

Supplemental Digital Content is available in the text.
Keywords: arteries, lymphangiogenesis, lymphatic vessels, mice, Polydom/Svep1, veins, zebrafish
Abstract
Rationale:
Lymphatic vessel formation and function constitutes a physiologically and pathophysiologically important process, but its genetic control is not well understood.
Objective:
Here, we identify the secreted Polydom/Svep1 protein as essential for the formation of the lymphatic vasculature. We analyzed mutants in mice and zebrafish to gain insight into the role of Polydom/Svep1 in the lymphangiogenic process.
Methods and Results:
Phenotypic analysis of zebrafish polydom/svep1 mutants showed a decrease in venous and lymphovenous sprouting, which leads to an increased number of intersegmental arteries. A reduced number of primordial lymphatic cells populated the horizontal myoseptum region but failed to migrate dorsally or ventrally, resulting in severe reduction of the lymphatic trunk vasculature. Corresponding mutants in the mouse Polydom/Svep1 gene showed normal egression of Prox-1+ cells from the cardinal vein at E10.5, but at E12.5, the tight association between the cardinal vein and lymphatic endothelial cells at the first lymphovenous contact site was abnormal. Furthermore, mesenteric lymphatic structures at E18.5 failed to undergo remodeling events in mutants and lacked lymphatic valves. In both fish and mouse embryos, the expression of the gene suggests a nonendothelial and noncell autonomous mechanism.
Conclusions:
Our data identify zebrafish and mouse Polydom/Svep1 as essential extracellular factors for lymphangiogenesis. Expression of the respective genes by mesenchymal cells in intimate proximity with venous and lymphatic endothelial cells is required for sprouting and migratory events in zebrafish and for remodeling events of the lymphatic intraluminal valves in mouse embryos.
The lymphatic vasculature serves key physiological roles during embryonic and adult life.1,2 It controls fluid homeostasis and retrieves water and macromolecules from the interstitium, it takes part in immune surveillance, and it is responsible for the uptake and transport of dietary lipids from the small intestine.
Editorial, see p 1216
Formation and maturation of the lymphatic vasculature has been studied in many vertebrate systems, including mice,3 Xenopus,4 and zebrafish.5,6 In mice, future lymphatic endothelial cells (LECs) egress from the cardinal vein (CV) as a network of loosely connected cells that express Prox-1 and then go on to form 2 initial lymphatic vessels, the primordial thoracic duct (TD) close to the CV and the more peripheral longitudinal lymphatic vessel.3 The migration of LECs depends on many secreted factors such as VEGF-C (vascular endothelial growth factor-C)7 and CCBE1 (collagen and calcium binding EGF domains 1),8 which act in concert with ADAMTS3 (a disintegrin and metalloproteinase with thrombospondin motifs 3)9,10 to generate mature and biologically active VEGF-C protein.11,12 VEGF-C in turn binds to VEGFR3 (vascular endothelial growth factor receptor-3) on venous and lymphatic endothelium. This system is evolutionarily conserved, and mutants in the respective genes lead to severely impaired lymphatic vasculature formation in both murine7–9 and zebrafish11,13 embryos. Later, once the primary lymphatic vascular network has been established, further morphological changes occur, resulting in the formation of capillaries that take up lymph from the interstitium, and collecting lymphatic vessels, which transport the contents of the lymphatic system back to the venous system. Mature collecting vessels are typically covered by smooth muscle cells, show basement membrane deposition, and contain intraluminal valves to prevent backflow of lymph.2 Much remains to be learned about the maturation steps toward fully functional lymphatic capillaries and collecting vessels, but the transcription factors Foxc2 (forkhead box protein C2) and NFATc1 (nuclear factor of activated T-cells, cytoplasmic 1) are known to be required for this process.14,15 Foxc2 mutant mouse embryos fail to form lymphatic valves.15 Human patients for lymphedema-distichiasis (Online Mendelian Inheritance in Man reference no. 153400) have been described to lack FOXC2 function.16 A somewhat milder phenotype is found in embryos with an endothelial-specific Integrin α9 deficiency, where valve leaflets are rudimentary, with disorganized fibronectin matrix.17
In zebrafish, the sprouting of venous and future lymphatic ECs occurs simultaneously, between 30 and 36 hours post-fertilization (hpf). Although venous sprouts connect to arterial intersegmental vessels (ISVs) and remodel them into veins, those sprouts that will form the trunk lymphatic vasculature migrate to the embryonic midline (the horizontal myoseptum [HMS]) and form a population of single cells, termed parachordal lymphangioblasts (PLs).13 PL cells populate the HMS region transiently, before they initiate a distinct migratory phase, causing them to move either dorsally to form the dorsal longitudinal lymphatic vessel or ventrally to form the TD. Migration in this phase always occurs along arteries.18–20 Later aspects of lymphatic vascular formation in zebrafish are less well defined, and the formation of valves, for example, has not been described.
Here, we report a novel gene function associated with lymphangiogenesis during early embryonic stages of both zebrafish and mouse. The extracellular protein Polydom/Svep1, which is not expressed by endothelial cells, has not previously been connected to lymphatic vessel formation, but is here shown to result in migratory defects and remodeling abnormalities in zebrafish and mouse mutant embryos, respectively.
Methods
Zebrafish ethyl-nitrosourea mutagenesis,13 the screening procedure,13 analysis of zebrafish mutants,13 and BAC (bacterial artificial chromosome) recombineering21 were performed as previously described. All remaining animal procedures, in situ hybridizations,3 and immunohistochemistry3 were performed as previously described or as detailed in the Online Data Supplement.
Results
Ly02-512 Zebrafish Mutants Display Defects of the Lymphatic Vasculature
In a forward genetic screen designed to uncover novel gene functions within the zebrafish lymphatic system,13 we identified a mutant, initially termed Ly02-512, which lacked all, or parts of, the TD and displayed edema around the eye, heart, and intestine at 5 days post-fertilization (dpf). Mutants developed a swim bladder and showed no signs of delayed development at 5 dpf (Figure 1A and 1B) but developed other defects such as brain and jaw abnormalities and heart defects (Figure 1A and 1B, and data not shown). A closer examination at 5 dpf demonstrated that in Ly02-512 mutant embryos the blood vasculature appeared normal and functional, with the exception of an increase in the number of arterial ISVs (highlighted in red), at the expense of venous ISVs (Figure 1B). This was clearly observed at 5 dpf in double transgenic embryos which express RFP (red fluorescent protein) under the control of the flt1enh promoter18 in arterial ISVs and GFP (green fluorescent protein) under the control of the pan-endothelial fli1a promoter.22 The TD, visualized using the same combination of transgenes (Figure 1C) or by highlighting the perfused blood vessels with rhodamine dextran angiography (Figure 1D), was absent in mutant embryos, although it could easily be identified in all sibling embryos as a thin vessel immediately ventral to the dorsal aorta (Figure 1C and 1D, arrows). However, the venous sprouting and TD defects in Ly02-512 mutants were not as severe when compared with ccbe1, vegf-c or flt4/vegfr-3 mutants, in which hardly any venous sprouts or TD are formed.11,13,23,24 Venous sprouts that did form in Ly02-512 mutants seemed to sprout correctly and formed normal intersegmental veins or morphologically normal PLs. Also, when quantifying the fate of venous sprouts, we could not find a difference in fate decisions for lymphovenous sprouts: there was no preference for venous sprouts to form definitive veins or PLs (see below).
Figure 1.
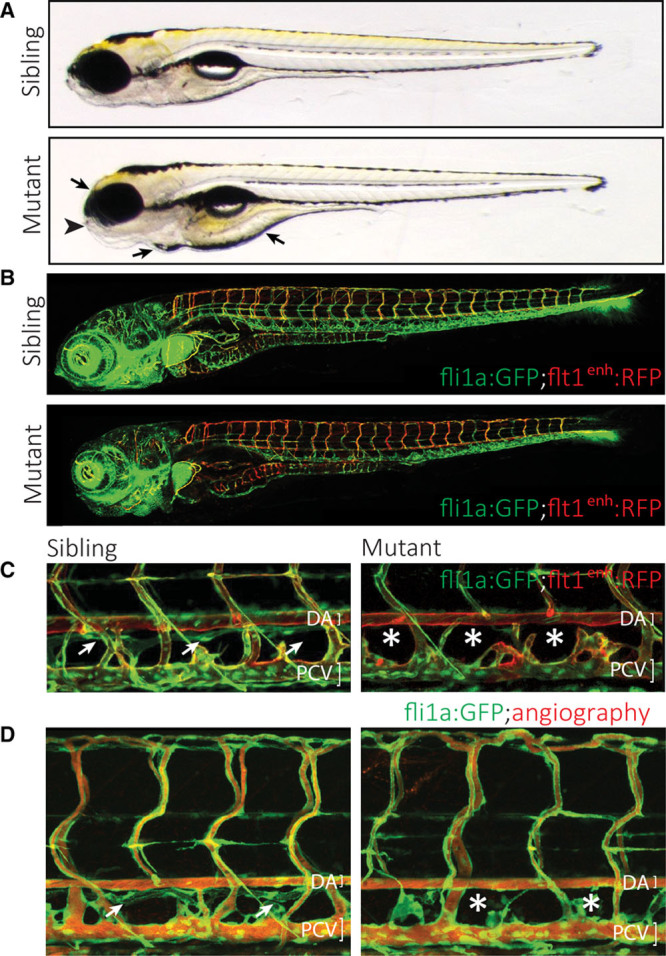
Characterization of a zebrafish mutant that affects the formation of the lymphatic vascular system. A, Gross morphology of the Ly02-512 mutant. The mutant embryos appear without major macroscopic defects or developmental delay, and form a swim bladder at 5 d post-fertilization (dpf). However, closer examination shows abnormalities in the jaw (arrowhead), and the embryo displays edema around the heart, eye, and intestine (arrows). B, Analysis of the vasculature in a fli1a:GFP; flt1enh:RFP transgenic background, highlighting arteries (red). Note the normal overall patterning of the blood vasculature despite the higher proportion of intersegmental arteries in the mutant embryo. C, Mutant embryos lack all or most aspects of the thoracic duct (TD), which in wild-type siblings is positioned just ventral to the dorsal aorta (DA; white arrows in C and D) but is absent in mutants (asterisks). D, Rhodamine dextran injection into the cardinal vein shows normal blood circulation in Ly02-512 mutants.
Ly02-512 Represents a Premature Stop Allele of the polydom/svep1 Gene
To identify the genetic lesion causing the phenotype, we used a positional mapping strategy using CA-repeat markers. Initial bulk segregant analysis placed the gene on linkage group 7 between markers z4.29 and z7.4 (Figure 2A), comprising a region of roughly 350 kb containing 12 genes (Zv9). Analysis of over 3500 mutant embryos and over 7000 meiosis revealed no further recombinants, which is why we applied a BAC rescue approach. We identified 3 independent BACs that were predicted to span the whole genomic region between the markers in question (Figure 2B) and generated independent transgenic lines with the rationale that one of the transgenic BAC alleles should be able to rescue the mutant phenotype. Although BACs CH73-220H7 and DKEY-108N6 failed to rescue, the BAC DKEY-8E16 reduced the number of phenotypically mutant embryos from the expected 25% to 4.1% (2/49; Figure 2C). Because 2 recombinants (2/7096) for z4.29 placed the region of interest to the right side of the marker, thereby excluding 5 of the 6 genes on BAC DKEY-8E16, the only remaining candidate gene on the BAC was polydom/svep1. Sequencing of all exons of the polydom/svep1 gene in mutant embryos revealed a premature stop codon in exon 14 of the gene (K836X; numbering based on a predicted protein starting 49 amino acids upstream of the annotation in Zv9, coded by 5′-flanking region and including a start methionine and a signal sequence).
Figure 2.
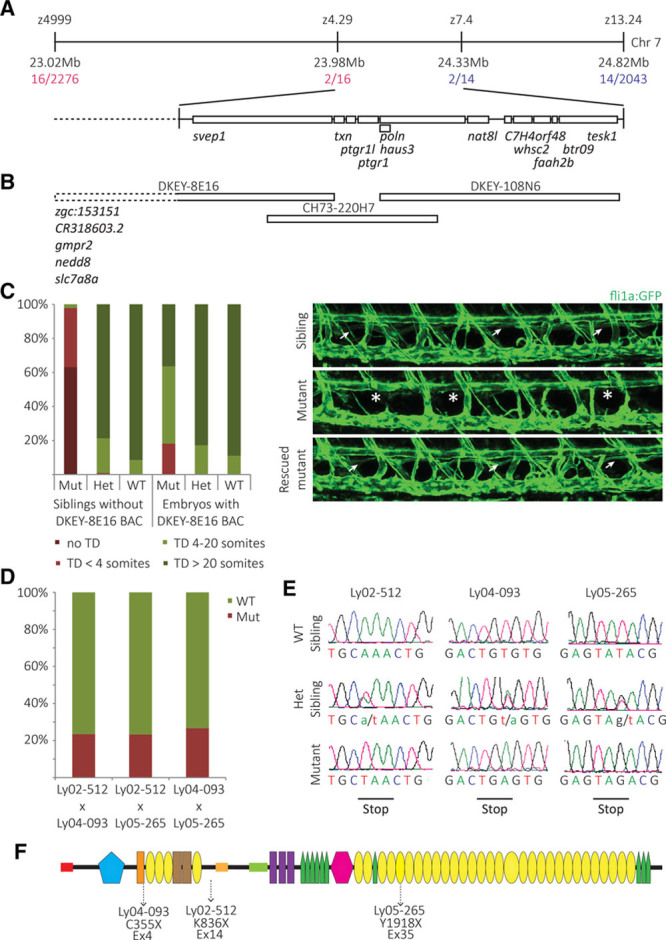
Ly02-512 encodes an allele of polydom/svep1. A, Positional cloning approach to identify the molecular lesion in Ly02-512 mutant embryos. Using polymorphic markers, Ly02-512 was mapped to a region between markers z4.29 and z.7.4 comprising ≈350 kb on linkage group 7. B, Three different BAC constructs that were independently inserted as transgenes into the Ly02-512 line containing 12 candidate genes within the linkage group. C, In Ly02-512 mutant embryos which contained the DKEY-8E16 BAC, the TD phenotype was rescued (P<0.001), suggesting DKEY-8E16 to contain the gene of interest. D, Two other mutant lines identified in the forward genetic screen, Ly04-093 and Ly05-265, failed to complement Ly02-512 and each other, suggesting that they represent alleles of the same gene. E, Sequencing of the 3 independent mutant alleles revealed 3 different nonsense substitutions, predicted to result in truncated versions of the Polydom/Svep1 protein. F, Schematic presentation of the Polydom/Svep1 protein domain structure and the positions of the truncations in the 3 mutant lines. Red rectangle: signal peptide; blue pentagon: von Willebrand factor type A domain; yellow ovals: SUSHI repeat; green pentagons: epidermal growth factor (EGF)–like and calcium-binding EGF-like domains; and pink hexagon: pentraxin domain.
Subsequently, 2 additional mutant alleles from the screen with similar phenotypes were found to fail to complement the original K836X allele, with a fully penetrant TD phenotype (ie, all mutant embryos lacked at least part of the TD) in all 3 cases (Figure 2D). Sequencing cDNA from mutant embryos demonstrated nonsense codons in the respective mutants, leading to a C355X conversion in Ly04-093 and a Y1918X conversion in Ly05-265 (Figure 2E). To exclude differential splice products that would produce transcripts without the affected exons, we performed reverse transcription-polymerase chain reaction (RT-PCR) analysis of sibling and mutant embryos from the Ly04-093 and the Ly02-512 alleles. RT-PCR and subsequent sequencing revealed no alternative splice products (Online Figure I). Injection of murine Svep1 mRNA was able to rescue the zebrafish mutant phenotype (Online Figure II). On the basis of (1) the genetic mapping results, (2) the successful rescue with BAC DKEY-8E16 and murine Svep1 mRNA, and (3) the identification of 3 independent mutant alleles, we concluded that the phenotype was caused by mutations in the zebrafish polydom/svep1 gene. Furthermore, a morpholino directed against the gene and an independently generated TALEN (transcription activator-like effector nuclease) allele (11 base pair deletion in the first exon; Morooka et al25) also result in an identical phenotype, strongly suggesting that all 4 mutant alleles represent loss-of-function situations. The protein product prediction of the gene comprised 3555 amino acids, contained a signal peptide but no predicted transmembrane domain, and harbored different domains (hence the name Polydom26) such as the ones that are abbreviated in the acronym SVEP27: Sushi, von Willebrand, epidermal growth factor, and pentraxin domain(s). Figure 2F provides a schematic representation of the domain structure of the predicted protein, with the position of the nonsense codons indicated.
The Migration of LECs Is Impaired in polydom/svep1 Zebrafish Mutants
Polydom/svep1 mutants were initially uncovered because of their aberrant lymphatic development, and we wanted to analyze the origin of the phenotype in more detail. Initially, we analyzed the number of secondary (venous) sprouts from the posterior CV (PCV). We counted secondary sprouts in phospholipase c γ1 (plcg1) morpholino-injected egg lays from polydom/svep1+/− parents. The interference with Plcg1 activity suppresses arterial development,28 allowing for easier appreciation of venous sprouting events in a fli1a:GFP transgenic background (Figure 3A). Polydom/svep1+/− embryos showed a small but statistically significant reduction of secondary sprouts compared with polydom/svep1+/+ siblings, whereas polydom/svep1−/− embryos only showed about 40% of the venous sprouts found in wild-type embryos (Figure 3F). This aspect of the phenotype was variable, and the effect on venous sprouting differed both between different clutches and within a single clutch of mutant embryos. Still, all mutant embryos show an increase in arteries over veins (Figure 3E and 3H).
Figure 3.
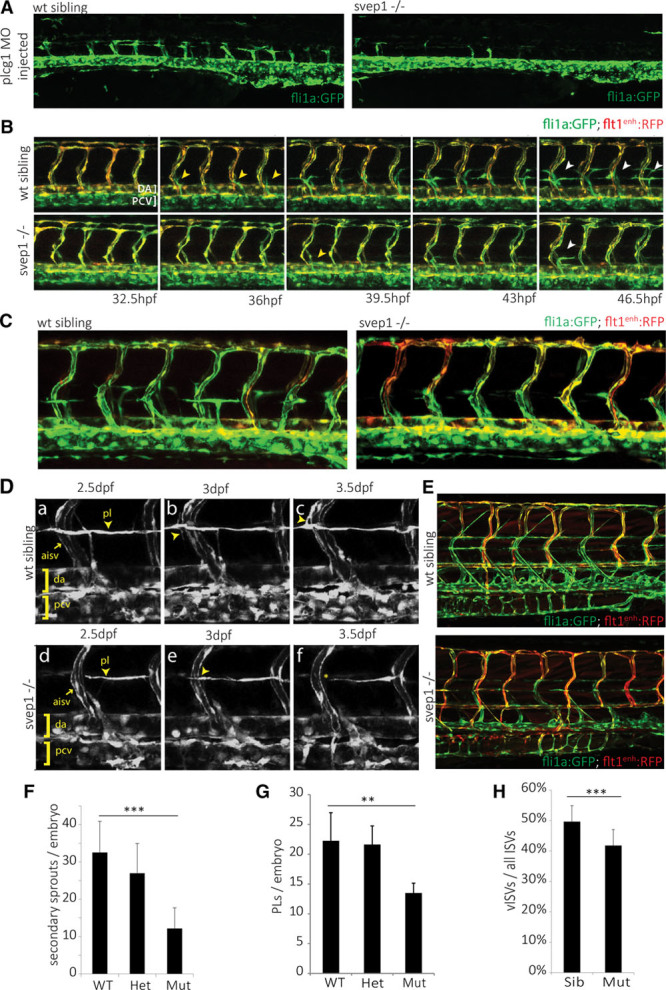
Polydom/svep1 mutants show reduced venous and lymphovenous sprouting. A and F, Quantification of sprouts from the posterior cardinal vein (PCV) in wild-type (wt) siblings and polydom/svep1 mutants, in plcg1 morphant embryos. Knockdown of plcg1 suppresses arterial formation, hence only venous structures can be observed in a fli1a:GFP transgenic background. Heterozygous embryos show a significant reduction in venous sprouting events from the PCV, and this is further exacerbated in mutant embryos at 54 h post-fertilization (hpf; wt siblings: n=29, heterozygous embryos: n=53, and polydom/svep1 mutants: n=26). B, Still frames from confocal time-lapse imaging of a wt sibling and polydom/svep1 mutant embryo in a fli1a:GFP; flt1enh:RFP double transgenic background are shown over the course of 32.5 to 46.5 hpf. Both the number of secondary sprouts from the PCV (yellow arrowheads) and parachordal lymphangioblast (PL) cells (white arrowheads) were reduced in mutant embryos. C and G, polydom/svep1 mutant embryos form a reduced number of PLs at the horizontal myoseptum (HMS) region. Confocal images of wt sibling and polydom/svep1 mutant embryos at 48 hpf in fli1a:GFP;flt1enh:RFP background and quantification of PLs at 54 hpf (wt siblings: n=8, heterozygous embryos; and n=11, polydom/svep1 mutants: n=6). D, PL cells at the level of the HMS fail to migrate along intersegmental arteries in the polydom/svep1 mutants. Still frames from confocal time-lapse imaging of a wt sibling (a–c) and a polydom/svep1 mutant embryo (d–f) in a fli1a:GFP transgenic background are shown over the course of 2.5 to 3.5 d post-fertilization (dpf). E and H, An increased number of arterial intersegmental vessels (ISVs) at the expense of venous ISVs in polydom/svep1 mutants is highlighted by flt1enh:RFP expression in fli1a:GFP background at 5 dpf (siblings: n=20, polydom/svep1 mutants: n=10). Values are presented as means±SD. **P<0.01; ***P<0.001.
We then examined the formation of PL cells, which in wild-type embryos form from those venous sprouts that do not stably connect to arterial ISVs. The formation of PL cells can be seen in Figure 3B and 3C, demonstrating robust formation of PLs at the HMS region between 32.5 and 46.5 hpf in wild-type sibling embryos. By contrast, in polydom/svep1 mutants, the number of PL cells forming was reduced (Figure 3B, 3C, and 3G). The population of PL cells in mutant embryos behaved normally about migration patterns along the horizontal midline region, but at 2.5 dpf, when in wild-type embryos PL cells started to migrate dorsally or ventrally along arterial ISVs (Figure 3D, a through c; Online Movie), in mutant embryos PL cells did not show the typical migratory behavior away from the HMS (Figure 3D, d through f), but rather remained in the midline region.
To substantiate whether PL cells at the level of the HMS are specified correctly, we performed Prox-1 antibody staining in situ at a point in time when PL cells populate the HMS region at 2 dpf. Fli1a:eGFP-positive PL cells were identified, and the proportion of cells simultaneously expressing Prox1 was quantified (Figure 4A and 4B). We found no difference in the proportion of Prox-1+ PL cells and conclude that the specification of PL cells is not deficient in svep1 mutant embryos.
Figure 4.
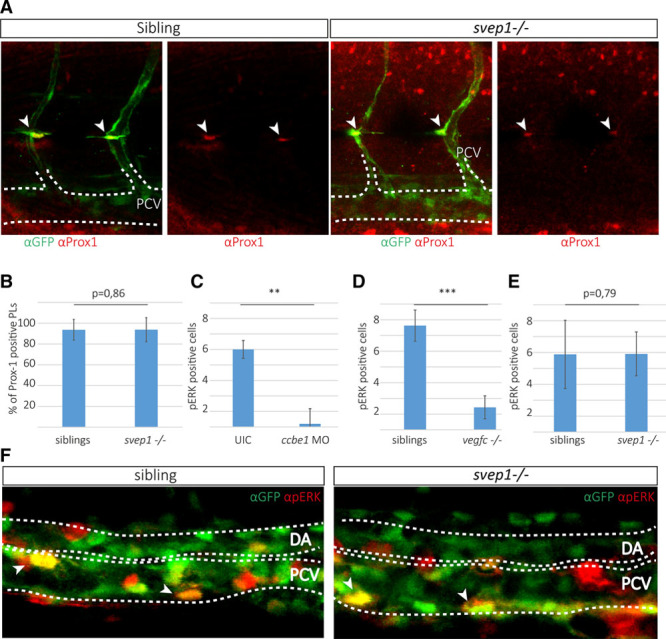
Posterior cardinal vein (PCV) cells express pERK and parachordal lymphangioblast (PL) cells express Prox-1 in svep1 mutant embryos. A, Partial maximal projection of antibody staining against Prox-1 (red) and fli1a:GFP (green) in embryos from an svep1+/−; fli1a:GFP incross at 48 h post-fertilization (hpf). Prox-1–positive PL cells are indicated by an arrowhead. B, Quantification of Prox-1–positive PL cells across 9 somites at the horizontal myoseptum (HMS) in siblings (96 out of 106 counted PLs are Prox-1 positive in 18 embryos) and svep1 mutants (28 out of 30 PLs are Prox-1 positive in 8 mutant embryos). C–E, pERK-positive cells were quantified in the PCV by scoring RFP and GFP coexpression (indicated by arrows) laterally across 6 somites in the trunk. C, In ccbe1 morpholino (MO) injected embryos (total number of 6 pERK-positive cells in 5 ccbe1 morphants; total number of 36 pERK-positive cells in 6 uninjected controls) and (D) vegf-chu6410 mutants (total number of 17 pERK-positive cells in 7 mutants and 67 pERK-positive cells in 8 siblings), the amount of pERK-positive cells is significantly reduced, whereas in svep1 mutants (E and F) no difference in pERK can be detected in the PCV (total number of 151 pERK-positive cells in 31 siblings compared with 48 pERK-positive cells in 9 svep1 mutants). F, Partial maximal projections of antibody staining against pERK (red) and fli1a:EGFP (green) in svep1+/−; fli1a:GFP incrosses show no difference in the amount of pERK-positive cells in the PCV at 32 hpf. Bar graphs show mean±SD. For statistical analysis, the Mann–Whitney test was applied in all panels.
Next, we analyzed whether Vegf-c/Vegfr-3 signaling is impaired in svep1 mutants. To this end, we measured the downstream signaling pathway of Vegfr-3 by immunofluorescence staining of phospho-ERK (phosphorylated extrecellular signal-regulated kinase) in the PCV at the time of venous sprouting.11 At 32 hpf, pERK staining was found in a subset of nuclei within the PCV endothelium. Whereas significant differences in pERK staining could be observed with ccbe1 morphants (Figure 4C) and vegf-C mutants (Figure 4D), we could not detect differences of pERK staining in svep1 mutant versus sibling embryos (Figure 4E and 4F). This suggests that Vegfr-3/pERK signaling is not affected in svep1 mutants or affected at considerably lower levels than in the absence of Ccbe1 and Vegf-c.
Next, we assessed the response of endothelial cells of svep1 mutant embryos to ectopically provided human VEGF-C expressed in the floor plate of zebrafish embryos. We could not observe a difference in Vegf-c signaling in svep1 mutants and siblings as demonstrated by the fact that at 2 dpf exogenous VEGF-C led to comparable levels of lymphovenous hypersprouting in sibling control and svep1 mutant embryos (Online Figure IIIA and IIIB). In line with this finding, double svep1/ccbe1 heterozygous zebrafish embryos did not show TD defects, indicating that Svep1 is not a component of the vegf-c/vegfr-3 pathway (Online Figure IIIC and IIID).
polydom/svep1 Is Expressed by Cells in Close Connection With Vessels, But Not by Endothelial Cells
To understand the polydom/svep1 expression pattern, we performed in situ hybridizations which demonstrated expression in multiple locations, including expression in the region of the PCV and the ISVs (Online Figure IV). To achieve cellular resolution in living embryos, we generated a svep1:Gal4FF transgenic line, introducing a Gal4FF cassette in the location of the predicted start codon of svep1 in the DKEY-8E16 BAC. Analyzing the expression in svep1:Gal4FF;UAS:eGFP transgenic embryos confirmed expression at multiple sites, including the epidermis, the cleithrum and operculum, and the trunk region (data not shown). We here restrict ourselves to the analysis of polydom/svep1 expression in regions with relevance to the vascular system. At 34 hpf, we found individual cells expressing polydom/svep1 in positions directly abutting the dorsal aorta and the PCV, as depicted in Figure 5A. Of note, in a transgenic kdrl:mCherry background, the population of polydom/svep1-positive cells (green) was not overlapping with endothelial cells (red), indicating expression by nonendothelial cells. The number of cells expressing the gene increased by 72 hpf, when a large number of polydom/svep1-positive cells surrounded the PCV (Figure 5B, 5F, and 5G, blue arrowhead). Given that PL cells migrate dorsally or ventrally almost exclusively along arterial ISVs, we examined whether polydom/svep1 expression is preferentially found around intersegmental arteries. We found no preference for arterial over venous ISVs, and in a flt1enh:RFP line which highlights arteries (red, Figure 5B and 5C), polydom/svep1-positive cells were found in an equal distribution around both venous and arterial ISVs. Again, as previously observed for endothelial cells of the PCV, polydom/svep1-positive cells were found in tight association with ISVs (Figure 5C), but there was no overlap between the 2 cell populations, further supporting the notion that endothelial cells do not express polydom/svep1. This was confirmed by RT-PCR of cultured human umbilical vein endothelial cells and LECs, in which SVEP1 mRNA could not be detected, whereas SVEP1 mRNA was abundant in the fibroblastic cell line VH32 (Online Figure VB).
Figure 5.
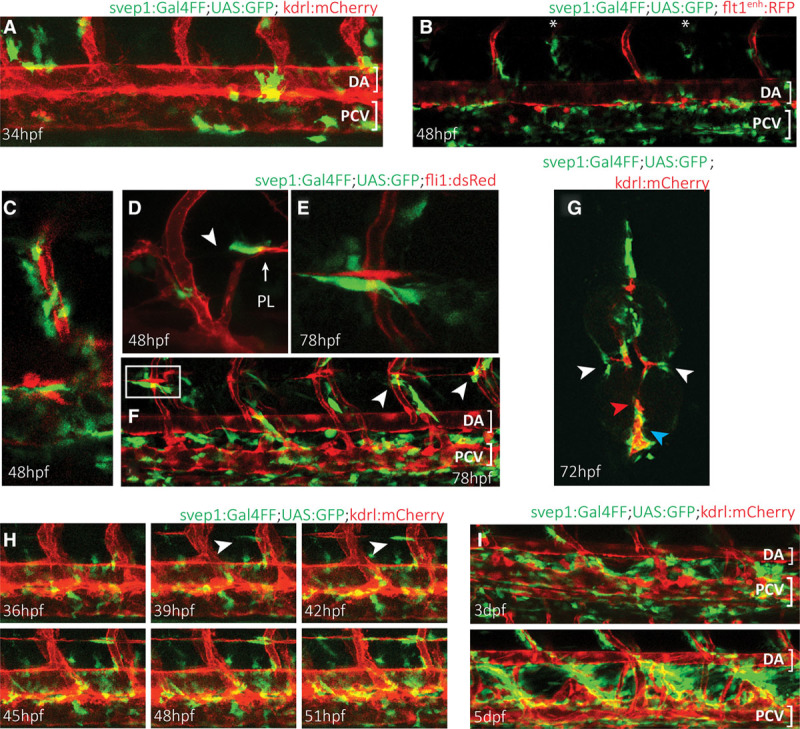
Zebrafish polydom/svep1 is expressed dynamically at regions of venous and lymphatic endothelial cell migratory activity. A, The first polydom/svep1 expression appears around 34 h post-fertilization (hpf) in nonendothelial cells along the dorsal aorta (DA) and posterior cardinal vein (PCV), as depicted by svep1:Gal4FF;UAS:GFP expression in a kdrl:mCherry transgenic background. B, By 48 hpf, the number of polydom/svep1-positive cells along the PCV has increased, and polydom/svep1-positive cells can abundantly be found in the immediate vicinity of both arterial (highlighted in red in flt1enh:RFP background) and venous (marked with asterisks) intersegmental vessels (ISVs). C, Higher magnification of an independent region at 48 hpf, demonstrating the tight connection between endothelial cells (red) and polydom/svep1-positive cells (green). D–F, Between 48 and 72 hpf, when parachordal lymphangioblast (PL) cells populate the horizontal myoseptum (HMS) region and start to migrate dorsally and ventrally along arterial ISVs, individual cells in the midline of the embryo start to express polydom/svep1. The polydom/svep1-expressing cells are in a immediate contact with migrating PL cells. G, Cross-section of a 72 hpf svep1:Gal4FF;UAS:GFP embryo in a kdrl:mCherry background. Note the close association of PL cells (red) and polydom/svep1-positive cells (green) in the HMS region (white arrow heads), and of PCV cells and polydom/svep1-positive cells (blue arrow head). The dorsal aorta (red arrow head) is not covered by polydom/svep1-positive cells at this time point any more. H, Still frames of a confocal time-lapse imaging of a svep1:Gal4FF;UAS:GFP;kdrl:mCherry embryo from 36 to 51 hpf. Polydom/svep1-positive cells (green) and PL cells (red) start appearing at the horizontal midline region around 39 hpf. Note the almost simultaneous appearance of both cell types. The full movie can be seen online. I, At 3 and 5 d post-fertilization (dpf), polydom/svep1 expression can be seen in between the DA and the PCV, exactly in the region, which the lymphatic cells of the future thoracic duct (TD) will populate.
Around 48 hpf, the first PL cells are identifiable in the HMS region, and we noticed that also polydom/svep1-positive cells were present in the region, usually closely abutting PL cells (Figure 5D). The close association of PL cells and polydom/svep1-expressing cells became even more evident at 72 to 78 hpf (Figure 5E through 5G). To determine whether PL cells populate the HMS early enough to possibly induce polydom/svep1 expression, we followed time-lapse sequences and observed that the expression of polydom/svep1 seemed to be independent of the presence of PL cells: at 39 hpf, expression of 1 or 2 polydom/svep1-expressing cells became apparent, and only a few hours later PL cells also started to populate the region (Figure 5H). Strikingly, there are always cells that express polydom/svep1 in the HMS region close to arteries at those points in time when PL cells need to make the critical transition to migrate dorsally or ventrally, suggesting that the presence of Polydom/Svep1 protein might be instrumental in governing this migratory step.
At later stages, when the TD formed in zebrafish embryos, polydom/svep1 expression was abundantly detected between the dorsal aorta and the PCV, that is, in the region of TD formation (Figure 5I). Neither at this point in time nor at earlier stages was it possible to unambiguously identify the nature of the cells that expressed polydom/svep1. We excluded muscle pioneer cells and neurons at 48 hpf (data not shown) and hence consider it most likely that mesenchymal cells are the cells producing Polydom/Svep1. This is consistent with in vitro expression data using mammalian cell lines (Online Figure VB) and in situ antibody staining in mouse embryos (Morooka et al25).
Polydom/Svep1 Is Required at Later Stages of Lymphatic Vascular Development and Valve Formation in Mice
To examine a possible evolutionarily conserved role of the Polydom/Svep1 protein, we generated knockout mice for the murine orthologue of the gene, based on the EUCOMM knockout first strategy (Figure 6A; see online information for details). Crossing of heterozygous carriers yielded homozygous mutant embryos (Figure 6B and 6C; Online Figures VI and VII), which were obtained at a normal Mendelian ratio (n=130 total, with 31 Polydom/Svep1+/+, 67 Polydom/Svep1+/−, and 32 Polydom/Svep1−/− from 17 litters at E14.5). Homozygous wild-type and heterozygous embryos were indistinguishable, but mutant embryos at E14.5 or older could often be recognized because of edema formation, which was somewhat variable in phenotypic strength (Online Figure VII). Mutant embryos at E18.5 always showed edema (Figure 6C).
Figure 6.
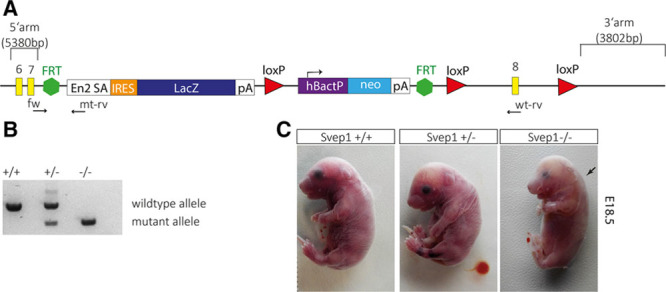
Characterization of Polydom/Svep1 function in mice. A, Targeting construct to create a Polydom/Svep1 knockin mouse. The LacZ construct disrupts the locus and is predicted to result in a protein truncated after amino acid 559. The primers used for genotyping are indicated as fw, mt-rv, and wt-rv. B, Genotyping using the PCR primers clearly distinguishes the wild-type from the mutant allele. C, E18.5 litters from heterozygous parents contained mutant and sibling embryos in normal Mendelian ratios. Heterozygous siblings are indistinguishable from wild-type embryos, but mutant embryos show a clear nuchal edema (arrow). FRT indicates flippase recognition target sites; fw, forward primer; loxP (locus of X-over P1), recognition sites for Cre recombinase; mt-rv, mutant reverse primer; and wt-rv, wild-type reverse primer.
To determine the exact onset and cause of the phenotype, we analyzed sibling and mutant E11.0, E12.5, and E13.5 embryos using whole mount antibody staining.3 Using VEGFR-3 antibodies to stain venous and lymphatic cells, and Prox-1 antibodies to highlight LECs, we did not observe any differences in formation of the first lymphatic structures between sibling and mutant E11.0 and E12.5 embryos. Egression of future LECs from the CV occurred normally in mutant embryos (Figure 7A through 7F), and formation of the primordial TD and the peripheral longitudinal lymphatic vessel progressed in mutant embryos in a manner indistinguishable from wild-type siblings. At E12.5, however, we noticed that the formation of the first lymphovenous contact site did not develop normally in mutant embryos. Whereas in sibling embryos, the endothelial layer of the CV and the cells of the primordial TD were observed in close juxtaposition, in mutant embryos these layers were spatially separated and clearly distinguishable (Figure 7G and 7H). In wild-type embryos, we observed an accumulation of Prox-1/Vegfr-3–positive cells at the lymphovenous connection site (Figure 7E, large arrow), whereas this typical clustering of cells did not occur in mutant embryos (Figure 7F, large arrow). Also in optical cross-sections (Figure 7G through 7J), the tight association of lymphatic and venous endothelium, a characteristic of the site of prospective valve formation in sibling embryos, was not observed in mutant embryos.
Figure 7.
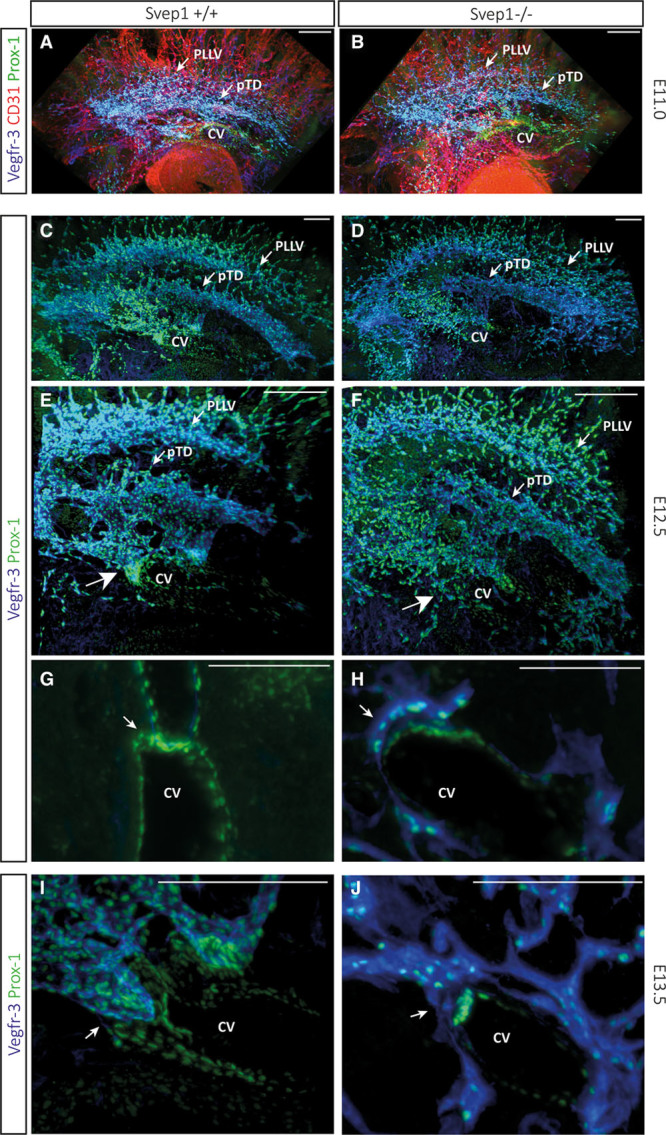
The first detectable abnormalities in Polydom/Svep1 mutant embryos occur at the level of lymphovenous valve formation. To determine the first phenotypic abnormalities in mutant embryos, we used whole-mount imaging on the ultramicroscope at E11.0, E12.5, and E13.5 embryos. Combined antibody staining highlighting CD31, Prox-1, and Vegfr-3 expression at E11.0 (A and B), and Prox-1 and Vegfr-3 at E12.5 (C–H) did not reveal any alterations between wild-type sibling and mutant embryos. In all cases examined, we could not detect differences in future lymphatic endothelial cells (LECs) egressing from the cardinal vein (CV), and the formation of the primitive primordial thoracic duct (pTD) and the peripheral longitudinal lymphatic vessel (pLLV) seemed unchanged. However, we did notice at E12.5 a difference at the level of the first lymphovenous connection: here, Prox-1–positive cells of the CV and Prox-1–positive LECs come together in tight association (G), but this association was not found in mutant embryos (H). I and J, Closer view of the lymphovenous contact site in volume reconstructions at E13.5. Whereas in wild-type embryos the dual contact sites are massive structures composed of multiple cells, in Polydom/Svep1 deficient embryos only few individual cells are actually in contact to the high Prox-1–positive expression domain inside the CV. Scale bars correspond to 100 μm in all panels.
Next, we analyzed lymphatic remodeling in E18.5 embryos by staining mesenteries for VEGFR-3, CD31, and Prox-1. In wild-type sibling embryos, remodeling of lymphatic vessels was apparent, including the formation of lymphatic valve structures that retained high levels of all 3 markers used, while these were downregulated elsewhere in collecting lymphatic vessels (Figure 8A through 8E). We noted that valve structures were absent in the mesentery of homozygous mutant embryos (Figure 8F through 8J). We also observed that the lymphatic vessels in mutants were not lumenized, were considerably smaller than in siblings, and retained higher levels of VEGFR-3, CD31, and Prox-1 expression (Figure 8A, 8B, 8F, and 8G).
Figure 8.
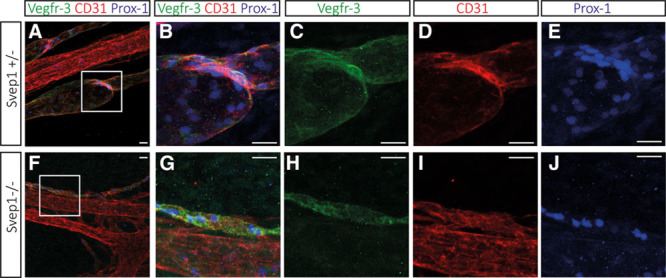
Polydom/Svep1 mutant embryos fail to remodel mesenteric lymphatic vessels at E18.5 and do not form valves. Lymphatic structures in the mesenteries of mutant embryos are closely associated with blood vessels as they are in siblings, but they are significantly smaller in size, appear nonluminized and do not contain valve structures. A–J, Whole-mount immunofluorescent staining of mesenteric vessels for Vegfr-3, CD31, and Prox-1 at E18.5 of a Svep1+/− and Svep1−/− embryo. At this time of the development, the maturation of lymphatic vessels is well underway, including the formation of lymphatic valve structures that retain high levels of Prox-1 protein. CD31 and VEGFR-3 levels are downregulated in the lymphatic vessels but remain high in the lymphatic valve regions. A, Overlay picture of VEGFR-3, CD31, and Prox-1 staining with the boxed region shown in higher magnification in B–E; F–J, Immunofluorescent staining of vessels in a E18.5 Svep1−/− embryo indicates that the size of the lymphatic vessel is dramatically decreased. Prox-1 expression remains high in the lymphatic endothelial cells (LECs), and lymphatic valves fail to form. F, Overlay picture of VEGFR-3, CD31, and Prox-1 staining with the boxed area shown in higher magnification in G–J. Scale bars correspond to 50 μm in all panels.
Discussion
Lymphangiogenesis and maturation of lymphatic vessels are essential to form a functional lymphatic vasculature.1,2 Here, we introduce the Polydom/Svep1 protein as a key factor for embryonic lymphangiogenesis and demonstrate that in both zebrafish and mouse mutants lack of the protein function results in defective lymphatic vasculature and lymphedema formation.
The formation of a functional lymphatic vascular plexus is a complex process that requires a series of sequential events. First, lymphatic endothelial sprouting occurs within the main embryonic vein. Second, cells that separate from the CV aggregate into lymphatic vascular structures, and these structures form a lumen. Third, in higher vertebrates, remodeling steps result in the formation of a hierarchical lymphatic vasculature, where collecting lymphatic vessels develop intraluminal valves and obtain pericyte coverage.14,15 The latter events have not been reported in teleosts, but earlier events such as sprouting from the embryonic CV seem to be genetically highly conserved: Prox-1 expression in future LECs is a marker for lymphatic fate,29–31 Vegf-c is the chemoattractant that is required for sprouting from the vein,5–7 and Vegfr3 constitutes the main receptor for Vegf-c within the venous and lymphatic endothelium.24,32,33 Given the significant evolutionary distances between the different phyla, it is not surprising that there are some differences: for example, zebrafish Prox-1 function is required maternally29 rather than zygotically,30 and lymphovenous sprouting in zebrafish is a simultaneous process rather than a sequential one as in mice. Nevertheless, the genetic analysis of key players in the process demonstrates a high degree of genetic conservation in respect to Prox-1, Vegf-c, Ccbe1, and Vegfr-3 function.7,8,11,13,24,29,30,34
We identified a zebrafish mutant that shows reduced venous sprouting. The degree of phenotypic severity varies, but there is a consistent and significant reduction of venous sprouting. This results in a decrease in venous ISV formation, in a consequential increase of arteries, and in a reduction of the number of PL cells in the HMS region. The PL cells that do succeed in populating the HMS region exhibit an additional phenotype: the PL cells fail to initiate migration along arteries in a dorsal or ventral direction and remain in the HMS region. Consequently, mutant embryos lack most, or all of the TD. This later aspect of the phenotype is reminiscent of PL migratory defects along arteries in embryos deficient of chemokine signaling,20 but in the absence of Cxcl12a/Cxcr4a/b signaling the initial sprouting from the PCV and the subsequent migration of PL cells to the HMS region occurs normally. We, therefore, consider it unlikely that Svep1 is involved in chemokine signaling.
Three mutant alleles of the polydom/svep1 gene were identified, and all 3 alleles encode nonsense mutations that are predicted to result in truncated proteins. The polydom/svep1 gene is predicted to encode a large secreted protein comprising 3555 amino acids, which is likely to be part of the extracellular matrix. The protein contains many different domains (hence the name Polydom),26,27 among which are Sushi repeats, a von Willebrand factor type A domain, epidermal growth factor, and epidermal growth factor–like calcium-binding repeats, and a pentraxin domain (hence the alternative name Svep1). None of these domains provide an intuitive explanation for the involvement of the protein in lymphangiogenesis. Polydom/Svep1 has been reported to bind strongly to Integrin α9β1,35 which in turn has been shown to play a critical role in valve morphogenesis in mice.17,35 We have confirmed that Integrin α9β1 and Svep1 protein are colocalized (Online Figure VIII) and turned to the zebrafish to test a possible genetic interaction. We generated a zebrafish integrin α9 mutant allele using the TALEN technology, but homozygous mutant embryos are viable and do not show a lymphatic phenotype (Online Figure IX). Also integrin α9 mutants that were heterozygous for polydom/svep1 show no phenotype. Furthermore, the EDDMMEVPY motif within the 21st CCP module, which constitutes the sequence binding most strongly to Integrin α9β1 protein,35 is lacking in the zebrafish Polydom/Svep1 protein. This, and the absence of a lymphatic phenotype in zebrafish integrin α9 mutants, makes it unlikely that Polydom/Svep1 acts exclusively through Integrin α9. Of course, other integrins might constitute binding partners for Polydom/Svep1, and we have analyzed integrin α4 and α5 mRNA distribution. Although the expression patterns of integrin α4 and α5 do not overlap with polydom/svep1 mRNA distribution in all aspects (Online Figure X), we are nevertheless currently generating mutant alleles for these genes as well. We have also considered the presence of RGD (arginine-glycine-aspartic acid) motifs in the Polydom/Svep1 protein, and indeed the zebrafish protein is predicted to contain 2 RGD domains. However, other vertebrate species do only contain one such motif (rat and human) or none (mouse, cat, guinea pig, and sheep). Furthermore, the sites of RGD motifs within the respective proteins are not conserved between different vertebrate species. We thus consider it unlikely that RGD motifs are functionally relevant.
To describe polydom/svep1 expression with cellular resolution and to make use of the unique imaging properties of early zebrafish embryos, we generated a transgenic line with a Gal4FF cassette inserted into the predicted start ATG position of the polydom/svep1 locus. Gene expression was predominantly found around the PCV, around ISVs, and in the HMS region. Whereas expression around the PCV is consistent with a venous sprouting defect in mutant embryos, the temporal and spatial expression pattern of few cells per somite at the HMS is particularly intriguing: expression commences just at the time before PL cells populate the region (Figure 5H), and in a position which coincides later with the area where PL cells migrate dorsally or ventrally. The defect in PL cell migration is a unique feature of the polydom/svep1 mutant, and the temporally and spatially restricted expression of the gene is likely to suggest that providing Polydom/Svep1 protein by a few cells is required to either instruct or allow PL cells to move ventrally or dorsally along arteries.
The nature of the cells expressing polydom/svep1 could not be unambiguously verified. Coexpression of the svep1:Gal4FF;UAS:GFP signal with any endothelial-specific reporter was never observed, suggesting nonendothelial expression. We have furthermore excluded muscle pioneer or neuronal cells as the source of Polydom/Svep1 protein and consider it, therefore, most likely that mesenchymal cells or pericytes produce Svep1. This is consistent with in vitro data of human cells and in situ lacZ expression data in mice, which also support a nonendothelial expression domain (Online Figure V). Furthermore, functional tests by Morooka et al25 did not yield a lymphatic phenotype in Tie2:Cre-mediated, endothelial-specific knockouts of Polydom/Svep1. Therefore, it is most likely that mesenchymal cells in both mouse and fish express Polydom/Svep1, and it will be interesting to see what regulates the highly specific expression pattern in the zebrafish HMS.
The mouse Polydom/Svep1 mutants present with edema formation. The phenotypic strength varies to some extent and is, in this respect, reminiscent of the variable venous sprouting phenotype in zebrafish mutants. At E18.5, the edema formation could be mild or severe (Online Figure VII). Nevertheless, all mutant embryos develop edema by E18.5. On the basis of the early venous sprouting defects in zebrafish mutants, a similar defect might have been predicted to occur in mouse mutants. However, whole-mount embryo analysis3 at E11.0 and E12.5 failed to provide evidence for early migratory defects in Prox-1–positive, future LECs. Rather, we noticed that the first defect in mutants appears at the first lymphovenous contact site at E12.5 and E13.5 (Figure 7G through 7J). Although in wild-type siblings a significant accumulation of Prox-1+ cells is observed in the region of lymphovenous juxtaposition, there are only few cells in mutants that aggregate in the correct location, and their association is not tight (Figure 7H and 7J). Of note, a similar phenotype has not been reported in integrin α9 mutants,14,17 rendering support to the notion that integrin α9 is unlikely to fully explain the Polydom/Svep1 phenotype.
During later stages of development, we focused on lymphatic maturation and remodeling events in the intestinal mesentery, as valve morphogenesis has been studied in this tissue in detail.14,17 Mutant Polydom/Svep1 embryos show only thin lymphatic structures associated with apparently normally developed arteries and veins, without an apparent lumen and without any valves. Hence, whereas in zebrafish embryos the lack of Polydom/Svep1 results in a reduced number of LECs to sprout from the CV and a failure to execute the correct migratory pattern at the level of the HMS, in mouse mutants early events occur normally. Rather, Polydom/Svep1 controls remodeling steps both early and late. This raises questions about the evolutionary implications of these different phenotypes, which manifest themselves in the same vascular bed. A better understanding of the molecular pathway which Polydom/Svep1 is a part of will be required to fully understand this phenomenon.
In summary, we here show the extracellular matrix protein Polydom/Svep1 to be required for key events during lymphatic vascular formation in fish and mice. Although there are differences in how the respective phenotypes emerge, intriguingly in both species, the protein is provided by nonendothelial cells close to the lymphatic vascular bed, and in both organisms the gene is essential for lymphatic vascular development.
Acknowledgments
We thank the Sekiguchi and Mochizuki laboratories for an extremely constructive collaboration in a collegial spirit. We thank Jeroen Korving for generating the mutant Polydom/Svep1 allele. Imaging was performed at the Hubrecht Imaging Center (HIC).
Sources of Funding
The work was supported by the CiM Cluster of Excellence (WWU Münster, Germany) and the DFG (Deutsche Forschungs-Gemeinschaft; SCHU 1228/3-1 to S.S.-M.; SFB656 and SFB629 to F.K.). An EMBO long-term fellowship, as well as a NWO (The Netherlands Organisation for Scientific Research) VENI grant, was awarded to T. Karpanen. N. Morooka received a travel fellowship from the Company of Biologists.
Supplementary Material
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- CV
- cardinal vein
- dpf
- days post-fertilization
- HMS
- horizontal myoseptum
- hpf
- hours post-fertilization
- ISVs
- intersegmental vessels
- LEC
- lymphatic endothelial cell
- PCV
- posterior cardinal vein
- PL
- parachordal lymphangioblast
- TD
- thoracic duct
- VEGF-C
- vascular endothelial growth factor-C
- VEGFR-3
- vascular endothelial growth factor receptor-3
In January 2017, the average time from submission to first decision for all original research papers submitted to Circulation Research was 13.77 days.
Current address T. Karpanen: Department of Cancer Immunology, Institute for Cancer Research, Oslo University Hospital Radiumhospitalet and K.G. Jebsen Center for Cancer Immunotherapy, Institute of Clinical Medicine, University of Oslo, Norway.
These authors contributed equally to this article.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.116.308813/-/DC1.
Novelty and Significance
What Is Known?
Lymphangiogenesis involves a complex array of embryonic cellular movements and, at later stages, an extensive set of maturation events to allow lymphatic vessels to exert their function.
In vertebrates, the development of the lymphatic system is evolutionarily conserved.
What New Information Does This Article Contribute?
The extracellular protein Polydom/Svep1 is essential for lymphatic vessel formation in fish and mice.
Zebrafish embryos mutant in svep1 show defective venous sprouting and aberrant migration behavior of future lymphatic endothelial cells.
Murine Polydom/Svep1 mutant embryos develop severe edema in utero, caused by defective lymphovenous connections, missing valve structures, and remodeling defects.
Lymphatic vessels play key roles in many physiological processes, but the genetic control of lymphatic vessel formation and lymphatic endothelial cell function is poorly understood. We show here that the large secreted Polydom/Svep1 protein, which has previously not been connected to lymphatic vessel formation or function, controls key steps of lymphatic development in zebrafish and mice. The combined analysis of zebrafish and mouse mutants demonstrates that the Polydom/Svep1 protein influences different aspects of lymphatic development in the respective species: zebrafish mutant embryos exhibit defects in early lymphatic endothelial cell migration, whereas mouse mutant embryos show defects at later stages of the development. This study introduces a new key gene function to the field of lymphatic endothelial cell biology, and demonstrates that Polydom/Svep1 is indispensable for different steps during lymphatic development.
References
- 1.Alitalo K. The lymphatic vasculature in disease. Nat Med. 2011;7:1371–1380. doi: 10.1038/nm.2545. doi: 10.1038/nm.2545. [DOI] [PubMed] [Google Scholar]
- 2.Schulte-Merker S, Sabine A, Petrova TV. Lymphatic vascular morphogenesis in development, physiology, and disease. J Cell Biol. 2011;193:607–618. doi: 10.1083/jcb.201012094. doi: 10.1083/jcb.201012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hägerling R, Pollmann C, Andreas M, Schmidt C, Nurmi H, Adams RH, Alitalo K, Andresen V, Schulte-Merker S, Kiefer F. A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. EMBO J. 2013;32:629–644. doi: 10.1038/emboj.2012.340. doi: 10.1038/emboj.2012.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ny A, Koch M, Schneider M, et al. A genetic Xenopus laevis tadpole model to study lymphangiogenesis. Nat Med. 2005;11:998–1004. doi: 10.1038/nm1285. doi: 10.1038/nm1285. [DOI] [PubMed] [Google Scholar]
- 5.Küchler AM, Gjini E, Peterson-Maduro J, Cancilla B, Wolburg H, Schulte-Merker S. Development of the zebrafish lymphatic system requires VEGFC signaling. Curr Biol. 2006;16:1244–1248. doi: 10.1016/j.cub.2006.05.026. doi: 10.1016/j.cub.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 6.Yaniv K, Isogai S, Castranova D, Dye L, Hitomi J, Weinstein BM. Live imaging of lymphatic development in the zebrafish. Nat Med. 2006;12:711–716. doi: 10.1038/nm1427. doi: 10.1038/nm1427. [DOI] [PubMed] [Google Scholar]
- 7.Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5:74–80. doi: 10.1038/ni1013. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- 8.Bos FL, Caunt M, Peterson-Maduro J, Planas-Paz L, Kowalski J, Karpanen T, van Impel A, Tong R, Ernst JA, Korving J, van Es JH, Lammert E, Duckers HJ, Schulte-Merker S. CCBE1 is essential for mammalian lymphatic vascular development and enhances the lymphangiogenic effect of vascular endothelial growth factor-C in vivo. Circ Res. 2011;109:486–491. doi: 10.1161/CIRCRESAHA.111.250738. doi: 10.1161/CIRCRESAHA.111.250738. [DOI] [PubMed] [Google Scholar]
- 9.Janssen L, Dupont L, Bekhouche M, Noel A, Leduc C, Voz M, Peers B, Cataldo D, Apte SS, Dubail J, Colige A. ADAMTS3 activity is mandatory for embryonic lymphangiogenesis and regulates placental angiogenesis. Angiogenesis. 2016;19:53–65. doi: 10.1007/s10456-015-9488-z. doi: 10.1007/s10456-015-9488-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roukens MG, Peterson-Maduro J, Padberg Y, Jeltsch M, Leppänen VM, Bos FL, Alitalo K, Schulte-Merker S, Schulte D. Functional dissection of the CCBE1 protein: a crucial requirement for the collagen repeat domain. Circ Res. 2015;116:1660–1669. doi: 10.1161/CIRCRESAHA.116.304949. doi: 10.1161/CIRCRESAHA.116.304949. [DOI] [PubMed] [Google Scholar]
- 11.Le Guen L, Karpanen T, Schulte D, Harris NC, Koltowska K, Roukens G, Bower NI, van Impel A, Stacker SA, Achen MG, Schulte-Merker S, Hogan BM. Ccbe1 regulates VEGFC-mediated induction of VEGFR3 signaling during embryonic lymphangiogenesis. Development. 2014;141:1239–1249. doi: 10.1242/dev.100495. doi: 10.1242/dev.100495. [DOI] [PubMed] [Google Scholar]
- 12.Jeltsch M, Jha SK, Tvorogov D, Anisimov A, Leppänen VM, Holopainen T, Kivelä R, Ortega S, Kärpanen T, Alitalo K. CCBE1 enhances lymphangiogenesis via A disintegrin and metalloprotease with thrombospondin motifs-3-mediated vascular endothelial growth factor-C activation. Circulation. 2014;129:1962–1971. doi: 10.1161/CIRCULATIONAHA.113.002779. doi: 10.1161/CIRCULATIONAHA.113.002779. [DOI] [PubMed] [Google Scholar]
- 13.Hogan BM, Bos FL, Bussmann J, Witte M, Chi NC, Duckers HJ, Schulte-Merker S. Ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nat Genet. 2009;41:396–398. doi: 10.1038/ng.321. doi: 10.1038/ng.321. [DOI] [PubMed] [Google Scholar]
- 14.Norrmén C, Ivanov KI, Cheng J, Zangger N, Delorenzi M, Jaquet M, Miura N, Puolakkainen P, Horsley V, Hu J, Augustin HG, Ylä-Herttuala S, Alitalo K, Petrova TV. FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J Cell Biol. 2009;185:439–457. doi: 10.1083/jcb.200901104. doi: 10.1083/jcb.200901104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrova TV, Karpanen T, Norrmén C, Mellor R, Tamakoshi T, Finegold D, Ferrell R, Kerjaschki D, Mortimer P, Ylä-Herttuala S, Miura N, Alitalo K. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med. 2004;10:974–981. doi: 10.1038/nm1094. doi: 10.1038/nm1094. [DOI] [PubMed] [Google Scholar]
- 16.Finegold DN, Kimak MA, Lawrence EC, Levinson KL, Cherniske EM, Pober BR, Dunlap JW, Ferrell RE. Truncating mutations in FOXC2 cause multiple lymphedema syndromes. Hum Mol Genet. 2001;10:1185–1189. doi: 10.1093/hmg/10.11.1185. [DOI] [PubMed] [Google Scholar]
- 17.Bazigou E, Xie S, Chen C, Weston A, Miura N, Sorokin L, Adams R, Muro AF, Sheppard D, Makinen T. Integrin-alpha9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev Cell. 2009;17:175–186. doi: 10.1016/j.devcel.2009.06.017. doi: 10.1016/j.devcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bussmann J, Bos FL, Urasaki A, Kawakami K, Duckers HJ, Schulte-Merker S. Arteries provide essential guidance cues for lymphatic endothelial cells in the zebrafish trunk. Development. 2010;137:2653–2657. doi: 10.1242/dev.048207. doi: 10.1242/dev.048207. [DOI] [PubMed] [Google Scholar]
- 19.Geudens I, Herpers R, Hermans K, et al. Role of delta-like-4/NOTCH in the formation and wiring of the lymphatic network in zebrafish. Arterioscler Thromb Vasc Biol. 2010;30:1695–1702. doi: 10.1161/ATVBAHA.110.203034. doi: 10.1161/ATVBAHA.110.203034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cha YR, Fujita M, Butler M, Isogai S, Kochhan E, Siekmann AF, Weinstein BM. Chemokine signaling directs trunk lymphatic network formation along the preexisting blood vasculature. Dev Cell. 2012;22:824–836. doi: 10.1016/j.devcel.2012.01.011. doi: 10.1016/j.devcel.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bussmann J, Schulte-Merker S. Rapid BAC selection for tol2-mediated transgenesis in zebrafish. Development. 2011;138:4327–4332. doi: 10.1242/dev.068080. doi: 10.1242/dev.068080. [DOI] [PubMed] [Google Scholar]
- 22.Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002;248:307–318. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- 23.Villefranc JA, Nicoli S, Bentley K, Jeltsch M, Zarkada G, Moore JC, Gerhardt H, Alitalo K, Lawson ND. A truncation allele in vascular endothelial growth factor c reveals distinct modes of signaling during lymphatic and vascular development. Development. 2013;140:1497–1506. doi: 10.1242/dev.084152. doi: 10.1242/dev.084152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hogan BM, Herpers R, Witte M, Heloterä H, Alitalo K, Duckers HJ, Schulte-Merker S. Vegfc/Flt4 signalling is suppressed by Dll4 in developing zebrafish intersegmental arteries. Development. 2009;136:4001–4009. doi: 10.1242/dev.039990. doi: 10.1242/dev.039990. [DOI] [PubMed] [Google Scholar]
- 25.Morooka N, Futaki S, Sato-Nishiuchi R, Nishino M, Totani Y, Shimono C, Nakano I, Nakajima H, Mochizuki N, Sekiguchi K. Polydom is an extracellular matrix protein involved in lymphatic vessel remodeling. Circ Res. 2017;120:1276–1288. doi: 10.1161/CIRCRESAHA.116.308825. doi: 10.1161/CIRCRESAHA.116.308825. [DOI] [PubMed] [Google Scholar]
- 26.Gilgès D, Vinit MA, Callebaut I, Coulombel L, Cacheux V, Romeo PH, Vigon I. Polydom: a secreted protein with pentraxin, complement control protein, epidermal growth factor and von Willebrand factor A domains. Biochem J. 2000;352(pt 1):49–59. [PMC free article] [PubMed] [Google Scholar]
- 27.Shur I, Socher R, Hameiri M, Fried A, Benayahu D. Molecular and cellular characterization of SEL-OB/SVEP1 in osteogenic cells in vivo and in vitro. J Cell Physiol. 2006;206:420–427. doi: 10.1002/jcp.20497. doi: 10.1002/jcp.20497. [DOI] [PubMed] [Google Scholar]
- 28.Lawson ND, Mugford JW, Diamond BA, Weinstein BM. Phospholipase C gamma-1 is required downstream of vascular endothelial growth factor during arterial development. Genes Dev. 2003;17:1346–1351. doi: 10.1101/gad.1072203. doi: 10.1101/gad.1072203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koltowska K, Lagendijk AK, Pichol-Thievend C, Fischer JC, Francois M, Ober EA, Yap AS, Hogan BM. VEGFC regulates bipotential precursor division and Prox1 expression to promote lymphatic identity in zebrafish. Cell Rep. 2015;13:1828–1841. doi: 10.1016/j.celrep.2015.10.055. doi: 10.1016/j.celrep.2015.10.055. [DOI] [PubMed] [Google Scholar]
- 30.van Impel A, Zhao Z, Hermkens DM, Roukens MG, Fischer JC, Peterson-Maduro J, Duckers H, Ober EA, Ingham PW, Schulte-Merker S. Divergence of zebrafish and mouse lymphatic cell fate specification pathways. Development. 2014;141:1228–1238. doi: 10.1242/dev.105031. doi: 10.1242/dev.105031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell. 1999;98:769–778. doi: 10.1016/s0092-8674(00)81511-1. [DOI] [PubMed] [Google Scholar]
- 32.Mäkinen T, Jussila L, Veikkola T, Karpanen T, Kettunen MI, Pulkkanen KJ, Kauppinen R, Jackson DG, Kubo H, Nishikawa S, Ylä-Herttuala S, Alitalo K. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med. 2001;7:199–205. doi: 10.1038/84651. doi: 10.1038/84651. [DOI] [PubMed] [Google Scholar]
- 33.Veikkola T, Jussila L, Makinen T, Karpanen T, Jeltsch M, Petrova TV, Kubo H, Thurston G, McDonald DM, Achen MG, Stacker SA, Alitalo K. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. 2001;20:1223–1231. doi: 10.1093/emboj/20.6.1223. doi: 10.1093/emboj/20.6.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gordon K, Schulte D, Brice G, Simpson MA, Roukens MG, van Impel A, Connell F, Kalidas K, Jeffery S, Mortimer PS, Mansour S, Schulte-Merker S, Ostergaard P. Mutation in vascular endothelial growth factor-C, a ligand for vascular endothelial growth factor receptor-3, is associated with autosomal dominant Milroy-like primary lymphedema. Circ Res. 2013;112:956–960. doi: 10.1161/CIRCRESAHA.113.300350. doi: 10.1161/CIRCRESAHA.113.300350. [DOI] [PubMed] [Google Scholar]
- 35.Sato-Nishiuchi R, Nakano I, Ozawa A, Sato Y, Takeichi M, Kiyozumi D, Yamazaki K, Yasunaga T, Futaki S, Sekiguchi K. Polydom/SVEP1 is a ligand for integrin α9β1. J Biol Chem. 2012;287:25615–25630. doi: 10.1074/jbc.M112.355016. doi: 10.1074/jbc.M112.355016. [DOI] [PMC free article] [PubMed] [Google Scholar]