


Abstract
We identify a novel activity of the RarA (also MgsA) protein of Escherichia coli, demonstrating that this protein functions at DNA ends to generate flaps. A AAA+ ATPase in the clamp loader clade, RarA protein is part of a highly conserved family of DNA metabolism proteins. We demonstrate that RarA binds to double-stranded DNA in its ATP-bound state and single-stranded DNA in its apo state. RarA ATPase activity is stimulated by single-stranded DNA gaps and double-stranded DNA ends. At these double-stranded DNA ends, RarA couples the energy of ATP binding and hydrolysis to separating the strands of duplex DNA, creating flaps. We hypothesize that the creation of a flap at the site of a leading strand discontinuity could, in principle, allow DnaB and the associated replisome to continue DNA synthesis without impediment, with leading strand re-priming by DnaG. Replication forks could thus be rescued in a manner that does not involve replisome disassembly or reassembly, albeit with loss of one of the two chromosomal products of a replication cycle.
INTRODUCTION
When a cellular replication fork encounters damage or barriers in the DNA template, it can stall or collapse (1–11). The damage/barriers may be DNA lesions, template strand breaks, actively transcribing RNA polymerases, or other tightly bound protein-DNA complexes (1–13). Failure to recognize, respond to, and resolve these replication fork encounters in a timely manner can threaten genomic integrity and cell viability. All cells have evolved diverse pathways to circumvent or directly repair replication fork impediments of all types (1–14). RecA family recombinases are major contributors to the reconstitution of active replication forks, particularly when the deleterious event generates a DNA double strand break (1–14). For example, if a replication fork encounters a discontinuity in the leading strand template, one arm of the fork could become separated, generating a double strand break. In bacteria, the most likely path for accurate repair of this event would involve RecA-mediated double strand break repair (DSBR) (1–7). In Escherichia coli, no pathway that could provide an alternative to DSBR in this situation, and lead to genome preservation, has been proposed to date. As one chromosome arm detaches, the DnaB helicase would find itself in a single strand gap, and (unless blocked or removed from the DNA) would translocate unproductively on the adjacent parental duplex DNA (15–18). Thus, DSBR of the broken fork in Figure 1 would presumably be accompanied by dismantling of the replisome.
Figure 1.

Replication fork encounters with leading strand template discontinuities. In bacteria, when a replication fork encounters a discontinuity on the leading strand template, it can collapse, forming a double-strand break and a single-strand gap. The replicative helicase, DnaB, is unable to continue unwinding the parental duplex due to the absence of a 3΄ single-stranded flap. In Escherichia coli, known pathways for restoring replication in this situation generally rely on RecA-mediated recombinational DNA repair.
However, there is another way—not previously considered—to enzymatically resolve this broken fork. If a flap could be generated at the end of the gap in the remaining contiguous DNA at the site of chromosome arm breakage, the DnaB and its associated replisome could continue unimpeded. Continued replication would feature re-priming of the leading strand template by DnaG as observed in vitro (19–21), but could result in net loss of one chromatid. In the current study, we present a protein with a flap-creating activity.
The RarA (Replication associated recombination gene/protein A) is part of what may be the most highly conserved family of DNA metabolism proteins, sharing 40% identity and 56–58% similarity with Mgs1 in yeast and WRNIP1 in mammals (22,23). The protein has also been called MgsA (Maintenance of genome stability A) (24) to better identify it with its yeast homolog. The RarA protein is also related to the clamp loader subunits of DNA polymerase III, encoded by dnaX, and the Holliday Junction helicase, RuvB (22). The proteins in this family are members of the clamp-loader clade of the AAA+ (ATPases associated with various cellular activities) superfamily, but function as tetramers (25). Members of the AAA+ superfamily are oligomeric enzymes that convert the chemical energy of ATP binding and hydrolysis to mechanical energy used to carry out various cellular processes, including functions associated with DNA replication (26–29).
Considerable work has been done with RarA/MgsA family proteins both in vivo and in vitro, but their function remains highly enigmatic. In vivo, all of the proteins co-localize with the replication fork throughout the cell cycle, both in the absence (23,30–33) and presence (34–36) of exogenous DNA damage. The localization is mediated by an interaction with SSB in bacteria (25,30) and ubiquitylated PCNA in eukaryotes (34,35,37,38). Knockout mutations tend to exhibit a modest phenotype, at most a slight growth defect and/or a tendency for hyperrecombination (22,39,40). However, combining a deletion of a rarA family gene with a defect in another gene involved in DNA repair or replication often results in an additively or synergistically enhanced phenotype (22–24,32,33,39–46). Overexpression of any RarA family protein results in severe genome instability and is lethal in E. coli strains lacking recA (24,32,40). Despite a wealth of in vivo experiments, the precise function of RarA family proteins remains unknown.
In vitro, numerous studies have shed limited light on the RarA protein. The apo X-ray crystal structure of the E. coli RarA protein revealed marked similarity between RarA and clamp loader proteins from various organisms (25). However, RarA adopts a unique homotetrameric configuration (25) rather than the typical heteropentameric form seen among clamp loaders and other AAA+ proteins (25,47,48). The RarA protein family exhibits a DNA-dependent ATPase activity that is stimulated by circular single-stranded DNA (25,40,43) and in one case (WRNIP1) by double-stranded DNA ends (49). Both Mgs1 and WRNIP1 bind ssDNA, ssDNA–dsDNA junctions, and forked DNA in the presence or absence of ATP (33,43). RarA homologs physically interact with the processivity clamp PCNA in yeast, and the lagging strand DNA polymerase δ in both yeast and humans (37,49,50). These interactions have been hypothesized to both localize Mgs1/WRNIP1 to sites of replication fork stress and to directly stimulate DNA polymerase δ activity at the replication fork. RarA also tightly binds the conserved C-terminus of the single-stranded DNA binding protein (SSB) in both E. coli and B. subtilis (25,30). This interaction is required for RarA to localize to the replication fork, which is a shared characteristic with other SSB-interacting proteins, including the branch migration helicase RecG, replication restart helicase PriA, and over a dozen additional proteins (30,51,52).
In this study, we further characterize the biochemical properties of the E. coli RarA protein using a combination of ATPase assays, DNA binding studies, and fluorescence spectroscopy. We reveal an ATPase-dependent strand separase activity operating at DNA ends. We speculate on one possible role for such an activity in a novel pathway for replication fork rescue.
We note that the protein acronym MgsA has gained some currency for this protein, even though the name RarA was proposed earlier (22,24). The term MgsA does provide a ready connection to the yeast Mgs1. However, in this report, we refer to the protein as RarA and recommend that this acronym be generally adopted, since the acronym mgsA is also used as an acronym for the gene encoding the E. coli protein methylglyoxal synthase.
MATERIALS AND METHODS
Protein expression and purification
Wild-type and R156A mutant RarA protein were purified as described previously (25) All proteins were carefully tested for endo- and exonuclease contamination using gel-based DNA degradation assays utilizing supercoiled and linear dsDNAs and circular and linear ssDNAs. No contaminating endo- or exonucleases were detected. Aliquots of purified proteins were thawed fresh from −80°C stocks prior to each experiment. RarA protein concentration was determined using the native extinction coefficient ε = 5.44 × 104 M−1 cm−1 (25).
DNA substrates
All DNA substrates used in this study were purchased from Integrated DNA Technologies (Coralville, IA). DNA substrates were annealed by heating equimolar amounts of each oligonucleotide (or a single oligonucleotide) in annealing buffer (10 mM Tris–Cl 80% cation, 1 mM EDTA, 50 mM NaCl) to 95°C for five minutes and cooling to room temperature over 2 h. DNA concentrations were calculated using A260 values measured using a Cary 300 UV-Vis spectrophotometer and extinction coefficients provided by IDT. All DNA substrates were stored at 4°C prior to usage in experiments. DNA concentrations are in all cases reported both in terms of total nucleotides and in total molecules.
All fluorescently labeled DNA substrates were HPLC-purified by IDT. Molecular beacon substrates (labeled with 6-carboxyfluorescein (FAM)) used in steady state fluorescence assays were constructed using a 5΄ 6-FAM fluorophore and a 3΄ 3-Dabcyl quencher. 2-Aminopurine (2-AP) labeled substrates were constructed by substituting a single 2-AP base for adenine. All sequences of the DNA oligonucleotides used in this study are shown in Table 1.
Table 1. Oligonucleotides used in this study. Hairpin-forming sequences are highlighted in bold.
| Name | Sequence | Study/Figure |
|---|---|---|
| oANP031 | 5΄-ACACAGAAGGAGACGG-3΄ | ATPase (Figure 1A) |
| oANP031_F | 5΄-/56-FAM/ACACAGAAGGAGACGG-3΄ | Fluorescence Polarization (Figure 4) |
| Oligo-25 | 5΄-GCAATTAAGCTCTAAGCCATCCGCAAAAATGACCTCTTATCAAAAGGA-3΄ | ATPase (Figure 1B) |
| Oligo-26 | 5΄-TCCTTTTGATAAGAGGTCATTTTTGCGGATGGCTTAGAGCTTAATTGC-3΄ | ATPase (Figure 1B) |
| oANP032 | 5΄-CCGTCTCCTTCTGTGT-3΄ | ATPase (Figure 1A), Fluorescence Polarization (Figure 4) |
| THS7 | 5΄-TTGGAGTTTGCTTCCGGTAACGGAAGCAAACTCCAA-3΄ | ATPase (Figures 1, and 3, Supplementary Figure S1) |
| THS7_FAM_Q | 5΄-/56-FAM/TTGGAGTTTGCTTCCGGTAACGGAAGCAAACTCCAA/3Dab/-3΄ | Fluorescence Intensity (Figure 6) |
| THS7 18nt 3΄ OH | 5΄-TTGGAGTTTGCTTCCGGTAACGGAAGCAAACTCCAATTTTTTTTTTTTTTTTTT-3΄ | ATPase (Figure 1) |
| THS7 18nt 5΄ OH | 5΄-TTTTTTTTTTTTTTTTTTTTGGAGTTTGCTTCCGGTAACGGAAGCAAACTCCAA-3΄ | ATPase (Figure 1) |
| THS9 0nt gap P | 5΄-/5Phos/GCGGAGTTACGTAACGGTAACGTTACGTAACTCCGCCGGAAGCATCGGCTTAGTAATAAGCCGATGCTTCCG-3΄ | ATPase (Figures 1 and 2) |
| THS9 3nt gap | 5΄-GCGGAGTTACGTAACGGTAACGTTACGTAACTCCGCTTTCGGAAGCATCGGCTTAGTAATAAGCCGATGCTTCCG-3΄ | ATPase (Figure 2) |
| THS9 8nt gap | 5΄-GCGGAGTTACGTAACGGTAACGTTACGTAACTCCGCTTTTTTTTCGGAAGCATCGGCTTAGTAATAAGCCGATGCTTCCG-3΄ | ATPase (Figure 2) |
| THS9 13nt gap | 5΄-GCGGAGTTACGTAACGGTAACGTTACGTAACTCCGCTTTTTTTTTTTTTCGGAAGCATCGGCTTAGTAATAAGCCGATGCTTCCG-3΄ | ATPase (Figure 2) |
| THS9 18nt gap | 5΄-GCGGAGTTACGTAACGGTAACGTTACGTAACTCCGCTTTTTTTTTTTTTTTTTTCGGAAGCATCGGCTTAGTAATAAGCCGATGCTTCCG-3΄ | ATPase (Figure 2) |
| THS9 18nt gap P | 5΄-/5Phos/GCGGAGTTACGTAACGGTAACGTTACGTAACTCCGCTTTTTTTTTTTTTTTTTTCGGAAGCATCGGCTTAGTAATAAGCCGATGCTTCCG-3΄ | ATPase (Figure 2) |
| THS9 18nt gap dd | 5΄-GCGGAGTTACGTAACGGTAACGTTACGTAACTCCGCTTTTTTTTTTTTTTTTTTGGGAAGCATCGGCTTAGTAATAAGCCGATGCTTCC/3ddC/-3΄ | ATPase (Figure 2) |
| THS9 23nt gap | 5΄-GCGGAGTTACGTAACGGTAACGTTACGTAACTCCGCTTTTTTTTTTTTTTTTTTTTTTTCGGAAGCATCGGCTTAGTAATAAGCCGATGCTTCCG-3΄ | ATPase (Figure 2) |
| THS19 | 5΄-TTGGAGTTTGCTTCTCAGCCGGTAACGGCTGAGAAGCAAACTCC/i2AmPr/A-3΄ | Fluorescence Intensity (Figure 6) |
| THS27 | 5΄-CCGGCGCCGGCGGCCGGTAACGGCCGCCGGCGCCGG-3΄ | ATPase (Figure 5) |
| THS28 | 5΄-AATTATAATTATTAATGTAAATTAATAATTATAATT-3΄ | ATPase (Figure 5) |
| THS29 | 5΄-ACGGCGCCGGCGGCCGGTAACGGCCGCCGGCGCCGT-3΄ | ATPase (Figure 5) |
| THS30 | 5΄-AAGGCGCCGGCGGCCGGTAACGGCCGCCGGCGCCTT-3΄ | ATPase (Figure 5) |
| THS31 | 5΄-AATGCGCCGGCGGCCGGTAACGGCCGCCGGCGCATT-3΄ | ATPase (Figure 5) |
| THS32 | 5΄-AATTCGCCGGCGGCCGGTAACGGCCGCCGGCGAATT-3΄ | ATPase (Figure 5) |
| 1T_FAM | 5΄-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTGACGCTGCCGAATTCTGGCTTGCTAGGACATCTTTGCCCACGTTGACCGC/36-FAM/-3΄ | Helicase (Supplementary Figure S2) |
| 1T | 5΄-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTGACGCTGCCGAATTCTGGCTTGCTAGGACATCTTTGCCCACGTTGACCGC-3΄ | Helicase (Supplementary Figure S2) |
| 42 | 5΄-GCGGTCAACGTGGGCAAAGATGTCCTAGCAAGCCAGAATTCGGCAGCGTC-3΄ | Helicase (Supplementary Figure S2) |
| 42T | 5΄-GCGGTCAACGTGGGCAAAGATGTCCTAGCAAGCCAGAATTCGGCAGCGTCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3΄ | Helicase (Supplementary Figure S2) |
ATPase activity assay
A coupled spectrophotometric assay was used to measure ATPase activity by RarA protein as described previously (53,54). All assays were carried out using a Varian Cary 300 dual-beam spectrophotometer equipped with a temperature-controlled 12-cell changer. The cell path length is 1 cm, and the bandwidth was 2 nm. All reactions were carried out at 37°C in 1× reaction buffer (25 mM Tris-acetate pH 7.5, 1 mM DTT, 3 mM potassium glutamate, 10 mM Mg(OAc)2, 5% (w/v) glycerol), an ATP regeneration system (10 U/ml pyruvate kinase, 3 mM phosphoenolpyruvate), a coupled detection system (10 U/ml lactate dehydrogenase, 3 mM NADH), 3 mM ATP, and the indicated amounts of RarA protein and DNA substrate. In some experiments, the potassium glutamate concentration was increased to 100 mM as noted. Vmax and apparent Km values were calculated by constructing Michaelis–Menten plots in Graphpad Prism software.
Fluorescence polarization assay
RarA protein was incubated with 1nM oANP031_FAM or annealed oANP031_FAM and oANP032 in 1× reaction buffer supplemented with indicated amounts of nucleotide cofactor at room temperature for 30 min. Fluorescence polarization was measured at 25°C using a Beacon 2000 fluorescence polarization system. The polarization values of experimental reactions were background corrected by subtracting the average polarization value of reactions containing only labeled DNA (for ssDNA, 36 mP; for dsDNA, 62.5 mP) from experimental polarization values. For all binding experiments, data were fit to a simple one-site specific interaction model and apparent Kd values were determined using Graphpad Prism software.
Steady-state fluorescence assays
Molecular beacon DNA substrate (50 nM) was incubated in 1× reaction buffer supplemented with 3 mM ATP and an ATP regeneration system (10 U/ml pyruvate kinase, 3 mM phosphoenolpyruvate) at 37°C for 5 min. Indicated amounts of wild-type or R156A RarA protein were then added to the reaction mixture. Fluorescence intensities were measured by exciting the reaction mixture at 494 nm and measuring emitted light at 521 nm in a QuantaMaster Model C-60/2000 spectrofluorimeter (Photon Technologies International). Measurements were taken at 37°C for a total of 500 s. Fluorescence intensity values were normalized by subtracting values from no protein control reactions. All experiments were completed at least three times and the fluorescence values averaged for each condition.
Stopped flow spectrofluorimetry
Stopped flow fluorescence experiments were conducted using a Kintek SF-300X stopped flow instrument. 2-Aminopurine substituted DNA substrates were pre-incubated with indicated amounts of RarA protein at room temperature for at least 10 min prior to loading into mixing syringes. Syringe A was loaded with DNA and RarA protein or no protein in stopped flow buffer (50 mM Tris-acetate pH 7.5, 6 mM potassium glutamate, 20 mM magnesium acetate, 10% glycerol). Syringe B was loaded with ATP, ATPγS, or no nucleotide in stopped flow buffer. Mixing in the flow cell resulted in final concentrations of DNA substrate (100 nM), RarA protein (as indicated), and NTP (500 μM). 2-Aminopurine was excited at 319 nm and emitted fluorescence was measured using a 341 nm long pass filter (Edmund Optics). Measurements were taken at 37°C for a total of 30 s. Fluorescence intensity values were background corrected by subtracting no protein control reactions from experimental values. All experiments were repeated at least three times and the fluorescence values averaged for each condition.
Helicase assay
DNA substrates used in helicase assays included a forked substrate (annealed oligonucleotides 1T_FAM and 42T) containing 50 base pairs of duplex DNA and 30 nucleotide 5΄ and 3΄ non-complementary overhangs, or a 5΄ flap substrate (annealed oligonucleotides 1T_FAM and 42) consisting of 50 base pairs of duplex DNA and a single 30 nucleotide 5΄ overhang. DNA substrate (30 nM in total molecules) was pre-incubated in helicase buffer (50 mM HEPES pH 7.0, 40 μg/ml BSA, 4 mM magnesium acetate, 2 mM ATP, 2 mM DTT) with or without SSB (800 nM) for 5 min at room temperature. Reactions were started by adding DnaB (1.8 μM), DnaC (12.0 μM) or RarA (600 nM) as indicated. Reactions were incubated at 37°C for 30 min and stopped using stop buffer (2.5% SDS, 100 mM EDTA, 500 nM unlabeled trap oligonucleotide 1T, 1 mg/ml proteinase K). After addition of stop buffer, reactions were incubated for another 30 min at 37°C. Boiled samples were boiled at 95°C for 15 min in a thermocycler. Reactions were loaded onto a 10% TBE gel and run at 150 V for 90 min. Gels were imaged using a GE Healthcare Typhoon FLA 9000.
RESULTS
RarA ATPase activity is stimulated by double-stranded DNA ends
Multiple previous reports on RarA and yeast Mgs1 (25,40,43) indicated that the ATPase activity of proteins in this family was stimulated by ssDNA. One report on WRNIP1 indicated that the ATPase was stimulated by duplex DNA ends (49). This could represent a distinction between protein family members. However, we note that in all the previous reports of RarA family protein stimulation by ssDNA, the ssDNA employed was a derivative of M13 phage DNA that has a considerable degree of secondary structure (55,56). We considered the possibility that the secondary structure may have led to a misinterpretation of results.
We examined the response of the RarA DNA-dependent ATPase activity to a series of DNA substrates with different structures. RarA exhibits a weak DNA-independent ATPase activity (0.8 ± 0.20 μM min−1 under the conditions of this experiment with 0.5 μM RarA; Figure 2A). A 16-nucleotide single-stranded DNA substrate (ssDNA; 0.5 μM molecules) with no secondary structure did not increase the ATPase rate (0.7 ± 0.10 μM min−1) when compared to DNA-independent reactions. However, when a 16-base pair duplex (dsDNA; 0.5 μM molecules) was included in the reaction, ATPase activity was strongly stimulated (68.0 ± 2.0 μM min−1) (Figure 2A). These results suggested double-stranded DNA was the actual primary substrate for RarA ATPase activity.
Figure 2.

Double-stranded DNA ends stimulate RarA ATPase activity. (A) The ATPase activity of RarA (0.5 μM) was measured in the presence of short single-stranded or double-stranded DNAs (0.5 μM in total molecules; 8 μM or 16 μM in nucleotides, respectively). (B) The ATPase activity of RarA (0.5 μM) was measured in the presence of various DNA substrates (48 μM nucleotides) including a 48-base pair duplex, M13mp8 linear double-stranded (lds) DNA, or pUC19 circular double-stranded (cds) DNA. (C) The rate of ATP hydrolysis mediated by RarA (0.5 μM) was measured in the presence of various duplex substrates (THS7, THS7 18nt 5΄ OH; THS7 18 nt 3΄ OH) containing different DNA end structures (0.1 μM molecules; 3.6-5.4 μM total nucleotides). Error bars represent one standard deviation from the mean.
To determine whether the interior or the ends of the dsDNA molecule were responsible for stimulating RarA ATPase activity, three DNA substrates were included in different ATPase reactions: circular double-stranded pUC19 DNA (cds DNA; 2686 bp), linear double-stranded M13mp8 DNA (lds DNA; 7229 bp), and a 48-base pair duplex. All reactions contained equimolar concentrations of DNA as measured in nucleotides (48 μM), but greatly differing concentrations of dsDNA ends. Circular double-stranded DNA (no dsDNA ends) did not significantly stimulate ATPase activity compared to a reaction with no DNA present (Figure 2B). Similarly, linear double-stranded DNA (0.018 μM dsDNA ends) only marginally increased RarA ATPase rate (Figure 2B). However, a reaction containing a 48-base pair duplex (1 μM dsDNA ends) strongly stimulated RarA ATPase activity when compared to all other substrates (Figure 2B). Figure 2A and B shows that double-stranded DNA ends maximally stimulated RarA ATPase activity, while closed-circular dsDNA and short ssDNA did not increase the rate of ATP hydrolysis relative to reactions lacking DNA.
RarA ATPase activity is affected by DNA end structure
To determine the effects of dsDNA end structure in stimulating RarA ATPase activity, we designed a DNA duplex hairpin substrate containing a single dsDNA end. This 36 nucleotide hairpin substrate consisted of a 16 base pair duplex with a four nucleotide 5΄-GTAA-3΄ hairpin. This hairpin sequence was selected due to its propensity to form a hairpin and its thermostability (57). We altered this dsDNA end structure with single-stranded DNA overhangs and examined their effects on RarA ATPase activity. In all cases, the concentration of DNA molecules, and thus free ends, was kept constant. A blunt end duplex maximally stimulated RarA ATPase activity (26.21 ± 1.80 μM min−1), followed by a dsDNA end with an 18-nucleotide 5΄ ssDNA overhang (19.79 ± 0.81 μM min−1) (Figure 2C). A duplex end modified with an 18-nucleotide 3΄ ssDNA overhang modestly stimulated RarA ATPase rate (6.71 ± 0.50 μM min−1) while a structure lacking exposed dsDNA ends (both ends capped in a hairpin structure) did not significantly stimulate ATPase activity when compared to a blunt end or 5΄ overhang substrate (Figure 2C). We note that both substrate specificity and ATPase rates are maintained in a reaction buffer with higher ionic strength (Supplementary Figure S1). From these data, we identified both a blunt end duplex and a duplex modified with a short ssDNA 5΄ overhang as the most effective substrates for stimulating RarA ATPase activity, although all three substrates were stimulatory.
Single-stranded DNA gaps stimulate RarA ATPase activity
Single-stranded DNA gaps are common structures found at stalled replication forks in vivo. As such, we designed DNA substrates containing one single-stranded DNA gap of varying size and tested their ability to stimulate RarA ATPase activity. As previously noted, a closed-circle DNA substrate does not significantly stimulate RarA ATPase activity. This situation is reprised here with a 72 nucleotide substrate containing 32 base pairs of duplex DNA, two four-nucleotide 5΄-GTAA-3΄ hairpins at either end, and no strand breaks (Figure 3). However, when a single nick is present in the same substrate (in the center), ATPase activity is notably increased (9.91 ± 0.47 μM min−1) (Figure 3). The rate of RarA ATP hydrolysis increases further with the size of the ssDNA gap, up to eight nucleotides (20.78 ± 1.65 μM min−1) (Figure 3). Substrates containing a gap larger than eight nucleotides did not further increase in the observed rates of ATP hydrolysis (Figure 3).
Figure 3.
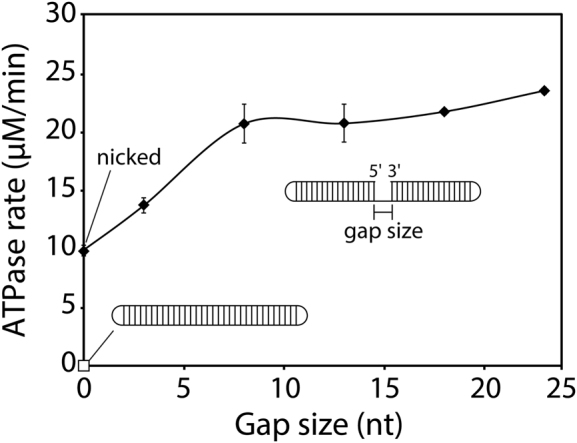
Single-stranded DNA gaps stimulate RarA ATPase activity. The rate of ATP hydrolysis was measured in the presence of RarA (0.5 μM) and gapped DNA hairpin substrates containing two 16 base pair duplexes separated by poly dT gaps of the indicated size and capped by two four-nucleotide hairpins (0.05 μM molecules, 3.6–4.75 μM total nucleotides). The white square represents a ligated closed-circle substrate while black diamonds represent substrates containing a gap ranging from a single nick to 23 nucleotides. Error bars represent one standard deviation from the mean.
RarA ATPase activity is equivalent at dsDNA ends and ssDNA gaps
To compare the stimulatory effect of a dsDNA end and ssDNA gap on RarA ATPase activity, a Michaelis-Menten kinetic analysis of reactions containing these substrates was conducted. ATP concentrations were held constant and in excess, while DNA substrate concentrations were increased and ATPase rates measured. In effect, the results measure RarA binding to DNA ends, as seen indirectly by ATPase stimulation. RarA ATPase activity in the presence of a blunt-end dsDNA substrate had a Vmax of 59.76 ± 2.04 μM min−1 and an apparent Km of 48.71 ± 6.21 nM (Figure 4A). Similarly, RarA ATPase activity in the presence of an 18 nucleotide ssDNA gap had a Vmax of 56.45 ± 2.64 μM min−1 and an apparent Km of 51.40 ± 8.03 nM (Figure 4B).
Figure 4.
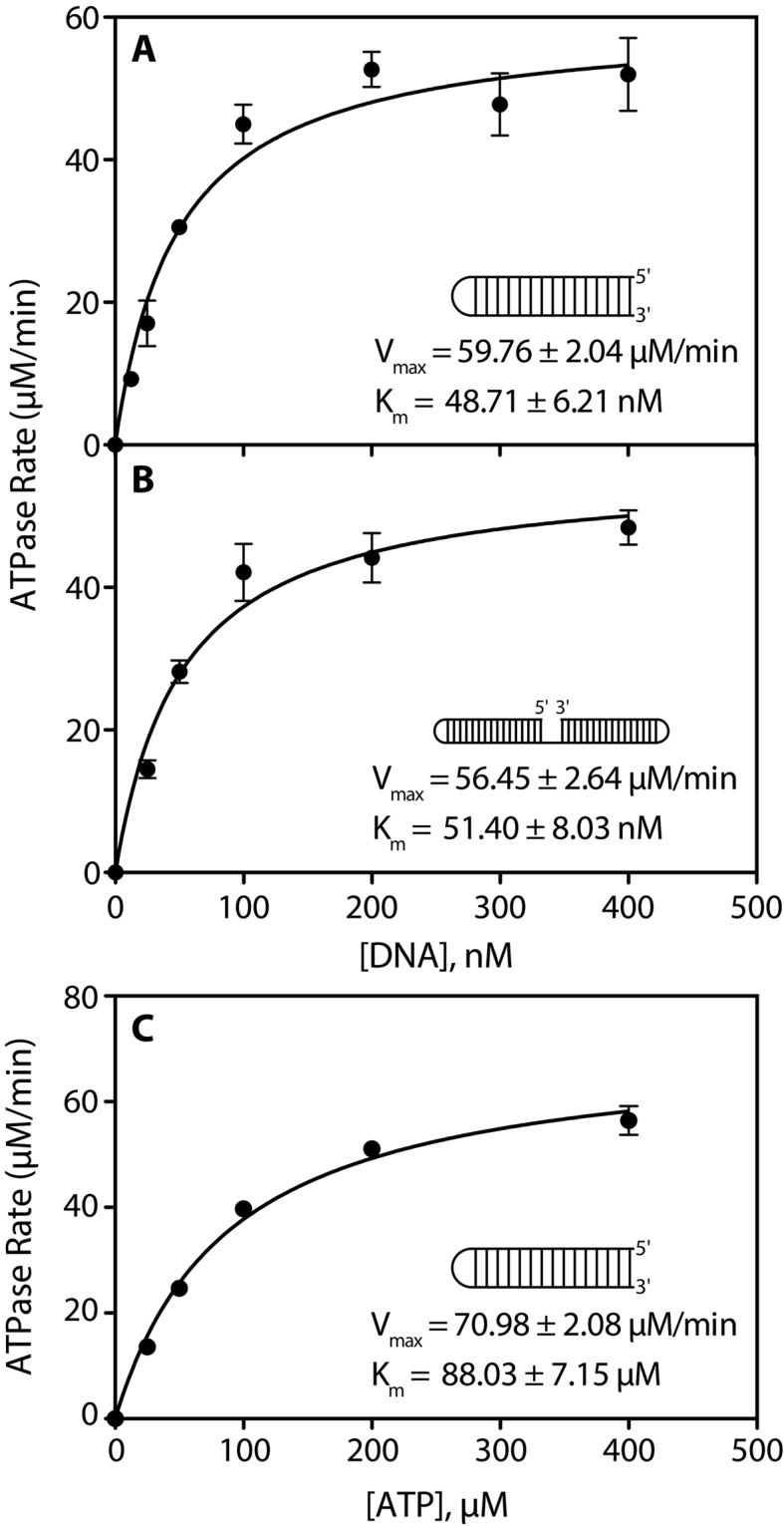
Michaelis–Menten kinetic analyses of RarA ATPase activity. The rate of ATP hydrolysis in the presence of different substrate concentrations was measured in the presence of 0.4 μM RarA protein. (A) ATP hydrolysis reactions were conducted in the presence of increasing concentrations of a 16 base pair blunt end DNA duplex hairpin substrate. (B) ATP hydrolysis reactions were conducted in the presence of increasing concentrations of a 90 nucleotide gapped DNA hairpin substrate containing two 16 base pair duplexes separated by an 18 nucleotide poly dT gap and capped with two four-nucleotide hairpins. (C) ATP hydrolysis reactions were conducted in the presence of blunt end duplex DNA substrate (0.4 μM molecules; 14.4 μM nucleotides) and increasing amounts of ATP. Each point is the average of at least three replicate experiments, with error bars representing one standard deviation from the mean ATPase rate.
Finally, the effect of ATP concentration on RarA ATPase activity was investigated by conducting a Michaelis–Menten kinetic analysis of reactions containing saturating amounts of DNA and increasing amounts of ATP. The measured Vmax for this reaction was 70.98 ± 2.08 μM min−1, the measured Km for the ATP substrate was 88.03 ± 7.15 μM, and the calculated kcat was 141.96 min−1 (Figure 4).
RarA binds double-stranded DNA in the presence of ATP, and to single-stranded DNA in the absence of nucleotide cofactor
We next examined DNA binding directly, and also investigated the effect of nucleotide cofactor on RarA binding to double-stranded and single-stranded DNA. Using a fluorescently-labeled 16 base pair duplex or 16 nucleotide single-stranded DNA substrate, equilibrium fluorescence polarization binding studies were conducted.
RarA exhibited a very weak affinity for dsDNA in the absence of nucleotide cofactor or in the presence of ADP, with an apparent dissociation constant (Kd,app) of > 10 μM (Figure 5A). However, when the minimally-hydrolyzable ATP analog ATPγS was included in binding reactions, RarA possessed a much greater affinity for dsDNA, with an apparent dissociation constant of 58 ± 3 nM (Figure 5A). Note that the Kd value with ATPγS compares very well with the apparent Km derived from ATPase values determined as DNA end concentration was varied (Figure 4A and B). Conversely, RarA exhibited a very weak affinity for ssDNA in the presence of ATPγS or ADP (Kd,app = > 10000 nM and 9000 ± 1000 nM, respectively), but a significant affinity in the absence of nucleotide cofactor (Kd,app = 890 ± 28 nM) (Figure 5B).
Figure 5.

RarA binds double-stranded DNA in the presence of ATPγS and single-stranded DNA in the absence of nucleotide cofactor. RarA binding affinity for dsDNA (A) and ssDNA (B) was measured using florescence polarization in the presence of various nucleotide cofactors. A table of calculated dissociation constants for each reaction condition is provided in (C). Error bars represent one standard deviation from the mean polarization value.
RarA ATPase activity is stimulated by AT-rich duplexes
We next sought to determine if the nucleotide sequence present at DNA ends affected the RarA ATPase activity. Using the previously described hairpin duplex substrate as a template, six substrates were designed. A duplex comprised entirely of G–C base pairs was designed and then modified to contain an increasing number of A–T base pairs from the dsDNA end inward to the hairpin. The resulting five DNA substrates, as well as a duplex comprised entirely of A-T base pairs, were included in ATPase reactions with RarA protein.
The GC-rich duplex stimulated RarA ATPase activity at a rate lower than the blunt end duplex substrate examined in Figure 2C (20.01 ± 1.80 μM min−1) (Figure 6). Conversion of the terminal-most base pair from G–C to A–T resulted in a significant increase in RarA ATPase activity (Figure 6). As more G–C base pairs were substituted with A–T base pairs in the substrate, the rate of ATP hydrolysis increased further. A duplex comprised entirely of A–T base pairs stimulated RarA ATPase activity maximally (35.24 ± 2.31 μM min−1) (Figure 6). This result suggested that RarA might engage in the catalysis of strand separation at DNA ends.
Figure 6.
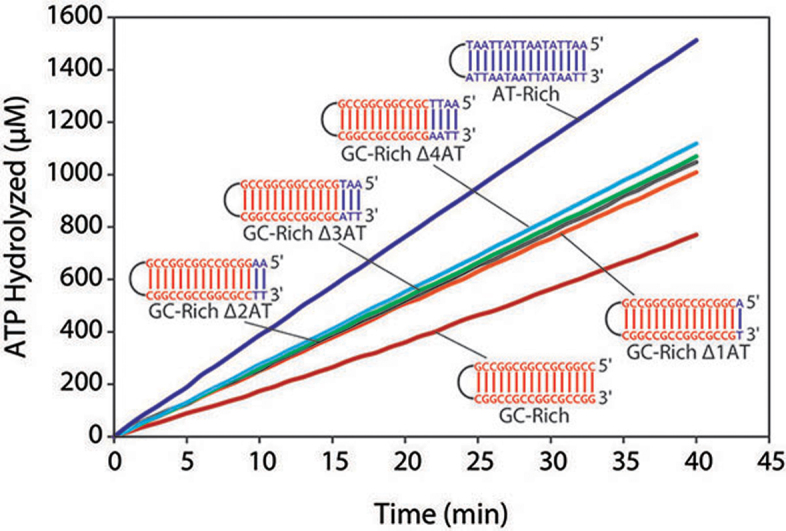
RarA ATPase rate is dependent on the GC-content of a DNA duplex. Six different DNA duplex substrates were designed with varying GC-content and included in RarA ATPase reactions. The rate of ATP hydrolysis was measured in the presence of these DNA substrates (0.1 μM molecules; 3.6 μM nucleotides) and RarA (0.4 μM).
RarA protein separates the strands of double-stranded DNA ends
To investigate the effect of RarA ATP hydrolysis on dsDNA ends, we used two assays. The first is an ensemble fluorescence intensity assay. We designed a hairpin duplex substrate (molecular beacon) modified with a 5΄ 6-carboxyfluorescein (6-FAM) fluorophore and a 3΄ 3-dabcyl quencher (Figure 7A and B). When the DNA is folded, the fluorescence of the 6-FAM fluorophore is quenched by the 3-dabcyl quencher. When the strands of the DNA substrate are separated, the fluorescence intensity increases. This DNA substrate was included in ATPase reactions with RarA protein while the fluorescence intensity of fluorescein was measured over time.
Figure 7.

RarA separates the strands of a double-stranded DNA duplex. (A) The fluorescence intensity of a 6-FAM/3-Dabcyl labeled DNA duplex was measured for 500 s after the addition of RarA in the presence of ATP. (B) The fluorescence intensity of the same DNA substrate was measured over the course of 500 s after addition of either wild-type RarA or RarA R156A variant (0.5 μM) in the presence of ATP. (C) RarA (0.4 μM) was pre-incubated with a 2-aminopurine labelled DNA substrate (0.1 μM molecules; 4.6 μM nucleotides). Fluorescence intensity of 2-aminopurine was measured for 30 seconds following addition of indicated nucleotide cofactors. (D) Varying concentration of RarA were pre-incubated with a 2-aminopurine labelled DNA substrate (0.1 μM molceules; 4.6 μM nucleotides). Fluorescence intensity of 2-aminopurine was measured for 30 seconds following addition of ATPγS. (E) RarA (0.4 μM) was added to a solution containing 2-aminopurine DNA substrate (0.1 μM molceules; 4.6 μM nucleotides) and ATPγS. In a previous experiment, ATPγS was added to a solution containing RarA (0.4 μM) and 2-aminopurine DNA substrate (0.1 μM molceules; 4.6 μM nucleotides). Fluorescence intensity of 2-aminopurine was measured over the course of 30 s in both experiments. All values represent the average of at least three replicate experiments.
Addition of RarA protein to reactions containing the labeled DNA substrate resulted in an increase in fluorescence intensity over time (Figure 7A). Increasing the amount of RarA included in these reactions resulted in an increase in overall fluorescence intensity in a protein concentration-dependent manner (Figure 7A).
RarA R156A is a variant protein lacking the arginine finger residue required by AAA+ proteins to hydrolyze ATP. When this variant protein was included in a reaction containing the 6-FAM/dabcyl-labeled DNA substrate, the increase in fluorescence intensity was dramatically less than that observed in a reaction containing wild-type protein (Figure 7B). These results suggest that ATP binding or hydrolysis is required for RarA to separate the strands of dsDNA ends. We note that the labels on this DNA substrate may interfere with RarA binding to the DNA ends to some degree.
To better characterize this strand separation activity, and to control for DNA end structure, we set up a second assay using a quite different DNA substrate and stopped-flow spectrophotometry. For these experiments, we designed another hairpin duplex substrate containing 21 base pairs. This substrate contained a 2-aminopurine at the penultimate position on the 3΄ end of the strand (Figure 7C and D). 2-aminopurine is a fluorescent base analog that mimics adenine both in its structure and its ability to base-pair with thymine (58–61). Additionally, its fluorescence intensity is naturally quenched when it is found in a duplex and unquenched when single-stranded (58–61). These properties made 2-aminopurine well suited for probing the interactions between RarA protein and dsDNA ends.
The fluorescence intensity of 2-aminopurine was monitored over a 30 second time period in reactions containing RarA protein and DNA upon addition of indicated nucleotide cofactor (Figure 7C). In a reaction lacking nucleotide cofactor, 2-aminopurine fluorescence marginally increased (Figure 7C). When ATP was included in the reaction, a modest sharp increase in fluorescence was observed in the first 150 ms, after which the fluorescence reached its peak (Figure 7C). Interestingly, when the minimally-hydrolyzable ATP analog ATPγS was included in the reaction, a large and steady increase in fluorescence was observed in the first 15 seconds (a shorter time scale than those seen in the assay presented in panels A and B), after which the fluorescence reached its peak (Figure 7C). However, when the ATPase-dead RarA R156A mutant was included in this reaction, fluorescence intensity only modestly increased (Figure 7C). Finally, we investigated whether this activity was RarA concentration-dependent. We included the indicated amounts of RarA protein in the presence of ATPγS and observed the fluorescence intensity over time. We observed an increase in both the rate of increase and overall fluorescence intensity when higher concentrations of RarA were included in these reactions (Figure 7D).
We note that the order of component addition was different between the method used in Figure 7A and B and the method used in Figure 7C and D. In the fluorimeter method (Figure 7A and B), reactions were initiated when RarA was added to a buffered solution of ATP and DNA substrate, while in the stopped-flow method (Figure 7C and D) RarA was pre-incubated with DNA and reactions were initiated upon addition of nucleotide cofactor. To ensure that the order of component addition did not significantly affect the results observed with the stopped-flow method, we performed a variation of the stopped-flow experiment where two orders of addition were directly compared. In this experiment (Figure 7E), the addition of DNA and ATPγS to RarA yielded a higher final signal, although the rate of approach to the endpoint was similar in both cases. This rate was in all cases considerably faster than that observed in the experiments employing the 6-FAM/dabcyl-labeled DNA substrate. Thus, while the order of component addition does not significantly affect the overall activity, the DNA end modification present in the experiments in Figure 7A and B did appear to affect the kinetics of this reaction.
DISCUSSION
This study provides three new insights into the highly conserved yet enigmatic RarA protein family. First, E. coli RarA is a DNA-dependent ATPase specifically stimulated by double-stranded DNA ends and single-stranded DNA gaps. The effects of DNA ends are documented in Figures 2–4. This is likely to be a general feature of this protein family. Previous reports of RarA and yeast Mgs1 protein ATPase stimulation by ssDNA likely reflects an interaction with the ends of the secondary structure that is prevalent in the M13-based DNA substrates employed in all cases (25,40,43), although we did not directly test Mgs1. ATPase stimulation by dsDNA ends has been demonstrated for WRNIP1 (49). Second, we characterized the RarA–DNA complex in various nucleotide-bound states. These experiments demonstrated that ATPγS-bound RarA possesses a high affinity for double-stranded DNA, while apo RarA possesses a substantial affinity for single-stranded DNA. As outlined below, these results begin to define an ATP hydrolytic cycle possessing multiple intermediates with different DNA affinities. Finally, we showed that RarA separates the strands at DNA duplex termini in the presence of ATPγS or ATP, creating ssDNA flaps. In addition to the direct demonstration in Figure 7, the effects of AT base pair content at the DNA end documented in Figure 6 also contributes to this conclusion. The ineffectiveness of a mutant RarA protein that does not hydrolyze ATP highlights the importance of ATP in this process. The strand separase function represents the first DNA substrate-based activity detected to date for this protein family. We note that RarA is not the only protein, even in E. coli, which can produce a fraying or strand separation at the ends of a duplex. For example, the RecBCD helicase can produce this effect at DNA ends (62,63), albeit presumably as a prelude to DNA unwinding.
The biochemical properties of RarA described here suggest to us a cellular role such as that depicted in Figure 8. When a replication fork encounters a discontinuity in the leading strand template, the leading strand arm would detach. If RarA rapidly created a flap at the end of the resulting gap on the lagging strand continuous arm, DnaB and its associated replisome could continue replication without a need for replisome disassembly and restart. Leading strand DNA synthesis could be re-primed by DnaG. This path would provide an alternative to RecA-mediated restoration of the replication fork. In bacteria lacking nonhomologous end-joining (NHEJ), this would generally lead to loss of one of the two chromatid products of replication, but would permit replication completion and survival of one complete genomic equivalent. In eukaryotes and bacteria possessing NHEJ, chromatid loss could be averted by ligation of the detached chromosome to the leading strand segment created by continued replication, creating a potentially efficient path for DNA damage tolerance.
Figure 8.

A model for RarA-mediated DNA flap creation at broken replication forks. RarA-mediated DNA flap creation may provide a pathway for DNA damage tolerance. Upon encounter with a leading strand discontinuity, RarA could create a suitable substrate for the replicative helicase DnaB to continue unwinding parental duplex DNA after a fork collapse. RarA is recruited to the fork through its interaction with SSB. When the leading strand arm detaches, ATP-RarA binds to the parental duplex, creating a single-stranded DNA flap. RarA hydrolyzes ATP, allowing it to dissociate and pass the flap to DnaB. This substrate is then competent for continued DnaB helicase activity, abrogating a need for replisome disassembly and restart.
The path outlined in Figure 8 provides an explanation for several published observations. It is consistent with the observed functional overlap between RecA and RarA reported by Shinagawa and colleagues (24), and provides a rationale for localization of RarA at the replication fork. This novel pathway might also explain many of the observations of phenotypic additivity or synergism between rarA family gene knockouts and mutations in other DNA metabolism functions (22–24,32,33,39–46). Recently, Fuchs, Pagès, and colleagues have directly observed loss of the detached chromatid when a bacterial replisome encounters an engineered template discontinuity in vivo and completes replication in the absence of RecA-mediated gap repair (64). The chromosome loss does not prevent cell division. The authors (64) framed the observation in the context of the downstream replication restart demonstrated by Marians and colleagues in vitro (19–21,65). In contrast, the model in Figure 8 would allow a continuation of replication with no replisome disassembly and restart.
However, we emphasize that the proposed pathway in Figure 8, if correct, may be only part of the RarA story. The structure of RarA resembles that of a clamp loader (25). Although no protein-loading function has yet been described for any RarA protein family member, such a function remains an intriguing possibility. The genetic observations associated with rarA, mgs1 and WRNIP1 as cited above are highly complex, and not all of them are readily accommodated by the model of Figure 8. Finally, the rate of replisome advancement, approaching 1000 nucleotides s–1, poses a potential kinetic challenge to the model. Pre-positioned by a relatively strong interaction with SSB (25,51), RarA would need to sense detachment of the leading strand chromatid arm. Then, it would need to (i) create a flap on a time scale of microseconds or (ii) engage with the replisome to effect stalling and allow time for flap creation.
RarA protein family members co-localize with the replication fork throughout the cell cycle, even in the absence of replication stress (23,30–35,66). RarA is required to recover from stalled replication forks accumulated in strains carrying a temperature-sensitive allele of the replicative polymerase, dnaE486 (42). The authors favored a model in which RarA made replication forks accessible to other DNA metabolism proteins, including the DNA damage-associated helicase RecQ. Similarly, yeast Mgs1 protein stimulates flap-specific endonuclease (Fen1) activity, but not through a direct interaction with Fen1. Mgs1 may thus remodel DNA substrates to make them better substrates for Fen1 activity (43). The work we report here suggests it might function by creating the flaps.
The ATPase activity of RarA provides many functional clues. We have previously shown that addition of SSB inhibits cssDNA-dependent RarA ATPase activity (25), perhaps because the SSB removes the stimulatory secondary structure of the M13-based ssDNA substrates used. RarA ATPase activity is stimulated by a single-stranded gapped DNA substrate, with the ideal gap size being eight nucleotides or greater (Figure 3). This may reflect some structural requirement for optimal access to the duplex at the end of the gap by a RarA tetramer. From the DNA binding study and the effects of different nucleotides on that binding, it is possible to begin to construct an ATP hydrolytic cycle. In that cycle, the ATP-bound form of the protein would bind most tightly to the duplex DNA (Figure 5) end and trigger strand separation (Figure 7).
Both DNA binding and flap creation activities improve in the presence of ATPγS when compared to ATP (Figures 5, and 7C). ATP binding is the key step for processes carried out by numerous AAA+ proteins, including the bacterial replication initiation protein DnaA (67), DNA polymerase III γ clamp loader complex (68), and the replicative helicase loader DnaC (69). Thus, like many AAA+ proteins, ATP hydrolysis may be coupled to release of RarA from the DNA substrate, the relatively weak but measurable binding of the apoprotein to ssDNA (Figure 5) notwithstanding. This hypothesis may explain the rapid equilibrium for DNA flap creation observed when ATP was used as a cofactor instead of ATPγS (Figure 7C). Additionally, no helicase activity has been observed for RarA on DNA substrates under experimental conditions known to stimulate its ATPase activity. An example of a trial to detect helicse activity is provided in Supplementary Figure S2. This suggests that RarA may hand off a remodeled substrate to another protein, such as a helicase or processivity clamp.
In conclusion, we have defined a novel biochemical function of the RarA protein. DNA strand separation at DNA ends so as to create flaps may function as we envision in Figure 8, or may play a role in recombinational DNA repair or some other process. The RarA protein functions at the replication fork, and close homologs of this protein function in all organisms. The detailed role of RarA and its protein family members in resolving the problems at stalled or broken forks should be elucidated with continued study.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Ananya Ray and Robert Landick for training and use of stopped-flow instrumentation and Christine Wolak and James Keck for training and use of fluorescence polarization instrumentation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of General Medical Sciences [GM32335-34 to M.M.C.]; Wharton and Steenbock Predoctoral Fellowships from the Department of Biochemistry, University of Wisconsin – Madison (to T.S.). Funding for open access charge: NIH [GM32335-34].
Conflict of interest statement. None declared.
REFERENCES
- 1. Cox M.M., Goodman M.F., Kreuzer K.N., Sherratt D.J., Sandler S.J., Marians K.J.. The importance of repairing stalled replication forks. Nature. 2000; 404:37–41. [DOI] [PubMed] [Google Scholar]
- 2. Cox M.M. Historical overview: Searching for replication help in all of the rec places. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8173–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kowalczykowski S.C. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 2000; 25:156–165. [DOI] [PubMed] [Google Scholar]
- 4. Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 1999; 63:751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuzminov A. DNA replication meets genetic exchange: chromosomal damage and its repair by homologous recombination. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8461–8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Michel B. Replication fork arrest and DNA recombination. Trends Biochem. Sci. 2000; 25:173–178. [DOI] [PubMed] [Google Scholar]
- 7. Michel B., Boubakri H., Baharoglu Z., LeMasson M., Lestini R.. Recombination proteins and rescue of arrested replication forks. DNA Repair. 2007; 6:967–980. [DOI] [PubMed] [Google Scholar]
- 8. Heller R.C., Marians K.J.. Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell Biol. 2006; 7:932–943. [DOI] [PubMed] [Google Scholar]
- 9. Klein H.L., Kreuzer K.N.. Replication, recombination, and repair: going for the gold. Mol. Cell. 2002; 9:471–480. [DOI] [PubMed] [Google Scholar]
- 10. Lopes M., Cotta-Ramusino C., Pellicioli A., Liberi G., Plevani P., Muzi-Falconi M., Newlon C.S., Foiani M.. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001; 412:557–561. [DOI] [PubMed] [Google Scholar]
- 11. Merrikh H., Zhang Y., Grossman A.D., Wang J.D.. Replication-transcription conflicts in bacteria. Nat. Rev. Microbiol. 2012; 10:449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cox M.M. The nonmutagenic repair of broken replication forks via recombination. Mutat. Res.-Fund. Mol. Mech. Mutagen. 2002; 510:107–120. [DOI] [PubMed] [Google Scholar]
- 13. Mirkin E.V., Mirkin S.M.. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007; 71:13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aguilera A., Garcia-Muse T.. Causes of genome instability. Ann. Rev. Genet. 2013; 47:1–32. [DOI] [PubMed] [Google Scholar]
- 15. Kaplan D.L. The 3΄-tail of a forked-duplex sterically determines whether one or two DNA strands pass through the central channel of a replication-fork helicase. J. Mol. Biol. 2000; 301:285–299. [DOI] [PubMed] [Google Scholar]
- 16. Kaplan D.L., O'Donnell M.. DnaB drives DNA branch migration and dislodges proteins while encircling two DNA strands. Mol. Cell. 2002; 10:647–657. [DOI] [PubMed] [Google Scholar]
- 17. Kaplan D.L., O'Donnell M.. Twin DNA pumps of a hexameric helicase provide power to simultaneously melt two duplexes. Mol. Cell. 2004; 15:453–465. [DOI] [PubMed] [Google Scholar]
- 18. Shin J.H., Jiang Y., Grabowski B., Hurwitz J., Kelman Z.. Substrate requirements for duplex DNA translocation by the eukaryal and archaeal minichromosome maintenance helicases. J. Biol. Chem. 2003; 278:49053–49062. [DOI] [PubMed] [Google Scholar]
- 19. Yeeles J.T., Marians K.J.. The Escherichia coli replisome is inherently DNA damage tolerant. Science. 2011; 334:235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yeeles J.T.P., Marians K.J.. Dynamics of leading-strand lesion skipping by the replisome. Mol. Cell. 2013; 52:855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yeeles J.T.P., Poli J., Marians K.J., Pasero P.. Rescuing stalled or damaged replication forks. Cold Spring Harbor Pers. Biol. 2013; 5:a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barre F.X., Soballe B., Michel B., Aroyo M., Robertson M., Sherratt D.. Circles: the replication-recombination-chromosome segregation connection. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8189–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sherratt D.J., Soballe B., Barre F.X., Filipe S., Lau I., Massey T., Yates J.. Recombination and chromosome segregation. Phil. Trans. Roy. Soc. London Ser. B-Biol. Sci. 2004; 359:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shibata T., Hishida T., Kubota Y., Han Y.W., Iwasaki H., Shinagawa H.. Functional overlap between RecA and MgsA (RarA) in the rescue of stalled replication forks in Escherichia coli. Genes Cells. 2005; 10:181–191. [DOI] [PubMed] [Google Scholar]
- 25. Page A.N., George N.P., Marceau A.H., Cox M.M., Keck J.L.. Structure and biochemical activities of Escherichia coli MgsA. J. Biol. Chem. 2011; 286:12075–12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davey M.J., Jeruzalmi D., Kuriyan J., O'Donnell M.. Motors and switches: AAA+ machines within the replisome. Nat. Rev. Mol. Cell Biol. 2002; 3:826–835. [DOI] [PubMed] [Google Scholar]
- 27. Erzberger J.P., Berger J.M.. Evolutionary relationships and structural mechanisms of AAA plus proteins. Ann. Rev. Biophys. Biomol. Struct. 2006; 35:93–114. [DOI] [PubMed] [Google Scholar]
- 28. Iyer L.M., Leipe D.D., Koonin E.V., Aravind L.. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004; 146:11–31. [DOI] [PubMed] [Google Scholar]
- 29. Ogura T., Wilkinson A.J.. AAA(+) superfamily ATPases: common structure-diverse function. Genes Cells. 2001; 6:575–597. [DOI] [PubMed] [Google Scholar]
- 30. Costes A., Lecointe F., McGovern S., Quevillon-Cheruel S., Polard P.. The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet. 2010; 6:e1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lau I.F., Filipe S.R., Soballe B., Okstad O.A., Barre F.X., Sherratt D.J.. Spatial and temporal organization of replicating Escherichia coli chromosomes. Mol. Microbiol. 2003; 49:731–743. [DOI] [PubMed] [Google Scholar]
- 32. Branzei D., Seki M., Onoda F., Enomoto T.. The product of Saccharomyces cerevisiae WHIP/MGS1, a gene related to replication factor C genes, interacts functionally with DNA polymerase delta. Mol. Genet. Genom. 2002; 268:371–386. [DOI] [PubMed] [Google Scholar]
- 33. Yoshimura A., Seki M., Kanamori M., Tateishi S., Tsurimoto T., Tada S., Enomoto T.. Physical and functional interaction between WRNIP1 and RAD18. Genes Genet. Syst. 2009; 84:171–178. [DOI] [PubMed] [Google Scholar]
- 34. Crosetto N., Bienko M., Hibbert R.G., Perica T., Ambrogio C., Kensche T., Hofmann K., Sixma T.K., Dikic I.. Human Wrnip1 is localized in replication factories in a ubiquitin-binding zinc finger-dependent manner. J. Biol. Chem. 2008; 283:35173–35185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saugar I., Parker J.L., Zhao S.K., Ulrich H.D.. The genome maintenance factor Mgs1 is targeted to sites of replication stress by ubiquitylated PCNA. Nucleic Acids Res. 2012; 40:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nomura H., Yoshimura A., Edo T., Kanno S.-i., Tada S., Seki M, Yasui A., Enomoto T.. WRNIP1 accumulates at laser light irradiated sites rapidly via its ubiquitin-binding zinc finger domain and independently from its ATPase domain. Biochem. Biophys. Res. Commun. 2012; 417:1145–1150. [DOI] [PubMed] [Google Scholar]
- 37. Hishida T., Ohya T., Kubota Y., Kamada Y., Shinagawa H.. Functional and physical interaction of yeast Mgs1 with PCNA: impact on RAD6-dependent DNA damage tolerance. Mol. Cell. Biol. 2006; 26:5509–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bish R.A., Myers M.P.. Werner helicase-interacting protein 1 binds polyubiquitin via its zinc finger domain. J. Biol. Chem. 2007; 282:23184–23193. [DOI] [PubMed] [Google Scholar]
- 39. Branzei D., Seki M., Onoda F., Yagi H., Kawabe Y., Enomoto T.. Characterization of the slow-growth phenotype of S-cerevisiae whip/mgs1 sgs1 double deletion mutants. DNA Repair. 2002; 1:671–682. [DOI] [PubMed] [Google Scholar]
- 40. Hishida T., Iwasaki H., Ohno T., Morishita T., Shinagawa H.. A yeast gene, MGS1, encoding a DNA-dependent AAA(+) ATPase is required to maintain genome stability. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8283–8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hishida T., Ohno T., Iwasaki H., Shinagawa H.. Saccharomyces cerevisiae MGS1 is essential in strains deficient in the RAD6-dependent DNA damage tolerance pathway. EMBO J. 2002; 21:2019–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lestini R., Michel B.. UvrD controls the access of recombination proteins to blocked replication forks. EMBO J. 2007; 26:3804–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim J.H., Kang Y.H., Kang H.J., Kim D.H., Ryu G.H., Kang M.J., Seo Y.S.. In vivo and in vitro studies of Mgs1 suggest a link between genome instability and Okazaki fragment processing. Nuc. Acids Res. 2005; 33:6137–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hayashi T., Seki M., Inoue E., Yoshimura A., Kusa Y., Tada S., Enomoto T.. Vertebrate WRNIP1 and BLM are required for efficient maintenance of genome stability. Genes Genet. Syst. 2008; 83:95–100. [DOI] [PubMed] [Google Scholar]
- 45. Kawabe Y.I., Seki M., Yoshimura A., Nishino K., Hayashi T., Takeuchi T., Iguchi S., Kusa Y., Ohtsuki M., Tsuyama T. et al. Analyses of the interaction of WRNIP1 with Werner syndrome protein (WRN) in vitro and in the cell. DNA Repair. 2006; 5:816–828. [DOI] [PubMed] [Google Scholar]
- 46. Yoshimura A., Seki M., Hayashi T., Kusa Y., Tada S., Ishii Y., Enomoto T.. Functional relationships between Rad18 and WRNIP1 in vertebrate cells. Biol. Pharm. Bull. 2006; 29:2192–2196. [DOI] [PubMed] [Google Scholar]
- 47. Bowman G.D., O'Donnell M., Kuriyan J.. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature. 2004; 429:724–730. [DOI] [PubMed] [Google Scholar]
- 48. Jeruzalmi D., O'Donnell M., Kuriyan J.. Crystal structure of the processivity clamp loader gamma (gamma) complex of E. coli DNA polymerase III. Cell. 2001; 106:429–441. [DOI] [PubMed] [Google Scholar]
- 49. Tsurimoto T., Shinozaki A., Yano M., Seki M., Enomoto T.. Human werner helicase interacting protein 1 (WRNIP1) functions as a novel modulator for DNA polymerase delta. Genes Cells. 2005; 10:13–22. [DOI] [PubMed] [Google Scholar]
- 50. Vijeh Motlagh N.D., Seki M., Branzei D., Enomoto T.. Mgs1 and Rad18/Rad5/Mms2 are required for survival of Saccharomyces cerevisiae mutants with novel temperature/cold sensitive alleles of the DNA polymerase delta subunit, Pol31. DNA Repair. 2006; 5:1459–1474. [DOI] [PubMed] [Google Scholar]
- 51. Shereda R.D., Kozlov A.G., Lohman T.M., Cox M.M., Keck J.L.. SSB as an Organizer/Mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008; 43:289–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lecointe F., Serena C., Velten M., Costes A., McGovern S., Meile J.C., Errington J., Ehrlich S.D., Noirot P., Polard P.. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO J. 2007; 26:4239–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lindsley J.E., Cox M.M.. Assembly and disassembly of RecA protein filaments occurs at opposite filament ends: relationship to DNA strand exchange. J. Biol. Chem. 1990; 265:9043–9054. [PubMed] [Google Scholar]
- 54. Morrical S.W., Lee J., Cox M.M.. Continuous association of Escherichia coli single-stranded DNA binding protein with stable complexes of RecA protein and single-stranded DNA. Biochemistry. 1986; 25:1482–1494. [DOI] [PubMed] [Google Scholar]
- 55. Gamper H.B., Cimino G.D., Hearst J.E.. Solution hybridization of cross-linkable DNA oligonucleotides to bacteriophage-M13 DNA - Effects of secondary structure on hybridization kinetics and equilibria. J. Mol. Biol. 1987; 197:349–362. [DOI] [PubMed] [Google Scholar]
- 56. Reckmann B., Grosse F., Urbanke C., Frank R., Blocker H., Krauss G.. Analysis of secondary structures in M13MP8 (+) single-stranded DNA by the pausing of DNA polymerase alpha. Eur. J. Biochem. 1985; 152:633–643. [DOI] [PubMed] [Google Scholar]
- 57. Antao V.P., Lai S.Y., Tinoco I.. A thermodynamic study of unusually stable RNA and DNA hairpins. Nucleic Acids Res. 1991; 19:5901–5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Eritja R., Kaplan B.E., Mhaskar D., Sowers L.C., Petruska J., Goodman M.F.. Synthesis and properties of defined DNA oligomers containing base mispairs involving 2-aminopurine. Nucleic Acids Res. 1986; 14:5869–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Law S.M., Eritja R., Goodman M.F., Breslauer K.J.. Spectroscopic and calorimetric characterizations of DNA duplexes containing 2-aminopurine. Biochemistry. 1996; 35:12329–12337. [DOI] [PubMed] [Google Scholar]
- 60. Raney K.D., Sowers L.C., Millar D.P., Benkovic S.J.. A fluorescence-based assay for monitoring helicase activity. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:6644–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ronen A. 2-Aminopurine. Mutat. Res. 1979; 75:1–47. [DOI] [PubMed] [Google Scholar]
- 62. Farah J.A., Smith G.R.. The RecBCD enzyme initiation complex for DNA unwinding: Enzyme positioning and DNA opening. J. Mol. Biol. 1997; 272:699–715. [DOI] [PubMed] [Google Scholar]
- 63. Wong C.J., Lucius A.L., Lohman T.M.. Energetics of DNA end binding by E. coli RecBC and RecBCD helicases indicate loop formation in the 3΄-single-stranded DNA tail. J. Mol. Biol. 2005; 352:765–782. [DOI] [PubMed] [Google Scholar]
- 64. Laureti L., Demol J., Fuchs R.P., Pages V.. Bacterial proliferation: keep dividing and don't mind the gap. PLoS Genet. 2015; 11:e1005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Heller R.C., Marians K.J.. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 2006; 439:557–562. [DOI] [PubMed] [Google Scholar]
- 66. Nomura H., Yoshimura A., Edo T., Kanno S., Tada S., Seki M., Yasui A., Enomoto T.. WRNIP1 accumulates at laser light irradiated sites rapidly via its ubiquitin-binding zinc finger domain and independently from its ATPase domain. Biochem. Biophys. Res. Commun. 2012; 417:1145–1150. [DOI] [PubMed] [Google Scholar]
- 67. Sekimizu K., Bramhill D., Kornberg A.. ATP activated DnaA protein in initiating replication of plasmids bearing the origin of the Escherichia coli chromosome. Cell. 1987; 50:259–265. [DOI] [PubMed] [Google Scholar]
- 68. Hingorani M.M., O'Donnell M.. ATP binding to the Escherichia coli clamp loader powers opening of the ring-shaped clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 1998; 273:24550–24563. [DOI] [PubMed] [Google Scholar]
- 69. Davey M.J., Fang L.H., McInerney P., Georgescu R.E., O'Donnell M.. The DnaC helicase loader is a dual ATP/ADP switch protein. EMBO J. 2002; 21:3148–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.