10–Csm complex through a direct physical interaction.
DISCUSSION
Here, we define the molecular requirements for crRNA maturation in the Cas10-Csm complex, and provide supporting evidence for a model in which CRISPR-associated proteins collaborate with non-Cas nucleases to catalyse the final maturation step (Figure 6). Csm3 is essential for complex assembly (23), with multiple copies forming a backbone that spans the length of the crRNA (24,26). Here, we show that charged amino acids, including K4 and D179, stabilise Csm3 self-interactions and therefore help increase the length of protection on crRNA 3΄-ends (Figure 2). A single copy of Csm5 caps the 3΄-end of complexes in related Type III-A systems (24,26), therefore placing Csm5 proximal to the site of maturation. We show that while Csm5 is devoid of detectable ribonuclease activity in vitro (Figure 3), it recruits and stimulates PNPase (Figures 4 and 5), which we speculate trims the 3΄-end. Both Csm3 and Csm5 are RNA binding proteins ((23) and Figure 3); however, only Csm5 is capable of protecting crRNAs from nonspecific degradation by the cellular nucleases RNase R and Cbf1 (Figure 4B and C and Supplementary Figure S4). Because a Csm5 knockout results in the accumulation of intermediate crRNAs (as opposed to nonspecific degradation), we hypothesize the recruitment of specific nucleolytic activity is the major contribution of Csm5 toward crRNA maturation, and its protective effect stabilises 3΄-ends once maturation has occurred. This model reconciles seemingly disparate observations in which a csm5 deletion eliminates maturation (15,19), while in a purified system Csm5 alone or in complex with Cas10–Csm is bereft of detectable nuclease activity ((23) and figures herein).
Figure 6.
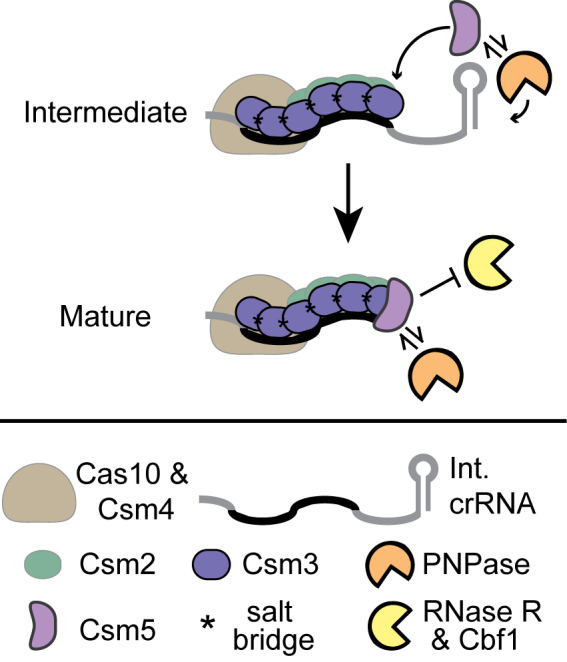
A model for crRNA maturation. Maturation occurs during Cas10–Csm complex assembly with intermediate crRNAs. Charged residues in Csm3 promote its self-interactions as it polymerizes on the crRNA. Csm5 facilitates crRNA maturation by recruiting PNPase to the 3΄-end of the crRNA. Once it joins the complex, Csm5 shields crRNAs from further nonspecific degradation by PNPase and other cellular nucleases.
We cannot rule out the possibility that PNPase might facilitate interference events downstream of crRNA maturation. The Csm5 E191A mutation abrogates both maturation and anti-plasmid immunity (Figure 3 and Supplementary Figure S2). The mechanism that leads to this phenotype is unclear. While we were unable to stably express this mutant for further characterization, we speculate that it might be defective in PNPase binding and/or stimulation, which could account for its failure to promote efficient maturation. The root cause of its impaired anti-plasmid immunity could stem from the defect in maturation and/or the absence of other downstream activities PNPase might have during the interference stage. In the presence of Csm5, PNPase robustly degrades both RNA and DNA (Figure 4E and G, respectively), two activities that might also enhance Cas10–Csm interference. All Types I and III CRISPR–Cas systems harbour one or more Csm5 homologs (i.e. Cas7 subunits) (10), raising the question of whether these might also link their respective effector complexes with the activity of PNPase. Indeed, PNPase is required for the function of a unique CRISPR-like element found in Listeria monocytogenes that consists of spacers integrated between structured repeats, but is completely devoid of cas genes (55). This CRISPR element, called RliB-CRISPR, exhibits small RNA processing and anti-plasmid immunity that is dependent on the presence of a Type I CRISPR–Cas system in trans—both of these activities also require PNPase. In addition to its central role in bulk RNA turnover as a member of the degradosome (51), PNPase is involved in small RNA processing and stability (56–58), and its DNase activity has been implicated in DNA repair pathways (53,59). This newly-identified interaction with the Type III-A CRISPR–Cas system now adds to the list of multiple and diverse roles for this enzyme.
In summary, our results elaborate on the mechanism of crRNA maturation in a Type III CRISPR–Cas system and establish a link between Cas10–Csm with PNPase, a highly-conserved multifunctional enzyme that plays diverse roles in RNA processing and turnover. Future work will explore further the relationship between non-Cas nucleases and CRISPR–Cas immunity in staphylococci.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to L.A. Marraffini for providing strains and constructive comments on the manuscript. We also thank J.W. Roberts for reviewing the manuscript, and M. Bonizzoni for critical discussions and unpublished experiments related to Csm5-PNPase interactions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [5K22AI113106-02 to A.H-A.]; University of Alabama. Funding for open access charge: National Institutes of Health [5K22AI113106-02 ].
Conflict of interest statement. None declared.
REFERENCES
- 1.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2.Brouns S.J.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J.H., Snijders A.P.L., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marraffini L.A., Sontheimer E.J.. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008; 322:1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bikard D., Hatoum-Aslan A., Mucida D., Marraffini L.A.. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe. 2012; 12:177–186. [DOI] [PubMed] [Google Scholar]
- 5.Godde J.S., Bickerton A.. The repetitive DNA elements called CRISPRs and their associated genes: evidence of horizontal transfer among prokaryotes. J. Mol. Evol. 2006; 62:718–729. [DOI] [PubMed] [Google Scholar]
- 6.Grissa I., Vergnaud G., Pourcel C.. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007; 8:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haft D.H., Selengut J., Mongodin E.F., Nelson K.E.. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005; 1:0474–0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marraffini L.A. CRISPR–Cas immunity in prokaryotes. Nature. 2015; 526:55–61. [DOI] [PubMed] [Google Scholar]
- 9.van der Oost J., Westra E.R., Jackson R.N., Wiedenheft B.. Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nat. Rev. Microbiol. 2014; 12:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J.J., Charpentier E., Haft D.H. et al. . An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shmakov S., Abudayyeh O.O., Makarova K.S., Wolf Y.I., Gootenberg J.S., Semenova E., Minakhin L., Joung J., Konermann S., Severinov K. et al. . Discovery and functional characterization of diverse Class 2 CRISPR–Cas systems. Mol. Cell. 2015; 60:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haurwitz R.E., Jinek M., Wiedenheft B., Zhou K., Doudna J.A.. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010; 329:1355–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carte J., Wang R., Li H., Terns R.M., Terns M.P.. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008; 22:3489–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nam K.H., Haitjema C., Liu X., Ding F., Wang H., Delisa M.P., Ke A.. Cas5d protein processes Pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg crispr-cas system. Structure. 2012; 20:1574–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatoum-Aslan A., Maniv I., Samai P., Marraffini L.A.. Genetic characterization of antiplasmid immunity through a type III-A CRISPR-cas system. J. Bacteriol. 2014; 196:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deltcheva E., Chylinski K., Sharma C.M., Gonzales K., Chao Y., Pirzada Z.A., Eckert M.R., Vogel J., Charpentier E.. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011; 471:602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fonfara I., Richter H., Bratovič M., Le Rhun A., Charpentier E.. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016; 532:517–521. [DOI] [PubMed] [Google Scholar]
- 18.Hale C., Kleppe K., Terns R.M., Terns M.P.. Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA. 2008; 14:2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatoum-Aslan a., Maniv I., Marraffini L.a.. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:21218–21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A.. Co-transcriptional DNA and RNA cleavage during type III CRISPR-cas immunity. Cell. 2015; 161:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldberg G.W., Jiang W., Bikard D., Marraffini L.a. Conditional tolerance of temperate phages via transcription-dependent CRISPR–Cas targeting. Nature. 2014; 514:633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang W., Samai P., Marraffini L.A.. Degradation of phage transcripts by CRISPR-associated RNases enables Type III CRISPR–Cas immunity. Cell. 2016; 164:710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatoum-Aslan A., Samai P., Maniv I., Jiang W., Marraffini L.A.. A ruler protein in a complex for antiviral defense determines the length of small interfering CRISPR RNAs. J. Biol. Chem. 2013; 288:27888–27897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Staals R.H.J., Zhu Y., Taylor D.W., Kornfeld J.E., Sharma K., Barendregt A., Koehorst J.J., Vlot M., Neupane N., Varossieau K. et al. . RNA targeting by the Type III-A CRISPR–Cas Csm complex of Thermus thermophilus. Mol. Cell. 2014; 56:518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makarova K.S., Aravind L., Wolf Y.I., Koonin E. V. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR–Cas systems. Biol. Direct. 2011; 6:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamulaitis G., Kazlauskiene M., Manakova E., Venclovas C., Nwokeoji A.O., Dickman M.J., Horvath P., Siksnys V.. Programmable RNA shredding by the Type III-A CRISPR–Cas system of Streptococcus thermophilus. Mol. Cell. 2014; 56:506–517. [DOI] [PubMed] [Google Scholar]
- 27.Charpentier E., Richter H., van der Oost J., White M.F.. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR–Cas adaptive immunity. FEMS Microbiol. Rev. 2015; 39:428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kreiswirth B.N., Lofdahl S., Betley M.J., O'Reilly M., Schlievert P.M., Bergdoll M.S., Novick R.P.. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983; 305:709–712. [DOI] [PubMed] [Google Scholar]
- 29.Gibson D.G., Young L., Chuang R.-Y., Venter J.C., Hutchison C.A., Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 30.Schagger H., Vonjagow G.. Blue native electrophoresis for isolation of membrane-protein complexes in enzymatically active form. Anal. Biochem. 1991; 199:223–231. [DOI] [PubMed] [Google Scholar]
- 31.Nijtmans L.G.J., Henderson N.S., Holt I.J.. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods. 2002; 26:327–334. [DOI] [PubMed] [Google Scholar]
- 32.Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.E.. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015; 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y.. The I-TASSER Suite: protein structure and function prediction. Nat. Methods. 2015; 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osawa T., Inanaga H., Sato C., Numata T.. Crystal structure of the crispr-cas RNA silencing cmr complex bound to a target analog. Mol. Cell. 2015; 58:418–430. [DOI] [PubMed] [Google Scholar]
- 35.Wong I., Lohman T.M.. A double-filter method for nitrocellulose-filter binding: Application to protein-nucleic acid interactions. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:5428–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka N., Schwer B.. Characterization of the NTPase, RNA-binding, and RNA helicase activities of the DEAH-box splicing factor Prp22. Biochemistry. 2005; 44:9795–9803. [DOI] [PubMed] [Google Scholar]
- 37.Ludwig M.R., Kojima K., Bowersock G.J., Chen D., Jhala N.C., Buchsbaum D.J., Grizzle W.E., Klug C.A., Mobley J.A.. Surveying the serologic proteome in a tissue-specific kras(G12D) knockin mouse model of pancreatic cancer. Proteomics. 2016; 16:516–531. [DOI] [PubMed] [Google Scholar]
- 38.Camacho-Carvajal M.M., Wollscheid B., Aebersold R., Steimle V., Schamel W.W.A.. Two-dimensional Blue Native/SDS gel electrophoresis of multi-protein complexes from whole cellular lysates. Mol. Cell. Proteomics. 2003; 3:176–182. [DOI] [PubMed] [Google Scholar]
- 39.Hrle A., Su A.a.H., Ebert J., Benda C., Randau L., Conti E.. Structure and RNA-binding properties of the type III-A CRISPR-associated protein Csm3. RNA Biol. 2013; 10:1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonnin R.A., Bouloc P.. RNA degradation in staphylococcus aureus: diversity of ribonucleases and their impact. Int. J. Genomics. 2015; 2015:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oussenko I.A., Sanchez R., Bechhofer D.H.. Bacillus subtilis YhaM , a member of a new family of 3 Ј -to-5 Ј exonucleases in Gram-positive bacteria. 2002; 184:6250–6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Z.F., Deutscher M.P.. Purification and characterization of the Escherichia coli exoribonuclease RNase R. Comparison with RNase II. J. Biol. Chem. 2002; 277:21624–21629. [DOI] [PubMed] [Google Scholar]
- 43.Vincent H.A., Deutscher M.P.. Substrate recognition and catalysis by the exoribonuclease RNase R. J. Biol. Chem. 2006; 281:29769–29775. [DOI] [PubMed] [Google Scholar]
- 44.Cheng Z.F., Deutscher M.P.. Quality control of ribosomal RNA mediated by polynucleotide phosphorylase and RNase R. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:6388–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaudhuri R.R., Allen A.G., Owen P.J., Shalom G., Stone K., Harrison M., Burgis T.A., Lockyer M., Garcia-Lara J., Foster S.J. et al. . Comprehensive identification of essential Staphylococcus aureus genes using Transposon-Mediated Differential Hybridisation (TMDH). BMC Genomics. 2009; 10:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ochoa S. Enzymic synthesis of polynucleotides. III. Phosphorolysis of natural and synthetic ribopolynucleotides. Arch. Biochem. Biophys. 1957; 69:119–129. [DOI] [PubMed] [Google Scholar]
- 47.Mohanty B.K., Kushner S.R.. Polynucleotide phosphorylase functions as both an exonuclease and a poly(A) polymerase in E. coli. Mol. Cell. Biol. 2001; 21:5408–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Condon C. RNA Processing and Degradation in Bacillus subtilis. Microbiol. Mol. Biol. Rev. 2003; 67:157–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grunberg-Manago M. Messenger RNA stability and its role in control of gene expression in bacteria and phages. Annu. Rev. Genet. 1999; 33:193–227. [DOI] [PubMed] [Google Scholar]
- 50.Sarkar D., Fisher P.B.. Polynucleotide phosphorylase: An evolutionary conserved gene with an expanding repertoire of functions. Pharmacol. Ther. 2006; 112:243–263. [DOI] [PubMed] [Google Scholar]
- 51.Roux C.M., DeMuth J.P., Dunman P.M.. Characterization of components of the Staphylococcus aureus mRNA degradosome holoenzyme-like complex. J. Bacteriol. 2011; 193:5520–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Numata S., Nagata M., Mao H., Sekimizu K., Kaito C.. CvfA protein and polynucleotide phosphorylase act in an opposing manner to regulate Staphylococcus aureus virulence. J. Biol. Chem. 2014; 289:8420–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cardenas P.P., Carrasco B., Sanchez H., Deikus G., Bechhofer D.H., Alonso J.C.. Bacillus subtilis polynucleotide phosphorylase 3΄-to-5΄ DNase activity is involved in DNA repair. Nucleic Acids Res. 2009; 37:4157–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi Z., Yang W.-Z., Lin-Chao S., Chak K.-F., Yuan H.S.. Crystal structure of Escherichia coli PNPase: central channel residues are involved in processive RNA degradation. RNA. 2008; 14:2361–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sesto N., Touchon M., Andrade J.M., Kondo J., Rocha E.P.C., Arraiano C.M., Archambaud C., Westhof É., Romby P., Cossart P.. A PNPase dependent CRISPR system in Listeria. PLoS Genet. 2014; 10:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saramago M., Domingues S., Arraiano M., Cook G.M.. The role of RNases in the regulation of small RNAs. Curr. Opin. Microbiol. 2014; 18:105–115. [DOI] [PubMed] [Google Scholar]
- 57.De Lay N., Gottesman S.. Role of polynucleotide phosphorylase in sRNA function in Escherichia coli. RNA. 2011; 17:1172–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andrade J.M., Pobre V., Matos A.M.. The crucial role of PNPase in the degradation of small RNAs that are not associated with Hfq. RNA. 2012; 18:844–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cardenas P.P., Carzaniga T., Zangrossi S., Briani F., Garcia-Tirado E., Dehò G., Alonso J.C.. Polynucleotide phosphorylase exonuclease and polymerase activities on single-stranded DNA ends are modulated by RecN, SsbA and RecA proteins. Nucleic Acids Res. 2011; 39:9250–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.