


Abstract
Non-coding RNA (ncRNA) genes play a major role in control of heterogeneous cellular behavior. Yet, their functions are largely uncharacterized. Current available databases lack in-depth information of ncRNA functions across spectrum of various cells/tissues. Here, we present FARNA, a knowledgebase of inferred functions of 10,289 human ncRNA transcripts (2,734 microRNA and 7,555 long ncRNA) in 119 tissues and 177 primary cells of human. Since transcription factors (TFs) and TF co-factors (TcoFs) are crucial components of regulatory machinery for activation of gene transcription, cellular processes and diseases in which TFs and TcoFs are involved suggest functions of the transcripts they regulate. In FARNA, functions of a transcript are inferred from TFs and TcoFs whose genes co-express with the transcript controlled by these TFs and TcoFs in a considered cell/tissue. Transcripts were annotated using statistically enriched GO terms, pathways and diseases across cells/tissues based on guilt-by-association principle. Expression profiles across cells/tissues based on Cap Analysis of Gene Expression (CAGE) are provided. FARNA, having the most comprehensive function annotation of considered ncRNAs across widest spectrum of human cells/tissues, has a potential to greatly contribute to our understanding of ncRNA roles and their regulatory mechanisms in human. FARNA can be accessed at: http://cbrc.kaust.edu.sa/farna
INTRODUCTION
For quite long time, the roles of many of the non-coding RNAs (ncRNA) such as micro RNAs (miRNAs) or long non-coding RNAs (lncRNAs) were not known and these were not perceived as essential as transcripts of protein-coding genes. Today, we know of the diverse roles miRNAs and lncRNAs have in critical cellular processes including control of gene expression, RNA splicing, RNA editing, or their involvement in various diseases (1). In this study, we will consider only miRNA and lncRNA as a number of them have been shown to exert key regulatory functions in numerous cellular processes (2–4). An illustrative example of such a miRNA is miR-503 implicated in several cancer types affecting reduction of cell proliferation through inducement of the G0/G1 cell cycle arrest by targeting CCND1 in both breast cancer (5) and endometrial cancer cell lines (6). miR-503 also directly inhibit CUGBP1 expression, thereby altering the expression of CUGBP1 target mRNAs, which causes increased sensitivity of intestinal epithelial cells to apoptosis (7) acting as a modulator of intestinal epithelial homoeostasis (7). Another example is human lncRNA Fendrr whose overexpression suppresses invasion and migration of gastric cancer cells in vitro, by down-regulating FN1 and MMP2/MMP9 expression (8). The mouse variant of this lncRNA, Fendrr, is shown to bind directly to PRC2 and TrxG/MLL complexes regulating heart and body wall development in mouse (9). Overall, insights about functions of these types of ncRNAs have stimulated interest in miRNA- and lncRNA-related research.
Since experimental elucidation of ncRNA functions is progressing slowly (10), in silico approaches for predicting ncRNA functions became increasingly important. Prediction methods are mainly based on guilt-by-association principle where a gene of interest with unknown or partially known functions is linked to other genes for which part of their functions is known, where links are based on shared or similar characteristics or behavior. The co-expression-based analysis is frequently used to infer function of ncRNA (11,12). However, similarly as with the other computational methods (13–16), co-expression-based analysis usually produces a significant number of false positive function assignments (10). Another widely-used approach employs targets of ncRNA to infer ncRNA functions from the known properties of these targets (13). The third generic approach relies on using properties of transcription factors (TFs) that control transcript activation in order to infer function of protein-coding (14) and ncRNA genes (15). It is shown that a single approach cannot detect all aspect of functional characteristics of a gene and since all these methods are complementary to each other (10,16) they can be combined to get a more complete picture of ncRNA functions.
As the function of many miRNAs and lncRNAs are not known in detail or frequently not known at all, many databases and tools have been actively developed to facilitate investigation of function of both miRNA and lncRNA and to infer potential functions of these transcripts. Some well-known databases and tools along this line include FAME (17), miR2GO (18), miRGator (12), miRò (13), miRBase (19), miRNAVISA (20), miRPath v3.0 (21), lncRNAWiki (22), lncRNAdb (22), LncRNADisease (23), lncRNA2Function (24), LncRBase (25), lncRNAtor (11), ChIPBase (15), starBase v2.0 (26), deepBase v2.0 (27) and NONCODE 2016 (28). The above-mentioned databases and tools provide important information about different aspects of ncRNAs, such as their association with the gene ontology (GO) terms, diseases, transcription factors, expression, etc. However, individually they: a/ provide function annotation only for small number of cells/tissues or b/ lack rich annotation with specific functions for large proportion of human miRNA or lncRNA, or c/ provide only part of such information for very small number of ncRNAs, or d/ provide only mechanistic information about ncRNAs, such as their length, strand, etc., without explicitly annotating functions of transcripts.
With all the above limitations in mind, we developed FARNA (Function Annotation of non-coding RNA), a knowledgebase that houses information related to inferred function of human miRNA and lncRNA in a cell/tissue-specific manner. In addition to function annotation, FARNA integrates ncRNA information related to expression, pathways and diseases in a large number of human tissues and primary cells. FARNA ranks annotated functions of ncRNA based on statistical enrichment of mapped terms from GO, pathways, diseases and parts of their regulatory networks that control activation of the ncRNA transcripts (Figure 1). In FARNA, we infer functions of an ncRNA transcript from the known functions of TFs and their associated transcription co-factors (TcoFs) that control the ncRNA transcript where the genes encoding these TFs and TcoFs co-express with the ncRNA transcript in a considered cell/tissue. The effect of TcoFs on transcriptional regulation and initiation, though indirect, is known to be significant in different cellular process (29,30).
Figure 1.

Brief description of the pipeline used to infer cell/tissue-specific functions of miRNA and lncRNA transcripts. First, FANTOM5 CAGE data for primary cells and tissues was collected. The miRNA, lncRNA, TF and TcoF transcripts that are expressed in different cells/tissues were used for further analysis in cell/tissue-specific manner. The TF binding site (TFBS) models from TRANSFAC and HOCOMOCO databases were used to predict TFBSs on promoter regions of miRNA and lncRNA transcripts. Only TFs and their associated TcoFs that are expressed in the considered cell/tissue together with the ncRNA transcript are used to infer statistically significant cell/tissue-specific functions of the transcript.
In a recent study (31), Yu et al. reported a large-scale tissue transcriptome comparison between Cap Analysis of Gene Expression (CAGE) and RNA-Seq (32) derived expression data in 22 tissues. The reported correlation coefficients between CAGE- and RNA-Seq-based expressions were quite high (e.g. 0.86, 0.87, 0.87, 0.88 for testis, placenta, pancreas and brain tissues, respectively). Also, Kawaji et al. (33) showed that among sequencing technologies HeliScope CAGE (34) and RNA-Seq are in the best agreement regarding expression (correlation coefficient was 0.88) in terms of linearity of measurements, reproducibility of quantification. As HeliScope CAGE protocol is used extensively to generate the CAGE data for FANTOM5 consortium (35), we used CAGE expression in the promoter regions of ncRNA as a proxy for the expression level of ncRNA and extended our functional annotation to cover the largest mapping of human tissues and primary cells for which CAGE data is publicly available to date. ncRNA genes demonstrate significant cell/tissue-specific expression (36,37) and therefore, we inferred cell/tissue-specific putative functions of RNA genes expressed in 119 tissues and 177 primary cells based on CAGE data generated by the FANTOM5 consortium. The potential ncRNA-associated functions are inferred as statistically significantly enriched. This has resulted in a rich annotation of functions for the considered ncRNAs. To the best of our knowledge, this is the first time that in combination with co-expression, both genes that encode for TFs and TcoFs that control activation of human miRNAs and lncRNAs are used for ncRNA function inference in a cell/tissue-specific manner for such a comprehensive number of tissues and primary cells.
MATERIALS AND METHODS
Inferring function of genes based on TFs that control them is a known approach (14). In FARNA, to infer functions of a transcript, we require that the transcript of the considered ncRNA and the transcripts of genes that encode for TFs and TcoFs likely controlling activation of the considered ncRNA transcript are all expressed in the considered cell/tissue. Each ncRNA transcript is then associated with a set T of TFs and TcoFs in a cell/tissue-specific manner and annotated functions of TFs and TcoFs from T are then used to infer statistically significant GO terms, pathways and diseases for an ncRNA transcript.
Selection of expressed transcripts in FANTOM5 samples
For our analyses, we used the robust CAGE peaks from FANTOM5 (http://fantom.gsc.riken.jp/5/datafiles/latest/extra/CAGE_peaks/). We used CAGE data from 119 human tissues and 177 human primary cells (in many cases data were collected from three individual donors). Following the description of (31), transcripts having expression value of at least 1 tag per million (TPM) (normalized TPM using the relative log expression (RLE) method in edgeR (38)) are considered as expressed in each tissue or primary cell. 9,025 alternative transcripts are used based on PROmiRNA algorithm (39) for miRNAs retrieved from miRBase v20 (19). For lncRNA, 23,898 transcripts from GENCODE (37) V19 were used. The one TPM threshold selects 14.87% primary miRNA transcripts and 8.89% lncRNA transcripts from different tissues/cells (Figure 2). We used transcripts of genes encoding for TFs and TcoFs from Refseq (40). The 5΄ end of lncRNA, TF and TcoF transcripts were considered to represent transcription start sites (TSSs). For the miRNA transcripts the TSSs were determined from PROmiRNA. CAGE tags were used to estimate expression level of transcripts. A CAGE tag was considered as associated with a transcript if it overlaps the promoter region that covers [–500 bp, +100 bp] around TSS of transcript on the same strand. When one promoter region is overlapping with multiple CAGE clusters (35), we select only one CAGE cluster based on the strongest signal.
Figure 2.

Expression of of RNA transcripts in different tissues/cells. Subfigure (A) shows scatter plot with contour overlay and (B) shows the box plot for the fraction of transcripts from miRNA and lncRNA having expression >1 TPM in different tissues/cells. Subfigure (C) and (D) highlight the distribution of expression for miRNA and lncRNA transcripts in several tissues. Vertical red dashed line highlights the one TPM (normalized TPM using the relative log expression (RLE) method in edgeR) threshold.
Association of transcripts with TFBS
For each transcript of ncRNA expressed in a particular cell/tissue, TFBSs were predicted on promoters of the transcript. To predict TFBSs we used TFBS models from both HOCOMOCO v10 (41) and TRANSFAC 2015 (42) databases. From HOCOMOCO, we used human TFBS models and mapped the corresponding matrix model on promoters the same way as in (43). If the mapped score of TFBS model is no less than the model score set at P-value = 0.0001 by HOCOMOCO, then we considered that position to be a potential TFBS hit. TFBS models from TRANSFAC 2015 were mapped with the MATCH program (42) on the promoter regions using minimum false-positive profiles of vertebrate high quality matrices.
Set of cell/tissue-specific TFs and TcoFs for each ncRNA transcript
For each ncRNA transcript in a specific cell/tissue, we first checked if it is expressed in that cell/tissue. If so, we associate with this transcript TFs based on their predicted TFBSs on the promoter of the transcript. We further associated these TFs with their known interacting high-confidence TcoFs from TcoF-DB (44), under the conditions that genes encoding these TFs and their associated TcoFs are expressed in the considered cell/tissue. All such associated TFs and TcoFs formed the set of regulatory elements for this transcript in that particular cell/tissue and are used to infer transcript functions.
Cell/tissue specificity score for ncRNA transcripts
We calculated the specificity score for expression of all ncRNA transcripts based on the method from (45,46). The tissue specificity score (τ) of an RNA transcript was calculated as follows:
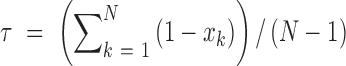 |
where N is the number of cells/tissues and xk is the expression level of transcript in the cells/tissues normalized by the maximum expression value. For example, if the specificity score is high in liver and the expression is also high in liver for an RNA transcript, then one may assume that this transcript exerts certain effects in liver.
Enrichment of GO, pathway, disease annotation
The Human GO annotation was taken from GO Consortium (47). ‘ELIM’ method (48) with Ontologizer (49,50) tool was used to perform GO enrichment analysis. Only GO terms that are statistically significant (false discovery rate FDR < 0.05) are included in the FARNA knowledgebase. For statistical analysis GO terms with less than five annotated protein-coding genes were excluded for the enrichment analysis as suggested in (24).In addition to the properties of the ncRNA transcripts described by the GO terms, we also identify statistically enriched pathways and diseases in which the transcript is likely involved. For identifying the implicated pathways we used the Reactome pathway repository (51), a curated and peer reviewed pathway database. KOBAS (KEGG Orthology Based Annotation System) 2.0 (52), which integrates OMIM (53), KEGG DISEASE (54), FunDO (55), GAD (56) and NHGRI GWAS Catalog (57) disease databases, is used to predict involvement of ncRNA transcripts in diseases. Enriched pathways are identified by calculating the p-value based on the hypergeometric distribution for each transcript using as the background all unique human TFs from both HOCOMOCO v10 and TRANSFAC 2015 and all TcoFs from TcoF DB. This P-value was then corrected for multiplicity testing using Benjamini–Hochberg method (58) to generate FDR values. For determining statistically enriched diseases, the background consisted of all human proteins from SwissProt. We kept only pathway and disease annotations that have FDR <0.05. Moreover, FARNA provides filters for selecting more stringent FDR corrections. Figure 1, briefly outlines the pipeline of FARNA.
RESULTS
FARNA is composed of four basic modules (Figure 3): Data sources, FARNA function association, FARNA DB and FARNA web interface. Here, we provide description of each module.
Figure 3.

Basic modules of FARNA. There are four basic modules in FARNA. Data Sources: The repository for all TFs, TcoFs, CAGE expression data from different cells/tissues, and transcripts for both miRNA and lncRNA. CAGE data was used to filter out low-expressed ncRNA transcripts, TFs, TcoFs. FARNA Associations: This module considers the association of TFs with TcoFs to RNA transcripts and identifies statistically enriched functions related to RNA transcripts in a cell/tissue-specific manner. FARNA DB: This module indexes all associated function annotation using Elasticsearch platform. FARNA Web Interface: Web interface for users to explore the FARNA annotated function and expression profile of RNA transcripts in different cells/tissues.
Data sources
This module contains information on miRNA promoters from PROmiRNA and lncRNA promoters from GENCODE. CAGE expression information on ncRNA transcripts from different cells/tissues is also stored. Information on all TFs and TcoFs from HOCOMOCO, TRANSFAC and TcoF-DB is also kept here.
FARNA function association
This module associates each ncRNA transcript with TFs and TcoFs in the cell/tissue-specific manner. Then, statistically significant GO terms, pathways and diseases are annotated for each ncRNA transcript based on their associated TFs and TcoFs. PROmiRNA for miRNAs and GENCODE for lncRNAs generate multiple transcripts per gene and these transcripts are analyzed and annotated separately. The following conditions have to be satisfied in order to assign inferred function to a target ncRNA transcript: (i) Each considered TF has binding sites in the promoter of the target ncRNA transcript as described earlier. (ii) Each TcoF is linked to a TF from 1/ if it is known that this TcoF binds this TFs (only the highest confidence interaction taken form TcoF DB). (iii) All such TFs and TcoFs and their target ncRNA are expressed in the considered cell/tissue. Only statistically enriched functions of such TFs and TcoFs in the considered cell/tissue are used to infer function of the target ncRNA in that cell/tissue based on the guilt-by-association principle.
FARNA DB and links to external resources
For each lncRNA transcripts we provided the corresponding Ensembl ID as its primary RNA ID, and also the corresponding gene IDs and gene names from Ensembl (59) linked to the related Ensembl resource. Similarly for miRNA, we used the miRBase names and provide also the corresponding gene IDs and names from Ensembl. For TFs and TcoFs we used UniProt (60) names linking them externally to UniProt entries. GO annotation IDs and descriptions are included and linked externally to AMIGO site (61). IDs and names of diseases and pathways from KOBAS and Reactome are included and externally linked to their respective entries.
FARNA web interface
Web interface of FARNA provides users with multiple options to explore the annotated functions. Users can search based on RNA IDs and names, GO terms, pathways, diseases, TFs and TcoFs. The search box provides autosuggestion and flexible search options, such as search for any specific term or a group of terms combined using OR, AND, etc., logical operators following the Elasticsearch query syntax (https://www.elastic.co/guide/en/elasticsearch/reference/current/query-dsl-query-string-query.html). For example, if users do not know the exact gene name, but have an idea that the name ends with 567, then the search with ENSG*567 can be done, or in order to query the role of MALAT1 in cancer, one can type ‘malat1 cancer’ or ‘malat1 AND cancer’ in the search box to find all relevant results from FARNA knowledgebase. Search is case-insensitive.
FARNA annotation data is stored in Elasticsearch based index (62). To facilitate exploration of the query results aggregation queries are used and the results are presented as attribute-filters on the query result pages with numbers indicating the size of the result subsets. Users can narrow the search results by selecting the available filters (e.g. attribute filters like tissues, cells, pathways, disease or specific GO category; FDR threshold filters for more stringent statistical significance of enrichment; RNA type selector: miRNA or lncRNA). Expression profiles for ncRNA transcripts across different cells/tissues are also highlighted. Users can sort the expression profiles based on expression values or cell/tissue names. As we have the annotation of inferred functions for each individual transcript of a gene, we also provided a pivot table for the ‘gene view’, which highlights the variation of annotations among different transcripts of a gene. Drag and drop functionality provided by the PivotTable library facilitates users to view the query results across multiple samples among multiple transcripts of a gene. Supplementary Figure S1 shows an example usage of the pivot table to check the annotations of MALAT1 in a tissue. Detailed usage instructions are provided at the FARNA web site.
Statistics of FARNA
For each ncRNA, there are more than one transcript provided by PROmiRNA (4.7 transcripts per miRNA) and GENCODE (1.72 transcripts per lncRNA). As we annotate functions of transcripts in different cells/tissues, we find transcripts could have different annotation depending on the cell/tissue. Also, since one gene could have multiple transcripts these transcripts could have different annotation even in the same cell/tissue (depending of the associated TFs and TcoFs). From Supplementary Figure S2, median value of box plot shows that in FARNA we were able to infer function in each cell/tissue for ∼2,000 lncRNA transcripts and ∼1,300 primary miRNA transcripts. In total, FARNA provides annotated functions for 2,734 miRNAs transcripts and 7,555 lncRNA transcripts, which are expressed (>1 TPM (normalized TPM using RLE method in edgeR)) in different human cells/tissues. Table 1 summarizes the annotation statistics of FARNA.
Table 1. Summary statistics for FARNA.
| Unique entries | miRNA | lncRNA |
|---|---|---|
| Transcripts | 2,734 | 7,555 |
| GO terms | 1,315 | 1,414 |
| Pathways | 179 | 186 |
| Diseases | 1,172 | 1,319 |
| Tissues | 119 | 119 |
| Primary cells | 177 | 177 |
This table summarizes the number of transcripts of miRNA and lncRNA for which FARNA provides function information in different cells and tissues. For these transcripts, the number of statistically significant (FDR < 0.05) unique GO terms, pathways and diseases are also highlighted.
To the best of our knowledge, FARNA is the first knowledgebase that contains rich annotation of inferred functions for a large number of human miRNA and lncRNA transcripts in a comprehensive number of cells/tissues. The annotation is provided per transcript. The functions include statistically enriched GO categories, pathways and diseases. FARNA displays GO, pathway and disease annotation enrichment at different user-selected level of statistical significance. In Table 2, we highlighted the extent of information available in FARNA and other similar databases based on their original publication or information presented on the respective web sites. Not all databases provide clear information regarding the statistics of their content.
Table 2. Basic features of databases that contain annotation of human miRNA or lncRNA transcripts/genes.
| Database | miRNA | lncRNA | GO | Pathway | Disease | TF | TcoF | Targets | CoE | CTFA |
|---|---|---|---|---|---|---|---|---|---|---|
| FARNA | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | - | ✓ | ✓ |
| lncRNAdb | - | ✓ | ✓ | ✓ | ✓ | - | - | - | ✓ | - |
| LncRNADisease | - | ✓ | - | - | ✓ | - | - | - | - | - |
| lncRNA2Function | - | ✓ | ✓ | ✓ | - | - | - | - | ✓ | - |
| lncRNAtor* | - | ✓ | ✓ | ✓ | - | - | - | ✓ | ✓ | ✓ |
| miRGator v3.0 | ✓ | - | ✓ | ✓ | ✓ | - | - | ✓ | ✓ | - |
| miRPath v3.0 | ✓ | - | ✓ | ✓ | - | - | - | ✓ | ✓ | - |
| miRò | ✓ | - | ✓ | - | ✓ | - | - | ✓ | - | - |
| ChIPBase | ✓ | ✓ | ✓ | ✓ | - | ✓ | - | - | - | - |
| FAME | ✓ | - | ✓ | ✓ | - | - | - | ✓ | ✓ | - |
| NONCODE 2016 | - | ✓ | ✓ | - | ✓ | - | - | - | ✓ | - |
| deepBase v2.0 | ✓ | ✓ | ✓ | ✓ | ✓ | - | - | - | ✓ | - |
This table compares the type of annotation provided by different database. ‘✓’ means presence and ‘-’ means absence. CoE: co-expression; CTFA: cell/tissue-specific function annotation. *: Resource with primary focus on cancer.
DISCUSSION
There is no extensive validated RNA annotation for a comprehensive number of cells/tissues, thus it is thus not possible to generate precision-recall (PR) curve or receiver operating curve (ROC) for RNAs in cell/tissue-specific manner (17,24,45). In the absence of a gold standard dataset for cell/tissue-specific functions of miRNA or lncRNA genes, it is difficult to measure to what extent the inferred functions by FARNA correspond to the true function of RNA genes. We therefore used indirect means of evaluating the predicted functions. We carried out a literature survey for well-studied ncRNA genes and highlight the FARNA predictions that are closest (17) to the reported function in different cells/tissues.
Example 1
miR-122 which is known to be down-regulated in liver diseases (63). HNF6 (64), HNF4A, CEBPA and FOXA2 (15) are shown to be liver enriched TFs and Nuclear factor-κB (NF-κB) is activated in response to several stresses and may cause liver damage (63). Suppression of miR-122 induced by HBV infection, leads to inactivation of IFN expression, which in turn enhances HBV replication, contributing to viral persistence and hepatocarcinogenesis (65). FARNA predictions show TFs HNF6, HNF4A, CEBPZ and FOXA2 associated to the miR-122 promoter region and that miR-122 is highly expressed in ‘normal’ liver tissue and hepatocyte cells. Moreover, FARNA returns several liver-related GO terms, pathways and diseases such as: GO:0001889 liver development, GO:0044255 cellular lipid metabolic process, GO:0038061 NIK/NF-kappaB signaling, GO:0033256 I-kappaB/NF-kappaB complex, REACT_25024 TRAF6 mediated induction of NFkB and MAP kinases upon TLR7/8 or 9 activation, REACT_13537 p75NTR signals via NF-kB, REACT_22258 Metabolism of lipids and lipoproteins, REACT_19241 Regulation of lipid metabolism by Peroxisome proliferator-activated receptor alpha (PPARalpha) and REACT_25359: RIG-I/MDA5 mediated induction of IFN-alpha/beta pathways in liver tissue.
Example 2
miRNAs from miR-200 family have been identified as biomarkers for epithelial-to-mesenchymal transition (EMT). In EMT epithelial cells morphologically and phenotypically transdifferentiate into mesenchymal cells in proliferative vitreoretinopathy (PVR) (66), renal fibrosis (67), cancers (68) and embryonic development (69). It has been shown that miR-200 family targets the E-cadherin transcriptional repressors zinc finger E-box binding homeobox 1 (ZEB1) and ZEB2 for EMT (70–72). Some other TFs that play a crucial role in EMT include SNAI1 and SNAI2 as initiation of EMT events have been associated with SNAI activation (73,74). It has further been demonstrated (73,74) that SNAI2 indirectly regulates ZEB1 and ZEB2 by regulating the miR-200 family transcripts, and in turn the miR-200 family regulates ZEB1 and ZEB2. ETS1, another TF, is a suggested upstream regulator of ZEB1 and ZEB2 (75) and it has also been identified as a biomarker of EMT. FARNA prediction associates TFs SNAI1, SNAI2, ZEB1 and ETS1 to miR-200a promoter region and shows that miR-200a is expressed in eye vitreous humor and skin palm tissue and in renal epithelial cells. FARNA additionally returns several related GO terms and disease annotations such as GO:0048596 embryonic camera-type eye morphogenesis, GO:0002088 lens development in camera-type eye, REACT_13776 p75 NTR receptor-mediated signaling (76), FunDO:(2152) Renal tubular acidosis, REACT_6966 Toll-Like Receptors Cascades (77), GO:0016605 PML body and FunDO:(1853) Eye cancer.
Example 3
The third example related to lncRNA, PCA3 (Prostate Cancer Antigen 3), which are reported to be expressed in human prostate tissue and further shown to be over-expressed in prostate cancer (78,79). EMT has also been demonstrated to play a critical role in the development of metastatic castration resistant prostate cancer (mCRPC) (80). FARNA prediction associates TFs SNAI1 and SNAI2 bind to the PCA3 promoter region and shows PCA3 to be strictly expressed in normal prostate and penis tissue. Schalken et al. reported DD3 (PCA3) possesses 4 TF binding sites, of which one binding site is a preferential target of a topologic transcription factor confirmed to be high-mobility group protein-I(Y) (HMGI-Y), with a subtle conformational change suggesting the recruitment of another TF, as yet unidentified (81). FARNA predicts HMGB1 and HMGB2 as co-factors associated with PCA3 transcription. FARNA additionally returns related GO and disease terms such as OMIM:(176807) Prostate cancer, GAD:(KOBAS:32174) Uterine prolapse, GO:2000134 negative regulation of G1/S transition of mitotic cell cycle, GO:0043518 negative regulation of DNA damage response, signal transduction by p53 class mediator, GO:0016605 PML body and GO:0051403 stress-activated MAPK cascade.
Example 4
Another lncRNA, RMST (rhabdomyosarcoma 2-associated transcript), has been reported to be expressed specifically in brain tissue and its increased expression has been demonstrated during neuronal differentiation, indicating a role in neurogenesis (82). More precisely, RMST physically interacts with SOX2, a TF known to regulate neural fate (82). FARNA shows SOX2 in the TF list of RMST, its expression in cerebellum adult tissue. It additionally returns related disease and GO terms such as GO:0038095 Fc-epsilon receptor signaling pathway, KEGG DISEASE:(KOBAS:22) Cancers of the nervous system. FARNA further shows that RMST expression is not entirely restricted to cerebellum adult tissue, but is also expressed in ductus deferens, seminal vesicle and adipose tissue. Thus, RMST may be involved in male reproductive system as well.
Example 5
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), also referred to as nuclear-enriched abundant transcript 2 (NEAT2), is highly abundant and is expressed in many healthy organs (83). Differentially expressed MALAT1 have now been linked to several cancer types including lung cancer (83), osteosarcoma (84), uterine endometrial stromal sarcoma (85), cervical cancer (86), hepatocellular carcinoma (HCC) (87), breast cancer (88), acute myeloid leukemia (89) and colorectal cancer (90), as well as viral infection or alcohol abuse (91). MALAT1 has been linked to a plethora of functions, as it was shown to promote cell motility of lung cancer cells (92), support proliferation and invasion of cervical cancer cells (93), function in trophoblast invasion during embryonic development (94), proliferation of vitreoretinopathy (95), pathogenesis of diabetes-related microvascular disease, diabetic retinopathy (96) and is associated with synaptogenesis (97).
FARNA-predicted MALAT1 functions that comply with these known ones include NHGRI GWAS Catalog:(KOBAS:4945) pulmonary function (interaction), REACT_163823 SUMOylation, REACT_163793 processing and activation of SUMO, REACT_25359 RIG-I/MDA5 mediated induction of IFN-alpha/beta pathways, KEGG DISEASE:(KOBAS:762) Cancers of the lung and pleura, KEGG DISEASE:(H00036) osteosarcoma, GAD:(KOBAS:32214) endometrial neoplasms, KEGG DISEASE:(H00048) hepatocellular carcinoma, FunDO:(1944) breast cancer, NHGRI GWAS Catalog:(KOBAS:2105) esophageal adenocarcinoma, OMIM:(601626) leukemia, acute myeloid, KEGG DISEASE:(H00004) chronic myeloid leukemia (CML), KEGG DISEASE:(H00001 and H00002) acute lymphoblastic leukemia, KEGG DISEASE:(KOBAS:172) cancers of haematopoietic and lymphoid tissues and GAD:(KOBAS:774) colorectal cancer.
As for the relation of MALAT1 and eye, the currently known involvement of MALAT1 is in proliferation of vitreoretinopathy (95) and pathogenesis diabetic retinopathy (96). FARNA shows MALAT1 expression in ‘eye vitreous humor’ and several types of ‘eye muscle’ tissue and predict ‘Toll-like receptors cascade’ pathways which acts as a part of immune system for infection in eye (98). FARNA returns disease related concepts such as FunDO:(1853) Eye cancer, NHGRI GWAS Catalog:(KOBAS:8905) Fuchs's corneal dystrophy and NHGRI GWAS Catalog:(KOBAS:5215) cataracts in type 2 diabetes.
Example 6
The lncRNA HOTAIR, originating from the HOXC locus, is reported overexpressed in several cancer types (99) including bladder cancer (100). HOTAIR has also been shown to be an important factor in the differentiation of skin (101). Chuong specifically reports the involvement of Homeobox genes in fetal wound healing (not in adults) and skin regional specificity (101). FARNA show HOTAIR expression in fibroblast skin walker warburg, smooth muscle cells bladder and smooth muscle cells umbilical vein and predicts several annotations from different sources that comply with known HOTAIR functioning such as GO:0009913 epidermal cell differentiation, REACT_111045 developmental biology, GAD:(KOBAS:2024) developmental, GO:0051241 negative regulation of multicellular organismal process, GO:0043066 negative regulation of apoptotic process, GO:0032481 positive regulation of type I interferon production, REACT_118764 ZBP1(DAI) mediated induction of type I IFNs, KEGG DISEASE:(KOBAS:172) cancers of haematopoietic and lymphoid tissues and FunDO:(1944) breast cancer.
FARNA also predicts annotation of function such as GO:0016514 SWI/SNF complex and GO:0006338 chromatin remodeling for HOTAIR that is reasonable as loss of the mammalian SWI/SNF complexes function has been associated with malignant transformation and it has also been demonstrated to mediate ATP-dependent chromatin remodeling processes that are critical for differentiation and proliferation (102).
Moreover, HOTAIR overexpression has also been shown to induce aberrant expression of HOX transcription factors (especially HOXD10, that regulate differentiation and tissue homeostasis). Heubach et al. reported that the effects of HOTAIR are strongly tissue-dependent and can even differ within one cancer type (103). They also showed that in 5637 cells (Homo sapiens urinary bladder grade II carcinoma), only HOXB8 was induced and HOX genes that were repressed include HOXD10, HOXA1 and HOXA11. The decreased expression of HOXD10 in 5637 cells was accompanied by slight increases in H3K27 and H3K9 methylation. Histone H3 lysine 9 (H3–K9) methylation has been shown to correlate with transcriptional repression (104,105) and deacetylation along with methylation of H3K9 coordinate chromosome condensation (106). FARNA predicted annotation of function that comply with these known HOTAIR functioning as well such as GO:1990619 histone H3-K9 deacetylation and GO:0006338 chromatin remodelling.
These examples demonstrate that FARNA annotations fit closely to and are supported by the known functions of several ncRNAs. Supplementary Table S1 highlights additional experimental evidences demonstrating the function of well-known miRNA and lncRNA collated from literature and FARNA predictions that support and complement the existing experimental evidence. There are also other ncRNAs for which there is no experimental evidence of its functions, such as for hsa-miR-4267. However, FARNA may help in some of these cases, as it shows that this miRNA is highly expressed in several ‘normal’ tissue types (esophagus, tonsil, small intestine, colon and tongue) and cells (corneal epithelial cells, smooth muscle cells pulmonary artery, urothelial cells, esophageal epithelial cells and amniotic membrane cells). These suggest prevalent, high expression of miR-4267 in digestive system related tissues/cells, thereby providing suggestions for future research.
Additionally, we also used semantic similarity based approach as an alterative indirect way to show that our annotation pipeline works well. Semantic similarity is widely used as a measure to assess performance of automated function prediction (107–109). We used ‘direct annotations’ from AMIGO for 76 human miRNAs (Supplementary Table S2). For the miRNAs that are annotated by both FARNA and FAME, the semantic similarity between FARNA and AMIGO is higher than the semantic similarity between FAME and AMIGO (Supplementary Figure S3). It was not possible to do the same for lncRNA as there is no equivalent resource that can be used for this purpose. To measure the semantic similarity, Lin's similarity measure (110) with Resnik information content (111) with best matching average (bma) option was used from ‘The semantic measures library and toolkit’ (112).
To explore the overlapped annotation between FARNA and some other databases, we selected FAME (for miRNA) and LncRNA2Function (for lncRNA) because they provide option to download their complete annotations. We found that these tools have good overlap of their annotations against FARNA. In order to find out overlap on the annotation between FARNA and these two databases, we applied the following. If an ncRNA is annotated by a GO term X in FARNA and if this matches exactly the annotation in the other database, or it matches a more general GO term (ISA ancestor relationship in GO hierarchy) of the other database, then we considered that the FARNA annotation is included in the annotation of the other database and vice versa. When we checked GO annotations of FARNA against high confidence GO annotations provided by FAME to compare the coverage of annotations by each other, we found that 58.78% annotations of FAME are covered by FARNA and 38.20% of annotations provided by FARNA are covered by FAME. For lncRNA, LncRNA2Function covers 65.02% of annotations provided by FARNA, while 30.90% annotations of LncRNA2Function are covered by FARNA (Supplementary Figure S4).
FUTURE DEVELOPMENTS AND UPDATE
In future, FARNA will be updated annually based on the availability of expression data from new cells and tissues. Also, we will update the annotation with new version of TFBS models and new versions of annotations from reference repositories. We also plan to extend FARNA to cover other species, specifically model organisms, such as plant Arabidopsis thaliana, fungi such as Aspergillus nidulans, Saccharomyces cerevisiae, or bacteria such as Escherichia coli, Bacillus subtilis or Synechocystis.
CONCLUSION
FARNA contains annotated functions for a large number of miRNA and lncRNA transcripts in different human cells/tissues. It also contains suitable search mechanism to interrogate the information contained. We believe that FARNA will be of broad interest to the researchers working on human miRNAs and lncRNAs.
Supplementary Material
ACKNOWLEDGEMENTS
The computational analysis for this study was performed on Dragon and Snapdragon compute clusters of Computational Bioscience Research Center at King Abdullah University of Science and Technology.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) [URF/1/1976-04]; KAUST base research funds (to V.B.B.). Funding for open access charge: KAUST [CCF URF 1976].
Conflict of interest statement. None declared.
REFERENCES
- 1.Mattick J.S., Makunin I.V.. Non-coding RNA. Hum. Mol. Genet. 2006; 15:R17–R29. [DOI] [PubMed] [Google Scholar]
- 2.Mercer T.R., Dinger M.E., Mattick J.S.. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009; 10:155–159. [DOI] [PubMed] [Google Scholar]
- 3.Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P. et al. . Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009; 458:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma L., Bajic V.B., Zhang Z.. On the classification of long non-coding RNAs. RNA Biol. 2013; 10:925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long J., Ou C., Xia H., Zhu Y., Liu D.. MiR-503 inhibited cell proliferation of human breast cancer cells by suppressing CCND1 expression. Tumour Biol. 2015; 36:8697–8702. [DOI] [PubMed] [Google Scholar]
- 6.Xu Y.Y., Wu H.J., Ma H.D., Xu L.P., Huo Y., Yin L.R.. MicroRNA-503 suppresses proliferation and cell-cycle progression of endometrioid endometrial cancer by negatively regulating cyclin D1. FEBS J. 2013; 280:3768–3779. [DOI] [PubMed] [Google Scholar]
- 7.Cui Y.H., Xiao L., Rao J.N., Zou T., Liu L., Chen Y., Turner D.J., Gorospe M., Wang J.Y.. miR-503 represses CUG-binding protein 1 translation by recruiting CUGBP1 mRNA to processing bodies. Mol. Biol. Cell. 2012; 23:151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu T.P., Huang M.D., Xia R., Liu X.X., Sun M., Yin L., Chen W.M., Han L., Zhang E.B., Kong R. et al. . Decreased expression of the long non-coding RNA FENDRR is associated with poor prognosis in gastric cancer and FENDRR regulates gastric cancer cell metastasis by affecting fibronectin1 expression. J. Hematol. Oncol. 2014; 7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grote P., Wittler L., Hendrix D., Koch F., Wahrisch S., Beisaw A., Macura K., Blass G., Kellis M., Werber M. et al. . The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell. 2013; 24:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen B.O., Vaid N., Musialak-Lange M., Janowski M., Mutwil M.. Elucidating gene function and function evolution through comparison of co-expression networks of plants. Front. Plant Sci. 2014; 5:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park C., Yu N., Choi I., Kim W., Lee S.. lncRNAtor: a comprehensive resource for functional investigation of long non-coding RNAs. Bioinformatics. 2014; 30:2480–2485. [DOI] [PubMed] [Google Scholar]
- 12.Cho S., Jang I., Jun Y., Yoon S., Ko M., Kwon Y., Choi I., Chang H., Ryu D., Lee B. et al. . MiRGator v3.0: a microRNA portal for deep sequencing, expression profiling and mRNA targeting. Nucleic Acids Res. 2013; 41:D252–D257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagana A., Forte S., Giudice A., Arena M.R., Puglisi P.L., Giugno R., Pulvirenti A., Shasha D., Ferro A.. miRo: a miRNA knowledge base. Database (Oxford). 2009; 2009:bap008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen H., Li H., Liu F., Zheng X., Wang S., Bo X., Shu W.. An integrative analysis of TFBS-clustered regions reveals new transcriptional regulation models on the accessible chromatin landscape. Sci. Rep. 2015; 5:8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang J.H., Li J.H., Jiang S., Zhou H., Qu L.H.. ChIPBase: a database for decoding the transcriptional regulation of long non-coding RNA and microRNA genes from ChIP-Seq data. Nucleic Acids Res. 2013; 41:D177–D187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radivojac P., Clark W.T., Oron T.R., Schnoes A.M., Wittkop T., Sokolov A., Graim K., Funk C., Verspoor K., Ben-Hur A. et al. . A large-scale evaluation of computational protein function prediction. Nat. Methods. 2013; 10:221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ulitsky I., Laurent L.C., Shamir R.. Towards computational prediction of microRNA function and activity. Nucleic Acids Res. 2010; 38:e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhattacharya A., Cui Y.. miR2GO: comparative functional analysis for microRNAs. Bioinformatics. 2015; 31:2403–2405. [DOI] [PubMed] [Google Scholar]
- 19.Kozomara A., Griffiths-Jones S.. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011; 39:D152–D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamanu T.K., Radovanovic A., Archer J.A., Bajic V.B.. Exploration of miRNA families for hypotheses generation. Sci. Rep. 2013; 3:2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vlachos I.S., Zagganas K., Paraskevopoulou M.D., Georgakilas G., Karagkouni D., Vergoulis T., Dalamagas T., Hatzigeorgiou A.G.. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res. 2015; 43:W460–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quek X.C., Thomson D.W., Maag J.L., Bartonicek N., Signal B., Clark M.B., Gloss B.S., Dinger M.E.. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2015; 43:D168–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen G., Wang Z., Wang D., Qiu C., Liu M., Chen X., Zhang Q., Yan G., Cui Q.. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013; 41:D983–D986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang Q., Ma R., Wang J., Wu X., Jin S., Peng J., Tan R., Zhang T., Li Y., Wang Y.. LncRNA2Function: a comprehensive resource for functional investigation of human lncRNAs based on RNA-seq data. BMC Genomics. 2015; 16(Suppl 3):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakraborty S., Deb A., Maji R.K., Saha S., Ghosh Z.. LncRBase: an enriched resource for lncRNA information. PLoS One. 2014; 9:e108010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H.. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng L.L., Li J.H., Wu J., Sun W.J., Liu S., Wang Z.L., Zhou H., Yang J.H., Qu L.H.. deepBase v2.0: identification, expression, evolution and function of small RNAs, LncRNAs and circular RNAs from deep-sequencing data. Nucleic Acids Res. 2016; 44:D196–D202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y., Li H., Fang S., Kang Y., Wu W., Hao Y., Li Z., Bu D., Sun N., Zhang M.Q. et al. . NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016; 44:D203–D208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coutts A.S., Weston L., La Thangue N.B.. A transcription co-factor integrates cell adhesion and motility with the p53 response. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:19872–19877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buaas F.W., Val P., Swain A.. The transcription co-factor CITED2 functions during sex determination and early gonad development. Hum. Mol. Genet. 2009; 18:2989–3001. [DOI] [PubMed] [Google Scholar]
- 31.Yu N.Y., Hallstrom B.M., Fagerberg L., Ponten F., Kawaji H., Carninci P., Forrest A.R., Hayashizaki Y., Uhlen M., Daub C.O.. Complementing tissue characterization by integrating transcriptome profiling from the Human Protein Atlas and from the FANTOM5 consortium. Nucleic Acids Res. 2015; 43:6787–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B.. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008; 5:621–628. [DOI] [PubMed] [Google Scholar]
- 33.Kawaji H., Lizio M., Itoh M., Kanamori-Katayama M., Kaiho A., Nishiyori-Sueki H., Shin J.W., Kojima-Ishiyama M., Kawano M., Murata M. et al. . Comparison of CAGE and RNA-seq transcriptome profiling using clonally amplified and single-molecule next-generation sequencing. Genome Res. 2014; 24:708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanamori-Katayama M., Itoh M., Kawaji H., Lassmann T., Katayama S., Kojima M., Bertin N., Kaiho A., Ninomiya N., Daub C.O. et al. . Unamplified cap analysis of gene expression on a single-molecule sequencer. Genome Res. 2011; 21:1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forrest A.R., Kawaji H., Rehli M., Baillie J.K., de Hoon M.J., Haberle V., Lassmann T., Kulakovskiy I.V., Lizio M., Itoh M. et al. . A promoter-level mammalian expression atlas. Nature. 2014; 507:462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roy S., Schmeier S., Arner E., Alam T., Parihar S.P., Ozturk M., Tamgue O., Kawaji H., de Hoon M.J., Itoh M. et al. . Redefining the transcriptional regulatory dynamics of classically and alternatively activated macrophages by deepCAGE transcriptomics. Nucleic Acids Res. 2015; 43:6969–6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G. et al. . The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012; 22:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marsico A., Huska M.R., Lasserre J., Hu H., Vucicevic D., Musahl A., Orom U., Vingron M.. PROmiRNA: a new miRNA promoter recognition method uncovers the complex regulation of intronic miRNAs. Genome Biol. 2013; 14:R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M. et al. . RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014; 42:D756–D763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Soboleva A.V., Kasianov A.S., Ashoor H., Ba-Alawi W., Bajic V.B., Medvedeva Y.A., Kolpakov F.A.. HOCOMOCO: expansion and enhancement of the collection of transcription factor binding sites models. Nucleic Acids Res. 2015; 4:D116–D125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matys V., Kel-Margoulis O.V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K. et al. . TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006; 34:D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alam T., Medvedeva Y.A., Jia H., Brown J.B., Lipovich L., Bajic V.B.. Promoter analysis reveals globally differential regulation of human long non-coding RNA and protein-coding genes. PLoS One. 2014; 9:e109443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schaefer U., Schmeier S., Bajic V.B.. TcoF-DB: dragon database for human transcription co-factors and transcription factor interacting proteins. Nucleic Acids Res. 2011; 39:D106–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu M.X., Chen X., Chen G., Cui Q.H., Yan G.Y.. A computational framework to infer human disease-associated long noncoding RNAs. PLoS One. 2014; 9:e84408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yanai I., Benjamin H., Shmoish M., Chalifa-Caspi V., Shklar M., Ophir R., Bar-Even A., Horn-Saban S., Safran M., Domany E. et al. . Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005; 21:650–659. [DOI] [PubMed] [Google Scholar]
- 47.Gene Ontology, C.. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015; 43:D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexa A., Rahnenfuhrer J., Lengauer T.. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006; 22:1600–1607. [DOI] [PubMed] [Google Scholar]
- 49.Bauer S., Grossmann S., Vingron M., Robinson P.N.. Ontologizer 2.0–a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics. 2008; 24:1650–1651. [DOI] [PubMed] [Google Scholar]
- 50.Grossmann S., Bauer S., Robinson P.N., Vingron M.. Improved detection of overrepresentation of Gene-Ontology annotations with parent child analysis. Bioinformatics. 2007; 23:3024–3031. [DOI] [PubMed] [Google Scholar]
- 51.Croft D., Mundo A.F., Haw R., Milacic M., Weiser J., Wu G., Caudy M., Garapati P., Gillespie M., Kamdar M.R. et al. . The Reactome pathway knowledgebase. Nucleic Acids Res. 2014; 42:D472–D477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie C., Mao X., Huang J., Ding Y., Wu J., Dong S., Kong L., Gao G., Li C.Y., Wei L.. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011; 39:W316–W322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hamosh A., Scott A.F., Amberger J.S., Bocchini C.A., McKusick V.A.. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005; 33:D514–D517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanehisa M., Goto S., Furumichi M., Tanabe M., Hirakawa M.. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010; 38:D355–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osborne J.D., Flatow J., Holko M., Lin S.M., Kibbe W.A., Zhu L.J., Danila M.I., Feng G., Chisholm R.L.. Annotating the human genome with Disease Ontology. BMC Genomics. 2009; 10(Suppl 1):S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Becker K.G., Barnes K.C., Bright T.J., Wang S.A.. The genetic association database. Nat. Genet. 2004; 36:431–432. [DOI] [PubMed] [Google Scholar]
- 57.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L. et al. . The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014; 42:D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McLaughlin M.J., Sainani K.L.. Bonferroni, Holm, and Hochberg corrections: fun names, serious changes to p values. PM R. 2014; 6:544–546. [DOI] [PubMed] [Google Scholar]
- 59.Cunningham F., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S. et al. . Ensembl 2015. Nucleic Acids Res. 2015; 43:D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.UniProt C. UniProt: a hub for protein information. Nucleic Acids Res. 2015; 43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carbon S., Ireland A., Mungall C.J., Shu S., Marshall B., Lewis S., Ami G.O.H. Web Presence Working, G. . AmiGO: online access to ontology and annotation data. Bioinformatics. 2009; 25:288–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gheorghe R., Hinman M., Russo R.. Elasticsearch in Action. 2015; NY: Manning Publications Company. [Google Scholar]
- 63.Sunami Y., Leithauser F., Gul S., Fiedler K., Guldiken N., Espenlaub S., Holzmann K.H., Hipp N., Sindrilaru A., Luedde T. et al. . Hepatic activation of IKK/NFkappaB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology. 2012; 56:1117–1128. [DOI] [PubMed] [Google Scholar]
- 64.Laudadio I., Manfroid I., Achouri Y., Schmidt D., Wilson M.D., Cordi S., Thorrez L., Knoops L., Jacquemin P., Schuit F. et al. . A feedback loop between the liver-enriched transcription factor network and miR-122 controls hepatocyte differentiation. Gastroenterology. 2012; 142:119–129. [DOI] [PubMed] [Google Scholar]
- 65.Gao D., Zhai A., Qian J., Li A., Li Y., Song W., Zhao H., Yu X., Wu J., Zhang Q. et al. . Down-regulation of suppressor of cytokine signaling 3 by miR-122 enhances interferon-mediated suppression of hepatitis B virus. Antiviral Res. 2015; 118:20–28. [DOI] [PubMed] [Google Scholar]
- 66.Chen Z., Shao Y., Li X.. The roles of signaling pathways in epithelial-to-mesenchymal transition of PVR. Mol. Vis. 2015; 21:706–710. [PMC free article] [PubMed] [Google Scholar]
- 67.Lovisa S., LeBleu V.S., Tampe B., Sugimoto H., Vadnagara K., Carstens J.L., Wu C.C., Hagos Y., Burckhardt B.C., Pentcheva-Hoang T. et al. . Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015; 21:998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Larue L., Bellacosa A.. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3΄ kinase/AKT pathways. Oncogene. 2005; 24:7443–7454. [DOI] [PubMed] [Google Scholar]
- 69.Kalluri R., Weinberg R.A.. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009; 119:1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park S.M., Gaur A.B., Lengyel E., Peter M.E.. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008; 22:894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Korpal M., Lee E.S., Hu G., Kang Y.. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008; 283:14910–14914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zeisberg M., Neilson E.G.. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 2009; 119:1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bell C.E., Watson A.J.. SNAI1 and SNAI2 are asymmetrically expressed at the 2-cell stage and become segregated to the TE in the mouse blastocyst. PLoS One. 2009; 4:e8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barrallo-Gimeno A., Nieto M.A.. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005; 132:3151–3161. [DOI] [PubMed] [Google Scholar]
- 75.Shirakihara T., Saitoh M., Miyazono K.. Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Mol. Biol. Cell. 2007; 18:3533–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang H., Wang R., Thrimawithana T., Little P.J., Xu J., Feng Z.P., Zheng W.. The nerve growth factor signaling and its potential as therapeutic target for glaucoma. Biomed. Res. Int. 2014; 2014:759473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eleftheriadis T., Pissas G., Liakopoulos V., Stefanidis I., Lawson B.R.. Toll-like receptors and their role in renal pathologies. Inflamm. Allergy Drug Targets. 2012; 11:464–477. [DOI] [PubMed] [Google Scholar]
- 78.Bussemakers M.J., van Bokhoven A., Verhaegh G.W., Smit F.P., Karthaus H.F., Schalken J.A., Debruyne F.M., Ru N., Isaacs W.B.. DD3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 1999; 59:5975–5979. [PubMed] [Google Scholar]
- 79.Neves A.F., Araujo T.G., Biase W.K., Meola J., Alcantara T.M., Freitas D.G., Goulart L.R.. Combined analysis of multiple mRNA markers by RT-PCR assay for prostate cancer diagnosis. Clin. Biochem. 2008; 41:1191–1198. [DOI] [PubMed] [Google Scholar]
- 80.Matuszak E.A., Kyprianou N.. Androgen regulation of epithelial-mesenchymal transition in prostate tumorigenesis. Expert Rev. Endocrinol. Metab. 2011; 6:469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schalken J.A., Hessels D., Verhaegh G.. New targets for therapy in prostate cancer: differential display code 3 (DD3(PCA3)), a highly prostate cancer-specific gene. Urology. 2003; 62:34–43. [DOI] [PubMed] [Google Scholar]
- 82.Ng S.Y., Bogu G.K., Soh B.S., Stanton L.W.. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol. Cell. 2013; 51:349–359. [DOI] [PubMed] [Google Scholar]
- 83.Ji P., Diederichs S., Wang W., Boing S., Metzger R., Schneider P.M., Tidow N., Brandt B., Buerger H., Bulk E. et al. . MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003; 22:8031–8041. [DOI] [PubMed] [Google Scholar]
- 84.Fellenberg J., Bernd L., Delling G., Witte D., Zahlten-Hinguranage A.. Prognostic significance of drug-regulated genes in high-grade osteosarcoma. Mod. Pathol. 2007; 20:1085–1094. [DOI] [PubMed] [Google Scholar]
- 85.Yamada K., Kano J., Tsunoda H., Yoshikawa H., Okubo C., Ishiyama T., Noguchi M.. Phenotypic characterization of endometrial stromal sarcoma of the uterus. Cancer Sci. 2006; 97:106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang Y., Wang T., Huang H.Q., Li W., Cheng X.L., Yang J.. Human MALAT-1 long non-coding RNA is overexpressed in cervical cancer metastasis and promotes cell proliferation, invasion and migration. J. Buon. 2015; 20:1497–1503. [PubMed] [Google Scholar]
- 87.Lin R., Maeda S., Liu C., Karin M., Edgington T.S.. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene. 2007; 26:851–858. [DOI] [PubMed] [Google Scholar]
- 88.Sun Y., Wu J., Wu S.H., Thakur A., Bollig A., Huang Y., Liao D.J.. Expression profile of microRNAs in c-Myc induced mouse mammary tumors. Breast Cancer Res. Treat. 2009; 118:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kern W., Grossmann V., Kohlmann A., Schnittger S., Haferlach C., Haferlach T.. A specific gene expression signature affecting the beta-catenin and notch signaling pathways and the downregulation of MALAT1 prove acute myeloid leukemia with limited differentiation (AML-LD) as a distinct entity with NPM1 mutation. Blood. 2009; 114:164–164. [Google Scholar]
- 90.Xu C., Yang M., Tian J., Wang X., Li Z.. MALAT-1: a long non-coding RNA and its important 3΄ end functional motif in colorectal cancer metastasis. Int. J. Oncol. 2011; 39:169–175. [DOI] [PubMed] [Google Scholar]
- 91.Kryger R., Fan L., Wilce P.A., Jaquet V.. MALAT-1, a non protein-coding RNA is upregulated in the cerebellum, hippocampus and brain stem of human alcoholics. Alcohol. 2012; 46:629–634. [DOI] [PubMed] [Google Scholar]
- 92.Tano K., Mizuno R., Okada T., Rakwal R., Shibato J., Masuo Y., Ijiri K., Akimitsu N.. MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett. 2010; 584:4575–4580. [DOI] [PubMed] [Google Scholar]
- 93.Guo F., Li Y., Liu Y., Wang J., Li Y., Li G.. Inhibition of metastasis-associated lung adenocarcinoma transcript 1 in CaSki human cervical cancer cells suppresses cell proliferation and invasion. Acta Biochim. Biophys. Sin. (Shanghai). 2010; 42:224–229. [DOI] [PubMed] [Google Scholar]
- 94.Tseng J.J., Hsieh Y.T., Hsu S.L., Chou M.M.. Metastasis associated lung adenocarcinoma transcript 1 is up-regulated in placenta previa increta/percreta and strongly associated with trophoblast-like cell invasion in vitro. Mol. Hum. Reprod. 2009; 15:725–731. [DOI] [PubMed] [Google Scholar]
- 95.Asato R., Yoshida S., Ogura A., Nakama T., Ishikawa K., Nakao S., Sassa Y., Enaida H., Oshima Y., Ikeo K. et al. . Comparison of gene expression profile of epiretinal membranes obtained from eyes with proliferative vitreoretinopathy to that of secondary epiretinal membranes. PLoS One. 2013; 8:e54191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu J.Y., Yao J., Li X.M., Song Y.C., Wang X.Q., Li Y.J., Yan B., Jiang Q.. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014; 5:e1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bernard D., Prasanth K.V., Tripathi V., Colasse S., Nakamura T., Xuan Z., Zhang M.Q., Sedel F., Jourdren L., Coulpier F. et al. . A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010; 29:3082–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu F.S., Hazlett L.D.. Toll-like receptors and the eye. Invest. Ophthalmol. Vis. Sci. 2006; 47:1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hajjari M., Salavaty A.. HOTAIR: an oncogenic long non-coding RNA in different cancers. Cancer Biol. Med. 2015; 12:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yan T.H., Lu S.W., Huang Y.Q., Que G.B., Chen J.H., Chen Y.P., Zhang H.B., Liang X.L., Jiang J.H.. Upregulation of the long noncoding RNA HOTAIR predicts recurrence in stage Ta/T1 bladder cancer. Tumour Biol. 2014; 35:10249–10257. [DOI] [PubMed] [Google Scholar]
- 101.Chuong C.M. Homeobox genes, fetal wound healing, and skin regional specificity. J. Invest. Dermatol. 2003; 120:9–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reisman D., Glaros S., Thompson E.A.. The SWI/SNF complex and cancer. Oncogene. 2009; 28:1653–1668. [DOI] [PubMed] [Google Scholar]
- 103.Heubach J., Monsior J., Deenen R., Niegisch G., Szarvas T., Niedworok C., Schulz W.A., Hoffmann M.J.. The long noncoding RNA HOTAIR has tissue and cell type-dependent effects on HOX gene expression and phenotype of urothelial cancer cells. Mol. Cancer. 2015; 14:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stewart M.D., Li J., Wong J.. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol. Cell. Biol. 2005; 25:2525–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schotta G., Ebert A., Krauss V., Fischer A., Hoffmann J., Rea S., Jenuwein T., Dorn R., Reuter G.. Central role of drosophila SU(VAR)3-9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J. 2002; 21:1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Park J.A., Kim A.J., Kang Y., Jung Y.J., Kim H.K., Kim K.C.. Deacetylation and methylation at histone H3 lysine 9 (H3K9) coordinate chromosome condensation during cell cycle progression. Mol. Cells. 2011; 31:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pesquita C., Faria D., Falcao A.O., Lord P., Couto F.M.. Semantic similarity in biomedical ontologies. PLoS Comput. Biol. 2009; 5:e1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Guo X., Liu R., Shriver C.D., Hu H., Liebman M.N.. Assessing semantic similarity measures for the characterization of human regulatory pathways. Bioinformatics. 2006; 22:967–973. [DOI] [PubMed] [Google Scholar]
- 109.Lei Z., Dai Y.. Assessing protein similarity with Gene Ontology and its use in subnuclear localization prediction. BMC Bioinformatics. 2006; 7:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lin D. ICML. 1998; 98:296–304. [Google Scholar]
- 111.Resnik P. Semantic similarity in a taxonomy: an information-based measure and its application to problems of ambiguity in natural language. J. Artif. Intell. Res. (JAIR). 1999; 11:95–130. [Google Scholar]
- 112.Harispe S., Ranwez S., Janaqi S., Montmain J.. The semantic measures library and toolkit: fast computation of semantic similarity and relatedness using biomedical ontologies. Bioinformatics. 2014; 30:740–742. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.