


Abstract
Adenosine deaminases that act on RNA (ADARs) carry out adenosine (A) to inosine (I) editing reactions with a known requirement for duplex RNA. Here, we show that ADARs also react with DNA/RNA hybrid duplexes. Hybrid substrates are deaminated efficiently by ADAR deaminase domains at dA-C mismatches and with E to Q mutations in the base flipping loop of the enzyme. For a long, perfectly matched hybrid, deamination is more efficient with full length ADAR2 than its isolated deaminase domain. Guide RNA strands for directed DNA editing by ADAR were used to target six different 2΄-deoxyadenosines in the M13 bacteriophage ssDNA genome. DNA editing efficiencies varied depending on the sequence context of the editing site consistent with known sequence preferences for ADARs. These observations suggest the reaction within DNA/RNA hybrids may be a natural function of human ADARs. In addition, this work sets the stage for development of a new class of genome editing tools based on directed deamination of 2΄-deoxyadenosines in DNA/RNA hybrids.
INTRODUCTION
Adenosine deaminases that act on RNA (ADARs) convert adenosine (A) to inosine (I) in duplex RNAs (1–3). Since I base pairs with cytidine (C), it functions like guanosine (G) in cellular processes such as splicing, translation and reverse transcription (1,4). The A to I modification is known to alter miRNA recognition sites, redirect splicing and change the meaning of specific codons (5–7). Two different enzymes carry out this form of RNA editing in humans; ADAR1 and ADAR2 (8). Dysregulated ADAR activity is associated with human disease (9–13). For instance, mutations in the ADAR1 gene are known to cause the autoimmune disease Aicardi-Goutieres Syndrome (AGS) (11,12). The ADAR proteins have a modular structure with double stranded RNA binding domains (dsRBDs) and a C-terminal deaminase domain (3). Double helical structure is required for ADAR substrates. Indeed, recent X-ray crystal structures of the human ADAR2 deaminase domain bound to substrate RNAs revealed a 20 bp binding site with extensive contacts in the minor groove near the editing site and in the two adjacent major grooves (14) (Figure 1A). In addition, these structures suggested the mechanism by which the reactive nucleotide gains access to the deaminase active site would require an A-form like double helix (14). Interestingly, these structures also identified five direct contacts to 2΄-hydroxyls in the minor groove near the editing site with only four of these common to the two different RNA sequences crystallized (14) (Figure 1B). These observations lead us to question whether 2΄-hydroxyl contacts are required for an ADAR reaction and, if not, could the reaction take place in the context of a DNA/RNA hybrid duplex which maintains an A-form helical conformation. This is an important question for multiple reasons. First, a recent literature report suggests overexpression of human ADAR1 can lead to dA to dG mutations in DNA yet no evidence has been provided for direct deamination in a DNA strand by an ADAR (15). Second, mutations in other AGS-related genes (e.g TREX1, RNAseH2 and SAMHD1) lead to an accumulation of DNA/RNA hybrids suggesting the ability to regulate DNA/RNA hybrid levels could be a common link among gene products mutated in this disease (16). Finally, the development of adenosine deaminases that act on DNA could lead to new genome editing tools based on dA to dI conversion creating specific dA to dG mutations in the DNA after replication. This is similar in concept to the recently reported dC to dU base editing systems involving cytidine deaminase–Cas9 fusion proteins and single guide RNAs (17,18). For these reasons, we chose to examine the reactivity of ADARs with DNA/RNA hybrid substrates.
Figure 1.

Interactions between hADAR2d and 2΄-hydroxyl groups. (A) Structure of hADAR2d bound to duplex RNA (14). The edited strand is colored salmon with the unedited strand colored slate. The editing site nucleotide is shown in red in its flipped out conformation. (B) 2΄-Hydroxyl contacts to the RNA substrate in the crystal structure (PDB: 5ED2).
MATERIALS AND METHODS
Protein overexpression and purification
hADAR1 deaminase domain (hADAR1d), hADAR1 deaminase domain E1008Q (hADAR1d E1008Q), hADAR2 deaminase domain (hADAR2d), hADAR2 deaminase domain E488Q (hADAR2d E488Q) and wild-type hADAR2 (hADAR2 wt) were expressed and purified as previously described (19). Protein concentrations were determined using BSA standards visualized by SYPRO Orange staining of SDS-polyacrylamide gels. Purified hADAR1d, hADAR1d E1008Q were stored in 50 mM Tris–HCl, pH 8.0, 200 mM KCl, 5 mM EDTA pH 8.0, 10% glycerol, 0.01% NP-40 and 1 mM DTT at −70°C. Purified hADAR2d, hADAR2d E488Q and hADAR2 wt were stored in 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 20% glycerol and 1 mM 2-mercaptoethanol at −70°C.
Oligonucleotide purification
Single-stranded RNA and DNA oligonucleotides were purified by denaturing polyacrylamide gel electrophoresis and visualized using UV shadowing. Bands were excised from the gel, crushed and soaked overnight at 4°C in 500 mM NH4OAc and 100 mM EDTA. Polyacrylamide fragments were removed using a 0.2-μm filter, followed by phenol-chloroform extraction and ethanol precipitation. The final solutions were lyophilized to dryness, re-suspended in nuclease-free water, quantified by absorbance at 260 nm and stored at −20°C. The oligonucleotides were later heated at 95°C for 5 min and then slowly cooled to room temperature in 10-mM Tris–HCl, 0.1 mM EDTA pH 7.5, 100 mM NaCl to allow them to hybridize.
Generation and deamination of internally 32P-labeled substrates
Oligonucleotides were purified as described above. The 3΄ 12-nt oligonucleotides of the top (edited) strand were radiolabeled with [γ-32P] ATP at the 5΄ end with T4 polynucleotide kinase as described previously (20). About 30 pmol of labeled 3΄ top strand 12 nt oligonucleotide was redissolved with 3 μl of 10 μM DNA splint, 2 μl of 20 μM 5΄ top strand 12 nt oligonucleotide, 0.5 μl of RNasin (1.6 units/μl), 2 μl of NEB T4 DNA ligase 10× buffer and 5 μl of water. This reconstituted solution was heated to 65°C for 5 min. After the solution was slowly cooled to room temperature, 1.5 μl of RNasin (1.6 units/μl), 5 μl of 4 mM ATP and 1 μl of T4 DNA ligase (400 U/μl) were added to the solution so that the final reaction volume was 20 μl. The reaction was incubated at 30°C for 2 h, then 2 μl of 40 μM trap DNA were added to the splint ligation reaction. The splint ligation products were purified as described above. Purified 32P labeled top strand was hybridized with the corresponding bottom strand 24 nt oligonucleotide as described above. Oligonucleotide sequences are shown in Supplementary Table S1. Deamination reactions were carried out as previously described (21) with following modifications. For the partially 2΄-deoxy-modified substrates, the final reaction volume was 10 μl. The final enzyme concentration was 300 nM. The final RNA concentration was 10 nM. The final reaction solution contains 16 mM Tris–HCl, pH 7.4, 3.3% glycerol, 1.6 mM EDTA, 0.003% NP-40, 60 mM KCl, 7.1 mM NaCl, 0.5 mM DTT, 160 units/ml RNasin and 1 μg/ml yeast tRNA. Reactions were quenched by adding 190 μl 95°C nuclease-free water followed by incubation at 95°C for 5 min. Deaminated products were purified by phenol–chloroform extraction and ethanol precipitation. The product-containing solution was lyophilized to dryness and suspended in 50 μl of 1× TE solution, followed by digestion with nuclease P1. The resulting 5΄-mononucleotides were resolved by thin-layer chromatography (TLC, Macherey-Nagel) (22). The TLC was visualized by exposure to storage phosphor imaging plates (Molecular Dynamics) on a Typhoon phosphorimager (Molecular Dynamics) and quantified by volume integration using ImageQuant software (Molecular Dynamics). Data were fitted to the equation: 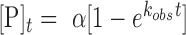 , where
, where 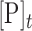 is the percent edited at time t, α is the fitted reaction end point, and kobs is the fitted rate constant using KaleidaGraph. Each experiment was carried out in triplicate, and the rate constant reported in the text are average values ± standard deviations. For the DD, DR, RD, RR substrates, deaminations were performed as above with following modifications. The final enzyme concentration was 250 nM. The final reaction solution for hADAR2d, hADAR2d E488Q and hADAR2 wt contain 17 mM Tris–HCl, pH 7.4, 4.2% glycerol, 1.6 mM EDTA, 0.003% NP-40, 60 mM KCl, 11.6 mM NaCl, 0.5 mM DTT, 160 units/ml RNasin and 1 μg/ml yeast tRNA. The final reaction solution for hADAR1d, hADAR1d E1008Q contain 12 mM Tris–HCl, pH 7.2, 3.3% glycerol, 1 mM EDTA, 0.002% NP-40, 40.5 mM potassium glutamate, 6.5 mM KCl, 6 mM NaCl, 0.5 mM DTT, 160 units/ml RNasin and 1 μg/ml yeast tRNA. The editing level for the corresponding zero time point was subtracted from each data point as background subtraction. Statistical significance between groups was determined by t tests using QuickCalcs (GraphPad Software). hADAR2 wt deamination was carried out twice, deamination with other proteins were carried out in triplicate.
is the percent edited at time t, α is the fitted reaction end point, and kobs is the fitted rate constant using KaleidaGraph. Each experiment was carried out in triplicate, and the rate constant reported in the text are average values ± standard deviations. For the DD, DR, RD, RR substrates, deaminations were performed as above with following modifications. The final enzyme concentration was 250 nM. The final reaction solution for hADAR2d, hADAR2d E488Q and hADAR2 wt contain 17 mM Tris–HCl, pH 7.4, 4.2% glycerol, 1.6 mM EDTA, 0.003% NP-40, 60 mM KCl, 11.6 mM NaCl, 0.5 mM DTT, 160 units/ml RNasin and 1 μg/ml yeast tRNA. The final reaction solution for hADAR1d, hADAR1d E1008Q contain 12 mM Tris–HCl, pH 7.2, 3.3% glycerol, 1 mM EDTA, 0.002% NP-40, 40.5 mM potassium glutamate, 6.5 mM KCl, 6 mM NaCl, 0.5 mM DTT, 160 units/ml RNasin and 1 μg/ml yeast tRNA. The editing level for the corresponding zero time point was subtracted from each data point as background subtraction. Statistical significance between groups was determined by t tests using QuickCalcs (GraphPad Software). hADAR2 wt deamination was carried out twice, deamination with other proteins were carried out in triplicate.
Preparation and deamination with 90 nt DNA + RNA hybrid substrates
The 90 nt DNA top strand and 24 nt RNA bottom strands were purchased from Integrated DNA Technology and purified as described above. The 90 nt DNA was PCR amplified with a T7 promoter-containing primer to generate a T7 RNA polymerase transcription template. Primer sequences are shown in Supplementary Table S1. PCR products were purified by agarose gel and extracted from the gel (QIAquick Gel Extraction Kit, Qiagen). The 93 nt RNA bottom strand was transcribed from this DNA template with MEGAscript® T7 Kit (ThermoFisher) and purified with polyacrylamide gel as described above. The 90 nt DNA was hybridized with corresponding RNAs. The bottom RNA to top DNA molar ratio was 3:1 for each hybridization. Deamination reactions were carried out as previously described (21) with following modifications. The final reaction volume was 10 μl. hADAR2d E488Q and hADAR2 wt were used for the reaction and final enzyme concentration was 250 nM. The final RNA concentration was 10 nM. The final reaction solution contains 17 mM Tris–HCl, pH 7.4, 4.2% glycerol, 1.6 mM EDTA, 0.003% NP-40, 60 mM KCl, 11.6 mM NaCl, 0.5 mM DTT, 160 units/ml RNasin and 1 μg/ml yeast tRNA. Reactions were quenched by adding 190 μl 95°C nuclease-free water followed by incubation at 95°C for 5 min. Reaction products were PCR amplified with extended primers using GoTaq® DNA Polymerase (Promega). Primer sequences are shown in Supplementary Table S1. PCR products were purified with DNA clean & concentrator (Zymo) and sequenced. The sequencing peak heights were measured with Chromas for calculating the editing level. Each experiment was carried out in triplicate. The editing level for the corresponding zero time point was subtracted from each data point as background subtraction. Statistical significance between groups was determined by t tests using QuickCalcs (GraphPad Software).
Deamination in the M13 genome
M13 genomic ssDNA (New England Biolabs) was hybridized with the corresponding guide RNAs. The guide RNA to genomic DNA molar ratio was 20:1 for each hybridization. Deamination reactions were carried out as previously described (21) with following modifications. The final reaction volume was 10 μl. hADAR1d E1008Q was used for the reaction and final enzyme concentration was 500 nM. The final RNA concentration was 2.8 nM. The final reaction solution contained 13 mM Tris–HCl, pH 7.2, 3.6% glycerol, 1.2 mM EDTA, 0.002% NP-40, 40.5 mM potassium glutamate, 12.5 mM KCl, 6 mM NaCl, 0.6 mM DTT, 160 units/ml RNasin and 1 μg/ml yeast tRNA. Reactions were quenched by adding 190 μl 95°C nuclease-free water followed by incubation at 95°C for 5 min. Target region of the reaction products were PCR amplified with primers using GoTaq® DNA Polymerase (Promega). Primer sequences are shown in Supplementary Table S1. PCR products were purified by agarose gel, extracted (QIAquick Gel Extraction Kit, Qiagen) and sequenced with the forward PCR primers. The sequencing peak heights were measured with Chromas for calculating the editing level. Each experiment was carried out in triplicate. The editing level for the corresponding zero time point was subtracted from each data point as background subtraction. Statistical significance between groups was determined by t tests using QuickCalcs (GraphPad Software).
RESULTS
The importance of 2΄-hydroxyl contacts to the ADAR deaminase domain
To determine if the 2΄-hydroxyl contacts observed in X-ray crystal structures of the human ADAR2 deaminase domain (hADAR2d) bound to substrate RNAs are required for an editing reaction, we prepared a chimeric substrate with each nucleotide contacted at its 2΄-hydroxyl replaced with the corresponding 2΄-deoxynucleotide. The substrate used in these experiments is similar to the human glioma factor 1 (hGli1)-derived substrate crystallized with hADAR2d that had five direct contacts to 2΄-hydroxyl groups including at each nucleotide in the UAGA sequence surrounding the editing site (underlined) and the nucleotide on the non-edited strand paired with the edited base (Figure 1B) (Figure 2, substrate a). We found that removal of the five 2΄-hydroxyl contacts slowed the rate of reaction at the editing site with hADAR2d by approximately 15-fold (Figure 2, substrate b). A similar rate was observed for a substrate with 2΄-deoxy substitutions only on the edited strand (Figure 2, substrate c). These results indicated that while the 2΄-hydroxyl contacts made by hADAR2d contribute to editing efficiency, they are not absolutely required for the reaction suggesting ADARs may react with DNA/RNA hybrids.
Figure 2.

Deamination kinetics for an RNA duplex and partially 2΄-deoxyribose-substituted substrates. (A) Sequences of deamination substrates. 2΄-Deoxynucleotides are labeled in red. Target sites are underlined and bolded. (B) Comparison of deamination product versus time for the three substrates with 300 nM hADAR2d. (C) Kinetic parameters for the deamination of RNA and partially 2΄-deoxy substrates.
Deamination of duplexes with different DNA/RNA strand combinations
To test for reactivity in DNA/RNA hybrids and compare this to reactions in similar all RNA or all DNA substrates, we prepared four new 24 bp duplexes each having the hGli1 substrate sequence but varying the backbone structure of the component strands (e.g. DNA or RNA) (Figure 3). We then measured editing activity at the position corresponding to the hGli1 editing site using internally 32P-labeled substrates and a standard thin layer chromatography assay (23). We tested hADAR2d and a mutant with enhanced editing activity (hADAR2d E488Q) (24). In addition, we also tested the human ADAR1 deaminase domain (hADAR1d) and its activated mutant (E1008Q) (25). Unsurprisingly, for each of the deaminase domains tested, the all RNA substrate (RR) was the most efficiently deaminated (Figure 3B–E) (underlining indicates substrate strand). Also, we observed no reaction in the all DNA substrate (DD) with any of the deaminase domains tested under any condition (Figure 3B–E). However, for both DNA/RNA hybrids (RD and DR), hADAR1d E1008Q and hADAR2d E488Q produced significant deamination (e.g. >40%) after a five-minute reaction time with complete editing observed at 120 min (Figure 3B–E). Lower reactivity was observed for the wild type deaminase domains with the hybrid substrates under these conditions. Indeed, observation of reaction of wild type hADAR2d in the hybrid substrates required a higher concentration of enzyme (Supplementary Figure S1).
Figure 3.

Comparison of deamination reactions with DNA/RNA hybrid, all RNA and all DNA substrates. (A) Sequences of deamination substrates. Strands colored red are DNA and strands colored black are RNA. Editing sites are underlined and bolded (DD: both strands DNA, DR: edited strand DNA, complementary strand RNA; RD: edited strand RNA, complementary strand DNA; RR: both strands RNA. Strands containing the edited A are underlined.). (B–E) Percent editing for deamination reactions at different time points with 250 nM of hADAR1d, hADAR1d E1008Q, hADAR2d and hADAR2d E488Q. Statistical significance between groups was determined by t tests. (***P-value ≤ 0.001, **P-value ≤ 0.01, *P-value ≤ 0.05). (F) Deamination reaction yield vs. time for 250 nM hADAR2 wt. Kinetic parameters (kobs): DD: kobs ≤ 0.001 min−1, DR: kobs = 0.02 min−1, RD: kobs = 0.02 min−1, RR: kobs ≥ 0.4 min−1 (n = 2, reported value is the average from two trials).
To determine the effects of dsRBDs on these reactions, we tested full length hADAR2 with the four 24 bp duplex substrates (RR, DD, RD and DR) (Figure 3F). Again, the RR substrate was deaminated most rapidly and no product was observed with the DD substrate. However, unlike the case of 250 nM wild type hADAR2d where little product was observed throughout the 2-h time course, this concentration of full length hADAR2 clearly produced deamination product in both DNA/RNA hybrids (Figure 3F). Thus, the presence of hADAR2's dsRBDs enhances reaction efficiency with hybrid substrates. While it is known that a duplex with two RNA strands is the preferred binding site for dsRBDs, dsRBD binding to DNA/RNA hybrids has been reported (26).
The effect of mismatches and length of DNA/RNA hybrid substrates
The 24 bp hGli1-derived duplex substrates each have an A-C mismatch at the editing site and an A-C mismatch at the 3΄ next nearest neighbor position (Figure 3A). To determine the role of these mismatches in the reaction of the DNA strand in a DNA/RNA hybrid and to test the effect of duplex length, we generated new substrate structures containing a longer DNA strand (90 nt) with the editing site hybridized to different RNAs that vary in sequence and in length (Figure 4). The longer DNA substrate strand allows for PCR amplification of the reaction products and Sanger sequencing to be used to assess editing on this strand (see Experimental section). Five different DNA/RNA hybrid substrates were prepared. Four have 24 nt RNA strands complementary to the sequence surrounding the editing site but vary the identity of the nucleotides paired with the editing site dA or 3΄ next nearest neighbor dA such that either a dA-C mismatch or a dA-U pair is formed at each site (Figure 4B–E). An additional substrate was formed with a complementary 93 nt RNA generating a 90 bp DNA/RNA hybrid duplex with a three-nucleotide overhang (Figure 4F). The reaction of full length, wild type hADAR2 was compared to that of the hADAR2d E488Q mutant at different times at three different positions in the DNA strand (Figure 4, sites A, B and C). Both full length hADAR2 and hADAR2d E488Q deaminate the dA at site B in the substrate bearing two A–C mismatches with the deaminase domain mutant showing higher levels of editing at the 3 min and 120 min time points (Figure 4B). Converting the dA-C mismatch at site C to an dA-U pair reduces reaction at this site (as expected) but had very little effect on editing at the B site (Figure 4C). In addition, a dA-C mismatch at site B significantly enhances editing at this site indicated by the very low B site editing levels observed in Figure 4D. Little editing is observed at either site for both proteins for the substrate with a fully complementary 24 nt RNA (Figure 4E). However, for the 90 bp DNA/RNA hybrid, full length hADAR2 reacts at all three sites A, B and C with site B the most efficiently edited (Figure 4F). No additional editing sites were observed for full length ADAR2 on this substrate (Supplementary Figure S2). Site A was not base paired in substrates with the 24 nt RNA strands and no editing was observed at site A in those substrates. Importantly, hADAR2d E488Q does not edit the fully matched hybrid duplex illustrating the importance of a dA-C mismatch in directing editing for this protein. This result also highlights the effect of hADAR2's dsRBDs in allowing editing in long, perfectly matched DNA/RNA hybrids. The presence of the RNA-binding N-terminal fragment containing the dsRBDs allows ADAR2 to edit the DNA strand of a long DNA-RNA hybrid without the requirement for an A-C mismatch at the editing site. It is likely the enhanced binding affinity afforded by the N-terminal fragment compensates for the lower reactivity of an A–U pair.
Figure 4.

Deamination in the DNA strand of DNA/RNA hybrid duplexes; effects of mismatches and duplex length. (A) Sequence surrounding three editing sites (A, B and C). Red color indicates DNA. Black corresponds to sequence of 24 nt RNA bottom strand with varying X and Y positions. Gray corresponds to 93 nt RNA bottom strand. (B–F) Percent editing for sites A, B and C in the different substrate structures shown with either hADAR2 wt or hADAR2d E488Q. Statistical significance between groups was determined by t tests. (***P-value ≤ 0.001, **P-value ≤ 0.01, *P-value ≤ 0.05).
Selective editing within the M13 bacteriophage ssDNA genome
The results described above indicated that RNA oligonucleotides could be used in combination with an ADAR or ADAR deaminase domain to direct editing at specific 2΄-deoxyadenosines in a DNA strand. To define further the scope and limitations of this reaction, we designed six different 24 nt guide RNAs complementary to different locations in the single stranded DNA genome of the M13 bacteriophage such that different 2΄-deoxyadenosines would be targeted for deamination by an ADAR deaminase domain in a DNA/RNA hybrid duplex (Figure 5A). Each RNA strand was designed to form a dA-C mismatch at the targeted site in the center of a 24 bp hybrid duplex. The six 2΄-deoxyadenosines have different nearest neighbor nucleotides so we could determine if preferences for the ADAR reaction in a DNA strand match those known for RNA strands (27). For these experiments, we used 500 nM hADAR1d E1008Q as the deaminase and allowed each reaction to proceed for two hours. With this approach, we were able to direct editing at each of the six targeted 2΄-deoxyadenosines (Figure 5B). The extent of editing observed varied among the target sites with the following yields: TAG site (94%), AAG site (81%), AAT site (43%), CAC site (53%), GAA site (27%) and the GAC site (19%). These trends closely match the known nearest neighbor preferences for hADAR1d in RNA substrates (27). Editing yield at difficult sites (e.g. GAC site) can be enhanced with additional enzyme and longer incubation times. Indeed, with two additions of 500 nM each of hADAR1 E1008Q over a total of four hours lead to 89% editing at the GAC site (Supplementary Figure S3). Editing for each target site was monitored by amplification and sequencing of ∼800 bp of the M13 genome surrounding that site. The only editing observed was at the six targeted 2΄-deoxyadenosines and one additional off-target site. This off target dA was edited to 15% yield and located adjacent to the targeted dA of the AAT site (Figure 5C).
Figure 5.

Selective editing of multiple sites in the ssDNA genome from M13 bacteriophage. (A) Single-stranded DNA and six target sites. Target sites are shown in red. Guide RNAs are shown in green. (B) Percent editing by 500 nM hADAR1d E1008Q at each site. (C) Off target site found adjacent to AAT target. Off target site is marked with an asterisk in sequence trace. Statistical significance between groups was determined by t tests (***P-value ≤ 0.001, **P-value ≤ 0.01, *P-value ≤ 0.05).
DISCUSSION
ADARs were first identified for their ability to unwind duplex RNA (28,29). This effect arises from the conversion of A-U pairs in a duplex substrate to less stable I–U pairs resulting in duplex denaturation (2). Early studies also showed that preincubation with an excess of duplex RNA but not single stranded RNA, double stranded DNA, single stranded DNA or tRNA, inhibited the unwinding reaction (29). Later, it was recognized that ADARs can also deaminate adenosines in duplex regions of more complex, folded RNAs (21,30–34). However, to our knowledge, there have been no previous reports describing the ability of an ADAR to edit the strands of DNA/RNA hybrid substrates. Our recently reported crystal structures of hADAR2d bound to duplex RNA showed the complex trapped at a point in the reaction with the reactive nucleotide flipped into the deaminase active site and suggested that base flipping by ADARs requires an A-form helix (14). DNA/RNA hybrids maintain an A-form like conformation so ADAR-induced base flipping might occur with such a substrate structure (35–37). However, the crystal structures also identified five direct contacts to 2΄-hydroxyl groups in the RNA substrates. The experiments described here with chimeric substrates bearing 2΄-deoxynucleotides at all the contact sites indicated that interactions with 2΄-hydroxyls are not absolutely required for deaminase activity (Figure 2).
The observation that ADARs can deaminate 2΄-deoxyadenosines in the DNA strands of a DNA/RNA hybrids has implications for understanding known ADAR properties. For instance, a recent report showed that overexpression of ADAR1 induced adenosine-targeted DNA mutations in a class switch recombination region (Ig-Sμ) in IgM B cells from ADAR1 transgenic mice and in the Ig-Sμ region as well as the c-Myc gene in wild type MEFs (15). This study suggested that ADAR1 is an inducer of somatic mutations like activation-induced deaminase (AID) but provided no mechanistic rationale for how ADAR1 expression could cause mutations in DNA. Since both class switch regions and the c-Myc gene are known to be genomic loci where DNA/RNA hybrids occur (in the form of R-loops) (38,39), ADAR1-induced DNA mutations at these sites could arise from reaction of the DNA strand of the hybrid duplex in an R-loop. 2΄-Deoxyinosine residues in DNA are subject to repair by endonuclease V (EndoV), an enzyme that cleaves the strand at the second phosphodiester bond 3΄ to the lesion (40,41). Additional enzymes are necessary to remove dI and complete the repair, but the subsequent steps of the repair of dI in dsDNA are poorly understood and completely unknown for dI in DNA/RNA hybrids (41,42). Overexpression of an ADAR may overwhelm dI repair pathways, allowing replication to render the dA to dG mutation permanent.
AGS is a severe childhood autoimmune disease that is characterized by overexpression of interferon α and increased innate immune response (11,12). This disease is caused by mutations in multiple genes whose protein products, including ADAR1, are all involved in nucleic acid metabolism (11). Recent studies suggest that the presence of increased levels of cytosolic double stranded RNA arising from defects in ADAR1 activity caused by AGS mutations leads to interferon induction (43,44). Interestingly, a common feature of cells isolated from patients with mutations in different AGS genes is the accumulation of DNA/RNA hybrid structures (16). It has been suggested that DNA/RNA hybrids represent a common immunogenic form of nucleic acids in AGS (16). It is possible that normal ADAR1 function leads to deamination and denaturation (or degradation triggered by EndoV) of DNA/RNA hybrids. ADAR1 mutations that disrupt deaminase activity could then lead to their accumulation. Further study of the possible role of DNA/RNA hybrids in ADAR-linked AGS is justified.
Our observation that the mutants of ADAR deaminase domains can efficiently edit DNA strands in DNA/RNA hybrids also has practical implications for the development of new genome editing tools. Recent years have seen an explosion in the number of new tools to manipulate the genomes of complex organisms, primarily by use of variants of the CRISPR–Cas9 system (45,46). While these tools are powerful, single point mutations introduced with these reagents require inefficient homology-directed repair (47,48). This has stimulated others to develop ‘base editing’ methods using Cas9–cytidine deaminase fusion proteins that can be directed to specific sites in the genome with a single guide RNA (17,18). While this approach has been shown to be effective for introducing dC to T mutations, the use of only cytidine deaminases for this purpose is limiting. Here, we show that an ADAR deaminase domain bearing an E to Q mutation in the enzyme's base flipping loop can be directed to edit specific dA-C mismatches in hybrid duplexes formed by the binding of 24 nt guide RNAs. Fusion of ADAR catalytic domains with nucleic acid binding domains, particularly hybrid binding domains, and activation with additional specific mutations are likely to enhance reactivity with DNA-RNA hybrids even further. It will be interesting to see if this RNA-guided ADAR reaction in DNA can be directed to specific locations in the genomes of complex organisms to induce single dA to dG mutations. Efficient and selective dA deamination in the M13 bacteriophage genome was possible here with the hADAR1 deaminase domain bearing a flipping loop mutation (hADAR1d E1008Q) and by targeting dA-C mismatches. The mutated residue is responsible for contacting the orphan base when the edited nucleotide occupies the deaminase active site (14). When this base is a C, a protonated E1008 side chain likely donates a hydrogen bond to N3 of C. The E1008Q mutant does not require protonation to hydrogen bond to N3 of C leading to an increase in editing activity. Off–target editing is minimized by using a relatively short 24 nt guiding RNA that is near the minimum length required for full contact to the deaminase domain. Since ADARs do not deaminate single strands, editing would not be expected outside the DNA/RNA hybrid duplex. Indeed, this is the case since no editing sites were observed in the regions of the M13 genome sequenced after the reaction besides those found within the region bound by the guide RNAs. Also, by positioning the targeted dA near the center of the 24 bp duplex, editing is restricted to an approximately four bp window in the center of the guide RNA–target DNA duplex. The ADAR catalytic domain would not fully engage the duplex for editing sites outside this region (14,49). The one off-target site observed is consistent with this hypothesis. The off-target dA is located immediately adjacent to the targeted dA of the AAT target site (Figure 5C). Furthermore, the off-target dA has a 5΄ T, the best 5΄ nearest neighbor for an ADAR editing site (27). It may be possible to reduce this rare type of off-target editing by introducing an unfavorable mismatch (e.g. dA-G) at this site in the DNA/RNA hybrid (50).
Overall, the ADAR-catalyzed editing of the DNA strands in DNA/RNA hybrids reported here expands the scope of possible biological functions of ADARs and points to potential applications in genome editing.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Y. Wang, A.I. Scott, J. Havel and K.J. Phelps for the kind gift of the proteins.
FUNDING
National Institutes of Health (NIH) [R01GM061115]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bass B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002; 71:817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bass B.L., Weintraub H.. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988; 55:1089–1098. [DOI] [PubMed] [Google Scholar]
- 3. Goodman R.A., Macbeth M.R., Beal P.A.. Samuel CE. Adenosine Deaminases Acting on RNA (ADARs) and A-to-I Editing. 2012; Berlin, Heidelberg: Springer Berlin Heidelberg; 1–33. [Google Scholar]
- 4. Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010; 79:321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Q., Hui H., Guo Z., Zhang W., Hu Y., He T., Tai Y., Peng P., Wang L.. ADAR1 regulates ARHGAP26 gene expression through RNA editing by disrupting miR-30b-3p and miR-573 binding. RNA. 2013; 19:1525–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rueter S.M., Dawson T.R., Emeson R.B.. Regulation of alternative splicing by RNA editing. Nature. 1999; 399:75–80. [DOI] [PubMed] [Google Scholar]
- 7. Yeo J., Goodman R.A., Schirle N.T., David S.S., Beal P.A.. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:20715–20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bass B.L., Nishikura K., Keller W., Seeburg P.H., Emeson R.B., O'Connell M.A., Samuel C.E., Herbert A.. A standardized nomenclature for adenosine deaminases that act on RNA. RNA. 1997; 3:947–949. [PMC free article] [PubMed] [Google Scholar]
- 9. Miyamura Y., Suzuki T., Kono M., Inagaki K., Ito S., Suzuki N., Tomita Y.. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am. J. Hum. Genet. 2003; 73:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang G., Shao M., Li Z., Gu Y., Du X., Wang X., Li M.. Genetic spectrum of dyschromatosis symmetrica hereditaria in Chinese patients including a novel nonstop mutation in ADAR1 gene. BMC Med. Genet. 2016; 17:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Livingston J.H., Crow Y.J.. Neurologic phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi–Goutières syndrome and beyond. Neuropediatrics. 2016; 47:355–360. [DOI] [PubMed] [Google Scholar]
- 12. Rice G.I., Kasher P.R., Forte G.M.A., Mannion N.M., Greenwood S.M., Szynkiewicz M., Dickerson J.E., Bhaskar S.S., Zampini M., Briggs T.A. et al. . Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012; 44:1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Slotkin W., Nishikura K.. Adenosine-to-inosine RNA editing and human disease. Genome Med. 2013; 5:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matthews M.M., Thomas J.M., Zheng Y., Tran K., Phelps K.J., Scott A.I., Havel J., Fisher A.J., Beal P.A.. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 2016; 23:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsuruoka N., Arima M., Yoshida N., Okada S., Sakamoto A., Hatano M., Satake H., Arguni E., Wang J.-Y.Y., Yang J.-H.H. et al. . ADAR1 protein induces adenosine-targeted DNA mutations in senescent Bcl6 gene-deficient cells. J. Biol. Chem. 2013; 288:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lim Y.W., Sanz L.A., Xu X., Hartono S.R., Chédin F.. Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi–Goutières syndrome. eLife. 2015; 4:e08007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R.. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016; 533:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishida K., Arazoe T., Yachie N., Banno S., Kakimoto M., Tabata M., Mochizuki M., Miyabe A., Araki M., Hara K.Y. et al. . Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016; 353:aaf8729. [DOI] [PubMed] [Google Scholar]
- 19. Mizrahi R.A., Phelps K.J., Ching A.Y., Beal P.A.. Nucleoside analog studies indicate mechanistic differences between RNA-editing adenosine deaminases. Nucleic Acids Res. 2012; 40:9825–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Phelps K.J., Ibarra-Soza J.M.M., Tran K., Fisher A.J., Beal P.A.. Click modification of RNA at adenosine: structure and reactivity of 7-ethynyl- and 7-triazolyl-8-aza-7-deazaadenosine in RNA. ACS Chem. Biol. 2014; 9:1780–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eifler T., Pokharel S., Beal P.A.. RNA-Seq analysis identifies a novel set of editing substrates for human ADAR2 present in Saccharomyces cerevisiae. Biochemistry. 2013; 52:7857–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Connell M.A., Keller W.. Purification and properties of double-stranded RNA-specific adenosine deaminase from calf thymus. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:10596–10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lehmann K.A., Bass B.L.. The importance of internal loops within RNA substrates of ADAR1. J. Mol. Biol. 1999; 291:1–13. [DOI] [PubMed] [Google Scholar]
- 24. Kuttan A., Bass B.L.. Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y., Havel J., Beal P.A.. A phenotypic screen for functional mutants of human adenosine deaminase acting on RNA 1. ACS Chem. Biol. 2015; 10:2512–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vukovic L., Koh H.R., Myong S., Schulten K.. Substrate recognition and specificity of double stranded RNA binding proteins. Biochemistry. 2014; 53:3457–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eggington J.M., Greene T., Bass B.L.. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011; 2:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bass B.L., Weintraub H.. A developmentally regulated activity that unwinds RNA duplexes. Cell. 1987; 48:607–613. [DOI] [PubMed] [Google Scholar]
- 29. Wagner R.W., Nishikura K.. Cell cycle expression of RNA duplex unwindase activity in mammalian cells. Mol. Cell. Biol. 1988; 8:770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niswender C.M., Sanders-Bush E., Emeson R.B.. Identification and characterization of RNA editing events within the 5-HT2C receptor. Ann. N.Y. Acad. Sci. 1998; 861:38–48. [DOI] [PubMed] [Google Scholar]
- 31. Sommer B., Köhler M., Sprengel R., Seeburg P.H.. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991; 67:11–19. [DOI] [PubMed] [Google Scholar]
- 32. Melcher T., Maas S., Herb A., Sprengel R., Seeburg P.H., Higuchi M.. A mammalian RNA editing enzyme. Nature. 1996; 379:460–464. [DOI] [PubMed] [Google Scholar]
- 33. Burns C.M., Chu H., Rueter S.M., Hutchinson L.K., Canton H., Sanders-Bush E., Emeson R.B.. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997; 387:303–308. [DOI] [PubMed] [Google Scholar]
- 34. Li J.B., Levanon E.Y., Yoon J.-K., Aach J., Xie B., LeProust E., Zhang K., Gao Y., Church G.M.. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009; 324:1210–1213. [DOI] [PubMed] [Google Scholar]
- 35. Xiong Y., Sundaralingam M.. Crystal structure of a DNA.RNA hybrid duplex with a polypurine RNA r(gaagaagag) and a complementary polypyrimidine DNA d(CTCTTCTTC). Nucleic Acids Res. 2000; 28:2171–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Conn G.L., Brown T., Leonard G.A.. The crystal structure of the RNA/DNA hybrid r(GAAGAGAAGC). d(GCTTCTCTTC) shows significant differences to that found in solution. Nucleic Acids Res. 1999; 27:555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davis R.R., Shaban N.M., Perrino F.W., Hollis T.. Crystal structure of RNA-DNA duplex provides insight into conformational changes induced by RNase H binding. Cell Cycle. 2015; 14:668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang Y., McBride K.M., Hensley S., Lu Y., Chedin F., Bedford M.T.. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol. Cell. 2014; 53:484–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu K., Chedin F., Hsieh C.-L.L., Wilson T.E., Lieber M.R.. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003; 4:442–451. [DOI] [PubMed] [Google Scholar]
- 40. Yao M., Hatahet Z., Melamede R.J., Kow Y.W.. Deoxyinosine 3΄ endonuclease, a novel deoxyinosine-specific endonuclease from Escherichia coli. Ann. N.Y. Acad. Sci. 1994; 726:315–316. [DOI] [PubMed] [Google Scholar]
- 41. Mi R., Alford-Zappala M., Kow Y.W., Cunningham R.P., Cao W.. Human endonuclease V as a repair enzyme for DNA deamination. Mutat. Res. 2012; 735:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee C.-C.C., Yang Y.-C.C., Goodman S.D., Yu Y.-H.H., Lin S.-B.B., Kao J.-T.T., Tsai K.-S.S., Fang W.-H.H.. Endonuclease V-mediated deoxyinosine excision repair in vitro. DNA Repair. 2010; 9:1073–1079. [DOI] [PubMed] [Google Scholar]
- 43. Liddicoat B.J., Piskol R., Chalk A.M., Ramaswami G., Higuchi M., Hartner J.C., Li J.B., Seeburg P.H., Walkley C.R.. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015; 349:1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mannion Niamh M., Greenwood S.M., Young R., Cox S., Brindle J., Read D., Nellåker C., Vesely C., Ponting Chris P., McLaughlin Paul J. et al. . The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014; 9:1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang H., La Russa M., Qi L.S.. CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 2016; 85:227–264. [DOI] [PubMed] [Google Scholar]
- 46. Doudna J.A., Charpentier E.. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 47. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. et al. . Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Phelps K.J., Tran K., Eifler T., Erickson A.I., Fisher A.J., Beal P.A.. Recognition of duplex RNA by the deaminase domain of the RNA editing enzyme ADAR2. Nucleic Acids Res. 2015; 43:1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wong S.K., Sato S., Lazinski D.W.. Substrate recognition by ADAR1 and ADAR2. RNA. 2001; 7:846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.