


Abstract
The emerging development of antibiotic resistant bacteria calls for novel types of antibacterial agents. In this work we examined the putative antibacterial effect of purine analogs in Listeria monocytogenes. We show that, among several tested purine analogs, only 6-N-hydroxylaminopurine (6-N-HAP) reduces the viability of the Gram-positive pathogen Listeria monocytogenes. As in Bacillus subtilis, 6-N-HAP terminates expression at guanine riboswitches in L. monocytogenes hence preventing expression of their downstream genes. However, we show that the bacteriocidal effect of the compound was unlinked to the terminated expression at the guanine riboswitches. When further examining the antimicrobial effect, we observed that 6-N-HAP acts as a potent mutagen in L. monocytogenes, by increasing the mutation rate and inducing the SOS-response. Also, addition of 6-N-HAP decreased virulence gene expression by reducing both the levels and activity of the virulence regulator PrfA.
INTRODUCTION
The problem of bacteria resistant to one or several antibiotics is evolving. It is therefore crucial to develop alternative types of antibacterial agents that could be used to treat bacterial infections. This is especially important when treating life-threatening diseases such as Mycobacterium tuberculosis, a bacterium killing more than one million people annually and showing a worrisome antibiotic resistance development (1). Purine analogs have been shown to be efficient combating cancer and viral infections, but has during the last years also proven effective against M. tuberculosis growth, although the molecular targets remain largely unknown (2). Also, purine analogs have been shown to block growth of Escherichia coli (3).
An attractive class of candidate antibacterial drugs are analogs to metabolites that are natural ligands of riboswitches (4–11). Riboswitches are RNA-elements directly binding specific metabolites that leads to altered expression of the downstream gene(s). Riboswitches harbor an aptamer domain responsible for the binding of a specific metabolite and a regulatory domain, which after metabolite binding leads to a specific regulatory outcome (12). In Gram-negative bacteria, binding of a metabolite generally leads to a blockage of translation initiation. In Gram-positive bacteria, binding of the metabolite in most cases leads to a premature transcription termination (12). Analogs of riboswitch-binding metabolites have previously been shown to function as antibacterial drugs in a bacteriocidal or bacteriostatic manner. These include analogs to lysine, thiamine pyrophosphate, flavin mononucleotide and guanine (13–21). An analog to guanine, 2,5,6-triaminopyrimidin-4-1 (PC1), was shown to block Staphylococcus aureus growth (20). PC1 was shown to bind to a guanine riboswitch, thereby blocking expression of guaA, encoding a GMP synthetase. Bacteria not having guaA under the control of a guanine riboswitch did not respond to PC1 (20). However, a recent study suggests that using purine analogs to target guanine riboswitches alone in S. aureus is insufficient to treat infections (22).
Other guanine analogs can also inhibit growth of bacteria: it was shown that 6-N-hydroxylaminopurine (6-N-HAP) could downregulate expression of genes lying downstream of a guanine riboswitch in Bacillus subtilis and inhibit bacterial growth (19).
We were interested to examine the putative antibacterial effect and identify a possible target of purine analogs in Listeria monocytogenes, a Gram-positive bacterial pathogen closely related to B. subtilis. In this work, we show that among the different purine analogs, only 6-N-HAP was able to block growth and reduce viability of L. monocytogenes. By specific chromosomal point-mutations, we conclude that 6-N-HAP targets the two guanine-riboswitches present in L. monocytogenes, but that the antimicrobial effect exerted by 6-N-HAP is not mediated through inhibition of genes downstream of the guanine riboswitches. Intriguingly, 6-N-HAP dramatically increases the mutation rate and induces the SOS-response. 6-N-HAP also represses L. monocytogenes virulence factor expression in a mechanism, at least partially, downregulating the amount and activity of the major virulence factor PrfA.
MATERIALS AND METHODS
Strain and growth media
L. monocytogenes strains were grown in minimal-medium (IMM) if not otherwise indicated (23). LB medium was used for overnight cultures (24). LB medium solidified with agar, LB agar plates was used for viable count. For construction of mutant strains, erythromycin (Em) was added at a concentration of 7 μg ml−1. For screening for rifampicin mutants 0.005 μg ml−1 rifampicin was added to the LB agar plates.
Chemicals and reagents
6-N-hydroxylaminopurine (MP Biomedicals) and 6-thioguanine (Sigma-Aldrich) was dissolved in dimethyl sulfoxide (DMSO). Guanine and 6-mercaptopurine (Sigma-Aldrich) was dissolved in 1M NaOH, and 2-aminopurine (Sigma Aldrich) was dissolved in phosphate buffered saline, respectively. Adenine (Sigma-Aldrich), cytosine (Sigma-Aldrich) and thymine (Sigma-Aldrich) were dissolved in 0.5 M HCl, H2O and 1 M NaOH, respectively. All chemicals were stored at 4°C.
Oligonucleotides
Supplementary Table S1 lists all oligonucleotides used in the study.
Growth assay
Overnight cultures of L. monocytogenes strains were grown in LB and diluted 100-fold in minimal-medium, IMM. Bacteria were grown to an OD600 = 0.2 when DMSO, NaOH or phosphate buffered saline (controls) or 100 μM of the specified compound were added to the culture. Growth of the cultures were followed by optical density (OD600) or viable count, at indicated time-points.
Viable count
Cultures from the growth assay were diluted and plated on LA-agar plates at 37°C overnight.
Isolation of RNA
Isolation of RNA was performed essentially as previously described in Loh et al. (25). Overnight cultures in LB were diluted 50-fold in minimal-media and incubated at 37°C with shaking. At OD600 = 0.25 compounds or solvent was added to a final concentration of 100 μM before the cultures were grown until OD600 = 0.6. The bacteria were collected by centrifugation in Eppendorf centrifuge 5430R at 4°C, 6000 g for 10 min and frozen at −80°C. Pelleted bacteria were resuspended in resuspension solution (10% glucose, 12.5 mM Tris pH 7.6, 5mM EDTA) and fresh EDTA (0.5 M). After transferring the samples to a beadbeater tube containing ∼0.4 g glassbeads and 0.5 ml acid phenol (pH 4.5) the cells were homogenized in a minibeadbeater (Biospect products) for 1 min and 15 s. The obtained mix was centrifuged for 5 min at 14000 rpm, 4°C (Beckman coulter) before the addition of 1 ml Trizol (Ambion) and 100 μl chloroform/isoamylalcohol (C:IAA) (24:1) to the aqueous phase. The samples were centrifuged for 5 min at 14000 rpm, 4°C. After centrifugation, two more C:IAA extractions were performed before precipitation of the RNA by addition of 0.7 volumes of isopropanol in freezer for ∼30 min. The sample was centrifuged for 20 min at 4°C, 14000 rpm. The dried RNA pellet was dissolved in 200 μl DEPC (HH) treated water. Samples were subjected to DNAseI treatment, 20U and incubated for 30 min at 37°C. The reaction was stopped by addition of phenol/chloroform/IAA (Ambion) and centrifuged for 5 min, 14000 rpm, 4°C. The aqueous phase was extracted with C:IAA before centrifugation. The purified RNA was pelleted by addition of 1/10 volumes of DEPC treated 3M NaOH, (pH 4.5) and 2.5 volumes of 99.5% ethanol, incubated in freezer (−20°C) for 30 min and pelleted by centrifugation, 14000 rpm (4°C for 20 min). The RNA was dissolved in 200 μl DEPC treated water. The extracted RNA was analyzed on 1.2% agarose gel to verify transcript integrity. The concentration of the RNA was measured on a Nanodrop (Nanodrop ND-1000 spectrophotometer).
Northern blotting
A total of 20 μg of RNA was separated on a 1% formaldehyde agarose gel and blotted onto a Hybond-N-membrane (GE healthcare). The membrane was hybridized with the indicated, PCR-amplified, DNA fragments labeled with α32P-dATP (PerkinElmer) using the Prime-a-gene Kit (Promega). Oligonucleotides used in PCR amplification are listed in Supplementary Table S1. The blot was exposed to a phosphor screen (Molecular Dynamics) and developed using a STORM device (Molecular Dynamics). Prior to re-hybridizations, membranes were stripped in 0.1% boiling sodium dodecyl sulphate (SDS).
Protein isolation and Western blotting
Proteins were isolated using buffer A as previously described in Netterling et al. (26). Overnight cultures in LB were washed three times in minimal-media, diluted and incubated at 37°C with shaking. At OD600 = 0.25 compounds or solvent were added to a final concentration of 100 μM. Bacteria were harvested after 3.5 h incubation and pellets were resupended in buffer A (200 mM KCl, 50 mM Tris-HCl [pH 8.0], 1 mM ethylenediaminetetraacetic acid (EDTA), 10% glycerol, 1 mM dithiothreitol). The bacteria were disrupted using 0.4 g of glass beads and a mini bead beater (Biospec products) for 70 s. After centrifugation (5 min, 4°C at 13000 rpm) supernatant was transferred to a new tube and centrifuged for 20 min (4°C at 13 000 rpm). Protein concentration was measured using a bradford assay. Five micrograms protein were used for sodium dodecyl sulphate-polyacrylamide gel electrophoresis using 12% SDS-polyacrylamide gels (Bio-Rad). After transfer onto polyvinylidene difluoride membranes using the Trans-Blot® Turbo™ Transfer System (Bio-Rad). Proteins were detected using the primary antibodies anti-PrfA (1:1500), anti-ActA (1:3000) and anti-LLO (1:3000) and the secondary antibody goat anti-rabbit HRP conjugate (Agrisera). Chemiluminescence was detected using the ECL™ Prime Western blotting System (GE Healthcare) and a LAS4000 image analyzer (Fuji).
Construction of the M1-M6 mutant strain
The M1-M6 mutant strain (resulting in anti-termination of both Listeria guanine riboswitches, Rli70 and Rli95) was constructed using a two-step protocol. In the first step, the Rli95-M1 mutant was constructed. The primer-pairs Rli-95-M1-F together with lmo1885D-NcoI and Rli95-M1-R together with Rli95U-EcoRI were used to amplify the riboswitch and its surrounding region from EGDe DNA. The primers included overlapping sequences giving the possibility to use the amplified products as templates for a new PCR using Rli95U-EcoRI and lmo1885D-NcoI as primers. This resulted in a 671 nt product that was digested with NcoI and EcoRI. After purification, the fragment was introduced into NcoI/EcoRI digested pMAD, and the resulting construct was used to transform the E. coli strain Novablue (Novagene) (27). The sequenced construct was electroporated into L. monocytogenes EGDe at 2.5 kV, 200 Ω and 25 μF. Transformants were selected at 30°C on BHI plates containing 7 μg ml−1 erythromycin (BHI-Em) and X-gal (50 μg ml−1). One colony was inoculated in BHI-Em broth at 42°C for 2 days, until the culture was confluent. One hundred microliters of the culture was plated onto BHI-Em plates and incubated at 42°C overnight. One colony was inoculated in BHI without Em overnight at 30°C. The temperature was then raised to 39°C for 3 h before plating on BHI containing X-Gal (50 μg ml−1). White colonies were analyzed by sequencing of PCR amplified fragments generated by using the lmo1888D-NcoI and Rli95U-EcoRI primers. In the second step, the Rli70M6 mutation was incorporated in to the chromosome following the same protocol as for Rli95M1, but using the primer pairs Rli70-M6-R together Rli70U-EcoRI and Rli70-M6-F together with lmo0573D-NcoI. The resulting construct was inserted into the Rli95-M1 strain and resulting colonies were verified by sequencing the chromosomal insert regions.
Screening for Rifampicin resistant mutants and determination of the mutation rate
Listeria monocytgenes EGDe was grown overnight in LB medium at 37°C. The overnight culture was washed three times with IMM, diluted 100- fold in IMM and incubated at 37°C. At an OD600 = 0.2 the culture was split into four flasks and DMSO (control: 6-N-HAP solvent), 100 μM 6-N-HAP, NaOH (control: guanine solvent) or 100 μM guanine were added. After an incubation for 10 h at 37°C serial dilutions of the cultures were plated on LB and LB rifampicin (0.005 μg ml−1) plates. After 1–3 days incubation at 37°C the colony forming units as well as rifampicin resistant mutants were counted and the mutation rate calculated. A region of the rpoB gene of a set of rifampicin resistant mutants was amplified using primer lmo0258_rpoB_fw and lmo0258_rpoB_rv and send for sequencing (GATC Biotech) using primer lmo0258_rpoB_fw as sequencing primer.
RESULTS
6-N-Hydroxylaminopurine (6-N-HAP) reduces Listeria monocytogenes growth and viability
It has previously been shown that cytotoxic purine analogs are able to inhibit growth of E. coli (3) and M. tuberculosis (2) and the guanine analogs 6-N-HAP and PC1 were shown to inhibit growth of B. subtilis and S. aureus (19,20). To examine if purine analogs are able to inhibit growth of the pathogenic bacterium L. monocytogenes, the wild-type strain was exposed to 100 μM of different purine analogs: 6-N-HAP, 6-thioguanine, 6-mercaptopurine and 2-aminopurine (Figure 1A). As a difference to E. coli, only 6-N-HAP could reduce growth of L. monocytogenes (Figure 1B). To further analyze the effect of 6-N-HAP in L. monocytogenes, bacterial cultures grown in minimal medium were exposed to different concentrations of 6-N-HAP. After 10 h of growth at 37°C, bacteria that were challenged with 6-N-HAP displayed a reduced growth compared to the DMSO-treated bacteria (Figure 1C). Viable count experiments suggest a bactericidal-like effect of 6-N-HAP at 100 μM, decreasing the number of growing bacteria ∼100-fold compared with the inoculum (Figure 1D).
Figure 1.

The antibacterial effect of purine analogs in L. monocytogenes. (A) Structures of the purines guanine, adenine and the purine analogs 6-N-hydroxylaminopurine (6-N-HAP), 2-aminopurine, 6-thioguanine and 6-mercaptopurine. (B) Growth of L. monocytogenes in the absence (-) or presence of 100 μM of different purine analogs (6-N-HAP; 6-thioguanine (TG); 6-mercaptopurine (MP) and 2-aminopurine (AP)). Bacteria were grown in minimal medium for 10 h, scored as OD600 and shown as mean values with standard deviations (n = 6). (C) Growth of wild-type L. monocytogenes in the absence (-) or presence of different concentrations of 6-N-HAP in minimal medium for 10 h and scored as OD600. Samples were compared to the control without added compound (dimethyl sulfoxide (DMSO)) and shown as mean values with standard deviations (n = 6) and statistical analysis (Student T-test (two-tailed, P < 0.05,*; P < 0.001,***)). (D) Measurement of viable wild-type L. monocytogenes in DMSO or in 100 μM 6-N-HAP in comparison with the original inoculum. After 10 h incubation colony forming units were determined. Samples were compared to the inoculum and shown as mean values with standard deviations (n = 3) and statistical analysis (Student's t-test (two-tailed, P < 0.001,***)).
6-N-HAP inhibits expression of genes downstream of two guanine riboswitches
B. subtilis harbors four guanine riboswitches controlling genes whose products are involved in purine transport as well as de novo purine synthesis (19). It has earlier been shown that guanine and 6-N-HAP binds to the guanine riboswitch xpt-pbuX in B. subtilis, thereby inducing transcriptional termination and in turn reducing expression of the downstream gene (Figure 2A) (19,28). We were therefore interested to examine if guanine and 6-N-HAP could force termination of transcription at guanine riboswitches also in L. monocytogenes. In contrast to B. subtilis, L. monocytogenes harbor only two guanine riboswitches regulating lmo0573 and lmo1885, respectively (Figure 2B). lmo0573 encodes a xanthine/uracil permease and lmo1885 encodes a xanthine phosphoribosyl transferase. lmo1884 that lies in the same operon as lmo1885 encodes a xanthine permease. Hence, unlike in B. subtilis, the guanine riboswitches in L. monocytogenes do not control expression of purine biosynthesis genes. Addition of 100 μM of guanine or 6-N-HAP led to a decreased expression of both lmo0573 and lmo1885 (Figure 2C–E), which is in line with previous findings in B. subtilis (19). The reduced expression of lmo0573 and lmo1885 was due to an increased riboswitch-mediated termination, since addition of guanine or 6-N-HAP increased the level of the guanine riboswitch fragments Rli70 and Rli95 (Supplementary Figure S1).
Figure 2.

6-N-HAP decrease expression of genes downstream of guanine riboswitches. (A) Schematic rationale for action of purines at purine riboswitches. Absence of purines results in the formation of a transcriptional anti-terminator allowing expression of downstream genes. Presence of purines force a terminator structure to be formed, prematurely terminating transcription of the downstream genes. (B) Schematic drawing of the genetic loci for the guanine riboswitches (Rli70 and Rli95) controlling expression of their downstream genes (lmo0573 and lmo1884/lmo1885, respectively). (C) Wild-type or M1-M6 double mutant strain were grown in minimal medium to an OD600 = 0.25 when guanine or 6-N-HAP (100 μM) were added for ∼1.5 generation before RNA extraction. Northern blot was hybridized with PCR-generated, radioactively labeled DNA probes complementary to lmo0573, lmo1885 and tmRNA (control), respectively. Arbitrary levels of (D) lmo0573 or (E) lmo1885/lmo1884, respectively, quantified from northern blots as in (C). Error bars show standard deviation (n = 3).
A strain carrying mutations allowing constitutive expression of the genes downstream of the guanine riboswitches is still sensitive to 6-N-HAP
The above results could suggest that 6-N-HAP acts at the guanine riboswitch. To further analyze a putative role of guanine and 6-N-HAP at the guanine riboswitches, base-substitution mutations were introduced in the riboswitches at the native site on the chromosome. Theoretically, such base-alterations would allow expression of the downstream genes also in presence of guanine/6-N-HAP by forcing the formation of an anti-terminator structure (Supplementary Figure S2) (28). Similar mutations verified a direct interaction between 6-N-HAP and a guanine riboswitch in B. subtilis (19). The resulting double-mutant strain (M1-M6) carried mutations in both guanine riboswitches in L. monocytogenes, lying in front of lmo0573 and lmo1885, respectively (Supplementary Figure S2). In presence of 100 μM guanine or 100 μM 6-N-HAP, transcription of the lmo0573 and lmo1885 genes could proceed in the M1-M6 strain, whereas transcription of these genes were terminated in the wild-type strain (Figure 2C–E). Also, the amount of Rli70 and Rli95 decreased in the presence of 6-N-HAP in the M1-M6 strain as compared to the wild-type strain (Supplementary Figure S1). These results were in line with results obtained in B. subtilis (19) and strongly implies that guanine and 6-N-HAP are able to induce transcriptional termination at wild-type guanine riboswitches.
Based on this, we were interested to examine if the abolished expression of the downstream genes was the reason for the inhibited growth of the wild-type strain in presence of 6-N-HAP. Since the two listerial riboswitches regulate genes encoding permeases and a phosphoribosyl transferase but not purine biosynthesis proteins, it is unlikely that the downregulation of the purine transport genes by 6-N-HAP would be sufficient to cause a growth defect in L. monocytogenes. If this nevertheless would be the case, the M1-M6 double mutant strain should be able to grow in presence of 6-N-HAP. However, presence of 6-N-HAP inhibited growth of the M1-M6 strain in a manner similar to the wild-type strain at both 10 μM and 100 μM (Figure 3A). A comparable outcome was observed by growth and viability experiments where the compound was added at early logarithmic growth phase and samples were taken at different time-points (Figure 3B, Supplementary Figure S3). The wild type and M1-M6 strains exhibit a similar reduction in the number of growing bacteria after 6-N-HAP treatment. Our results therefore suggest that 6-N-HAP mediated transcriptional termination at the two guanine riboswitches is not sufficient to cause an antibacterial effect in L. monocytogenes but rather likely caused by an action at other targets. Our results also imply that the antibacterial activity of 6-N-HAP could be triggered by different mechanisms in B. subtilis and L. monocytogenes.
Figure 3.

The antibacterial effect of 6-N-HAP is not mediated through the guanine riboswitches. (A) Growth of wild-type or M1-M6 double mutant strains in the absence (-) or presence of 10 μM or 100 μM of 6-N-HAP in minimal medium for 10 h and scored as OD600. Samples were compared to the DMSO control and shown as mean values with standard deviations (n = 9) and statistical analysis (Student's t-test (two-tailed, P < 0.001,***)). (B) Survival of wild type or M1-M6 double mutant strain in the absence (DMSO) or presence of 100 μM of 6-N-HAP. Colony forming units were determined at the indicated time points. Shown are the mean values with standard deviations (n = 2).
The antibacterial effect of 6-N-HAP in L. monocytogenes can be restored by adenine
We decided to further investigate the target of 6-N-HAP in L. monocytogenes. To test if the antimicrobial effect of 6-N-HAP was dose-dependent, L. monocytogenes was exposed to 0, 10, 100 or 500 μM of 6-N-HAP. Although presence of 10 μM 6-N-HAP had the weakest effect on bacterial viability, the survival difference of this culture compared to bacteria exposed to either 100 or 500 μM of 6-N-HAP was only marginal (Figure 4A). Due to the structural similarity of 6-N-HAP to guanine and adenine we further investigated if the antimicrobial effect of 6-N-HAP could be restored by addition of purines and pyrimidines. The pyrimidines cytosine and thymine did not influence the 6-N-HAP-mediated growth defect (Figure 4B), whereas the 6-N-HAP-induced growth defect was slightly but reproducibly reversed by guanine (Figure 4B). To our surprise, the addition of adenine completely abolished the antimicrobial effect of 6-N-HAP (Figure 4B). To test the possibility that adenine would play a role at guanine riboswitches, expression of lmo0573, lying downstream of Rli70, was examined. Addition of adenine did not reduce lmo0573 expression, rather a slight upregulation was observed. This result clearly shows that Rli70 is strictly responding to guanine and not adenine (Supplementary Figure S4).
Figure 4.

The influence of purines and pyrimidines on the antibacterial effect of 6-N-HAP in L. monocytogenes. (A) Growth of the wild-type strain in the presence of different 6-N-HAP concentrations. DMSO, 10, 100 or 500 μM of 6-N-HAP were added to L. monocytogenes grown to an OD600 of 0.15 and incubated for 1.5 generations. Bacterial survival was scored as colony forming units. Samples were compared to the inoculum and shown as mean values with standard deviations (n = 3). (B) Growth of L. monocytogenes in the absence and presence of 100 μM of adenine (A), cytosine (C), guanine (G), thymine (T) or 6-N-HAP as well as 6-N-HAP in combination with one of each nucleoside. The bacteria were grown in minimal medium to an OD600 = ∼0.15 before solvents, nucleosides or 6-N-HAP were added. The growth was scored as OD600 after 10 h. Samples were compared to the wild type grown in minimal medium and shown as mean values with standard deviations (n = 3). Differences between 6-N-HAP and 6-N-HAP + A or 6-N-HAP + G were compared using Student's t-test (two-tailed, P < 0.005,**, P < 0.01,*)).
The notion that addition of guanine and especially adenine could reverse the growth-repressive effect of 6-N-HAP, prompted us to speculate whether the compound was interfering with purine biosynthesis. Besides the decreased expression of the genes lmo0573 and lmo2254 encoding purine permeases in the presence of 6-N-HAP (Supplementary Figure S4), we did not observe a clear effect of 6-N-HAP on the expression of the major purine biosynthesis operon purEKBCSLQFMNHD (data not shown).
The growth inhibitory effect of 6-N-HAP is caused by its mutagenic properties
6-N-HAP has previously been shown to act as a potent mutagen in both pro- and eukaryotes (3,29–31). It could therefore be hypothesized that the 6-N-HAP mediated killing of L. monocytogenes would be due to increased mutagenesis. Initially, we believed that this was not the case in L. monocytogenes based on the following two findings: (i) Other mutagenic guanine-analogs do not inhibit L. monocytogenes growth (Figure 1B) and (ii) The antimicrobial effect seems not to be dose-dependent since we observed a similar viability at 10 μM as in 500 μM 6-N-HAP (Figure 4A). However, while working with 6-N-HAP-treated bacteria we observed that the colony sizes after 6-N-HAP treatment differed among clones, possibly indicating an increased level of mutagenesis in the presence of the analog (data not shown). We therefore decided to further investigate a potential mutagenic effect of 6-N-HAP in L. monocytogenes. If 6-N-HAP increases the mutation frequency we would expect an elevated spontaneous mutation rate throughout the chromosome. To test this hypothesis, the occurrence of spontaneous rifampicin resistance was used as a read-out system for the mutation rate. The antibiotic rifampicin (rif) binds to the β-subunit of the DNA-dependent RNA polymerase and inhibits its enzymatic activity (32). Resistance against rifampicin can be achieved by a single amino acid exchange of the rifampicin binding site of the β-subunit (32,33). We hypothesized that the presence of a mutagenic agent such as 6-N-HAP, should increase the occurrence of mutations in the rpoB gene encoding the β-subunit. To test this, L. monocytogenes was grown in the presence of 100 μM guanine or 100 μM 6-N-HAP for 10 h. Colony forming units and rifampicin resistant mutants were determined by plating on plain LB plates or LB plates containing rifampicin. As a difference to guanine, a drastically increased number of rifampicin resistant bacteria could be observed after treatment with 6-N-HAP (Figure 5A). Almost a 1000-fold higher occurrence of rifampicin resistant mutants were observed in bacteria treated with 6-N-HAP compared to DMSO-treated bacteria (Figure 5A).
Figure 5.

6-N-HAP increases the mutation rate in L. monocytogenes. (A) Screening for spontaneous mutations leading to rifampicin (Rif) resistance after 6-N-HAP treatment. L. monocytogenes wild-type was grown in minimal medium to an OD600 = 0.15 before guanine (100 μM) or 6-N-HAP (100 μM) were added and incubated for 10 h at 37°C. Mean values of the ratio of rifampicin resistant mutant to colony forming units are shown with standard deviations (n = 3). (B) Characterization of mutations that occurred in the rpoB gene of L. monocytogenes after 6-N-HAP treatment. Base- substitutions in the rpoB gene of nine different RifR colonies isolated after 6-N-HAP treatment (No. 2–10) and one RifR colony isolated after DMSO treatment (D). Shown is the nucleotide exchange that occurred as well as the resulting amino acid exchange with its position in the amino acid sequence.
To further examine the mutations, a part of the rpoB gene (rifampicin target site) of a set of rifampicin sensitive as well as rifampicin resistant mutants were sequenced. As expected, the rifampicin sensitive strains did not harbor any mutations within the sequenced region. Three out of four spontaneous rifampicin resistant mutants isolated after DMSO-treatment and one resistant mutant isolated after 6-N-HAP treatment did not carry a mutation in the sequenced rpoB region, possibly due to changes in uptake, inactivation or modification of rifampicin (34–36). Sequencing of nine additional rifampicin resistant mutants isolated after 6-N-HAP treatment as well as one rifampicin resistant mutant isolated after DMSO treatment, revealed single nucleotide exchange mutations in the rpoB gene, resulting in an amino acid exchange (Figure 5B). All of this mutations were transitions from either C → T (7 mutants) or G →A (3 mutants). These results suggest that 6-N-HAP reduces L. monocytogenes viability by increasing the level of mutagenesis.
6-N-HAP induces the SOS response
In bacteria, lesions in DNA induce the SOS response, a bacterial system involved in DNA damage repair (37). Conditions that lead to an accumulation of single stranded DNA trigger the SOS response. Expression of the SOS response genes is controlled by the proteins RecA and LexA. The transcriptional regulator LexA represses the expression of the SOS response genes. Single stranded DNA resulting from DNA damage activates the RecA protein which in turn stimulates the autoproteolytic activity of LexA and results in expression of the SOS response genes. In addition to LexA and RecA, the listerial SOS response regulon encompass the translesion DNA polymerases DinB (polymerase IV; Lmo1975), UmuCD (polymerase V, UmuC/Lmo2676) and the excinuclease UvrBA (38). We were interested to examine if exposure to 6-N-HAP triggers the SOS response in L. monocytogenes as has been observed in other bacteria (39). Therefore, the expression of components of the SOS response were examined. 6-N-HAP treatment increases the expression of lmo1975 and lmo2676 indicating that exposure of L. monocytogenes to 6-N-HAP indeed induces the SOS response (Figure 6) (40,41).
Figure 6.
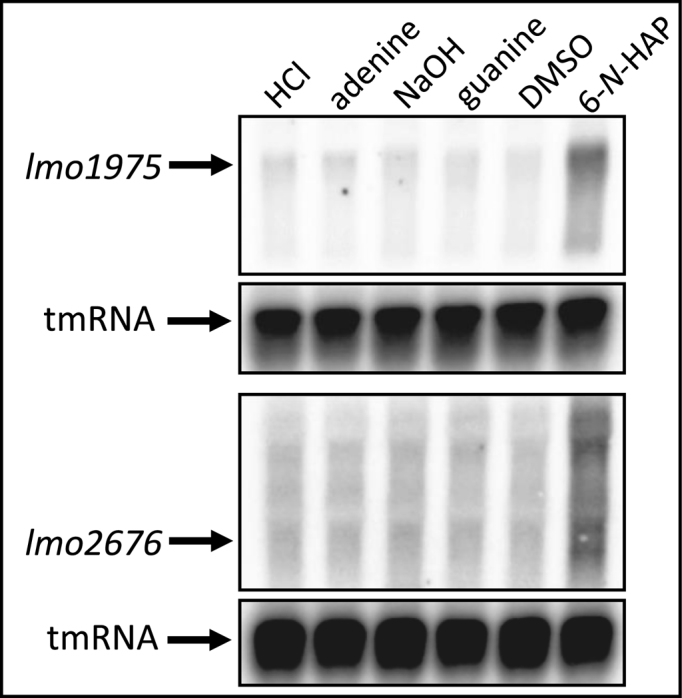
6-N-HAP induces the SOS response. The wild-type strain was grown in minimal medium to an OD600 = 0.25 when 100 μM adenine, guanine or 6-N-HAP were added for ∼1.5 generation before RNA extraction. PCR-generated, radioactively labeled DNA probes were used to detect lmo1975, lmo2676 or tmRNA (control).
6-N-HAP reduces expression of L. monocytogenes virulence genes
We have previously shown that a riboswitch metabolite analog, roseoflavin (analog to riboflavin), was able to inhibit growth of L. monocytogenes by downregulating expression of a riboflavin transporter (17). However, in that work, we also observed an increased virulence gene-expression and cell-infection capacity of L. monocytogenes treated with roseoflavin which is troublesome in the prospect of developing novel antibacterial compounds. A purine analog, 2,5,6-traminopyrimidin-4-1 (PC1), inhibited S. aureus infection in a mechanism where the analog possibly acted through a purine riboswitch (20). We therefore set to analyze the virulence gene-expression in presence of 6-N-HAP. Strikingly, 6-N-HAP dramatically reduced expression of hly and actA in in both the wild-type and the M1-M6-strain (Figure 7A). hly encodes the haemolysin LLO, which is essential for Listerial escape from the intracellular phagosome (42), whereas actA encodes ActA, a protein mediating intracellular movement by recruiting the Arp2/3 complex (43). In agreement with the Northern blot, the levels of the LLO and ActA proteins were reduced after bacterial exposure to 6-N-HAP for 3.5 h (Figure 7B). The 6-N-HAP mediated reduction of LLO and ActA protein levels was however not as pronounced as the effect on hly and actA mRNA levels, probably due to a slower turnover of proteins than mRNAs at the time of compound addition (Figure 7A and B). Expression of hly and actA is controlled by PrfA, a transcriptional activator of the Crp/Fnr family of regulators that control L. monocytogenes virulence gene expression. Expression of prfA is initiated at three different promoters (44). Expression from two of them results in a monocistronic prfA transcript, whereas expression from the third produces a bicistronic plcA-prfA transcript. All of the prfA-promoters are controlled differentially: One of the two promoters producing a monocistronic messenger is expressed constitutively, through the vegetative sigma-factor σ70, whereas the other is controlled by the stress-sigma-factor σB (44). The bicistronic plcA-prfA messenger requires post-translationally active PrfA protein for expression, creating a positive feed-back loop (45). Presence of 6-N-HAP reduced expression of the PrfA-regulated plcA-prfA messenger in both the wild type and the M1-M6 strain, which is in agreement with the decreased PrfA-dependent hly and actA expression observed with 6-N-HAP (Figure 7A). Addition of 6-N-HAP induced expression from the σB–dependent prfA promoter (Figure 7A), indicating that 6-N-HAP triggers σB-activity. Despite this, it appeared that 6-N-HAP decreased expression of the PrfA protein (compare Figure 7A and B, Supplementary Figure S5).
Figure 7.
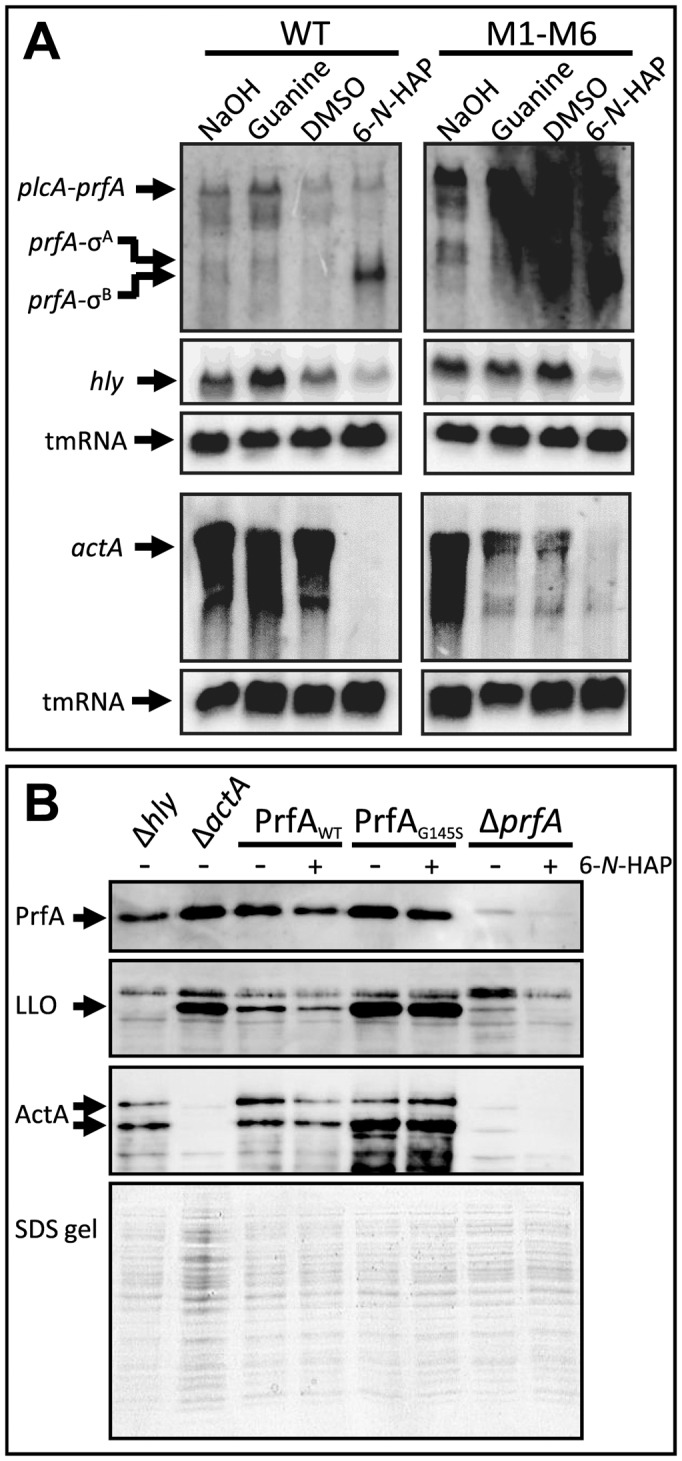
6-N-HAP decreases L. monocytogenes virulence gene expression. (A) The wild-type or the M1-M6 double mutant strains were grown in minimal medium to an OD600 = 0.25 when guanine or 6-N-HAP (100 μM) were added for ∼1.5 generation before RNA extraction. Northern blot was hybridized with PCR-generated, radioactively labeled DNA probes complementary to prfA, hly, actA or tmRNA (control), respectively. (B) The indicated strains were grown in minimal medium to an OD600 = 0.25. 6-N-HAP (100 μM, +) were added to strains carrying a wild-type PrfA (PrfAWT), a constitutively active PrfA (PrfAG145S) or a strain lacking PrfA, respectively. As a control, DMSO was added to the other cultures (-). After 3.5 h, the proteins were extracted before western blot using PrfA, LLO and ActA antibodies, respectively. Total protein samples are included as a loading control.
PrfA require a co-factor, glutathione, for maximal activity (46). However, amino-acid substitutions in PrfA can make it constitutively active, bypassing the need of glutathione for maximal virulence factor expression (46). To further examine the effect of 6-N-HAP on PrfA activity, we used a constitutively active PrfA protein (PrfAG145S) expressed from its native site on the chromosome. The PrfAG145S protein has a stabilized helix–turn–helix motif that increases its DNA-binding affinity compared with the wild type (42). After 3.5 h exposure, 6-N-HAP did not affect LLO and ActA levels in a strain expressing PrfAG145S protein as observed in the PrfAWT background (Figure 7B, Supplementary Figure S5). These results indicate that a constitutively active PrfA protein might overcome the repressive effect of 6-N-HAP on virulence gene expression, despite reduced levels of PrfA.
DISCUSSION
In this work, we have studied the effect of guanine analogs on growth/survival of the Gram-positive pathogen Listeria monocytogenes. Guanine analogs have been shown to block growth of M. tuberculosis and E. coli (2,3). Our results show that of the tested analogs, only 6-N-hydroxylaminopurine (6-N-HAP) has an antimicrobial effect in Listeria monocytogenes (Figure 1). It has previously been shown that addition of 6-N-HAP blocks growth of Bacillus subtilis (19). 6-N-HAP was shown to directly bind to the xpt guanine riboswitch and block expression of the downstream reporter gene (19). Analogs to metabolites that bind riboswitches are attractive candidates when trying to develop novel types of antibacterial drugs. The underlying mechanism of their action is simple, the analog (normally not metabolized) should bind the riboswitch and thereby preserve it in a form preventing expression of the downstream genes.
We observed that 6-N-HAP represses expression of genes lying downstream of two guanine riboswitches in L. monocytogenes by premature transcriptional termination (Figure 2 and Supplementary Figure S1). However, the toxic effect of 6-N-HAP was not mediated through blocked expression of the genes downstream of the guanine riboswitches, since a strain carrying chromosomal mutations in the riboswitches allowing constitutive expression of these genes still were sensitive to 6-N-HAP (Figures 2 and 3, Supplementary Figures S1–S3). Although the toxic effect of 6-N-HAP was not caused by blocked expression of genes downstream the guanine riboswitches in L. monocytogenes, such a mechanism might still be responsible for or contribute to the bacteriocidal effect in B. subtilis. There are differences between B. subtilis and L. monocytogenes regarding the function of the genes downstream guanine riboswitches. Indeed, B. subtilis harbors five purine (adenine and guanine) riboswitches whereas L. monocytogenes only harbors two (guanine). Furthermore, the guanine riboswitches in B. subtilis regulate genes that are involved in purine transport and also in de novo purine biosynthesis, indicating a more pivotal role of the riboswitches and their downstream genes in B. subtilis. Presumably, the downregulation of purine transporters and a phosphoribosyl transferase by targeting the riboswitches is not sufficient to cause a growth defect in L. monocytogenes. It remains to be experimentally determined to which extent the antibacterial effect of 6-N-HAP in B. subtilis is due to inhibited expression of the genes downstream of the guanine riboswitches, and/or if the effect is unlinked to these targets as shown herein for L. monocytogenes (e.g. mutagenesis).
An alternative hypothesis for the antibacterial action of 6-N-HAP would be that it acts as a mutagen, being incorporated into DNA or RNA as has been suggested earlier (3,29–31). We did not observe a dose-dependent cytotoxic effect of 6-N-HAP in L. monocytogenes (i.e. more 6-N-HAP would cause more mutations, thereby decreasing Listeria viability) (Figure 4A). This could be due to a decreased expression of the genes lmo0573 and lmo2254 (encoding purine permeases), in the presence of 6-N-HAP (Supplementary Figure S4). Hence, expression of the purine permeases is blocked at high 6-N-HAP concentrations, thereby reducing further uptake of 6-N-HAP. These two permeases together with Lmo1845 are homologous to the E. coli nucleobase permease YjcD (Lmo0573: 35% identity and 3e−68 BLAST homology score; Lmo2254: 34% identity and 6e−63 BLAST homology score and Lmo1845: 29% identity and 7e−37 BLAST homology score, respectively). It was previously shown that YjcD is important for uptake of the purine analogs 6-N-HAP, 2-amino-HAP, 6-mercaptopurine, 6-thioguanine and 2-aminopurine in E. coli (3). This suggests a similar uptake mechanisms for purine analogs in L. monocytogenes. Nevertheless, it is unknown why only 6-N-HAP and none of the other tested purine analogs repress growth of L. monocytogenes (Figure 1B). It is believed that following uptake, purine analogs get converted to (deoxy)nucleoside triphosphates and are incorporated into DNA or RNA that induce mutations due to the ambivalent coding capacity of the analogs (31). Presence of 6-N-HAP did indeed increase the mutation rate ∼1000-fold (Figure 5A). 6-N-HAP has previously been shown to harbor ambiguous base pairing probabilities (47). By replacing guanine in DNA, 6-N-HAP could pair either with cytosine or thymine during replication, and thereby increase the chance of mutations (47). In agreement with such a mechanism, only transitions (A↔G or C↔T) were observed in the rpoB genes of rifampicin mutants obtained in presence of 6-N-HAP (Figure 5B). As would be expected of an increased mutation rate, the exposure of L. monocytogenes to 6-N-HAP causes an upregulation of the SOS response (Figure 6). It could be hypothesized that the increased expression of the error-prone DNA polymerases IV and V further increases the mutation rate in L. monocytogenes (Figure 6) (38). The mechanism by how 6-N-HAP increases the mutation rate in L. monocytogenes needs further attention.
Intriguingly, the antimicrobial effect of 6-N-HAP could be completely rescued by addition of adenine, whereas guanine only partially recovered the inhibitory growth effect of 6-N-HAP. Presumably, adenine competes with 6-N-HAP uptake, conversion to (deoxy)nucleoside triphosphate and/or incorporation into the DNA. The strong recovery effect of adenine on cells also exposed to 6-N-HAP suggests that an increased pool of adenine and guanine would better compete with 6-N-HAP for the incorporation into the DNA. In this context, it is interesting to note that B. subtilis strains developing resistance against 6-N-HAP showed mutations in the pbuE adenine riboswitch and not in a guanine riboswitch (19).
In addition, we observed that the presence of 6-N-HAP decreased expression of several virulence factors required for a successful L. monocytogenes infection. Surprisingly, the level of the PrfA-protein, the transcriptional activator essential for virulence factor expression (45), was decreased in presence of 6-N-HAP, by a mechanism unlinked to PrfA-activity (the levels both PrfAWT and PrfAG145S were reduced in presence of 6-N-HAP, Figure 7B). Furthermore, our results indicate that 6-N-HAP affects the activity of PrfA. We reasoned that if 6-N-HAP affects any step in the activation of PrfA, the repressive effect of 6-N-HAP should be bypassed in a strain with a constitutively active PrfA protein. In agreement with that, we observed similar LLO and ActA levels in the presence and absence of 6-N-HAP in a strain expressing constitutively active PrfA. This indicates that 6-N-HAP affects one or several steps prior to PrfA activation and that a constitutively active PrfA protein might overcome the 6-N-HAP-mediated repression virulence gene expression. However, it needs to be considered that the dramatically higher amount of virulence proteins in the PrfAG145S strain compared to the wild-type strain and a slower turnover of proteins might result in a delayed manifestation of a potential 6-N-HAP-mediated repressive effect.
The decreased virulence factor expression in the presence of 6-N-HAP was not mediated through its action at the guanine riboswitches or decreased expression of the downstream genes, since a similar pattern of regulation was also observed in a strain having constitutive expression of lmo0573 and lmo1885 (Figure 7A).
Previously, we observed that a riboflavin analog, roseoflavin, showing an antibacterial property stimulated PrfA activity (17), making it less appropriate as a possible antibacterial drug. Interestingly, 6-N-HAP is antibacterial and inhibits virulence gene expression making it a possible scaffold for further development of antibacterial drugs, although care has to be undertaken to avert possible mutagenic effects in humans.
In summary, we observed that 6-N-HAP acts on several levels in L. monocytogenes. It repress expression of genes downstream of guanine riboswitches, it increase the mutation frequency, it induce the SOS-response and it also inhibit virulence gene expression.
Supplementary Material
ACKNOWLEDGEMENTS
J.J. was supported by Umeå University, the Swedish Research Council grants K2011-56X-15144-08-6 and 621-2012-2451 and an ERC starting grant no 260764 - RNAntibiotics. S.S.K. was supported with a research fellowship (KR 4664/1-1) by the German Research Foundation (DFG). The authors thank M. Mansjö for valuable technical assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Umeå University; Swedish Research Council [K2011-56X-15144-08-6 and 621-2012-2451 to J.J.]; ERC [260764 - RNAntibiotics]; German Research Foundation (DFG) research fellowship [KR 4664/1-1]. Funding for open access charge: Swedish Research Council [K2011-56X-15144-08-6 and 621-2012-2451 to J.J.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Nguyen L. Antibiotic resistance mechanisms in M. tuberculosis: an update. Arch. Toxicol. 2016; 90:1585–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pathak A.K., Pathak V., Seitz L.E., Suling W.J., Reynolds R.C.. 6-Oxo and 6-thio purine analogs as antimycobacterial agents. Bioorg. Med. Chem. 2013; 21:1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kozmin S.G., Stepchenkova E.I., Chow S.C., Schaaper R.M.. A critical role for the putative NCS2 nucleobase permease YjcD in the sensitivity of Escherichia coli to cytotoxic and mutagenic purine analogs. mBio. 2013; 4:e00661–e00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blount K.F., Breaker R.R.. Riboswitches as antibacterial drug targets. Nat. Biotechnol. 2006; 24:1558–1564. [DOI] [PubMed] [Google Scholar]
- 5. Lea C.R., Piccirilli J.A.. 'Turning on' riboswitches to their antibacterial potential. Nat. Chem. Biol. 2007; 3:16–17. [DOI] [PubMed] [Google Scholar]
- 6. Breaker R.R. Riboswitches: from ancient gene-control systems to modern drug targets. Future Microbiol. 2009; 4:771–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Long Q., Ji L., Wang H., Xie J.. Riboflavin biosynthetic and regulatory factors as potential novel anti-infective drug targets. Chem. Biol. Drug Des. 2010; 75:339–347. [DOI] [PubMed] [Google Scholar]
- 8. Deigan K.E., Ferre-D'Amare A.R.. Riboswitches: discovery of drugs that target bacterial gene-regulatory RNAs. Acc. Chem. Res. 2011; 44:1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guan L., Disney M.D.. Recent advances in developing small molecules targeting RNA. ACS Chem. Biol. 2012; 7:73–86. [DOI] [PubMed] [Google Scholar]
- 10. Penchovsky R., Stoilova C.C.. Riboswitch-based antibacterial drug discovery using high-throughput screening methods. Expert Opin. Drug Discov. 2013; 8:65–82. [DOI] [PubMed] [Google Scholar]
- 11. Mulhbacher J., St-Pierre P., Lafontaine D.A.. Therapeutic applications of ribozymes and riboswitches. Curr. Opin. Pharmacol. 2010; 10:551–556. [DOI] [PubMed] [Google Scholar]
- 12. Winkler W.C., Breaker R.R.. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 2005; 59:487–517. [DOI] [PubMed] [Google Scholar]
- 13. Blount K.F., Wang J.X., Lim J., Sudarsan N., Breaker R.R.. Antibacterial lysine analogs that target lysine riboswitches. Nat. Chem. Biol. 2007; 3:44–49. [DOI] [PubMed] [Google Scholar]
- 14. Sudarsan N., Cohen-Chalamish S., Nakamura S., Emilsson G.M., Breaker R.R.. Thiamine pyrophosphate riboswitches are targets for the antimicrobial compound pyrithiamine. Chem. Biol. 2005; 12:1325–1335. [DOI] [PubMed] [Google Scholar]
- 15. Lee E.R., Blount K.F., Breaker R.R.. Roseoflavin is a natural antibacterial compound that binds to FMN riboswitches and regulates gene expression. RNA Biol. 2009; 6:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ott E., Stolz J., Lehmann M., Mack M.. The RFN riboswitch of Bacillus subtilis is a target for the antibiotic roseoflavin produced by Streptomyces davawensis. RNA Biol. 2009; 6:276–280. [DOI] [PubMed] [Google Scholar]
- 17. Mansjo M., Johansson J.. The riboflavin analog roseoflavin targets an FMN-riboswitch and blocks Listeria monocytogenes growth, but also stimulates virulence gene-expression and infection. RNA Biol. 2011; 8:674–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pedrolli D.B., Matern A., Wang J., Ester M., Siedler K., Breaker R., Mack M.. A highly specialized flavin mononucleotide riboswitch responds differently to similar ligands and confers roseoflavin resistance to Streptomyces davawensis. Nucleic Acids Res. 2012; 40:8662–8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim J.N., Blount K.F., Puskarz I., Lim J., Link K.H., Breaker R.R.. Design and antimicrobial action of purine analogues that bind Guanine riboswitches. ACS Chem. Biol. 2009; 4:915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mulhbacher J., Brouillette E., Allard M., Fortier L.C., Malouin F., Lafontaine D.A.. Novel riboswitch ligand analogs as selective inhibitors of guanine-related metabolic pathways. Plos Pathog. 2010; 6:e1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Howe J.A., Wang H., Fischmann T.O., Balibar C.J., Xiao L., Galgoci A.M., Malinverni J.C., Mayhood T., Villafania A., Nahvi A. et al. Selective small-molecule inhibition of an RNA structural element. Nature. 2015; 526:672–677. [DOI] [PubMed] [Google Scholar]
- 22. Kofoed E.M., Yan D., Katakam A.K., Reichelt M., Lin B., Kim J., Park S., Date S.V., Monk I.R., Xu M. et al. De novo guanine biosynthesis but not the riboswitch-regulated purine salvage pathway is required for Staphylococcus aureus Infection in vivo. J. Bacteriol. 2016; 198:2001–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Phan-Thanh L., Gormon T.. A chemically defined minimal medium for the optimal culture of Listeria. Int. J. Food Microbiol. 1997; 35:91–95. [DOI] [PubMed] [Google Scholar]
- 24. Bertani G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 1951; 62:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loh E., Dussurget O., Gripenland J., Vaitkevicius K., Tiensuu T., Mandin P., Repoila F., Buchrieser C., Cossart P., Johansson J.. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell. 2009; 139:770–779. [DOI] [PubMed] [Google Scholar]
- 26. Netterling S., Bareclev C., Vaitkevicius K., Johansson J.. RNA helicase important for Listeria monocytogenes Hemolytic activity and virulence factor expression. Infect. Immun. 2016; 84:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arnaud M., Chastanet A., Debarbouille M.. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 2004; 70:6887–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mandal M., Boese B., Barrick J.E., Winkler W.C., Breaker R.R.. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell. 2003; 113:577–586. [DOI] [PubMed] [Google Scholar]
- 29. Barrett J.C. Induction of gene mutation in and cell transformation of mammalian cells by modified purines: 2-aminopurine and 6-N-hydroxylaminopurine. Proc. Natl. Acad. Sci. U.S.A. 1981; 78:5685–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pavlov Y.I., Noskov V.N., Lange E.K., Moiseeva E.V., Pshenichnov M.R., Khromov-Borisov N.N.. The genetic activity of N6-hydroxyadenine and 2-amino-N6-hydroxyadenine in Escherichia coli, Salmonella typhimurium and Saccharomyces cerevisiae. Mutat. Res. 1991; 253:33–46. [DOI] [PubMed] [Google Scholar]
- 31. Kozmin S.G., Schaaper R.M., Shcherbakova P.V., Kulikov V.N., Noskov V.N., Guetsova M.L., Alenin V.V., Rogozin I.B., Makarova K.S., Pavlov Y.I.. Multiple antimutagenesis mechanisms affect mutagenic activity and specificity of the base analog 6-N-hydroxylaminopurine in bacteria and yeast. Mutat. Res. 1998; 402:41–50. [DOI] [PubMed] [Google Scholar]
- 32. Wehrli W. Rifampin: mechanisms of action and resistance. Rev. Infect. Dis. 1983; 5(Suppl 3): S407–S411. [DOI] [PubMed] [Google Scholar]
- 33. Morse R., O'Hanlon K., Virji M., Collins M.D.. Isolation of rifampin-resistant mutants of Listeria monocytogenes and their characterization by rpoB gene sequencing, temperature sensitivity for growth, and interaction with an epithelial cell line. J. Clin. Microbiol. 1999; 37:2913–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dabbs E.R., Yazawa K., Tanaka Y., Mikami Y., Miyaji M., Andersen S.J., Morisaki N., Iwasaki S., Shida O., Takagi H. et al. Rifampicin inactivation by Bacillus species. J. Antibiot. 1995; 48:815–819. [DOI] [PubMed] [Google Scholar]
- 35. Chandrasekaran S., Lalithakumari D.. Plasmid-mediated rifampicin resistance in Pseudomonas fluorescens. J. Med. Microbiol. 1998; 47:197–200. [DOI] [PubMed] [Google Scholar]
- 36. Tribuddharat C., Fennewald M.. Integron-mediated rifampin resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1999; 43:960–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Janion C. Inducible SOS response system of DNA repair and mutagenesis in Escherichia coli. Int. J. Biol. Sci. 2008; 4:338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van der Veen S, van Schalkwijk S., Molenaar D., de Vos W.M., Abee T., Wells-Bennik M.H.. The SOS response of Listeria monocytogenes is involved in stress resistance and mutagenesis. Microbiology. 2010; 156:374–384. [DOI] [PubMed] [Google Scholar]
- 39. Burgis N.E., Brucker J.J., Cunningham R.P.. Repair system for noncanonical purines in Escherichia coli. J. Bacteriol. 2003; 185:3101–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tiensuu T., Andersson C., Ryden P., Johansson J.. Cycles of light and dark co-ordinate reversible colony differentiation in Listeria monocytogenes. Mol. Microbiol. 2013; 87:909–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Utratna M., Cosgrave E., Baustian C., Ceredig R., O'Byrne C.. Development and optimization of an EGFP-based reporter for measuring the general stress response in Listeria monocytogenes. Bioeng. Bugs. 2012; 3:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hamon M., Bierne H., Cossart P.. Listeria monocytogenes: a multifaceted model. Nat. Rev. Microbiol. 2006; 4:423–434. [DOI] [PubMed] [Google Scholar]
- 43. Cossart P. Actin-based motility of pathogens: the Arp2/3 complex is a central player. Cell. Microbiol. 2000; 2:195–205. [DOI] [PubMed] [Google Scholar]
- 44. Monk I.R., Gahan C.G., Hill C.. Tools for functional postgenomic analysis of Listeria monocytogenes. Appl. Environ. Microbiol. 2008; 74:3921–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Scortti M., Monzo H.J., Lacharme-Lora L., Lewis D.A., Vazquez-Boland J.A.. The PrfA virulence regulon. Microbes Infect. 2007; 9:1196–1207. [DOI] [PubMed] [Google Scholar]
- 46. Reniere M.L., Whiteley A.T., Hamilton K.L., John S.M., Lauer P., Brennan R.G., Portnoy D.A.. Glutathione activates virulence gene expression of an intracellular pathogen. Nature. 2015; 517:170–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pavlov Y.I., Suslov V.V., Shcherbakova P.V., Kunkel T.A., Ono A., Matsuda A., Schaaper R.M.. Base analog N6-hydroxylaminopurine mutagenesis in Escherichia coli: genetic control and molecular specificity. Mutat. Res. 1996; 357:1–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.