

Supplemental Digital Content is Available in the Text.
Key Words: daclatasvir, sofosbuvir, HIV/HCV coinfection, real-world data, compassionate use, advanced liver disease
Abstract
Background:
Efficacious, well-tolerated, direct antiviral agents have drastically changed the prognosis of hepatitis C virus (HCV) disease, but real-world data for oral treatments are limited in key populations such as HIV/HCV coinfection with advanced liver disease. Daclatasvir (DCV) efficacy and safety was assessed in the French “Autorisation Temporaire d'Utilisation” (ATU) program, providing DCV ahead of market authorization to patients with advanced HCV disease without other treatment options.
Methods:
This was a subanalysis of HIV/HCV coinfected ATU patients treated with DCV plus sofosbuvir (SOF). Recommended duration was 24 weeks; addition of ribavirin (RBV) and/or shorter treatment was at the physician's discretion. The primary efficacy analysis was sustained virologic response at posttreatment week 12 (SVR12; modified intention-to-treat). Safety was assessed by spontaneous adverse event reporting.
Results:
The efficacy population (N = 407) was mostly cirrhotic (72%, of whom 18% were decompensated), HCV treatment–experienced (82%), and infected with genotypes 1 (69%), 3 (12%), or 4 (19%). Median CD4 was 555 cells/mm3; 95% had HIV RNA <50 copies/mL. Most (74%) were treated for 24 weeks; 14% received RBV. SVR12 was 92% overall (95% confidence interval: 88.6% to 94.0%); 90% (86.4% to 93.2%) in patients with cirrhosis; 95% (88.9% to 97.5%) in patients without cirrhosis. SVR12 was consistent across HCV genotypes and antiretroviral regimens. Among 617 patients with safety data, 7 discontinued for an adverse event and 10 died.
Conclusions:
DCV+SOF±RBV achieved high SVR12 and was well tolerated in this large real-world cohort of HIV/HCV coinfected patients with advanced liver disease.
INTRODUCTION
The risk of hepatitis C virus (HCV) infection is estimated to be 6 times higher for HIV-positive individuals than for the HIV-negative population,1 and although the prevalence of HIV/HCV coinfection varies widely by geography and demography,1,2 it is consistently high among people who inject drugs (PWID).1 Thus, HCV coinfection is common among HIV-infected individuals—particularly where injection drug use contributes significantly to HIV epidemiology—with typical HCV prevalence estimates of ∼16% observed in HIV-infected cohorts from France3 and the United States.2
HIV infection accelerates HCV-associated liver fibrosis, most notably in those with more advanced immunodeficiency, resulting in high rates of end-stage liver disease and shorter survival after hepatic decompensation events.4–6 Despite significantly improved life expectancy in HIV infection, liver disease remains a major non-AIDS cause of mortality among coinfected patients.7 Effective treatment of HCV in HIV/HCV coinfection is therefore a public health priority, particularly for those with more advanced HCV or HIV disease.
Historical uptake of HCV treatment based on pegylated interferon (pegIFN) and ribavirin (RBV) was low among coinfected patients,8,9 due to poor efficacy and tolerability10–12 and a high frequency of adherence-limiting comorbidities in this population. The development of pegIFN-free oral regimens of direct-acting antivirals (DAAs) greatly improved the efficacy and tolerability of HCV treatment in coinfection.13–17 However, data from clinical DAA studies are of limited generalizability to the broader coinfected population. Treatment-limiting pharmacokinetic interactions with DAAs remain a significant issue with some types of combination antiretroviral (ARV) therapy (cART),18,19 as does the risk of interaction between some DAAs and oral opioids in PWID on drug substitution treatment.20 Switching cART regimens to avoid DAA–ARV interactions may be possible, but risks loss of HIV control, especially in those with previous ARV experience.21 The complex medical needs and lifestyles of many HIV/HCV coinfected patients typically result in their exclusion from clinical efficacy studies, which, together with restrictions on permitted ARVs, has resulted in highly stratified recruitment estimated to exclude 60%–94% of the real-world coinfected population.22
Daclatasvir (DCV), a pan-genotypic inhibitor of HCV NS5A,23 and sofosbuvir (SOF),24 a pan-genotypic inhibitor of NS5B, both have limited ARV drug interactions, usually manageable by straightforward dose adjustments for DCV.25 In the phase 3 ALLY-2 study, which had the broadest inclusion criteria among recent DAA coinfection studies,22 DCV+SOF showed high efficacy (97% sustained virologic response) and good tolerability in patients receiving a wide range of cART regimens.13 Real-world cohorts can enhance clinical study data with findings from much broader patient sets, including those ineligible for clinical studies. Early access programs, which provide promising new drugs ahead of their market authorization to patients with urgent need, are a potentially rich source of such data. More than 7000 patients were referred under early access initiatives for DCV,26 with the largest being the French “Autorisation Temporaire d'Utilisation” (ATU) program that treated ∼4000 HCV-infected patients with advanced liver disease with DCV+SOF, with or without RBV. We present herein an analysis of DCV+SOF±RBV efficacy and safety in HIV/HCV coinfected ATU patients with severe liver disease.
METHODS
Patients
ATU program patients coinfected with HIV-1 and HCV were included. Eligible patients for the ATU were adults with chronic HCV infection, no alternative treatment options, and an indication for treatment due to advanced liver disease (physician-assessed F3 or F4 fibrosis and/or severe extrahepatic HCV manifestations), HCV recurrence after liver transplant, and/or an indication for liver or kidney transplant.
Determination of Fibrosis and Cirrhosis
Cirrhosis status was determined through a hierarchical algorithm (Supplemental Digital Content, Table 1, http://links.lww.com/QAI/A979) applied to information provided in the Treatment Access Request (TAR) form. The algorithm considered the patient's fibrosis stage (F0–F4) as reported according to any assessment method, any available transient elastography data, and stage of disease. A reported fibrosis stage of F4 was considered compatible with the definition of cirrhosis; where the stage was <F4 or missing, the patient was also considered cirrhotic if an elastography result ≥14.5 kPa was reported. If elastography data were missing or inconsistent with the reported fibrosis stage, the stage of disease reported in the physician's assessment of ATU eligibility was used.
Patients with cirrhosis were further categorized on the basis of Child–Pugh class as compensated (Child–Pugh A) or decompensated (Child–Pugh B or C).
Treatment Dose and Duration
Recommended treatment was DCV 60 mg plus SOF 400 mg once daily for 24 weeks. RBV could be added and/or a shorter treatment duration undertaken at physician's discretion. A reduced DCV dose (30 mg daily) was recommended for patients receiving ritonavir-boosted atazanavir or other potent inhibitors of cytochrome P450 3A4 (CYP3A4) or P-glycoprotein, and a dose increase (90 mg) recommended with efavirenz or other moderate inducers of CYP3A4. Potent inducers of CYP3A4 or P-glycoprotein were contraindicated. DCV was not recommended for pregnant women or women of childbearing potential not using effective contraception.
Program Management
The ATU program was not a clinical study, and treatment was undertaken according to standard clinical practice. In accordance with the French regulations, the ATU cohort was approved by the French authorities and TAR forms for individual patients submitted to the program sponsor (Bristol-Myers Squibb) by their attending physicians. On granting of a TAR, the patient's hospital pharmacy could order DCV directly from the sponsor. SOF was not provided.
The provision of outcome data was voluntary. Attending physicians were invited to return completed visit forms to the sponsor at treatment initiation (day 0), treatment weeks 2, 4, 8, 12, 16, 20, and 24 (as appropriate), posttreatment weeks 4, 12 (PT12), and 24, and at treatment discontinuation. Forms reporting pregnancy or adverse events (AEs) were provided by physicians as appropriate. No clarification was requested for the AE data provided.
Data sharing with the databases of the French national prospective cohort of patients with HIV/HCV coinfection (ANRS CO13 HepaVIH) was undertaken for patients enrolled in both this cohort and the ATU to improve the quality and robustness of the results.
Program Assessment
Laboratory assessments were made locally. For each visit form, quantitative HCV-RNA data were provided along with the assay used and its lower limit of quantitation (LLOQ). An outcome of “quantifiable” (>LLOQ) or “unquantifiable” (≤LLOQ) was then assigned. If a qualitative result was reported, HCV-RNA was considered unquantifiable if target RNA was reported as undetected.
Safety was evaluated as frequencies of serious AEs, all AEs, and discontinuations due to AEs. The physician was responsible for AE reporting. Standard pharmacovigilance practice was used, imputing AEs of unreported causality as treatment related.
Analysis Populations and Endpoints
The treated population comprised patients with at least 1 completed visit form and/or AE report, while the primary efficacy population consisted of the subset who had more than 1 day of treatment and detectable HCV-RNA at baseline. The primary efficacy outcome was SVR12, defined as unquantifiable HCV-RNA at PT12. The primary analytic approach was a modified intention-to-treat (mITT) assessment that excluded patients without PT12 virologic data because of loss to follow-up or discontinuation for reasons unspecified or other than predefined treatment failure.
Treatment failure was defined as absence of SVR12 for virologic or specific nonvirologic reasons. Virologic failure comprised virologic breakthrough (quantifiable HCV-RNA on-treatment from week 2 after an unquantifiable measure), or relapse [unquantifiable HCV-RNA at end of treatment (EOT) but quantifiable at PT12], or undefined virologic failure—quantifiable HCV-RNA at all on-treatment/follow-up visits or at all on-treatment visits for patients with no posttreatment data. Nonvirologic treatment failure comprised missing HCV-RNA at PT12 due to treatment discontinuation for AEs or death on or after treatment.
An observed-values sensitivity analysis was also performed, which excluded nonvirologic treatment failures.
Statistical Analysis
Intermittent missing data were imputed as the worse of the flanking outcomes, except for missing PT12 data, which were back-imputed from the next available measurement.
Treatment duration was derived from documented start and end dates or inferred from the pharmacovigilance database or the day 0/last on-treatment visit dates if missing. Treatment duration was analyzed as 12 or 24 weeks based on treatment length between the derived start and end dates: those treated for ≤14 weeks were analyzed as 12 weeks, and >14 weeks as 24 weeks. Sensitivity subgroup analyses were undertaken for actual durations: <10, 10 to <14, 14 to <20, and ≥20 weeks. Comparisons between baseline characteristics were made using 2-tailed t tests or Wilcoxon rank sum tests for continuous variables, and χ2 or Fisher's exact tests for categorical variables.
RESULTS
Patients
Between ATU cohort initiation in March 2014 and closure in October 2014, 669 HIV/HCV coinfected patients treated with DCV were enrolled by 265 physicians. From these, 617 records were available for safety assessments and 407 for mITT efficacy (Supplemental Digital Content, Fig. 1, http://links.lww.com/QAI/A979).
Baseline characteristics for the mITT efficacy population are shown in Table 1. Patients were primarily cirrhotic (72%, of whom 18% were decompensated) and infected with HCV genotype (GT) 1 (69%; 53% GT 1a, 14% GT 1b), GT 3 (12%), or GT 4 (19%). Most were treatment experienced (82%), of whom almost all had previously received pegIFN/RBV with (23%) or without (77%) an NS3 protease inhibitor (PI). Most previous failures were for null or partial response (76%) or relapse (23%). Model for end-stage liver disease (MELD) scores were >10 in 47% of patients with available data, baseline albumin <35 g/L in 25%, and 55% had baseline HCV-RNA ≥6 log10 IU/mL. All patients with data (n = 393) were receiving cART with a wide variety of PIs, nonnucleoside reverse transcriptase inhibitors, and integrase inhibitors; 9% had a baseline CD4 cell count <200 cells/mm3, and 95% had plasma HIV RNA <50 copies/mL at baseline. Eight patients (2%) were HIV/HCV/HBV coinfected.
TABLE 1.
Baseline Characteristics
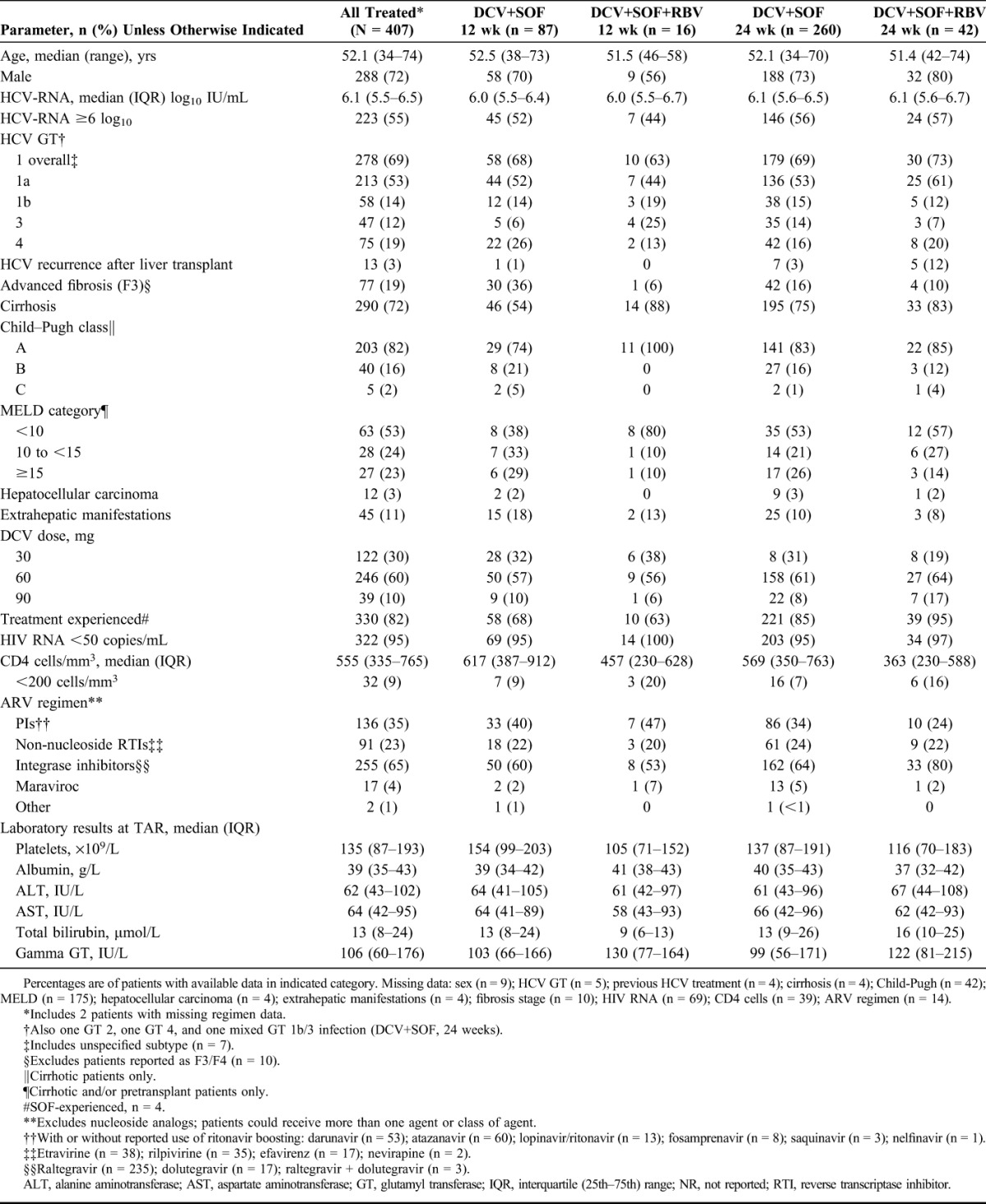
Baseline characteristics between the mITT efficacy population and the 183 intention-to-treat patients excluded from the primary analysis for missing data not related to treatment failure were comparable (Supplemental Digital Content, Table 2, http://links.lww.com/QAI/A979). Compared with the mITT population, the only differences with P < 0.05 among excluded patients were lower RBV use (7% vs 14% receiving RBV), a shorter duration of actual treatment among those with a derived duration of 12 weeks (13% treated <10 weeks vs 4% in mITT patients) or 24 weeks (10% vs 3% treated 14 to <20 weeks), and a higher use of nucleoside-sparing HIV treatment (17% vs 10%).
The median duration of treatment in the mITT population was 168 days (range 11–215). Most patients (86%) received DCV+SOF without RBV, of whom 75% received an analyzed duration of 24 weeks. Of the 14% who received RBV, most (72%) were in the 24-week group. In the 24-week group, those who received RBV had more advanced baseline markers of both HCV and HIV disease (Supplemental Digital Content, Table 3, http://links.lww.com/QAI/A979) as shown by more posttransplant HCV recurrence (12% vs 3% of those not receiving RBV), more hepatic encephalopathy (8% vs 1%), higher gamma GT (median 138 vs 96 IU/mL), and lower CD4 cell counts (median 363 vs 569 cells/mm3, with 47% vs 25% <350 cells/mm3), and were more likely to be receiving HIV integrase inhibitors (80% vs 64%; all comparisons P < 0.05). Trends (P < 0.1) were also noted for more ascites, a higher proportion with previous HCV treatment, more total bilirubin >60 μmol/L, and lower median albumin among patients who received RBV.
Virologic Response
SVR12 outcomes are shown in Table 2 for all patients, and broken down by cirrhosis status. Among all treated patients, SVR12 (mITT) was 92% (95% without cirrhosis; 90% with cirrhosis). Among patients who received DCV+SOF without RBV for 24 weeks, SVR12 was 96% overall (98% without cirrhosis; 95% with cirrhosis), and 100% both with and without cirrhosis in the smaller group who received DCV+SOF+RBV for 24 weeks.
TABLE 2.
Sustained Virologic Response and Treatment Failure by Derived Treatment Regimen and Cirrhosis Status
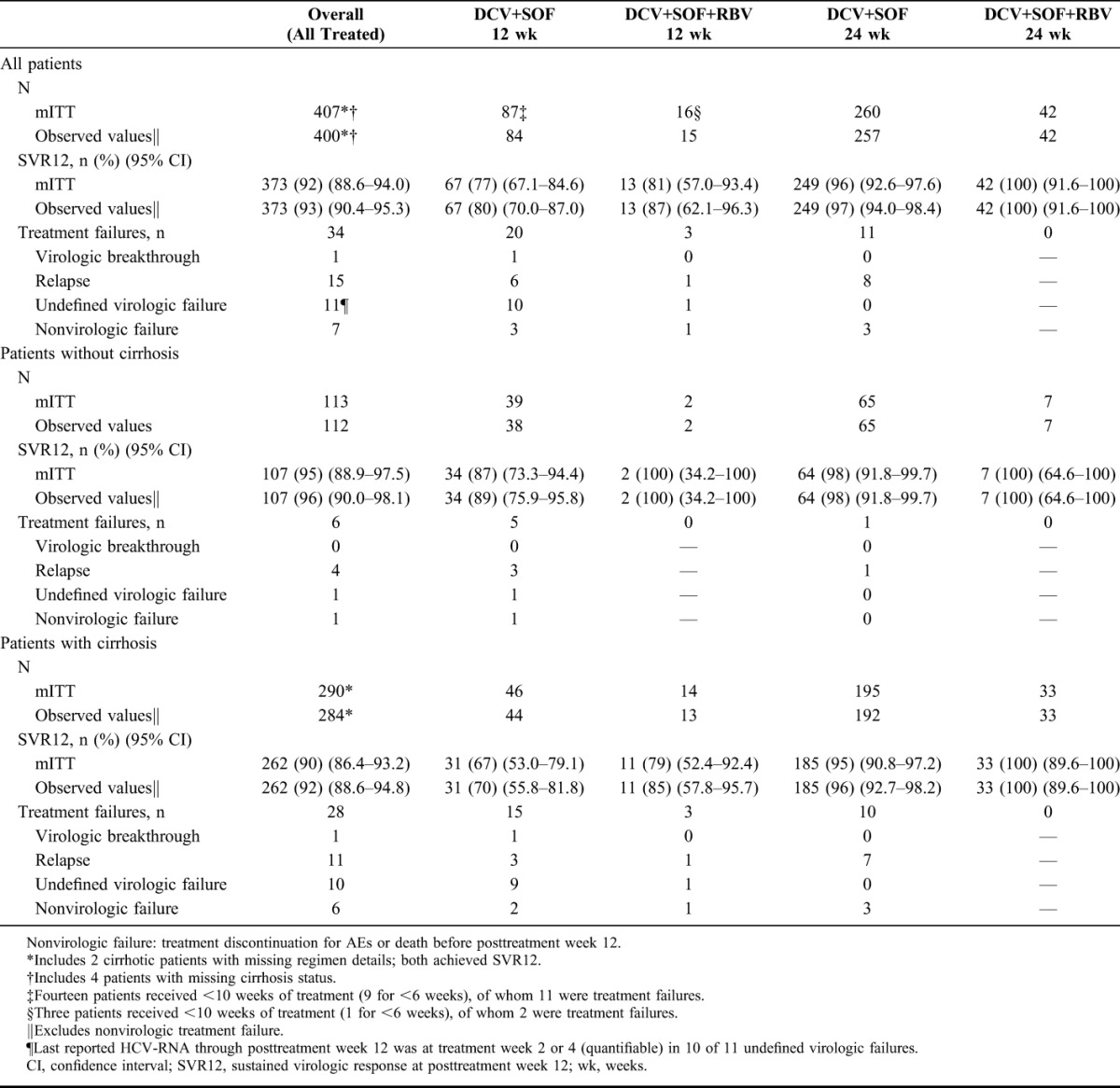
SVR12 was numerically lower among the 103 patients analyzed as having received 12 weeks of treatment [78% (80/103) with or without RBV], driven primarily by a high proportion (17%; 17/103) with very short (<10 weeks) actual durations (Fig. 1). Among this subgroup with short treatment, the incidence of both nonvirologic treatment failure [12% (2/17)] and undefined virologic failure for missing HCV-RNA data after week 2 or 4 (9 of 10 undefined failures) was substantially higher than among patients treated for longer. Among 13 treatment failures in patients with less than 10 weeks of treatment, 8 (62%) were treated for ≤6 weeks.
FIGURE 1.
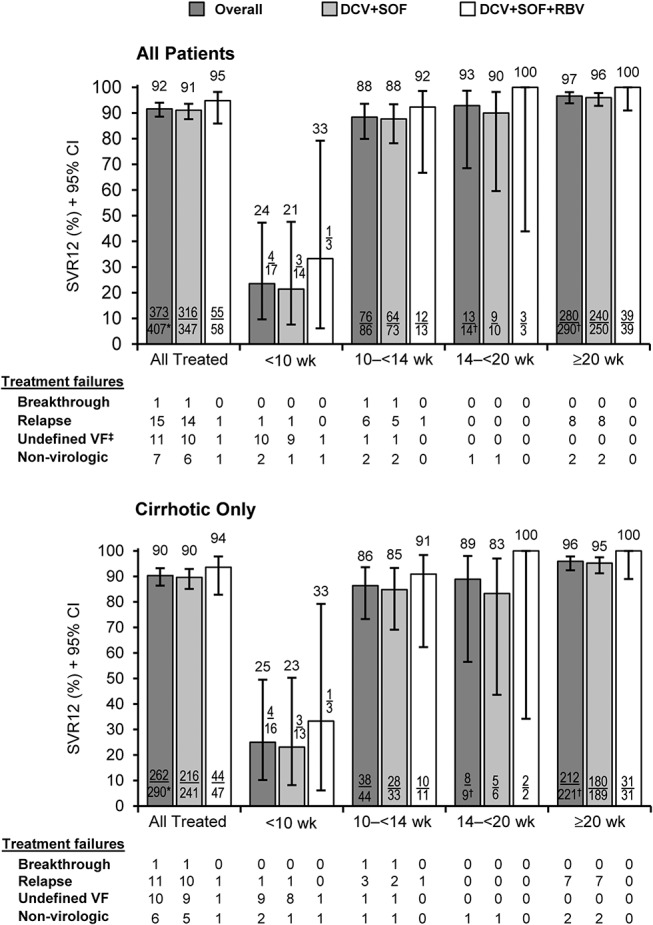
Sustained virologic response (mITT) according to actual duration of treatment. CI, confidence interval; SVR12, sustained virologic response at posttreatment week 12; VF, virologic failure; wk, weeks. Missing regimen details: *n = 2 and †n = 1. ‡Ninety-one percent (10/11) of undefined VFs were patients whose last reported HCV-RNA through posttreatment week 12 was a detectable measure at treatment week 2 or 4.
In patients with compensated (Child–Pugh A) cirrhosis treated for 24 weeks, SVR12 was 95% (134/141) without RBV and 100% (22/22) with RBV (Supplemental Digital Content, Table 4, http://links.lww.com/QAI/A979). Only 4 patients with decompensated cirrhosis (Child–Pugh B or C) received RBV; among the 33 decompensated patients treated for 24 weeks with or without RBV, SVR12 was 94% (31/33).
In the 368 patients with baseline CD4 data, SVR12 (mITT) was 81% (26/32; 95% CI: 64.7%–91.1%) in those <200 cells/mm3 vs 92% (309/336; 95% CI: 88.6%–94.4%) in those ≥200 cells/mm3.
SVR12 was consistent across HCV GTs and broadly comparable across cART regimens (Fig. 2). A slightly lower SVR12 was observed among patients receiving PIs, driven primarily by higher rates of nonvirologic failure [4% (6/136) vs <1% (1/257)] and undefined virologic failure due to missing data after treatment week 2 or 4 [5% (7/136) vs 2% (4/257)] than those not receiving PIs. Restricting the denominator to virologic breakthroughs or relapses resulted in SVR12 rates of 93% (115/123) for those taking PIs vs 97% (244/252) for those who were not.
FIGURE 2.
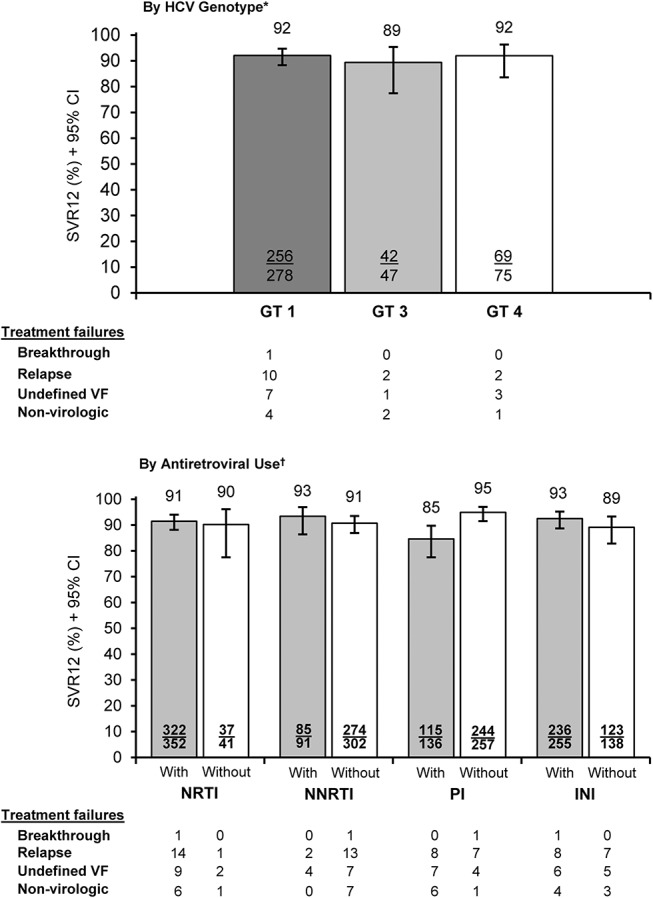
Sustained virologic response (mITT) by HCV GT and use of ARV drug classes. CI, confidence interval; INI, integrase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; SVR12, sustained virologic response at posttreatment week 12; VF, virologic failure; wk, weeks. *Excludes 1 GT 2 (achieved SVR12), 1 GT 6 (SVR12), and 5 missing GT (4 SVR12) patients. †Excludes 14 patients without ARV usage data (all SVR12).
Treatment Failure
Thirty-four patients in the mITT population did not achieve SVR12 for virologic (n = 27) or nonvirologic (n = 7) failure. Virologic failures comprised one breakthrough, 15 relapses, and 11 undefined failures (10 for missing HCV-RNA after a quantifiable result at treatment week 2 or 4). Nonvirologic failures consisted of 5 deaths and 2 discontinuations for AEs, detailed below.
Individual characteristics of the 34 patients with treatment failure are shown in Supplemental Digital Content, Table 5, http://links.lww.com/QAI/A979, and aggregated baseline characteristics for patients with virologic or nonvirologic failure and those with SVR12 in Supplemental Digital Content, Table 6, http://links.lww.com/QAI/A979. Overall, patients with treatment failure, particularly nonvirologic failure, showed more advanced indicators of liver and/or HIV disease than those who achieved SVR12, with trends toward more decompensated liver disease, higher MELD scores, more baseline laboratory abnormalities, and lower CD4 cells.
Evolution of Liver Disease and HIV-Associated Parameters
Paired data at baseline and PT12 were available for Child–Pugh class in 42 patients and MELD score in 21 patients. Improved Child–Pugh results at PT12 were observed in 14% (6/42) of patients (5 class B to class A, and 1 class C to class A); 83% remained unchanged, and one deteriorated (class A to class B). For MELD score, 19% (4/21) had a reduction in score category at PT12 from 10–<15 to <10 (n = 1), or from ≥15 to 10–<15 (n = 1) or to <10 (n = 2); 67% (14/21) had an unchanged category, and 14% (3/21) had worsened.
CD4 cell count was stable between baseline and EOT in 153 patients with paired data, with a median change of 9 cells/mm3 and an interquartile range of −10 to 29 cells/mm3. Of patients with paired HIV RNA data, 93% (141/152) of those who had <50 copies/mL at day 0 were <50 copies/mL at EOT, and 97% (150/154) of those <200 copies/mL at day 0 remained <200 copies/mL at EOT.
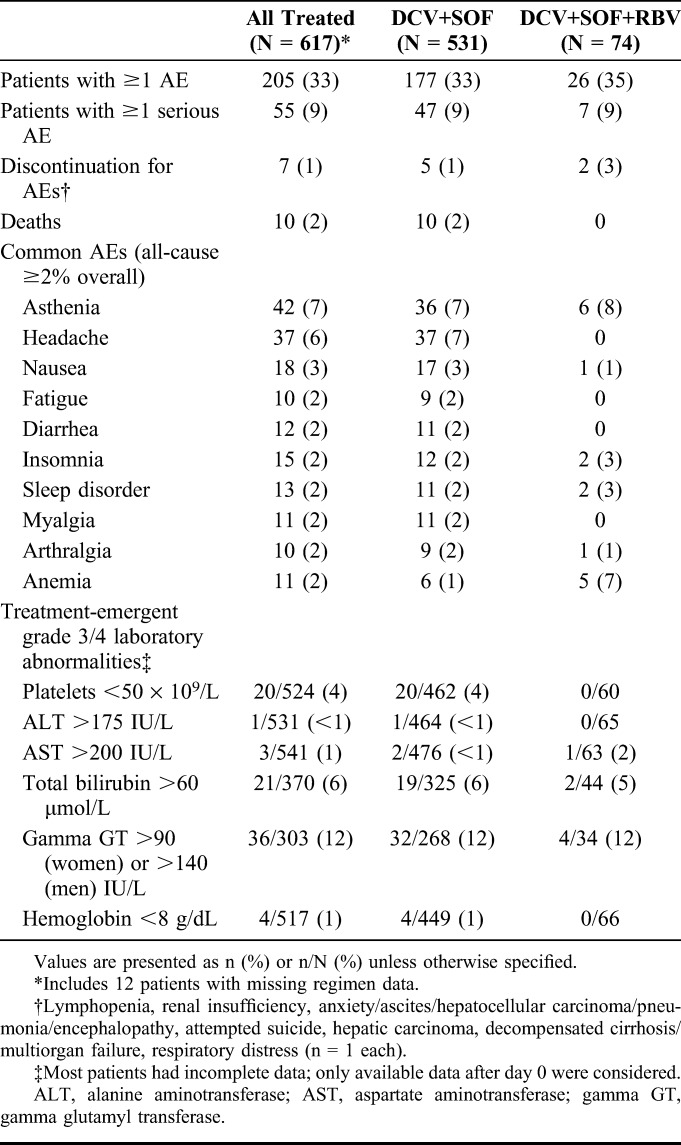
Safety
On-treatment AEs in the overall safety population (N = 617) are summarized in Table 3. Fifty-five patients (9%) experienced one or more serious AEs (Supplemental Digital Content, Table 7, http://links.lww.com/QAI/A979), and 26 (4%) experienced one or more AEs of severity grade 3 or 4 (Supplemental Digital Content, Table 8, http://links.lww.com/QAI/A979). There were 10 on- or off-treatment deaths, mostly for causes consistent with complications of advanced liver disease (Supplemental Digital Content, Table 9, http://links.lww.com/QAI/A979); one (decompensated cirrhosis in a patient who also had multiorgan failure) was considered possibly related to HCV or HIV treatment by the physician, and 2 (multiorgan failure plus septic shock plus intestinal obstruction, and hepatic carcinoma) were imputed as treatment-related for unreported causality. The remaining 7 deaths were not considered treatment related. Five deaths were classed as nonvirologic treatment failures. There were 7 discontinuations for AEs, of which 3 were subsequently fatal (hepatic carcinoma, decompensated cirrhosis/multiorgan failure, respiratory distress) and 4 nonfatal—lymphopenia, renal insufficiency (both reported as related to treatment), attempted suicide (imputed as treatment-related for missing causality), and anxiety/ascites/hepatocellular carcinoma/pneumonia/encephalopathy (not related to treatment). Two nonfatal AEs leading to discontinuation were classed as nonvirologic treatment failures.
TABLE 3.
On-Treatment Safety Summary (All Treated Patients)
DISCUSSION
As a regimen, DCV+SOF±RBV has several features of relevance to HIV-HCV coinfection. It is a pan-genotypic combination active against HCV GT 3—considered the most difficult GT to treat and against which many of the current DAAs have reduced or absent antiviral activity—with minimal potential as a perpetrator of drug–drug interactions and no significant impact on ARV drug exposures.25 Although DCV is a cytochrome P450 3A4 (CYP 3A4) substrate,27 administration of DCV and SOF as separate agents confers the flexibility to accommodate CYP 3A4–active ARVs such as efavirenz or boosted atazanavir through DCV dose adjustment. By contrast, some fixed-dose coformulation regimens for HCV are contraindicated or not recommended for use with efavirenz—eg, EPCLUSA (velpatasvir + SOF)28 and ZEPATIER (elbasvir + grazoprevir)29—or with boosted PIs (eg, ZEPATIER29) because of alterations in exposure to one or more regimen components.
This large real-world analysis evaluated DCV+SOF±RBV in a mostly cirrhotic and treatment-experienced cohort of HIV/HCV coinfected patients receiving a broad range of cART. Most (74%) were treated for the program-recommended 24 weeks, which was also the median time on treatment. Overall, the SVR12 rate (mITT) was 92%. In those treated for 24 weeks, SVR12 was 96%–100% and broadly comparable with or without compensated or decompensated cirrhosis. These SVR12 rates are similar to 12-week treatment studies of several DAA regimens—including DCV+SOF—in coinfected patients with less advanced disease.13,15–17 Excluding patients with very short (<10 weeks) durations of actual treatment, SVR12 in the 12-week analysis groups was 88%–92%. The incremental benefit of RBV use was small and confounded by low numbers and nonrandomized treatment allocation. The regimen was well tolerated with or without RBV, with only 1% (7/617) treatment discontinuations for fatal or nonfatal AEs, and 10 (2%) deaths, primarily from causes consistent with advanced liver disease.
Clinical data for DAA regimens in coinfected patients with advanced liver disease are sparse, particularly for decompensated cirrhosis. These real-world data are encouraging, and suggest that SVR12 rates >90% are achievable with or without RBV in decompensated patients given DCV+SOF, irrespective of HCV GT or cART regimen. These data also help address the current lack of longer duration (>16 weeks) clinical trial data for DCV+SOF±RBV. The high SVR12 rate in cirrhotic patients after 24 weeks of DCV+SOF is relevant for patients who are RBV intolerant, or for whom RBV might be considered inadvisable, such as those with relevant comorbidities or cirrhosis, or older or more clinically advanced HIV coinfected patients already receiving multiple therapeutic agents.
These ATU data are consistent with smaller cohorts of HIV/HCV coinfected patients with advanced liver disease: 92% SVR12 (48/52) was observed in a subgroup of coinfected patients treated with DCV+SOF±RBV in a Europe-wide DCV compassionate use program,30 of whom 95% had cirrhosis, and just under half were Child–Pugh class B or C.31 Similarly, 93% SVR12 was observed in 189 cirrhotic patients (8% Child–Pugh B or C) treated with various DAA regimens, including DCV+SOF±RBV, for 12 or 24 weeks in the French ANRS CO13 HepaVIH coinfection cohort.32 The data are also consistent with >90% SVR12 observed in clinical DAA studies in predominantly noncirrhotic HCV monoinfected33–37 and HIV/HCV coinfected patients,13–17 and similarly with slightly lower rates seen in HCV monoinfection with decompensated liver disease.38–40
As with all real-world data, there are limitations to these analyses. Treatment allocation and duration was at the physician's discretion, and the subsequent group imbalances made it impossible to fully assess the contribution of RBV or treatment duration to outcome. Data collection was nonstandardized and based on local practice, resulting in intersite reporting variability and substantial missing data. Reporting was also voluntary, leading to potential reporting bias. Data are limited for HIV-associated parameters. Neither DCV nor SOF has any clinically relevant interactions with maintenance opioids,41,42 and it is highly likely that many patients of this cohort would have been current or former PWID receiving opioid substitution, for whom clinical data are sparse.43,44 Thus, it is a limitation that data on use of injection drugs and maintenance opioids were not captured. Finally, safety data were based on pharmacovigilance reporting, and it is likely that AEs were underreported.
Despite these limitations, this cohort represents the largest real-world assessment of HCV treatment efficacy yet reported in unselected patients with HIV/HCV coinfection. These patients had very limited treatment options, and their advanced disease, lifestyle characteristics, and broad range of cART regimens would have made many ineligible for randomized studies. The data indicate that HIV/HCV coinfection with decompensated liver disease does not preclude the probability of a high response rate to DCV+SOF treatment, with or without RBV.
In conclusion, therefore, DCV+SOF±RBV was efficacious and well tolerated in this real-world HIV/HCV coinfected cohort with advanced liver disease, and this regimen is an appropriate option in this context.
ACKNOWLEDGMENTS
The authors thank the physicians and associated health care professionals involved in the ATU program, the patients, and their families. Editorial assistance with this manuscript was provided by Nick Fitch, PhD, CMPP (ArticulateScience, LLC, London, UK), and funded by Bristol-Myers Squibb.
Footnotes
Presented in part as an interim analysis at the eighth IAS Conference on HIV Pathogenesis, Treatment and Prevention; 19–22 July, 2015; Vancouver, Canada.
K.L. has received institutional grant support from Janssen and Gilead, and consulting fees from Gilead, AbbVie, Janssen, and Merck Sharp & Dohme (MSD). H.F. reports obtaining personal fees from AbbVie, Gilead, MSD, Janssen and Bristol-Myers Squibb (BMS) for advisory boards, presentations, invitations to national and international meetings, and participation in therapeutic trials. C.D. reports obtaining personal fees from Gilead, ViiV Healthcare and MSD for board membership, consultancy services, and speaker services, respectively, and travel/accommodation or meeting expenses from Gilead, ViiV Healthcare, MSD, and BMS. S.M. reports obtaining personal fees from AbbVie, Gilead, and BMS for speaker bureaus or local meetings, and invitations to national or international meetings. E.R. reports obtaining personal fees from Gilead for board membership. T.A. reports obtaining personal fees from AbbVie, BMS Gilead, Janssen, and MSD for presentations, invitations to national and international meetings, and participation in therapeutic trials. M.A.V. reports obtaining personal fees for consulting/educational lectures and travel grants from Janssen, Gilead, ViiV Bristol-Myers Squibb and MSD/ Schering-Plough. P.M. reports obtaining personal fees from Gilead, MSD, and BMS, for consultancy and/or board memberships. S.H. reports obtaining travel/accommodation and/or meeting expenses, and stock options in Gilead, Janssen, BMS, MSD, Pfizer, AbbVie, and Astellas. D.B. reports obtaining personal fees from Gilead, BMS, and MSD. G.-P.P. reports obtaining personal fees for consultation and/or board membership from BMS, Gilead, and Janssen, and for speaker services from BMS, Gilead, Janssen, MSD, and Astellas. J.C. reports obtaining personal fees for board membership or speaker services from AbbVie, Gilead, and BMS. H.A. reports obtaining personal fees from BMS for board membership, and from BMS, AbbVie, Janssen, and Gilead, for speaker services, plus invitations to national and international meetings. S.D. reports obtaining personal fees for board membership from Gilead, MSD, and AbbVie. A.L. reports obtaining personal fees for speaker/moderator services from BMS, ViiV Healthcare, MSD, and Gilead. P.D.T. reports obtaining personal fees from BMS, Gilead, MSD, Janssen, and ViiV Healthcare, for advisory boards, presentations, and meeting participation. V.L. reports obtaining personal fees from advisory boards and speakers' bureaus for AbbVie, BMS, and Gilead, and from consulting and speakers' bureaus for Janssen and Merck. V.D.L. reports obtaining personal fees from BMS, AbbVie, Janssen and Merck, and personal fees and grants from Gilead. P.S. reports obtaining personal fees from BMS, Gilead, Janssen, AbbVie, MSD, and Mayoly-Spindler. L.F., R.A., Y.B., A.F., and Y.Z. are all employees of BMS. D.S.C. reports obtaining personal fees from BMS and Gilead for advisory board participation, and from BMS, Gilead, MSD, Janssen, and ViiV Healthcare for presentations and meeting participation. The remaining authors have no funding or conflicts of interest to disclose. K.L. and H.F. are joint first authors.
This was a compassionate use program authorized by a regulatory authority to allow physicians early access to daclatasvir for specific patients in need. No financial support was provided by BMS.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
REFERENCES
- 1.Platt L, Easterbrook P, Gower E, et al. Prevalence and burden of HCV co-infection in people living with HIV: a global systematic review and meta-analysis. Lancet Infect Dis. 2016;16:797–808. [DOI] [PubMed] [Google Scholar]
- 2.Sherman KE, Rouster SD, Chung RT, et al. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the US adult AIDS clinical trials group. Clin Infect Dis. 2002;34:831–837. [DOI] [PubMed] [Google Scholar]
- 3.Cacoub P, Dabis F, Costagliola D, et al. Burden of HIV and hepatitis C co-infection: the changing epidemiology of hepatitis C in HIV-infected patients in France. Liver Int. 2015;35:65–70. [DOI] [PubMed] [Google Scholar]
- 4.Graham CS, Baden LR, Yu E, et al. Influence of human immunodeficiency virus infection on the course of hepatitis C virus infection: a meta-analysis. Clin Infect Dis. 2001;33:562–569. [DOI] [PubMed] [Google Scholar]
- 5.Thein HH, Yi Q, Dore GJ, et al. Natural history of hepatitis C virus infection in HIV-infected individuals and the impact of HIV in the era of highly active antiretroviral therapy: a meta-analysis. AIDS. 2008;22:1979–1991. [DOI] [PubMed] [Google Scholar]
- 6.McGovern BH. Hepatitis C in the HIV-infected patient. J Acquir Immune Defic Syndr. 2007;45(suppl 2):S47–S56. [DOI] [PubMed] [Google Scholar]
- 7.Smith CJ, Ryom L, Weber R, et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): a multicohort collaboration. Lancet. 2014;384:241–248. [DOI] [PubMed] [Google Scholar]
- 8.Reiberger T, Obermeier M, Payer BA, et al. Considerable under-treatment of chronic HCV infection in HIV patients despite acceptable sustained virological response rates in a real-life setting. Antivir Ther. 2011;16:815–824. [DOI] [PubMed] [Google Scholar]
- 9.Young J, Potter M, Cox J, et al. Variation between Canadian centres in the uptake of treatment for hepatitis C by patients coinfected with HIV: a prospective cohort study. CMAJ Open. 2013;1:E106–E114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung RT, Andersen J, Volberding P, et al. Peginterferon Alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIV-coinfected persons. N Engl J Med. 2004;351:451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torriani FJ, Rodriguez-Torres M, Rockstroh JK, et al. Peginterferon Alfa-2a plus ribavirin for chronic hepatitis C virus infection in HIV-infected patients. N Engl J Med. 2004;351:438–450. [DOI] [PubMed] [Google Scholar]
- 12.Gatti F, Nasta P, Matti A, et al. Treating hepatitis C virus in HIV patients: are side effects a real obstacle? AIDS Rev. 2007;9:16–24. [PubMed] [Google Scholar]
- 13.Wyles DL, Ruane PJ, Sulkowski MS, et al. Daclatasvir plus sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med. 2015;373:714–725. [DOI] [PubMed] [Google Scholar]
- 14.Rockstroh JK, Nelson M, Katlama C, et al. Efficacy and safety of grazoprevir (MK-5172) and elbasvir (MK-8742) in patients with hepatitis C virus and HIV co-infection (C-EDGE CO-INFECTION): a non-randomised, open-label trial. Lancet HIV. 2015;2:e319–e327. [DOI] [PubMed] [Google Scholar]
- 15.Naggie S, Cooper C, Saag M, et al. Ledipasvir and sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med. 2015;373:705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sulkowski MS, Eron JJ, Wyles D, et al. Ombitasvir, paritaprevir co-dosed with ritonavir, dasabuvir, and ribavirin for hepatitis C in patients co-infected with HIV-1: a randomized trial. JAMA. 2015;313:1223–1231. [DOI] [PubMed] [Google Scholar]
- 17.Wyles D, Brau N, Kottilil S, et al. Sofosbuvir/velpatasvir fixed dose combination for 12 weeks in patients co-infected with HCV and HIV-1: the phase 3 ASTRAL-5 Study [abstract]. J Hepatol. 2016;64(suppl 2):S188–S189. [Google Scholar]
- 18.Poizot-Martin I, Naqvi A, Obry-Roguet V, et al. Potential for drug-drug interactions between antiretrovirals and HCV direct acting antivirals in a large cohort of HIV/HCV coinfected patients. PLoS One. 2015;10:e0141164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinello M, Dore GJ, Skurowski J, et al. Antiretroviral use in the CEASE cohort study and implications for direct-acting antiviral therapy in human immunodeficiency virus/hepatitis C virus coinfection. Open Forum Infect Dis. 2016;3:ofw105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gervasoni C, Peri AM, Cattaneo D, et al. Simeprevir-induced severe withdrawal syndrome in an HIV/HCV coinfected patient on long-term maintenance methadone therapy. Eur J Clin Pharmacol. 2015;71:1027–1028. [DOI] [PubMed] [Google Scholar]
- 21.Eron JJ, Young B, Cooper DA, et al. Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trials. Lancet. 2010;375:396–407. [DOI] [PubMed] [Google Scholar]
- 22.Saeed S, Strumpf EC, Walmsley SL, et al. How generalizable are the results from trials of direct antiviral agents to people coinfected with HIV/HCV in the real world? Clin Infect Dis. 2016;62:919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao M, Nettles RE, Belema M, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sofia MJ, Bao D, Chang W, et al. Discovery of a β-d-2'-deoxy-2'-α-fluoro-2'-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem. 2010;53:7202–7218. [DOI] [PubMed] [Google Scholar]
- 25.Bifano M, Hwang C, Oosterhuis B, et al. Assessment of pharmacokinetic interactions of the HCV NS5A replication complex inhibitor daclatasvir with antiretroviral agents: ritonavir-boosted atazanavir, efavirenz and tenofovir. Antivir Ther. 2013;18:931–940. [DOI] [PubMed] [Google Scholar]
- 26.Bristol-Myers Squibb. Data on File (DACL-047). New York, NY: Bristol-Myers Squibb; 2016. [Google Scholar]
- 27.Bristol-Myers Squibb Pharma EEIG. Daklinza EU Summary of Product Characteristics. Uxbridge, UK: Bristol-Myers Squibb Pharma EEIG; 2014. [Google Scholar]
- 28.Gilead Sciences International Ltd. EPCLUSA EU Summary of Product Characteristics. Cambridge, UK: Gilead Sciences International Ltd; 2016. [Google Scholar]
- 29.Merck Sharp & Dohme Ltd. ZEPATIER EU Summary of Product Characteristics. Hoddesdon, UK: Merck Sharp & Dohme Ltd; 2016. [Google Scholar]
- 30.Welzel TM, Petersen J, Herzer K, et al. Daclatasvir plus sofosbuvir, with or without ribavirin, achieved high sustained virologic response rates in patients with HCV infection and advanced liver disease in a real-world cohort. Gut. 2016;65:1861–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rockstroh JK, Ingiliz P, Petersen J, et al. Daclatasvir plus sofosbuvir, with or without ribavirin, in real-world patients with HIV-HCV coinfection and advanced liver disease. Antivir Ther. 2016. 10.3851/IMP3108. [DOI] [PubMed] [Google Scholar]
- 32.Sogni P, Gilbert C, Lacombe K, et al. All-oral direct-acting antiviral regimens in HIV/hepatitis C virus-coinfected patients with cirrhosis are efficient and safe: real-life results from the prospective ANRS CO13-HEPAVIH cohort. Clin Infect Dis. 2016;63:763–770. [DOI] [PubMed] [Google Scholar]
- 33.Afdhal N, Reddy KR, Nelson DR, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med. 2014;370:1483–1493. [DOI] [PubMed] [Google Scholar]
- 34.Afdhal N, Zeuzem S, Kwo P, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2014;370:1889–1898. [DOI] [PubMed] [Google Scholar]
- 35.Ferenci P, Bernstein D, Lalezari J, et al. ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med. 2014;370:1983–1992. [DOI] [PubMed] [Google Scholar]
- 36.Zeuzem S, Ghalib R, Reddy KR, et al. Grazoprevir-elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: a randomized trial. Ann Intern Med. 2015;163:1–13. [DOI] [PubMed] [Google Scholar]
- 37.Feld JJ, Jacobson IM, Hezode C, et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med. 2015;373:2599–2607. [DOI] [PubMed] [Google Scholar]
- 38.Curry MP, O'Leary JG, Bzowej N, et al. Sofosbuvir and velpatasvir for HCV in patients with decompensated cirrhosis. N Engl J Med. 2015;373:2618–2628. [DOI] [PubMed] [Google Scholar]
- 39.Charlton M, Everson GT, Flamm SL, et al. Ledipasvir and sofosbuvir plus ribavirin for treatment of HCV infection in patients with advanced liver disease. Gastroenterology. 2015;149:649–659. [DOI] [PubMed] [Google Scholar]
- 40.Poordad F, Schiff ER, Vierling JM, et al. Daclatasvir with sofosbuvir and ribavirin for HCV infection with advanced cirrhosis or post-liver transplant recurrence. Hepatology. 2016;63:1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garimella T, Wang R, Luo WL, et al. Assessment of drug-drug interactions between daclatasvir and methadone or buprenorphine-naloxone. Antimicrob Agents Chemother. 2015;59:5503–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gilead Sciences International Ltd. Sovaldi EU Summary of Product Characteristics. Cambridge, UK: Gilead Sciences International Ltd; 2014. [Google Scholar]
- 43.Lalezari J, Sullivan JG, Varunok P, et al. Ombitasvir/paritaprevir/r and dasabuvir plus ribavirin in HCV genotype 1-infected patients on methadone or buprenorphine. J Hepatol. 2015;63:364–369. [DOI] [PubMed] [Google Scholar]
- 44.Grebely J, Dore GJ, Altice F, et al. C-EDGE CO-STAR: efficacy of elbasvir/grazoprevir in HCV-infected persons who inject drugs receiving opioid agonist therapy. Proceedings and Abstracts of HepDART 2015: Frontiers in Drug Development for Viral Hepatitis, Wailea, HI; 6–10 December 2015:112 [Abstract 124].