


Abstract
Advances in transcriptomics have led to the discovery of a large number of long intergenic non-coding RNAs (lincRNAs), which are now recognized as important regulators of diverse cellular processes. Although originally thought to be non-coding, recent studies have revealed that many lincRNAs are bound by ribosomes, with a few lincRNAs even having ability to generate micropeptides. The question arises: how widespread the translation of lincRNAs may be and whether such translation is likely to be functional. To better understand biological relevance of lincRNA translation, we systematically characterized lincRNAs with ribosome occupancy by the expression, structural, sequence, evolutionary and functional features for eight human cell lines, revealed that lincRNAs with ribosome occupancy have remarkably distinctive properties compared with those without ribosome occupancy, indicating that translation has important biological implication in categorizing and annotating lincRNAs. Further analysis revealed lincRNAs exhibit remarkable cell-type specificity with differential translational repertoires and substantial discordance in functionality. Collectively, our analyses provide the first attempt to characterize global and cell-type specific properties of translation of lincRNAs in human cells, highlighting that translation of lincRNAs has clear molecular, evolutionary and functional implications. This study will facilitate better understanding of the diverse functions of lincRNAs.
INTRODUCTION
Long intergenic non-coding RNAs (lincRNAs) are an abundant class of endogenous RNA molecules that are transcribed from intergenic regions of the genome. Although originally defined as non-coding RNAs, accumulating evidence has revealed that lincRNAs play important roles in many cellular processes (1–3). The aberrant expression of lincRNAs has been associated with a wide variety of human diseases such as cancer, aging and ocular disorders (4–6), making them attractive candidates for biomarkers and therapeutic targets.
Notably, despite receiving remarkable attention in recent years, the biological roles of the majority of lincRNAs remain largely unknown. Due to the diverse functions and molecular mechanisms, lincRNAs are far more complex than initially thought. Previous studies have suggested they may act as signals, decoys, guides and scaffolds to regulate the expression of either neighbouring genes in cis or distant genes in trans (7). In recent years, advances in genomic technologies have made comprehensive understanding of lincRNA functions feasible (8). It is now possible, for example, to directly identify genomic localization of lincRNAs using chromatin isolation by RNA purification (ChIRP), to dissect biochemical partners using capture hybridization analysis of RNA targets (Chart) and to investigate biological functions using clustered regularly interspaced short palindromic repeat (CRISPR) (9–11).
Recently developed ribosome profiling allows us to globally monitor translation of transcripts by measuring RNAs associated with 80S ribosomes in cells (12,13). Many studies using ribosome profiling have shown apparent ribosome occupancy inside and outside of protein-coding regions, including lincRNA regions (14–17). Although the density of ribosomes in lincRNA regions is lower than that of protein-coding regions, several previous studies have suggested that many lincRNAs may undergo active translation and this translation closely resembles that observed at the 5΄ leaders of protein-coding genes (14–15,17). Beyond these, more recently, emerging evidence has shown the existence of short peptides encoded by small open reading frames (sORFs) on lincRNAs (18–20), revealing that lincRNAs could be an important source of new peptides (16) and even orchestrate biological processes through encoded micro-peptides (21,22). These findings add a new layer of complexity in understanding the functions of lincRNAs. Nevertheless, ribosome profiling also provides a valuable way to characterize functions of translation in lincRNAs that cannot be revealed by RNA-sequencing (RNA-seq). The question then arises: how widespread the translation of lincRNAs may be and whether such translation is likely to be functional. Furthermore, as the application of ribosome profiling continues increasing, a large amount of data has been generated (23,24), affording a unique opportunity to appreciate translation implications of lincRNAs for different cell types. Given the cell-type specificity of lincRNAs observed at the transcriptional level (25–29), it is anticipated that they also display cell-type specificity at the translational level. Therefore, a comprehensive characterization of lincRNAs with and without ribosome occupancy across different cell types may facilitate better understanding of complex functions of lincRNAs.
In this study, we systematically characterized lincRNAs with ribosome occupancy for eight human cell lines. The integrative analysis of data collected from ribosome profiling and RNA-seq showed that the majority of well-transcribed lincRNAs did not show ribosome occupancy. In total 1332 (28%) out of 4709 well-transcribed lincRNAs showed ribosome occupancy in at least one cell line, where only 19 (1.42%) were evidenced by all the eight cell lines. We systematically characterized the expression, structural, sequence, evolutionary and functional features of lincRNAs with ribosome occupancy (ribo-lincRNAs) and compared them with lincRNAs without ribosome occupancy (nonribo-lincRNAs), as well as protein-coding genes. We found that ribo-lincRNAs have remarkably distinctive properties compared with nonribo-lincRNAs or protein coding genes, indicating that translation has important biological implication in categorizing and annotating lincRNAs. Further analysis revealed that lincRNAs exhibit a high degree of cell-type specificity with differential translational repertoires. Moreover, functional analysis revealed substantial discordance in potential functionality between lincRNAs with and without ribosome occupancy. Collectively, Our analysis provide the first attempt to characterize global and cell-type specific properties of translation of lincRNAs, highlighting that translation of lincRNAs has clear molecular, evolutionary and functional implications with remarkable cell-type specificity.
MATERIALS AND METHODS
Data processing
The original ribosome profiling and RNA-seq data were downloaded from the sequence read archive (SRA) (30) as described in detail in Table 1. For all the ribosome profiling and RNA-seq data, the adapters were clipped by Cutadapt (v1.8.1). Low-quality read ends with quality score of <20 were trimmed and reads with length of <20 were discarded by Sickle (v1.33). The trimmed ribosome profiling reads mapped to the human rRNA and tRNA reference were further removed. The remaining reads were then aligned to the human genome (GENCODE v23) using Tophat2 (v2.0.11). Gene expression levels were estimated using fragments per kilobase of transcript per million fragments mapped (FPKM) by Cufflinks (v2.1.1).
Table 1. Summary of RNA-Seq and ribosome profiling data used in the study.
| Cell-type | Description | Treatment | Accession | Reference |
|---|---|---|---|---|
| BJ | human BJ fibroblast cell line | CHX | SRA093551 | (71) |
| HEK293T | human embryonic kidney 293T cell line | CHX | SRA237056 | (72) |
| HeLa | human cervical cancer cell line | CHX | SRA099816 | (73) |
| hES | human embryonic stem cell line | CHX | SRA189363 | (74) |
| LCL | Human lymphoblastoid cell line | CHX | SRA198298 | (75) |
| PC3 | human prostate cancer cell line | CHX | SRA049772 | (76) |
| RPE | human retinal pigment epithelial cell line | CHX | SRA259601 | (77) |
| U2OS | human osteosarcoma cell line | CHX | SRA246366 | (78) |
CHX: Cycloheximide.
Identifying transcribed lincRNAs
LincRNAs with expression level higher than certain threshold were considered to be well-transcribed. To determine the threshold, a half-Gaussian distribution of log2(FPKM) values for each RNA-seq data was fitted by kernel density estimation using kde function in R. The half-Gaussian was then mirrored to a full Gaussian distribution. Three standard deviation below the mean of the distribution was defined as the minimum value of the expression, described by Hart et al. (31). To obtain a reliable list of transcribed lincRNAs for each cell line, only those above the threshold in replicated experiments were considered for further analysis.
Localization analysis
Nuclear and cytoplasmic RNA-seq data for the human cervical cancer cell line (HeLa) and Human lymphoblastoid cell line (LCL) were obtained from the ENCODE (29). The raw data were pre-processed with the same procedures as above. Nuclear-cytoplasmic FPKM ratios were then calculated for each lincRNA. Based on the nuclear-cytoplasmic ratio, lincRNAs with well-transcribed both in nuclear and cytoplasmic fractions were further divided into nuclear (with ratio >2) and cytoplasmic lincRNAs (with ratio <0.5).
Calculating RNA folding free-energy
The free-energy of secondary structure formation for a given RNA sequence was calculated by RNAfold (v1.8.5), which uses the nearest-neighbor thermodynamic model to predict the minimum free-energy of RNA sequences (32).
Conservation analysis
PhyloP base-wise conservation score based on Multiz alignments of 100 vertebrate species was retrieved from the UCSC Genome Browser (33). The fractional base-wise conservation metric, measured by the fraction of significantly conserved bases (phyloP, P < 0.01), was used to nominate evolutionary conservation of each lincRNA transcript.
PolyA feature analysis
The polyA site and polyA signal manually annotated by HAVANA were obtained from GENCODE (v23). Only those lincRNAs with sequence elements of polyadenylation including the polyA site and polyA signal were considered to have polyA feature.
Transposable element analysis
Transposable elements (TEs) in the human genome sequences were detected by RepeatMasker (v4.0.6) with default parameters, ‘-species human’ flag and the RepeatMasker libraries version 20150807 (http://www.girinst.org/server/RepBase/). The fraction of repetitive elements in each lincRNA was determined based on the RepeatMasker outputs.
Identifying actively translated sORFs
The actively translated sORFs were determined by RibORF (34), which combines alignment of ribosomal A-sites, three-nucleotide periodicity and uniformity across codons. Only those sORFs longer than 6 aa with a start codon followed by an in-frame stop codon within the lincRNA transcripts, high percentage of maximum entropy (PME > 0.6) and predicted probability > 0.5, were defined as actively translated regions.
Mass spectrometry data analysis
A large-scale proteome data through SWATH MS-based experiments using pan-human library was obtained from the PRIDE database with accession number PXD000953 (35). All MS data were analyzed using Mascot (v2.3.0) against a custom-made database, combining sequences from UniProt with sequences derived from lincRNA transcripts, using carbamidomethyl as a fixed modification, oxidation as a variable modification, mass tolerance of 50 ppm (precursor ion) and 0.1 Da (fragment ion). After peptide searching, peptide hits were filtered at the 1% false discovery rate (FDR) level using the target-decoy strategy.
Cell-type specificity analysis
For each lincRNA, its transcriptional or translational specificity was determined byτindex (36) as follows:
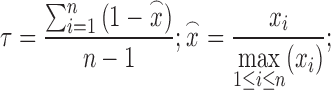 |
(1) |
where n represents the number of cell lines and xi represents the FPKM value of the lincRNA in the ith cell line. It varies on a scale from 0 to 1, with 0 indicating to be ubiquitous and 1 indicating to be specific.
Differential translation analysis
The DESeq2 package (37) was used to detect lincRNAs with changes in the translational and transcriptional levels between different cell lines. Only those lincRNAs with at least two-fold change and P-value < 0.05 after Benjamini–Hochberg correction for multiple testing were determined to be significantly differentiated. To determine the relationship of translational and transcriptional regulation, we adopted a strategy previously used (38), where differential lincRNAs were classified into three categories: (i) lincRNAribo_unique, defined as those exhibiting significant differences in translational but not transcriptional level; (ii) lincRNAboth, defined as those exhibiting significant differences in both transcriptional and translational levels; and (iii) lincRNARNA_unique, defined as those exhibiting significant differences in transcriptional but not translational level.
Inferring putative biological functions of lincRNAs
For each lincRNA, correlation of expression between the lincRNA and protein-coding genes across all samples were measured using Pearson's correlation coefficient. Significant correlation of lincRNA and protein-coding genes were determined for pairs having a P-value below 0.05 after Benjamini–Hochberg correction for multiple testing. All 825 gene ontology (GO) sets, retrieved from the C5_BP collection of Molecular Signatures Database (MSigDB, v5.1) (39), were tested for over-representation of its co-expressed protein-coding genes. Only those GO terms with adjusted P-value by FDR below 0.05 were determined to be statistically significant.
Calculating translational efficiency
Translational efficiency for each lincRNA was calculated as the ratio of normalized read density (FPKM) of ribosome profiling over normalized read density (FPKM) of RNA-seq (14).
Determining degree of overlapping
The degree of overlap between lincRNAs was measured based on the Jaccard coefficient (JC) between sets of enriched GO terms, as follows:
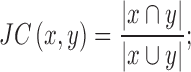 |
(2) |
where x and y represent two different sets of GO terms. It varies on a scale from 0 to 1, with 0 indicating no overlap and 1 indicating complete overlap.
Measuring functional similarity
The functional similarity between lincRNAs was measured based on the semantic similarity between sets of enriched GO terms. For any given pair of lincRNAs, their functional similarity was calculated using the bioconductor package, GOSemSim, with Wang's method (40).
RESULTS
Identification of lincRNAs with ribosome occupancy by ribosome profiling
To systematically characterize lincRNAs with ribosome occupancy, we retrieved all the human ribosome profiling data from the SRA database (30). We adopted four filtering criteria to select datasets, where only those meeting all of the following criteria were considered for further analysis: (i) they included parallel RNA-seq and ribosome profiling measurements; (ii) they were vehicle-treated or served as controls in the experiments; (iii) these experiments had biological replicates with high degree of reproducibility, where correlation coefficient of replicates is at least 0.9 (Supplementary Figure S1); (iv) the peak of the footprint size distribution of ribosome profiling reads ranged between 29 and 32 bp (Supplementary Figure S2). Finally, eight different human cell lines were included in this study, including three cancer cells (HeLa, PC3 and U2OS), one cancer-stem like cell (HEK293T), one assimilated cell (LCL) and three primary/embryonic cells (BJ, hES and RPE) (Table 1).
A consensus analysis workflow was applied to all samples. LincRNAs with low expression levels were excluded from subsequent analysis (see ‘Materials and Methods’ section for details). Since previous studies have shown that some sequencing reads from ribosome profiling experiments could originate from aspecific ribosome binding (17,41). Therefore, only those lincRNAs being actively translated by the ribosomes are defined as ribo-lincRNAs, where ribosome occupancy must (i) show three-nucleotide periodicity, and (ii) be relatively evenly distributed, measured by PME. The PME value that reflects the degree of localization of sequence reads was used to further distinguish true protected RNA regions by the ribosomes from aspecific ribosome binding, as suggested by a recent publication (42). After filtration, a total of 4709 annotated lincRNAs were transcribed detectably in at least one cell line, of which 1332 showed ribosome occupancy (Supplementary Table S1). Separately for each cell line, the number of ribo-lincRNAs ranged from 113 (BJ) to 561 (U2OS), although the majority of well-transcribed lincRNAs did not show ribosome occupancy (Figure 1A). Notably, some well-characterized lincRNAs such as ‘MALAT1’, ‘NEAT1’ and ‘PVT1’ showed obvious occupancy in Ribo-seq data from multiple cell lines.
Figure 1.

General characteristics of lincRNAs and protein-coding genes. (A) Percentage of lincRNAs occupied by ribosomes in each cell line. The number of ribo-lincRNAs for each cell line is given at the top of each bar. (B) Density plots of the expression levels; (C) Box-and-whisker plots of transcript lengths; (D) Distributions of exon numbers; (E) Cumulative distribution plots of minimum free energy; (F) Violin plots of the base-wise conservation fraction; (G) Distributions of TE-derived sequences in exons; (H) Distributions of GC-content. Error bars represent interquartile range; (I) Fractions of transcripts containing polyA features, shown here for a representative cell line (HeLa). To avoid duplication of presentation, other cell lines are shown in Supplementary Figures S3–8 and Tables S2–8.
LincRNAs with ribosome occupancy exhibit relatively high expression and apparent cytoplasmic localization
We compared the expression patterns of ribo-lincRNAs, nonribo-lincRNAs and protein-coding genes. In agreement with previous findings (43,44), lincRNAs were generally expressed at lower levels than protein-coding genes (Figure 1B). However, ribo-lincRNAs showed significantly higher expression than nonribo-lincRNAs, with at least 1.75-fold increase in the median expression levels (Mann–Whitney U test, P-value < 2.2e-16). Interestingly, many ribo-lincRNAs seemed to have a tendency to resemble protein-coding genes regarding expression pattern. The same trends were observed in all the eight cell lines (Supplementary Figure S3 and Table S2), which is highly unlikely to happen by random chance (Binomial test, P-value < 1.0e-22), indicating that it is a general property of ribo-lincRNAs having higher expression levels than nonribo-lincRNAs. Notably, distinct expression patterns between ribo-lincRNAs and nonribo-lincRNAs may reflect differences in spatial and temporal regulation paradigms.
Based on the nuclear and cytoplasmic RNA-seq data from the ENCODE, we further examined the subcellular localization of these two classes of lincRNAs. Among the subset of lincRNAs for which subcellular localization could be determined in the HeLa cell line, ribo-lincRNAs were significantly enriched in the cytoplasm (Fisher's exact test, P-value = 9.7e-05). Although nonribo-lincRNAs were not enriched in the nuclear or cytoplasm, they showed significantly higher nuclear-cytoplasmic ratios compared to ribo-lincRNAs (Mann–Whitney U test, P-value = 0.0032), suggesting that nonribo-lincRNAs tend to localize in the nucleus. The similar results were observed for the LCL cell line, with a significant enrichment in the cytoplasm for ribo-lincRNAs (Fisher's exact test, P-value = 7.4e-05) and a tendency toward the nuclear localization for nonribo-lincRNAs (Mann–Whitney U test, P-value = 0.0003; Supplementary Figure S4). These results provided further evidence for accessibility of ribo-lincRNAs to the translation machinery.
LincRNAs with ribosome occupancy exhibit increasing structural complexity
We next investigated whether there are inherent differences between ribo-lincRNAs and nonribo-lincRNAs in several genomic structural features. In contrast to protein-coding genes, lincRNAs generally showed shorter transcript length and fewer exons (Figure 1C). Compared to nonribo-lincRNAs, ribo-lincRNAs had significantly longer transcript length, with at least 1.55-fold increase in the median length (Mann–Whitney U test, P-value < 1.2e-03). In addition, as shown in Figure 1D, ribo-lincRNAs had significantly more exons, with at least 1.50-fold increase in the median number of exons (Mann–Whitney U test, P-value < 2.6e-09). Remarkably, the significance of structural differences was observed in all the eight cell lines (Supplementary Figure S5 and Table S3). Overall, these results suggested that lincRNAs with ribosome occupancy have potentials to fold into complex shapes and may provide greater versatility in target recognition. As expected, ribo-lincRNAs were further observed to have significantly lower minimum folding free energy than nonribo-lincRNAs in at least seven out of eight cell lines (Kolmogorov–Smirnov test, P-value < 0.05; Supplementary Figure S6 and Table S4), indicated by the left shifts in cumulative distributions (Figure 1E). This result demonstrated that lincRNAs with ribosome occupancy are likely to be more structured.
LincRNAs with ribosome occupancy exhibit elevated evolutionary conservation
Evolutionary feature has been widely used as an indicator of the functional significance. LincRNAs with important molecular functions are likely subject to selective pressure (17). We therefore examined the extent of evolutionary conservation of ribo-lincRNAs and nonribo-lincRNAs, based on base-wise conservation scores across 100 vertebrates calculated by PhyloP (33). As shown in Figure 1F, we observed that lincRNAs were generally less conserved than protein-coding genes in all the eight cell lines (Mann–Whitney U test, P-value < 2.2e-16), consistent with previous findings (44,45). However, in contrast to nonribo-lincRNAs, ribo-lincRNAs showed higher levels of evolutionary conservation. Statistically significant differences were observed in six out of eight cell lines (Mann–Whitney U test, P-value < 0.05; Supplementary Figure S7 and Table S5), which is unlikely to occur by random chance (Binomial test, P-value < 0.05). Higher evolutionary conservation of ribo-lincRNAs may imply their relevance to biological functions that are providing high structural constraints under natural selection pressure. This relevance was further evidenced by TEs that are a major factor driving lincRNA evolution and biological function (46–48), as shown in the next section.
LincRNAs with ribosome occupancy exhibit discernible genomic features
We next characterized the TE composition and GC-content of lincRNAs. As shown in Figure 1G, we observed significant depletion of TE-derived sequences in ribo-lincRNAs compared to nonribo-lincRNAs (Mann–Whitney U test, P-value < 2.0e-03). Interestingly, Alu elements were also significantly depleted from ribo-lincRNAs in all the eight cell lines (Mann–Whitney U test, P-value < 0.05; Supplementary Figure S8A and Table S6). Given that protein-coding genes were severely depleted for TEs (48), this observation provided evidence that some lincRNAs may function through encoded products. Also, ribo-lincRNAs generally exhibited higher GC-content than nonribo-lincRNAs (Mann–Whitney U test, P-value < 0.05; Figure 1H), except for the BJ and RPE, with marginal significance for the hES and PC3, although lincRNAs typically had lower GC-content than protein-coding genes (Mann–Whitney U test, P-value < 3.5e-11; Supplementary Figure S8B and Table S7). This explained the rationale behind the consensus lower minimum folding free energy of ribo-lincRNAs, considering that RNA sequences with high GC-content often fold into low-energy structures. Beyond these characteristics, the factor influencing ribosome engagement, such as 3΄ polyadenylation, was further examined. Taking advantage of poly(A) features manually annotated by HAVANA, we observed that lincRNA, especially for ribo-lincRNAs, were significantly less polyadenylated than protein-coding genes (Figure 1I). Interestingly, polyadenylation could also distinguish ribo-lincRNAs from nonribo-lincRNAs, with statistically significant differences in at least six out of of eight cell lines (Fisher's exact test, P-value < 0.05; Supplementary Figure S8C and Table S8). Taken together, these analyses showed disparity between these two classes of lincRNAs, demonstrating the resolving power of the genomic features to lincRNAs with and without ribosome occupancy.
Coding potential of lincRNAs with ribosome occupancy
We next asked whether ribo-lincRNAs have the potential to encode peptides. To this end, we first examined possible products of ribo-lincRNAs in all three frames and scanned each of them against databases of UniProt protein sequences and Pfam protein domains using blastp (49) and hmmscan (50). In total, 334 ribo-lincRNAs were found to contain regions with homology to known proteins or domains, showing evidence of protein-coding potential. Given the recent evidence emerging on functional peptides derived from sORFs, we then systematically searched translatable sORFs in ribo-lincRNAs using RibORF (34). Applying rigorous filtering criteria (see ‘Materials and Methods’ section for details), we identified translatable sORFs for 233 out of 1332 ribo-lincRNAs (Supplementary Table S9). Notably, although sORFs were presented in only a subset of ribo-lincRNAs, many of these had multiple footprints found in multiple cell lines (Figure 2A), indicating active translation and putatively functional significance.
Figure 2.

Peptide-coding potential assessment for sORFs in lincRNAs. (A) An example of ribosome footprint profiles on the SNHG8 transcript. The exon structure is shown with gray rectangles on the right side of the arrow. sORFs are shown with rectangles in dark red and initiation sites are indicated by thin lines. (B) Evolutionary conservation of ribo-lincRNAs containing-sORFs with peptide evidence and those without peptide evidence. (C) Expression behaviors of lincRNAs with sORFs and short protein-coding genes (<100 aa), shown here for a representative cell line (HeLa). Other cell lines are shown in Supplementary Figure S9.
Next we further sought to determine whether the peptide products emanating from lincRNAs could be detected by mass spectrometry. Integrative analysis with human proteomic data from the PRIDE database revealed peptide evidence for 18 out of those ribo-lincRNAs containing sORFs (Supplementary Table S10). Notably, although several lincRNAs such as ‘ENST00000445430’, ‘ENST00000626089’ and ‘ENST00000628917’ showed peptide evidence in multiple cell lines, it was difficult to distinguish them to pseudogene ‘SDHAP2’ as well as protein-coding gene ‘SDHA’ due to sharing the same peptides together into one protein group. Here we further explored several possibilities that may explain this low validation by mass spectrometry. First, ribo-lincRNAs with peptide evidence were found to have stronger conservation than those without peptide evidence (Student's t-test, P-value = 0.0155; Figure 2B). Given that higher levels of sequence conservation generally lead to stabilization of proteins (51), this finding suggested that the majority of ribo-lincRNAs having lower conservation are less likely to produce stable peptides. Second, ribo-lincRNAs with sORFs were generally expressed at much lower levels than short protein-coding genes (<100 aa) in at least seven out of the eight cell lines (Figure 2C and Supplementary Figure S9). This finding suggested that even though ribo-lincRNAs have the ability to generate peptides, peptide products will escape detection due to their low abundance. Taken together, these results provided further evidence that lincRNAs with ribosome occupancy can encode peptides, although not all translation events will produce stable peptides.
Characterization of cell-type specific translation pattern
Several previous studies have shown that lincRNAs exhibit notably higher degree of cell-type specificity than protein coding genes at the transcriptional level (28,29). We next asked to what extent lincRNAs show cell-type specificity at the translational level. We also assessed whether the degree of specificity differs between the translational and transcriptional levels.
For each cell line, on average, 21% of well-transcribed lincRNAs exhibited ribosome occupancy, with a minimum of 9% for the BJ and a maximum of 31% for the U2OS. However, among those ribo-lincRNAs, only 1.42% (19 lincRNAs) were commonly found in all the eight cell lines, in contrast to more than 53% (710 lincRNAs) were uniquely found in only one cell line (Figure 3A). Of these ribo-lincRNAs showing unique occupancy in a single cell line, nearly two-thirds were well expressed in multiple cell lines (Supplementary Figure S10). This is more likely to reflect a potential pattern of ribosome-mediated translational specificity rather than an aftereffect of transcriptional specificity. We further determined their cell-type specificity by usingτindex (36), and found that ribo-lincRNAs had significantly higher cell-type specificity at the translational level than the transcriptional level (Mann–Whitney U test, P-value < 2.2e-16; Figure 3B). Because these data from eight cell lines were retrieved from different studies, we asked whether the cell-type specificity was confounded by possible technical batch effects (52). We considered different studies as a surrogate and used F-statistic to test association by stratifying measurements by the surrogate. Under 5% of FDR control level, no lincRNA was found with susceptibility to the technical batch effects, confirming our findings.
Figure 3.

Translation pattern of lincRNAs in human cell lines. (A) Overlap of the number of ribo-lincRNAs in different cell lines. (B) Cell-type specificity of ribo-lincRNAs at the translational and transcriptional levels, measured by usingτindex. (C) Overall translational efficiencies of ribo-lincRNAs in different cell lines. (D) Translational efficiencies of overlapping ribo-lincRNAs (19) in each cell line. The eight cell lines are divided into two types: cancerous and non-cancerous, as shown by the dotted line.
We next examined the translational efficiency of well transcribed-lincRNAs in each cell line. As shown in Figure 3C, the majority (>60%) of lincRNAs exhibited relatively low levels of translational efficiency (<1) and different cell types exhibited distinct translational efficiency, showing striking differences in the distributions of translational efficiency among all pairwise comparisons between the eight cell lines (Kolmogorov–Smirnov test, P-value < 7.5e-03). Even if only focusing on those overlapping ribo-lincRNAs among the eight cell lines, we still observed inter-cell type differences in the translational efficiency (Figure 3D), indicating that their relative contribution toward each cell line are obviously different. Distinct translational efficiency may also reflect differences in flexibility of translation versus transcription in modulating activity of lincRNAs.
Cell-type specific translation regulation
To understand differences in lincRNA translation between different cell types, we determined the significant changes in lincRNAs translation between all pairwise comparisons of the eight cell lines with the DESeq2 package (37). Differential lincRNAs were observed among different cell types (Figure 4A). The differences between cancerous cell lines were larger than those between cancerous and non-cancerous cell lines, and more larger than those between non-cancerous cell lines (average number of differential lincRNAs of 219, 169 and 113, respectively) (Figure 4B). Here HEK293T was classified as a cancerous cell line, considering that it exhibits cancer stem cell-like features (53). In particular, we identified 527 ribo-lincRNAs, representing 40% of all the ribo-lincRNAs, showing significant translational changes in any cell line when compared to either of other cell lines. The ribo-lincRNAs with significant translational changes had significantly higher translational efficiency than those without significant translational changes (Mann–Whitney U test, P-value < 0.05 except for the BJ; Figure 4C). These differential ribo-lincRNAs with greater translational efficiencies suggested increased flexibility of the spectrum of control of translation. These results demonstrated that the widespread differential translation of lincRNAs exists among different cell types, reinforcing inter-cell type differences.
Figure 4.

Scope and characteristics of lincRNAs translation in different cell lines. (A) Overlap of differential translation lincRNAs between different cell lines. Notably, 2-fold change and FDR < 0.05 are used as the determination of differential translation. (B) Comparison of average number of differential translation lincRNAs in different cell types. Blue and red colors represent cancerous and non-cancerous cell lines, respectively. (C) Translational efficiencies of differential and non-differential translation ribo-lincRNAs in each cell line. (D) Scatter plot showing the differences in regulation levels between cancerous and non-cancerous cell lines for different categories of lincRNAs. Red, yellow and blue slopes demonstrate translational regulation for lincRNAribo_unique, lincRNAboth and lincRNARNA_unique, respectively.
In addition, we performed the same differential analysis between cancerous and non-cancerous cell lines, and detected hundreds of significant differential lincRNAs, including 157 lincRNAribo_unique, 141 lincRNAboth and 478 lincRNARNA_unique (see ‘Materials and Methods’ section for details). We then quantified the global contribution of translational regulation to differences in lincRNA usage by calculating the slope between Ribo-seq and RNA-seq fold changes (38). As shown in Figure 4D, co-occurrence of translational and transcriptional regulation was prevalent in lincRNAboth, with approximately equal magnitude and the same direction (slope = 0.91), suggesting the coordination of transcription and translation. Different from the coordination pattern in lincRNAribo_unique, lincRNAboth and lincRNARNA_unique showed discordant patterns separately with enhanced translational regulation and transcriptional regulation. Moreover, lincRNAribo_unique had a significantly higher slope than lincRNAboth (Likelihood ratio test, P-value < 3.1e-10) and lincRNARNA_unique (Likelihood ratio test, P-value < 2.2e-16). Overall, these results demonstrated that translational control is a distinct regulatory system, uncoupled from transcription, shaping the translational landscape.
Functional divergence of lincRNAs across different cell types
To gain further insights into the biological roles of lincRNAs, we computationally inferred their functions by commonly used guilt-by-association analysis, wherein the potential functions of lincRNAs could be predicted from the known protein-coding genes to which they are significantly correlated in expression (FDR < 0.05). Out of 4709 well-expressed lincRNAs, 944 (20.05%) were assigned biological functions (FDR < 0.05), including 254 ribo-lincRNAs and 690 nonribo-lincRNAs. Among those enriched GO terms, more than 30% (54) were associated with <6 lincRNAs (Figure 5A). In contrast, more than 29% (55) were associated with more than 100 lincRNAs and those GO terms primarily participated in some fundamental biological functions (Figure 5B), including those involved in cellular metabolism, gene regulation, response to stimulus and signal transduction. Interestingly, on average, 18% ribo-lincRNAs in each cell line had functional annotations, as opposed to only 12% for nonribo-lincRNAs, indicating that lincRNAs with ribosome occupancy tend to be functional. However, further analysis revealed generally higher levels of functional overlap among nonribo-lincRNAs than among ribo-lincRNAs (mean JC of 0.80 versus 0.63), suggesting functional convergence among nonribo-lincRNAs (Figure 5C). The GO-based semantic similarity analysis showed that five out of eight cell lines had similar degrees of functional overlap between ribo-lincRNAs and nonribo-lincRNAs, while BJ showed relatively lower degree of functional overlap and HEK293T and LCL had higher degrees (Figure 5D). Moreover, different cell types also exhibited functional divergence in lincRNAs with ribosome occupancy. The functional overlap between cancerous cell lines were higher than those between cancerous and non-cancerous cell lines, and more higher than those between non-cancerous cell lines (mean JC of 0.70, 0.63 and 0.50, respectively). Taken together, these results indicated the existence of functional differences not only between lincRNAs with and without ribosome occupancy but also between different cell types.
Figure 5.

Characterization and functional features of lincRNAs. (A) Distribution of occurrences of the significant GO terms assigned to the well-expressed lincRNAs. (B) Major functional categories associated with multiple lincRNAs (more than 100). Complete list of functional categories is shown in Supplementary Table S11. (C) The functional overlap maps for ribo-lincRNAs and nonribo-lincRNAs. Red, blue and white colors represent the overlap of GO terms among ribo-lincRNAs, among nonribo-lincRNAs and between ribo-lincRNAs and nonribo-lincRNAs, respectively. (D) Functional similarity between ribo-lincRNAs and nonribo-lincRNAs in each cell line, measured by using GO semantic similarity.
DISCUSSION
Ribosome profiling allows direct measurements of the RNA fragments protected by ribosomes and quantitative readouts of the regions being translated (12,13). The discovery of non-canonical translation events on lincRNAs by ribosome profiling suggests that different lincRNAs may employ radically different mechanisms of action (56). In this study, our integrative analysis of ribosome profiling, RNA-seq and mass spectrometry data reveals distinctive properties of lincRNAs with and without ribosome occupancy regarding their expression, structure, sequence, evolutionary and functional features. Further comparative analysis of lincRNAs with ribosome occupancy in different cell types reveals that they exhibit a high degree of cell-type specificity with differential translational repertoires. To our knowledge, this is the first attempt to characterize global and cell-type specific properties of translational landscape of lincRNAs in human cells.
Compared to those without ribosome occupancy, lincRNAs with ribosome occupancy generally tend to be expressed at higher levels, to have multi-exonic structures and to exhibit stronger sequence conservation. Increasing structural complexity may not only enhance the capacity for adaptability and versatility but also enhance their stability, as further demonstrated by relatively low folding free energy and high GC-content. Preferential cytoplasmic localization leads to increased availability of the translation machinery. Meanwhile, the question of which of the potential lincRNAs are actually translated, to some extent, can be reduced to the question of the extent to which cytoplasmic lincRNAs are translated. Elevated evolutionary conservation may further endow them with functional constraints. A depletion of TE content in lincRNAs with ribosome occupancy is also consistent with constraint in the evolution. All such properties have begun to shed light on which lincRNAs are translated.
It should be noted that nonribo-lincRNAs had higher levels of polyadenylation than ribo-lincRNAs, which is somewhat surprising. Several previous studies have also shown that a significant fraction of the nucleus-localized lncRNAs are stable transcripts and are spliced and polyadenylated (44,57,55). Consistent with this, we also observe that polyadenylated lincRNAs exhibited higher stability than those non-polyadenylated lincRNAs by analyzing minimal free energy (Mann–Whitney U test, all P-values < 0.05 for all the eight cell lines). Thus, one possible reason that many nonribo-lincRNAs are polyadenylated is that they are likely to be stable and passively retained in the nucleus, which prevents them from accessing the cytoplasmic translation machinery.
Although more ribo-lincRNAs resemble protein-coding genes than nonribo-lincRNAs in several features, they are clearly different from protein-coding genes, hinting that they may act as intermediary entities between canonical coding and bona fide non-coding. This possibility can be supported by several lines of evidence: (i) lincRNAs can be translated but in a non-canonical mode (17–20); (ii) the encoded products are generally unstable and rapidly degraded (34,58); and (iii) at least some products encoded by sORFs, if not all, can exist in stable functional micro-peptides (21,22). It has also been proposed that these lincRNAs may act as bifunctional RNAs that are generally non-coding, but under specific circumstances, enclosed sORFs can be translated (54,59–61). Therefore, even though some lincRNAs appear translated in ribosome profiling data, they are usually largely invisible in mass spectrometry (18,20,62–65). Nevertheless, the discovery of sORFs and their encoded micropeptides has made a significant step toward our understanding of lincRNA genes. The discovery of functionally verified micropeptides such as ‘MRLN’ (21) and ‘DWORF’ (22) further emphasizes the functional potential of lincRNA translation.
Many lincRNAs, unlike canonical protein coding-genes, do not have a predominant ORF that is translated instead they often contain multiple sORFs with more dispersed translation (15). A finding common to many recent ribosome profiling studies is the widespread use of non-canonical initiation sites, such as non-AUG start codons (66,67). For some other initiation events of lincRNAs, the apparent elevation of non-canonical translation, under various extracellular cues like stress stimuli, will advance our understanding. Moreover, micro-peptides encoded by sORFs are often deemed to lack an N-terminal signal sequence and released into the cytoplasm immediately after translation. They may exert distinct molecular functions from those of the secreted small peptides that are translated as large precursors with signal sequences at the N-terminus and then translocated into the secretory machinery, where they undergo extensive modification or processing, eventually becoming bioactive peptides (68,69).
To date, the translatome of lincRNAs in various cell types remains poorly understood. Our comparative analysis reveals that the translated lincRNAs have higher cell-type specificity at the translational level than the transcriptional level. Different cell types possess different lincRNA profiles and exhibit different fractions of translated lincRNAs. In particular, cancerous cell lines tend to have a higher translated fraction than non-cancerous cell lines (mean fraction of 23 versus 18%). Cancerous cell lines also tend to have relatively higher median translational efficiency than non-cancerous cell lines (see Figure 3D). Remarkably, the observation of discordance in the translational efficiency between different cell types suggests the existence of extensive cellular controls, such as post-transcriptional and translational regulations. Indeed, extensive differential translated lincRNAs are observed among different cell types. In particular, the translational differences between cancerous cell lines are more severe than those between non-cancerous cell lines (see Figure 4B). All such disparities in the translational repertoire are more likely to reflect the inherent biological differences between different cell types. Cell type-specific properties of lincRNAs may be used for both potential biomarkers and therapeutic targets (5). An appreciation of the roles of translated lincRNAs will offer new avenues of research into translational regulatory mechanisms and development of therapeutic interventions by either mimicking their functions or inhibiting their activities.
Functional analysis further provides a glimpse into the potential functions of lincRNAs with ribosome occupancy. They are found to participate in diverse biological processes, ranging from cellular metabolism to cellular signalling. Different cell types exhibit functional divergence. LincRNAs with ribosome occupancy from cancerous cell lines generally share more functions than those from non-cancerous cell lines (mean JC of 0.70 versus 0.50). Although our computational analysis based on ribosome profiling may provide important hints into functionality of translation in lincRNAs, it is still unclear to what extent these lincRNAs are of functional importance. Additionally, it should be noted that despite the guilt-by-association strategy is frequently used for inferring potential functions of lincRNAs (28,43,70), determination of the precise function of lincRNAs and experimental validation still remain challenging.
In conclusion, our results highlight that translation of lincRNAs has clear molecular, evolutionary and functional implications. Also, translated lincRNAs show remarkable cell-type specificity at the translational level with different translational repertoires. This study will facilitate better understanding of lincRNA functions. Future work will be needed to distinguish functional and non-functional peptides encoded by lincRNAs and to determine the precise roles of bioactive peptides originating from lincRNAs.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank all the members of Zhi Xie's lab and the anonymous reviewers to provide helpful suggestions for improving the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31471232]; Science and Technology Planning Projects of Guangdong Province [2014B030301040]; Major Program of Science and Technology of Guangzhou [201607020001]; Joint Research Fund for Overseas Natural Science of China [3030901001222 to Z.X.]; China Postdoctoral Science Foundation [2015M582459, 2015M582460 to H.W.W., S.Q.X.]. Funding for open access charge: National Natural Science Foundation of China [31471232]; Science and Technology Planning Projects of Guangdong Province [2014B030301040]; Joint Research Fund for Overseas Natural Science of China [3030901001222 to Z.X.]; China Post-doctoral Science Foundation [2015M582459, 2015M582460 to H.W.W., S.Q.X.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Geisler S., Coller J.. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013; 14:699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fatica A., Bozzoni I.. Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 2014; 15:7–21. [DOI] [PubMed] [Google Scholar]
- 3.Mercer T.R., Mattick J.S.. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013; 20:300–307. [DOI] [PubMed] [Google Scholar]
- 4.Huarte M. The emerging role of lncRNAs in cancer. Nat. Med. 2015; 21:1253–1261. [DOI] [PubMed] [Google Scholar]
- 5.Sahu A., Singhal U., Chinnaiyan A.M.. Long noncoding RNAs in cancer: from function to translation. Trends Cancer. 2015; 1:93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li F., Wen X., Zhang H., Fan X.. Novel insights into the role of long noncoding RNA in ocular diseases. Int. J. Mol. Sci. 2016; 17:478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K.C., Chang H.Y.. Molecular mechanisms of long noncoding RNAs. Mol. Cell. 2011; 43:904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu C., Spitale R.C., Chang H.Y.. Technologies to probe functions and mechanisms of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015; 22:29–35. [DOI] [PubMed] [Google Scholar]
- 9.Chu C., Qu K., Zhong F.L., Artandi S.E., Chang H.Y.. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell. 2011; 44:667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simon M.D., Wang C.I., Kharchenko P.V., West J.A., Chapman B.A., Alekseyenko A.A., Borowsky M.L., Kuroda M.I., Kingston R.E.. The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:20497–20502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu P.D., Lander E.S., Zhang F.. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014; 157:1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ingolia N.T. Ribosome profiling: new views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014; 15:205–213. [DOI] [PubMed] [Google Scholar]
- 14.Ingolia N.T., Lareau L.F., Weissman J.S.. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011; 147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chew G.L., Pauli A., Rinn J.L., Regev A., Schier A.F., Valen E.. Ribosome profiling reveals resemblance between long non-coding RNAs and 5΄ leaders of coding RNAs. Development. 2013; 140:2828–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruiz-Orera J., Messeguer X., Subirana J.A., Alba M.M.. Long non-coding RNAs as a source of new peptides. Elife. 2014; 3:e03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingolia N.T., Brar G.A., Stern-Ginossar N., Harris M.S., Talhouarne G.J., Jackson S.E., Wills M.R., Weissman J.S.. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014; 8:1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bazzini A.A., Johnstone T.G., Christiano R., Mackowiak S.D., Obermayer B., Fleming E.S., Vejnar C.E., Lee M.T., Rajewsky N., Walther T.C. et al. . Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014; 33:981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews S.J., Rothnagel J.A.. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014; 15:193–204. [DOI] [PubMed] [Google Scholar]
- 20.Mackowiak S.D., Zauber H., Bielow C., Thiel D., Kutz K., Calviello L., Mastrobuoni G., Rajewsky N., Kempa S., Selbach M.. Extensive identification and analysis of conserved small ORFs in animals. Genome Biol. 2015; 16:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson D.M., Anderson K.M., Chang C.L., Makarewich C.A., Nelson B.R., McAnally J.R., Kasaragod P., Shelton J.M., Liou J., Bassel-Duby R. et al. . A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 2015; 160:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson B.R., Makarewich C.A., Anderson D.M., Winders B.R., Troupes C.D., Wu F., Reese A.L., McAnally J.R., Chen X., Kavalali E.T. et al. . A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 2016; 351:271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie S.Q., Nie P., Wang Y., Wang H., Li H., Yang Z., Liu Y., Ren J., Xie Z.. RPFdb: a database for genome wide information of translated mRNA generated from ribosome profiling. Nucleic Acids Res. 2016; 44:D254–D258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michel A.M., Fox G., A M.K., De Bo C., O'Connor P.B., Heaphy S.M., Mullan J.P., Donohue C.A., Higgins D.G., Baranov P.V.. GWIPS-viz: development of a ribo-seq genome browser. Nucleic Acids Res. 2014; 42:D859–D864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iyer M.K., Niknafs Y.S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T.R., Prensner J.R., Evans J.R., Zhao S. et al. . The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015; 47:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ranzani V., Rossetti G., Panzeri I., Arrigoni A., Bonnal R.J., Curti S., Gruarin P., Provasi E., Sugliano E., Marconi M. et al. . The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat. Immunol. 2015; 16:318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma W., Ay F., Lee C., Gulsoy G., Deng X., Cook S., Hesson J., Cavanaugh C., Ware C.B., Krumm A. et al. . Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes. Nat. Methods. 2014; 12:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P. et al. . Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009; 458:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F. et al. . Landscape of transcription in human cells. Nature. 2012; 489:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leinonen R., Sugawara H., Shumway M.. The sequence read archive. Nucleic Acids Res. 2010; 39:D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hart T., Komori H.K., LaMere S., Podshivalova K., Salomon D.R.. Finding the active genes in deep RNA-seq gene expression studies. BMC Genomics. 2013; 14:778–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clote P., Ferre F., Kranakis E., Krizanc D.. Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA. 2005; 11:578–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A.. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010; 20:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji Z., Song R., Regev A., Struhl K.. Many lncRNAs, 5΄UTRs, and pseudogenes are translated and some are likely to express functional proteins. Elife. 2015; 4:e08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenberger G., Koh C.C., Guo T., Rost H.L., Kouvonen P., Collins B.C., Heusel M., Liu Y., Caron E., Vichalkovski A. et al. . A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data. 2014; 1:140031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yanai I., Benjamin H., Shmoish M., Chalifa-Caspi V., Shklar M., Ophir R., Bar-Even A., Horn-Saban S., Safran M., Domany E. et al. . Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005; 21:650–659. [DOI] [PubMed] [Google Scholar]
- 37.Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schafer S., Adami E., Heinig M., Rodrigues K.E., Kreuchwig F., Silhavy J., van Heesch S., Simaite D., Rajewsky N., Cuppen E. et al. . Translational regulation shapes the molecular landscape of complex disease phenotypes. Nat. Commun. 2015; 6:7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu G., Li F., Qin Y., Bo X., Wu Y., Wang S.. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010; 26:976–978. [DOI] [PubMed] [Google Scholar]
- 41.Guttman M., Russell P., Ingolia N.T., Weissman J.S., Lander E.S.. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013; 154:240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ji Z., Song R., Huang H., Regev A., Struhl K.. Transcriptome-scale RNase-footprinting of RNA-protein complexes. Nat. Biotechnol. 2016; 34:410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cabili M.N., Trapnell C., Goff L., Koziol M., Tazon-Vega B., Regev A., Rinn J.L.. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011; 25:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G. et al. . The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012; 22:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Necsulea A., Soumillon M., Warnefors M., Liechti A., Daish T., Zeller U., Baker J.C., Grutzner F., Kaessmann H.. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014; 505:635–640. [DOI] [PubMed] [Google Scholar]
- 46.Johnson R., Guigo R.. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA. 2014; 20:959–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapusta A., Kronenberg Z., Lynch V.J., Zhuo X., Ramsay L., Bourque G., Yandell M., Feschotte C.. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013; 9:e1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelley D., Rinn J.. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012; 13:R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 50.Eddy S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011; 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sullivan B.J., Nguyen T., Durani V., Mathur D., Rojas S., Thomas M., Syu T., Magliery T.J.. Stabilizing proteins from sequence statistics: the interplay of conservation and correlation in triosephosphate isomerase stability. J. Mol. Biol. 2012; 420:384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leek J.T., Scharpf R.B., Bravo H.C., Simcha D., Langmead B., Johnson W.E., Geman D., Baggerly K., Irizarry R.A.. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010; 11:733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Debeb B.G., Zhang X., Krishnamurthy S., Gao H., Cohen E., Li L., Rodriguez A.A., Landis M.D., Lucci A., Ueno N.T.. Characterizing cancer cells with cancer stem cell-like features in 293T human embryonic kidney cells. Mol. Cancer. 2010; 9:180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ulveling D., Francastel C., Hube F.. When one is better than two: RNA with dual functions. Biochimie. 2010; 93:633–644. [DOI] [PubMed] [Google Scholar]
- 55.Clark M.B., Johnston R.L., Inostroza-Ponta M., Fox A.H., Fortini E., Moscato P., Dinger M.E., Mattick J.S.. Genome-wide analysis of long noncoding RNA stability. Genome Res. 2012; 22:885–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Housman G., Ulitsky I.. Methods for distinguishing between protein-coding and long noncoding RNAs and the elusive biological purpose of translation of long noncoding RNAs. Biochim. Biophys. Acta. 2016; 1859:31–40. [DOI] [PubMed] [Google Scholar]
- 57.Kapranov P., Cheng J., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermuller J., Hofacker I.L. et al. . RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007; 316:1484–1488. [DOI] [PubMed] [Google Scholar]
- 58.Quinn J.J., Chang H.Y.. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2015; 17:47–62. [DOI] [PubMed] [Google Scholar]
- 59.Dinger M.E., Gascoigne D.K., Mattick J.S.. The evolution of RNAs with multiple functions. Biochimie. 2011; 93:2013–2018. [DOI] [PubMed] [Google Scholar]
- 60.Nam J.W., Choi S.W., You B.H.. Incredible RNA: dual functions of coding and noncoding. Mol. Cell. 2016; 39:367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crappé J., Van Criekinge W., Menschaert G.. Little things make big things happen: a summary of micropeptide encoding genes. EuPA Open Proteomics. 2014; 3:128–137. [Google Scholar]
- 62.Banfai B., Jia H., Khatun J., Wood E., Risk B., Gundling W.E. Jr, Kundaje A., Gunawardena H.P., Yu Y., Xie L.. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res. 2012; 22:1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Slavoff S.A., Mitchell A.J., Schwaid A.G., Cabili M.N., Ma J., Levin J.Z., Karger A.D., Budnik B.A., Rinn J.L., Saghatelian A.. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol. 2012; 9:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prabakaran S., Hemberg M., Chauhan R., Winter D., Tweedie-Cullen R.Y., Dittrich C., Hong E., Gunawardena J., Steen H., Kreiman G. et al. . Quantitative profiling of peptides from RNAs classified as noncoding. Nat. Commun. 2014; 5:5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aspden J.L., Eyre-Walker Y.C., Phillips R.J., Amin U., Mumtaz M.A., Brocard M., Couso J.P.. Extensive translation of small open reading frames revealed by poly-ribo-seq. Elife. 2014; 3:e03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mitchell S.F., Parker R.. Modifications on translation initiation. Cell. 2015; 163:796–798. [DOI] [PubMed] [Google Scholar]
- 67.Wang X.Q., Rothnagel J.A.. 5΄-untranslated regions with multiple upstream AUG codons can support low-level translation via leaky scanning and reinitiation. Nucleic Acids Res. 2004; 32:1382–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hashimoto Y., Kondo T., Kageyama Y.. Lilliputians get into the limelight: novel class of small peptide genes in morphogenesis. Dev. Growth Differ. 2008; 50(Suppl. 1):S269–S276. [DOI] [PubMed] [Google Scholar]
- 69.Arnison P.G., Bibb M.J., Bierbaum G., Bowers A.A., Bugni T.S., Bulaj G., Camarero J.A., Campopiano D.J., Challis G.L., Clardy J. et al. . Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2012; 30:108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Khalil A.M., Guttman M., Huarte M., Garber M., Raj A., Rivea Morales D., Thomas K., Presser A., Bernstein B.E., van Oudenaarden A. et al. . Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:11667–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rooijers K., Loayza-Puch F., Nijtmans L.G., Agami R.. Ribosome profiling reveals features of normal and disease-associated mitochondrial translation. Nat. Commun. 2013; 4:2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sidrauski C., McGeachy A.M., Ingolia N.T., Walter P.. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife. 2015; 4:e05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stumpf C.R., Moreno M.V., Olshen A.B., Taylor B.S., Ruggero D.. The translational landscape of the mammalian cell cycle. Mol. Cell. 2013; 52:574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Werner A., Iwasaki S., McGourty C.A., Medina-Ruiz S., Teerikorpi N., Fedrigo I., Ingolia N.T., Rape M.. Cell-fate determination by ubiquitin-dependent regulation of translation. Nature. 2015; 525:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cenik C., Cenik E.S., Byeon G.W., Grubert F., Candille S.I., Spacek D., Alsallakh B., Tilgner H., Araya C.L., Tang H. et al. . Integrative analysis of RNA, translation, and protein levels reveals distinct regulatory variation across humans. Genome Res. 2015; 25:1610–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hsieh A.C., Liu Y., Edlind M.P., Ingolia N.T., Janes M.R., Sher A., Shi E.Y., Stumpf C.R., Christensen C., Bonham M.J. et al. . The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012; 485:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tanenbaum M.E., Stern-Ginossar N., Weissman J.S., Vale R.D.. Regulation of mRNA translation during mitosis. Elife. 2015; 4, doi:10.7554/eLife.07957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Elkon R., Loayza-Puch F., Korkmaz G., Lopes R., van Breugel P.C., Bleijerveld O.B., Altelaar A.M., Wolf E., Lorenzin F., Eilers M. et al. . Myc coordinates transcription and translation to enhance transformation and suppress invasiveness. EMBO Rep. 2015; 16:1723–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.