


Abstract
Many viruses use a powerful terminase motor to pump their genome inside an empty procapsid shell during virus maturation. The large terminase (TerL) protein contains both enzymatic activities necessary for packaging in such viruses: the adenosine triphosphatase (ATPase) that powers DNA translocation and an endonuclease that cleaves the concatemeric genome at both initiation and completion of genome packaging. However, how TerL binds DNA during translocation and cleavage remains mysterious. Here we investigate DNA binding and cleavage using TerL from the thermophilic phage P74-26. We report the structure of the P74-26 TerL nuclease domain, which allows us to model DNA binding in the nuclease active site. We screened a large panel of TerL variants for defects in binding and DNA cleavage, revealing that the ATPase domain is the primary site for DNA binding, and is required for nuclease activity. The nuclease domain is dispensable for DNA binding but residues lining the active site guide DNA for cleavage. Kinetic analysis of DNA cleavage suggests flexible tethering of the nuclease domains during DNA cleavage. We propose that interactions with the procapsid during DNA translocation conformationally restrict the nuclease domain, inhibiting cleavage; TerL release from the capsid upon completion of packaging unlocks the nuclease domains to cleave DNA.
INTRODUCTION
Most double-stranded DNA viruses package their genomes using an adenosine triphosphate (ATP)-dependent motor to pump DNA into an empty capsid protein shell. As DNA fills the shell, internal pressure builds due to confinement of the highly charged DNA. Therefore, these motors have evolved to become some of the most powerful bio-motors known (1,2). For this reason, there is much interest in engineering packaging motors for delivery of nucleic acid therapeutics and as functionalized nano-devices. Moreover, genome packaging motors from herpes viruses are the targets of various Food and Drug Administration approved anti-viral drugs (3–9).
There are two distinct families of packaging motors for membrane-free dsDNA viruses: the terminase family and the Phi29-family motors (10). Here we focus on the more common terminase packaging apparatus, which has been studied in many viral systems (11–18). Terminase motors consist of the portal, large terminase (TerL) and small terminase (TerS) proteins, each of which assembles into a homomeric ring (19,20). Genome packaging by terminases can be broadly summarized as a five-step process (21) (Figure 1). (Step 1) First, the motor recognizes the concatemeric viral genome, primarily through TerS binding (22–26). (Step 2) Next, TerL cleaves the DNA at a specific site and binds to the portal complex. (Step 3) TerL uses ATP hydrolysis to translocate DNA (27–29) through the portal ring into the capsid (30). (Step 4) Upon completing the translocation of at least one genome-length of DNA, TerL switches its enzymatic activity from translocation to cleavage (31). This cleavage occurs either after encapsidating exactly one genome length (termed ‘unit-length packaging’) or after the capsid is completely filled with DNA, resulting in slightly more than one-genome length being packaged (termed ‘headful packaging’). (Step 5) Finally the terminase subunits are released from the capsid for maturation of another virus, while portal binds to the tail proteins to complete a mature, infectious virion (13). Although the sequence of these events has been well studied, the structural mechanism for each step is largely unknown. In particular, how the motor holds DNA during either translocation or cleavage remains obscure.
Figure 1.

Schematic of a generic genome packaging reaction catalyzed by a terminase enzyme.
The TerL protein is the catalytic engine of the packaging apparatus, harboring the two enzymatic activities of the motor: the adenosine triphosphatase (ATPase) that drives DNA translocation, and the endonuclease that cleaves genome concatemers at both initiation and termination of packaging (32,33). The terminase motor is capable of generating high force (stall force up to ∼60 pN) and high speeds (up to ∼2000 bp/s) (2,34). However, several distinct structural mechanisms have been proposed for both the force generation reaction and DNA cleavage (7,18,35–41). Even more mysterious is the mechanism of nuclease activity regulation; current models for nuclease regulation include TerL auto-inhibition (38,42), catalytic regulation via competition between fast DNA translocation and slow DNA cleavage (37,43), and inhibition mediated by TerS (37,39,42,44). Careful dissection of TerL structure and mechanism is necessary to discern between competing models for TerL activity and regulation.
TerL contains two domains: a C-terminal nuclease domain of a RNaseH fold (7,18,35,38–39,41) and an N-terminal ATPase domain of the ASCE (additional strand, conserved glutamate) superfamily (45). The TerL protein forms an oligomeric ring (46) that binds to the procapsid (35). The oligomeric state for large terminases have been shown to be either tetrameric for lambda (47–49), while the distinct family of Phi29 motors are more controversial as to their oligomeric state (50–53). The C-terminal tail of TerL is thought to interact with the portal and/or the procapsid (32,46,54–59), although an alternate arrangement in which the N-terminal ATPase domain contacts the portal has also been proposed (35,60). Our group recently proposed a model for the TerL ring in which the ATPase domains form a ring with intersubunit contacts contributing to ATP hydrolysis and a small inner pore for binding DNA (46). In this model, the nuclease domains are positioned on the periphery of the TerL ring so that they can use their C-terminal tails to interact with the portal ring. Because the ATPase domains line the inner pore of the ring, this model suggests that the ATPase domains are the primary point of contact for DNA during DNA translocation. However, this model did not explain how DNA cleavage occurs. How does the nuclease domain contribute to DNA binding? Does the interaction surface change during the cleavage reaction?
Here we report enzymatic and structural characterization of TerL DNA binding and nuclease function. We use TerL from the thermophilic phage P74-26 (TerLP74-26) (61) due to its high expression, solubility and stability. Employing a thermophilic terminase affords us a unique opportunity to separately evaluate DNA binding and cleavage, as the latter function only occurs at elevated temperatures. We show that both tight DNA binding and cleavage are nucleotide dependent. Our analysis of cleavage kinetics reveals that dual strand cleavage is fast, suggesting that multiple nuclease domains collaborate to cut both strands of the double helix. We also report the structure of the P74-26 nuclease domain, which we use to map the contributions of individual residues to both DNA binding and cleavage. Our data indicate that the ATPase domain is the primary determinant of DNA binding and that the nuclease domain is dispensable for DNA binding. We integrate our results to propose a mechanism for how TerL switches between DNA translocase and nuclease modes.
MATERIALS AND METHODS
Protein expression and purification
Both the isolated ATPase domain (1–256) and full-length P74-26 TerL mutants were expressed and purified as previously described (46). The nuclease domain (residues 256–485) was subcloned from our previously described pET24a full-length construct and was overexpressed identically to the above constructs (46). Cells were lysed in a cell disruptor and pelleted. The lysate was applied to a 10-ml His-Trap column (GE Healthcare) preequilibrated in buffer A (500 mM NaCl, 20 mM Imidazole, 50 mM Tris pH 8.5, 5 mM βME, 10% (v/v) glycerol). The column was washed with buffers A, followed by buffer A’ (150 mM NaCl, 20 mM Imidazole, 50 mM Tris pH 7.5, 5 mM βME, 10% (v/v) glycerol). Protein was eluted with buffer B (150 mM NaCl, 250 mM Imidazole, 50 mM Tris pH 8.5, 5 mM βME, 10% (v/v) glycerol). Eluate was dialyzed into buffer QA (125 mM NaCl, 25 mM Tris pH 7.5, 2 mM DTT, 10% (v/v) glycerol) and the tag was cleaved with prescission protease overnight. Dialysate was loaded onto a 10-ml Q column (GE Healthcare) preequilibrated with buffer QA. The column was then washed with buffer QA. Protein was eluted by applying a 0–100% (v/v) gradient of buffer QA to buffer QB (1 M NaCl, 25 mM Tris pH 7.5, 2 mM DTT, 10% glycerol (v/v)). Eluate was injected onto an S200 HR26/60 (GE Healthcare) column preequilibrated with gel filtration buffer (125 mM NaCl, 25 mM Tris pH 7.5, 4 mM DTT), and eluted in overlapping peaks consistent with dimer (58.2 kDa from gel filtration, 57.4 kDa calculated mass) and monomer (∼30 kDa from gel filtration, 28.7 kDa calculated mass). Eluted protein was concentrated to ∼20 mg/ml and flash frozen in liquid nitrogen.
Crystallization, structure determination and refinement
Native crystals formed in hanging drops containing 20 mg/ml TerL Nuclease domain mixed 2:1 with buffer containing 0.23 M sodium phosphate monobasic/potassium phosphate dibasic pH 6.2 and 2.5 M sodium chloride and 4 mM dTMP. Crystals were plunged into cryoprotectant containing 0.28 M sodium phosphate monobasic/potassium phosphate dibasic pH 6.2, 4 M sodium chloride and 2.5 mM dTMP before being flash frozen in liquid nitrogen. Data were collected at the Advanced Light Source at SIBYLS beamline 12.3.1 at wavelength 1.000 Å. Heavy atom derivative crystals were obtained by incubating native crystals with 3 mM potassium hexachloroplatinate 24 h prior to flash freezing. Cryoprotectant for heavy atom derivative crystals contained 3.4 mM potassium hexachloroplatinate. Derivative crystal data were collected at the Advanced Photon Source GM/CA CAT beamline 23ID-B at wavelength 0.855 Å in inverse beam mode. All diffraction data were processed with HKL3000 (62). Platinum bound to Met 265 allowed SAD phasing (63) of the 2.7 Å derivative crystal dataset using the PHENIX autosol pipeline (64). Native dataset anisotropic diffraction data were corrected with the UCLA Diffraction Anisotropy Server (65) and phases were extended to 2.6 Å resolution. Model building and structure refinement were performed with COOT (66) and PHENIX (67). The structure was deposited in the RCSB (PDB code 5TGE).
DNA binding and nuclease digestion
ADP-Beryllium Fluoride (ADP-BeF3) was formed by incubating 50 mM Tris pH 8.5, 150 mM potassium chloride, 1 mM DTT, 1 mM ADP, 10 mM sodium fluoride, 4 mM beryllium chloride and 10 mM magnesium chloride for 2 h prior to usage. TerL and ADP-BeF3 were mixed and incubated for 5 min prior to addition of 150 ng of plasmid pET28a (final concentration of 30.3 μM base pairs). Upon DNA addition, samples were incubated at room temperature (DNA binding) or 60°C (nuclease digestion) for 30 min unless otherwise indicated. During kinetics experiments, addition of cold 25 mM ethylenediaminetetraaceticacid (EDTA) (final) and rapid cooling in an ice bath quenched cleavage. Unless otherwise noted, all DNA cleavage samples were quenched with 1.5% (w/v) sodium dodecyl sulphate (SDS) (final) to prevent TerL's DNA binding activity from perturbing DNA migration through the gel. Standard 1.5% (w/v) agarose Tris-Acetate EDTA pH 8.0 gels were used, with the exception of isolated ATPase domain DNA-binding assay. The pH of the running buffer and gel were raised to pH 8.5 to account for the ATPase domain PI of 8.1. Gels were imaged on an LAS 3000. Gel densitometry was performed using ImageJ (68).
To analyze and rank TerL variant DNA binding, the lanes of all variants were first aligned relative to the DNA ladder on each gel. Migration distance of the most intense DNA bands relative to wild-type migration distance was assessed to rank TerL variants into three general categories. Bands that migrated roughly equivalent to wild-type were designated as unaffected. Likewise bands that migrate equivalent to free plasmid were designated ‘severe defect’. Intermediate migration distances were designated ‘moderate defect’.
For monitoring DNA cleavage, variants were considered to have wild-type levels of digestion if there was no longer any relaxed or linear plasmid bands remaining. Variants were ranked as ‘moderate defect’ if the supercoiled band had been cut, but relaxed or linear bands remained. Variants were ranked as ‘severe defect’ if a supercoiled band remains and there was no significant smearing of degraded, lower molecular weight fragments. Each variant was tested in at least duplicate.
Fitting of the kinetic data was performed using the GraphPad Prism software (GraphPad Inc.) using equations described in Freifelder et al. (69) and Cowan et al. (70). Our Freifelder-Trumbo analysis utilized the relative fraction, based on densitometry analysis, of supercoiled, relaxed, and linearized plasmid bands to calculate the ratio of single strand nicks to linearization events per molecule during early time points. The fraction of linearized plasmid DNA, ƒL, was used to calculate linearization events (nlinear) with Equation (1):
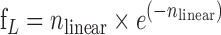 |
(1) |
The fraction of supercoiled DNA, ƒSc, was used to calculate the single strand nicks (nnick) with Equation (2):
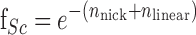 |
(2) |
Simulated data for purely single strand cleavage was calculated from Equation (3) where h represents the maximum base pair distance between two nicks on opposing DNA strands that results in linearization, and L is the total DNA base pairs in the plasmid:
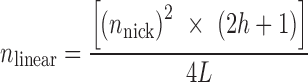 |
(3) |
If a plasmid has been nicked once, we calculated the probability, Rn, of no additional nicks occurring on the opposing DNA strand within a taboo zone with 2h width, that would result in linearization. The taboo zone, b, is expressed as a fraction of the total plasmid, 2h/L. This probability, Equation (4), yields the percentage of plasmid molecules that have been relaxed by a single nick, but have not yet been linearized given the number of nicks, n and taboo zone fraction size, b.
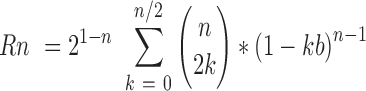 |
(4) |
The full time course of DNA cleavage was globally fit to the nuclease model described in Cowan et al. (70). Supercoiled, relaxed and linearized plasmid band intensities were fit to the following three equations, respectively:
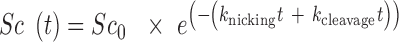 |
(5) |
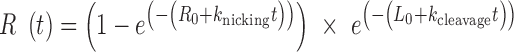 |
(6) |
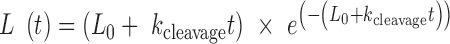 |
(7) |
Where Sc, R and L are the fractions of supercoiled, relaxed, and linearized plasmid and Sc0, R0 and L0 are the initial fractions of those species. knicking and kcleavage are the nicking and dual-strand cleavage rates, respectively.
Isolation of P23-45 Phage genomic DNA
An initial stock of phage P23-45 was kindly provided by the Severinov laboratory. A fresh culture of Thermus thermophilus HB8 was grown to an OD600 of ∼1.0 in growth medium (0.8% (w/v) Tryptone, 0.4% (w/v) Yeast Extract, 0.3% (w/v) NaCl, 1 mM MgCl2, and 0.5 mM CaCl2) (71). A total of 150 μl of fresh culture was combined with 100 μl P23-45 phage stock at a concentration of 106 Plaque Forming Units per ml (PFU/ml) and incubated at 65°C for 10 min. This mixture was then inoculated into 20 ml of fresh growth medium and incubated for 4–6 h at 65°C. The culture was spun at 4000 × g for 20 min to remove cell debris. Supernatant (>109 PFU/ml) was then treated with DNase I (final concentration, 2 Units/ml) and incubated at 30°C for 30 min. Genomic DNA was extracted from P23-45 phage stocks using the Phage DNA Isolation Kit (Norgen Biotek Corp) according to the manufacturer's protocol.
RESULTS
TerLP74-26 displays robust nuclease activity
We previously established that TerLP74-26 binds DNA at room temperature in the presence of ADP•BeF3 using an electrophoretic mobility shift assay (EMSA) (46). Because P74-26 phage is a thermophile, we hypothesized that the relatively low temperature (∼20°C) of our previous DNA binding experiments ‘masked’ the underlying endonuclease activity. Indeed, raising the temperature stimulates TerLP74-26's nuclease activity, with robust cleavage of plasmid DNA at ∼40°C that accelerates at 60°C (Figure 2A). TerLP74-26 must also be locked in an ‘ATP-bound’ state by a non-hydrolyzable ATP analog to efficiently cleave DNA; we only observe DNA cleavage with TerLP74-26 pre-loaded with ADP•BeF3 and no cleavage in the apo or ADP-loaded states (Figure 2B). Incubation with ATP does not result in DNA cleavage because robust TerL ATPase activity (46) rapidly converts all available ATP to ADP. Therefore, TerLP74-26 needs to be locked in an ‘ATP-bound’ state for productive cleavage, indicating a strong linkage between the ATPase and nuclease activities.
Figure 2.

Characterization of TerLP74-26 DNA-binding and nuclease activity. (A) Elevated temperature enhances TerLP74-26 nuclease activity. Plasmid DNA migrates slowly when mixed with 15 μM TerLP74-26 (left panel, no SDS) at low temperatures, indicating TerLP74-26 primarily binds DNA at these temperatures. DNA cleavage occurs at higher temperatures indicated by the low molecular weight smearing. Addition of SDS (right panel) reveals minimal cleavage at room temperature with increased intensity of the relaxed (R) and linear (L) plasmid bands, and concomitant decrease in the supercoiled (Sc) band. At temperatures ≥40°C, we observe robust cleavage that increases as the temperature is raised. (B) At room temperature, DNA weakly binds to TerLP74-26 in the apo state or when incubated with ADP or ATP. Locking TerL into an ‘ATP-bound’ state with the non-hydrolyzable analog ADP•BeF3 results in tight DNA binding. At 60°C TerL cleaves DNA, but only in the presence of ADP•BeF3. Buffer control samples containing ADP•BeF3 do not exhibit perturbed plasmid migration (final lane at each temperature). (C) Mutation of D294, the conserved nuclease active site residue necessary for metal coordination, to alanine results in a severe loss of nuclease activity (10 μM protein) without affecting TerLP74-26's affinity for DNA.
To exclude the possibility of a co-purified contaminant being responsible for the observed nuclease activity, we mutated an absolutely conserved metal-coordinating active site residue (D294) to alanine. TerLP74-26 is expected to have a two-metal coordinated active site as is found in related nucleases (7,37–39,42,72–73). D294 is the best candidate for mutagenesis because this residue coordinates one of the metal ions in the active site (7,35,37–39). The D294A variant displays an almost complete loss of cleavage activity, while displaying EMSA band shifts characteristic of wild-type DNA binding (Figure 2C). These results illustrate that the nuclease activity is due to TerLP74-26 and not a contaminant. Therefore, isolated TerLP74-26 retains the three critical activities necessary for terminase function: ATPase activity, DNA-binding activity (46) and DNA cleavage.
To our knowledge, TerLP74-26 is the only known large terminase that can both bind and cleave DNA as a full-length protein. The isolated ATPase domain of TerLT4 is competent to bind DNA, whereas the full-length protein shows no significant affinity for DNA (74). Other full-length TerL proteins exhibit significant in vitro nuclease activity (7,37,39,41,75), although DNA-binding activity is undetectable. Therefore both DNA binding and cleavage can be separately dissected with TerLP74-26.
We next investigated specificity of the DNA binding and cleavage activities of TerLP74-26. As we have shown previously, TerLP74-26 binds DNA with no sequence specificity, as all bands in a 1 kb-ladder are shifted (46). When incubated at 60°C, TerLP74-26 degrades all bands in the 1kb-ladder, as well as negatively supercoiled plasmid, linearized plasmid and other linear fragments (Supplementary Figure S1). Thus, TerLP74-26 both binds and cleaves DNA with no discernible sequence specificity. TerL proteins from other headful packaging phages cleave DNA with no sequence specificity, indicating that TerLP74-26 is similar to most other TerL proteins in this respect.
To investigate the linkage between DNA binding and cleavage, we measured the TerLP74-26 concentration dependence for both activities. DNA binding and cleavage reactions were performed at different temperatures, but otherwise with identical reaction conditions and an identical plasmid substrate. Strikingly, we observe that DNA binding and DNA cleavage exhibit nearly identical dependence on TerL concentration (Figure 3A and B). At TerLP74-26 concentrations ≤2 μM (one TerL monomer for every 15.2 DNA base pairs), no DNA binding or cleavage is observed within the 30 min of the reaction. However, at >2 μM, TerLP74-26 exhibits significant binding at room temperature, with higher TerLP74-26 concentrations resulting in slower DNA migration. Likewise, at 60°C TerLP74-26 concentrations higher than 2 μM result in substantial fragmentation of the plasmid over the 30-min time course. Increasing incubation time to 16 h at 60°C results in substantial, but incomplete DNA cleavage at 2 μM TerLP74-26, with minor cleavage at 1.5 μM (Supplementary Figure S2). The coincident TerL dependencies of DNA binding and cleavage activities indicate that these two functions are tightly linked. Moreover, the steepness of the activity transition suggests that a cooperative process drives both DNA binding and cleavage. Band smearing and multiple TerLP74-26 binding sites per plasmid prevent accurate measurement and fitting to calculate a Kd. However, binding occurs roughly at concentrations consistent with a Kd in the low μM range. The affinity of TerLP74-26 for DNA is therefore similar to that measured for the Lambda phage TerL protein (Kd ∼3–4 μM) (49), indicating that TerLP74-26 is consistent with known terminase enzyme function.
Figure 3.

DNA binding and cleavage exhibit identical protein concentration dependence. (A) At room temperature, full length TerLP74-26 binds DNA at concentrations greater than 2 μM (no SDS added to the samples). DNA migrates slower, indicating greater binding, as the TerLP74-26 concentration is raised. (B) At 60°C, TerLP74-26 cleaves DNA proportional to the degree of DNA binding observed at room temperature. Tighter binding (slower migration) at room temperature corresponds with more complete DNA digestion at 60°C.
Kinetic analysis of TerLP74-26 cleavage
To further investigate the mechanism of TerL nuclease activity, we followed the kinetics of plasmid cleavage. We chose a TerLP74-26 concentration of 5 μM due to the potent activity observed during the 30-min reaction. By measuring the intensities of the supercoiled, relaxed, and linear plasmid bands over a 10-min time course, we can distinguish between single-strand versus dual-strand cleavage (see below). To accurately quantify each band, we added SDS to our gel-loading buffer to prevent TerLP74-26's DNA binding activity from perturbing DNA migration. We observe a rapid loss of supercoiled DNA (t1/2∼20 s), whereas the relaxed and linearized plasmid bands increase and then decrease in intensity (Figure 4A). The relaxed plasmid increases in intensity (peak ∼30 s), and is then degraded until it is undetectable at ∼480 s. Linearized DNA increases with no observable lag until ∼60 s, and then is degraded to form a smear, indicating more substantial fragmentation.
Figure 4.

Kinetics of TerLP74-26 nuclease activity. (A) A representative gel with 5 μM TerLP74-26 incubated with plasmid at 60°C for different durations. Band intensities for supercoiled (Sc), relaxed (R) and linear (L) species were measured by densitometry of four replicates for Freifelder-Trumbo analysis and kinetic fitting. (SDS present in loading buffer.). (B) Freifelder-Trumbo (69) analysis of the untreated plasmid (black circle), 5 s timepoint (orange diamond) and 10 s timepoint (orange square) calculated with Equations (1–2). The simulated single strand nickase curves were calculated with Equation (3) for h = 16 bp (black hexagons) or 50 bp (black triangles). (C) Nuclease data were globally fit by a kinetic model (Equations 5–7) in which TerL can catalyze both nicking and dual strand cleavage (70). See ‘Materials and Methods’ section for more details. Residuals to the fit are shown in Supplementary Figure S3C.
The rapid rise in the linearized fraction of DNA suggests TerLP74-26 employs a dual-strand cleavage mechanism. The plasmid banding pattern closely resembles a mixed, dual-strand and single-strand cutting mechanism of DNaseI (76). In the presence of magnesium, DNaseI can only nick DNA. However, in the presence of manganese DNaseI can also cleave both strands, as well as produce single nicks. Campbell et al. used a similar plasmid digestion assay to characterize DNaseI activity (76). They observed a rapid rise in the fraction of linearized DNA in presence of manganese, as DNaseI's dual-strand cleavage activity quickly linearized the DNA. However, in the presence of magnesium, there is a significant lag in the appearance of linearized plasmid, because double-strand breaks only occur when DNaseI produces two nicks nearby each other on opposing strands. Because our plasmid size (5372 bp) is similar to that of Campbell et al. (5224 bp), the density of nicks and thus the cleavage patterns are comparable between the two experiments. We observe qualitatively similar cleavage patterns for TerLP74-26 plasmid digestion as for DNaseI•Mn2+ (Figure 4A), with no lag observed for the appearance of linearized plasmid. We therefore hypothesized that TerLP74-26 both nicks and directly cleaves both DNA strands.
Quantifying the fractions of supercoiled, relaxed, and linearized DNA supports a dual strand cleavage mechanism. Freifelder-Trumbo analysis uses the proportion of supercoiled, relaxed, and linearized plasmid species to distinguish between dual strand cleavage versus the accumulation of a sufficient number of random nicks to result in eventual linearization (69). This analysis has previously determined the cleavage mechanism for other nucleases or DNA degrading small molecules (77–81). Because Freifelder-Trumbo analysis is limited to time points with low levels of fragmentation, we used only early time points (up to 10 s) for our Freifelder-Trumbo analysis where plasmid DNA has not yet been measurably fragmented (Supplementary Figure S3A). The number of DNA linearization events (nlinear) and nicks (nnick) and were calculated from Equations (1) and (2) respectively. We simulated curves from a purely single-stranded cutting enzyme with Equation (3), with L base pairs in the plasmid (5372 bp) and h, the maximum base pair distance between two nicks on opposing DNA strands that results in linearization. The analyzed time points exhibit a proportion of double strand breaks that is >400 fold higher than that predicted for a random, purely single strand cutting mechanism for h = 16 bp (69) (Figure 4B). Therefore, we conclude that TerLP74-26 has significant dual strand cleavage activity.
Because plasmid fragmentation limited us to a few data points for Freifelder-Trumbo analysis, we sought to verify our conclusions by investigating possible sources of error in our calculations. First, intercalation of ethidium bromide into supercoiled DNA differs from that of relaxed and linear DNA, often requiring a correction factor to adjust for a diminished fluorescent signal (82,83). Although we could not calculate the correction factor for our plasmid substrate, increasing the supercoiled signal intensity by a factor of 1.4, similar to other observations, does not perturb the results or affect our conclusion of dual strand cleavage (Supplementary Figure S3B and Table S1). Second, we adjusted the distance between two single strand nicks on opposing strands (h in Equation (3)) required for a double-strand break. Although linearization due to nicks 50 bp apart is physically unreasonable, increasing the maximum distance between opposing strand nicks from the standard 16 (69) to 50 bp does not change the result that TerLP74-26 linearizes plasmid DNA two-orders of magnitude faster than predicted for a pure nicking mechanism (Figure 4B and Supplementary Table S1).
Because Freifelder-Trumbo analysis indicates TerL can directly cleave both DNA strands, we further investigated the nuclease mechanism by fitting the full TerLP74-26 experimental data to a mechanism of mixed single-strand and dual-strand cleavage. We use a set of equations derived by Cowan et al. to describe this situation (70). The data were fit to Equations (5–7) (70). This model assumes dual strand cleavage is rapid enough that single strands cannot sufficiently accumulate as to cause substantial linearization. The Freifelder-Trumbo analysis supports this assumption as the linear to relaxed proportions of DNA are well above a simulated pure single strand cleaving mechanism. The fitted parameters include the nicking rate (knicking) and dual-strand cleavage rate (kcleavage), as well as the initial fractions of the supercoiled (S0), relaxed (R0) and linearized (L0) plasmid bands.
The high quality fit of the data (R2 for global fit = 0.986; Figure 4C; Supplementary Figure S3C and Table S2) to the dual-strand linearization model confirms that TerLP74-26 can catalyze dual strand cleavage. The nicking rate (0.0408 ± 0.0025 s−1) is roughly twice that of the dual strand cleavage rate (0.0197 ± 0.0007 s−1). These values suggest a plasmid would only accumulate two nicks on average for every linearization event. Importantly, after making an initial nick to relax a plasmid, Equation (4) allows us to calculate how many random single strand breaks a nicking enzyme would have to make for a 50% population of linearized plasmid (70). Given a taboo zone of 16 bp (69) on either side of the initial nick, plasmids would need to accumulate 23 nicks to form a 50% mixture of both relaxed and linearized plasmid molecules. Furthermore, if we increase the taboo zone to 50 bp on either side of the initial nick, as we did in the Freifelder-Trumbo analysis above, 50% of the plasmid would be linearized only after 13–14 nicks. Thus, the amount of nicks required for a single strand cutter to linearize 50% of a relaxed plasmid is more than an order of magnitude greater than our observed rate of nicks per linearization event. Only a dual-strand cleavage mechanism can account for the observed rapid linearization.
Because we observe rapid plasmid cleavage, we sought to determine whether a larger and physiologically relevant substrate would exhibit substantially different kinetics. A digestion time course with the genome of related phage P23-45 (92% identical between nucleotide sequence of the P23-45 and P74-26 genomes; 99.8% identity between amino acid sequence of P23-45 and P74-26 TerL proteins (71)) showed a similar rate of fragmentation (Supplementary Figure S3D). Only dual strand cleavage explains the rapid digestion of the phage genome. A pure nicking mechanism would result in significantly slower fragmentation of the 85 kb genome substrate relative to the 5.4 kb plasmid. Dual-strand cleavage however would rapidly fragment the genome as observed, further supporting our conclusion of dual-strand cleavage activity. Moreover, this result suggests that the DNA sequence alone cannot alter TerLP74-26 mediated cleavage. Altogether, the evidence of dual-strand cleavage has important impacts on how the nuclease functions in the TerL protein (see ‘Discussion’ section).
Although our analysis of cleavage kinetics indicates that TerL performs both dual strand cleavage and single-strand nicking, this simple model may not perfectly describe the mechanism. The Freifelder-Trumbo analysis and fitting to the model described by Cowan et al. assume that binding and cleavage is random. We observe a weak banding pattern in the fragmented plasmid (Figure 4A and Supplementary Figure S2) suggesting TerL may have a mild preference for binding and/or cleaving at specific points in the plasmid. However, TerLP74-26 binds (46) and cleaves numerous different substrates with no observable sequence specificity (Supplementary Figure S1). Given that TerL therefore binds and cleaves all tested DNA sequences, we assume that plasmid binding and cleavage remains mostly random. Given the ratio of linearized to relaxed molecules is >400 fold higher than a pure single strand cutter, and the goodness of the fit to the kinetic model, if there is a minor sequence preference, its impact is minimal.
Structure of TerLP74-26 nuclease domain
To gain insight into the structural mechanism of DNA cleavage by TerLP74-26, we solved the structure of the TerLP74-26 nuclease domain (hereafter, TerLP74-26-ND) to 2.6 Å resolution (Figure 5A). We obtained experimental phases from single-wavelength anomalous diffraction of a platinum derivative (see ‘Materials and Methods’ section) (Table 1). The overall fold of TerLP74-26 -ND is similar to those of other terminase nuclease domains (7,35,38–41), with an average Cα RMSD of 2.0 Å (for individual Cα RMSDs, see Supplementary Table S3).
Figure 5.

The TerLP74-26 nuclease domain structure. (A) Overall features of the TerLP74-26 nuclease domain structure. The electron density is missing for both a flexible loop at residues 350–352 (Gly-Val-Gly; dotted lines) and for C-terminal residues 450–485. Potential metal-coordinating active site residues (294, 300, 347, 428 and 429) are represented with sticks. A β-hairpin unique to the terminase family extends away from the nuclease domain. (B) Zoomed view of the nuclease active site shows the acidic residues for metal coordination. (C) The RNase H configuration of DNA bound the TerLP74-26-ND was created by aligning an RNase H structure bound to a RNA/DNA hybrid duplex (85) to the TerLP74-26-ND using default parameters in Chimera (87). The RNA:DNA duplex clashes (red boxes) with TerLP74-26-ND. There are severe clashes in the regions around metal-coordinating residue 347 and the β-hairpin. (D) The RuvC configuration of DNA bound to TerLP74-26-ND was generated using the structure of RuvC resolvase bound to a Holliday junction (86). Clashing is minimal, occurring at G424 and the side chains of R421 and R425. The flexibility of these residues suggests they may change conformation to accommodate DNA binding.
Table 1. Crystallographic data and refinement statistics.
| Data Collection | |
| Space group | P 43 21 2 |
| Wavelength | 1 |
| Resolution range | 47.51-2.60 |
| Unit cell angles (°) | 71.37 71.37 127.32 |
| Unit cell dimensions (Å) | 90 90 90 |
| Total reflections | 299 519 |
| Unique reflections | 10 480 (862) |
| Multiplicity | 28 (28.8) |
| Completeness % | 98 (98) |
| Mean I/sigma I | 23.3 (6.8) |
| Wilson B factor | 31.9 |
| R-merge | 0.089 (0.638) |
| R-meas | 0.090 (0.650) |
| R-pim | 0.017 (0.121) |
| CC1/2 | 0.999 (0.999) |
| CC* | 1.000 (0.999) |
| Refinement | |
| R-work % | 21.8 |
| R-free % | 24.7 |
| RMS bonds | 0.003 |
| RMS angles | 0.52 |
| Ramachandran favored % | 94 |
| Ramachandran outliers % | 0 |
| Rotomer outliers % | 0.62 |
| Clashscore | 4.05 |
| Average B | 38 |
Several high-resolution structures of the large terminase nuclease domain for highly related phage, G20C, are solved in the accompanying article from Xu et al. The protein sequences for TerLG20C-ND and TerLP74-26-ND are nearly identical, and differ only at residue 315 (G20C A315 versus P74-26 V315). TerLG20C-ND crystallized in three crystal forms that are distinct from TerLP74-26-ND. Overall, the structures of TerLG20C-ND and TerLP74-26-ND complement one another to provide key insight into TerL nuclease structure and function.
The active site of TerLP74-26 -ND contains several metal-coordinating residues that are conserved across the terminase family. Because no divalent cations were added during purification and crystallization, we do not observe any metal coordination in the active site. As expected from structures of other TerL nuclease domains (7,35,38–41), we observe D294 in the heart of the active site accompanying several other metal-coordinating residues (D294, D300, D347, D428, D429) in TerLP74-26 (Figure 5A and B). The accompanying article by Xu et al. discusses specific residue interactions during metal coordination in detail for TerLG20C-ND. The positions of acidic residues in TerLP74-26 most closely resemble the arrangement observed in phage T4/RB49 (Supplementary Figure S4), which binds metal with residues equivalent to TerLP74-26 D294, D347 and D429 (35,37).
Beyond the active site residues, there is remarkably little sequence conservation within the nuclease domain across the TerL family. The relative lack of conservation may reflect the fact that different viruses use varied strategies for cleaving DNA (21,84). Because of this lack of sequence conservation, identifying how DNA accesses the nuclease active site has been particularly challenging.
To address how DNA is positioned in the TerL nuclease active site, we compared the TerL nuclease domain to distantly related nuclease structures for which there are structures of substrates bound. In particular, the structure of human RNaseH bound to an RNA:DNA hybrid (85) and a more recent structure of T. thermophilus RuvC resolvase bound to a Holliday junction (86) provide two different possibilities for the DNA orientation in the nuclease active site. By superposing (87) these two structures with that of TerLP74-26-ND, we can model potential DNA interaction modes. Superposition of the RNaseH:RNA–DNA structure positions the DNA helix along a surface that extends from a flexible loop at residues 350–352, across the active site toward the N-terminus of the nuclease domain (Figure 5C). However, as has been noted previously (38–39,41), this positioning clashes with a β-hairpin that is present in all TerL proteins but is absent in other known members of the RNaseH superfamily of nucleases (38). Therefore, the β-hairpin must considerably flexible in order to accommodate DNA in the RNaseH configuration. Because of this substantial clash, the β-hairpin has been proposed to play an auto-regulatory role in controlling nuclease activity (38). In contrast, superposition of RuvC suggests an orthogonal DNA orientation (Figure 5D). Importantly, the β-hairpin does not produce a significant clash with modeled DNA but would instead provide a surface for cradling the DNA as it crosses the active site. Thus, the two RNaseH and RuvC models predict different roles for the β-hairpin: the RuvC model predicts that the β-hairpin assists in DNA cleavage while the RNaseH model predicts that the β-hairpin inhibits cleavage.
To further investigate the role of the nuclease domain in terminase function, we used the TerLP74-26 nuclease structure to identify residues that may be important for DNA binding and DNA cleavage. We selected conserved or semi-conserved basic residues that are predicted to contact DNA, based on previous predictions of DNA binding surfaces (7,18,35,38–39,41) and our comparisons with RNaseH and RuvC. Combined with variants in the ATPase domain that we previously generated to study ATP hydrolysis and DNA binding (46), our panel includes 23 point mutations across both domains (Supplementary Table S4). We also used our isolated TerLP74-26 ATPase domain (TerLP74-26-AD) and TerLP74-26-ND constructs to examine the overall role of each domain in TerL function. By separately measuring DNA-binding and nuclease cleavage for each variant, we provide critical insight into how DNA is bound and cleaved during viral genome packaging.
The ATPase domain is the primary DNA binding region
To assess how DNA binds to TerLP74-26 we first focused on the ATPase domain, as we previously observed a complete loss of DNA binding from the R101E mutation (46). R101 is in a patch of basic residues along one surface of the ATPase domain that we predict forms the DNA binding surface within the pore of the assembled TerL ring (46). Here we extend this analysis to other residues across the surface of the ATPase domain, including other residues in the ‘basic patch’. Three of the mutations in the basic patch (R102A, R104E and R128A) also display a complete loss of DNA binding, while the final basic patch variant (R121E) does not significantly affect binding (Figure 6A; Supplementary Figures S5 and S6A). To verify that these DNA-binding effects are due to specific disruption of the DNA binding interface, we tested variants with mutations predicted to be outside of the pore of the TerL ring (R58A and R170A). Neither of these mutations severely affects DNA binding affinity (Figure 6A). These results support our previous conclusion that this basic patch is critical for gripping DNA.
Figure 6.

Mapping of residues important for DNA binding and cleavage. (A) Mutation of ATPase domain (left panel) basic patch residues severely inhibits DNA binding, whereas nuclease domain variants do not severely impact binding (right panel). DNA binding was assessed as described in the ‘Materials and Methods’ section. Variants were separated into three categories, as indicated by color. The ATPase domain is shown in the context of our ATPase ring model (46) to illustrate how mutational effects match the ring topology (green spheres indicate ADP•BeF3 in the ATPase active site). Mutations that severely inhibit binding (orange) tend to be in the pore of the ATPase model (left panel inset) including the basic patch (R101E, R102A, R104E or R128A). Interfacial residues (R139A and R235A) also inhibit binding. (Mutational mapping of the ATPase ring model can be viewed in greater detail in Supplementary Figure S6.) The nuclease structure is shown with DNA in the RuvC configuration. Nuclease domain variants fail to significantly inhibit DNA binding. (B) Mapping of residues important for DNA cleavage. Variants were ranked as described in ‘Materials and Methods’ section. Residues in the ATPase that are important for binding are likewise critical for DNA cleavage (left panel). DNA binding is therefore a prerequisite for effective cleavage. Nuclease domain residues predicted to interact with DNA in the RuvC configuration (K297, K377 and R421) are critical for nuclease activity, suggesting that DNA binds in a similar orientation. Nuclease metal coordinating variant D294A serves as a negative control. Ranking of each residue's contribution to DNA binding and cleavage is shown in Supplementary Table S4.
Because TerLP74-26 needs to be locked into an ATP-bound state to tightly grip DNA (Figure 2B), we next investigated how mutations in or near the ATPase active site affect DNA binding. R39 is a conserved residue in the P-loop of the active site and directly contacts the γ-phosphate group of ATP (46). R139 is the trans-acting arginine finger that is critical for ATP hydrolysis (46). We also tested several residues (R228, R229, R235, R236, R245) in the Lid subdomain, a region that caps the active site and changes conformation upon ATP hydrolysis and release (Figure 6A) (46). The R228A, R229A and R236A variants have no apparent effect on DNA binding, while the R39A and R245A variants exhibit a moderate decrease in DNA binding. Only the R139A and R235A variants display severe defects in DNA binding. Because the trans-acting arginine finger R139 and Lid subdomain residue R235 are important for both ATP hydrolysis and interactions between adjacent ATPase subunits (46), we hypothesize that the DNA binding defects observed with these variants is due to the severe loss of both ATP binding and/or ring assembly.
We next focused on the role of the nuclease domain in DNA binding. We mutated residues in the active site D294A and K377A), the β-hairpin (Y410A, R412A and R421E), and other regions that have been predicted in other structural studies of phages T4 (35) and Sf6 (18) to bind DNA (K297A, K372A, K399A and R406A). Interestingly, none of the mutations in the nuclease domain severely impact DNA binding (Figure 6A). Variants K297A and K377A display a moderate loss of affinity for DNA. Similarly, the mutations in the β-hairpin display modest defects. Neither R412 nor R421 are conserved in the β-hairpin, but structures of terminase nuclease domains often exhibit basic residues in similar locations (35,38–40), suggesting that basic residues may play some role in function. A third β-hairpin mutation (Y410A), designed to disrupt the β-hairpin structure, only moderately affected DNA binding. Overall these results indicate that the nuclease domain is not a primary determinant for high affinity DNA binding.
Because our panel of point mutants highlights the importance of the ATPase domain in binding DNA, we next investigated whether isolated domains bind DNA. TerLP74-26-AD binds DNA at similar concentrations as full-length TerLP74-26 (Figure 7A and Supplementary Figure S7). Interestingly, TerLP74-26-AD binds DNA independent of nucleotide, indicating that the nuclease domain is important for the ATP-dependent regulation of DNA binding. In contrast, TerLP74-26-ND does not detectably bind DNA, even at concentrations >30-fold higher than the approximate Kd for full-length TerLP74-26 binding (Figure 7B). Therefore, the ATPase domain is necessary and sufficient for TerL to bind DNA, an event that is a prerequisite for nuclease activity.
Figure 7.

The ATPase domain is necessary and sufficient for DNA binding. (A) The isolated TerLP74-26-AD binds DNA with the same affinity as full length TerLP74-26 independent of ADP•BeF3. A total of 15 μM TerLP74-26-AD slows migration of a significant portion of plasmid DNA in the apo form or the presence of ADP. In the presence of ADP•BeF3, DNA migration slows with TerLP74-26-AD concentrations above 2 μM with decreasing migration proportional to the rise in protein concentration. Coomassie staining confirms the presence of TerLP74-26-AD co-migrating with DNA (Supplementary Figure S7). (B) The isolated TerLP74-26-ND neither binds nor cleaves DNA, even at concentrations 30-fold higher than where we observe binding and cleavage for full-length TerLP74-26. DNA binding appears necessary for effective cleavage.
Identifying the requirements for nuclease activity
We next examined our panel of variants to determine the role of individual residues on the DNA cleavage reaction. We find a strong correlation between DNA binding and nuclease activity across all ATPase mutants. Mutations that abrogate DNA binding likewise inhibit DNA cleavage, while mutations in the ATPase domain that do not disrupt DNA binding have no effect on nuclease activity. Specifically, mutations in the ATPase domain's basic patch (R101E, R102A, R104E and R128A) or the active site (R39A, R139A, R235A) show a severe loss of nuclease function (Figure 6B; Supplementary Figures S5 and S6B). These results support our finding that the isolated TerLP74-26-ND fails to bind and cleave DNA. Therefore binding and cleavage hinge on the ATPase domain's ability to bind DNA.
Interestingly, a subset of nuclease domain mutants disrupts DNA cleavage without severely impairing DNA binding. We observe a severe loss of nuclease activity in the K297A, K377A and R421E variants, and a moderate decrease in activity in the K372A variant. K297A, K377A and R421E are the only variants in full-length TerLP74-26 where DNA binding remains relatively unperturbed yet nuclease activity is severely impacted. As mentioned previously, the RuvC and RNaseH binding modes predict very different behavior for several of the variants, particularly those with mutation in the β-hairpin. The RuvC binding mode predicts a favorable role for the β-hairpin in DNA cleavage, while the RNaseH mode predicts that the β-hairpin plays an auto-inhibitory role. Overall, cleavage defects in these variants are consistent with the DNA contacts predicted by the RuvC-like model of DNA binding (Figure 5D). In particular, R421 is on the face of the β-hairpin predicted to interact favorably with DNA in the RuvC binding mode; this residue is necessary for DNA cleavage activity (Figure 6B). In contrast, Y410 and R412 are on the opposite face of the β-hairpin and both are dispensable for nuclease activity. Regardless of whether the RuvC or RNaseH binding modes are correct for TerL nuclease engagement, our results demonstrate that cleavage depends on two factors, (i) the ability of TerL to bind DNA as dictated primarily by the ATPase domain, and, (ii) a specific set of nuclease domain residues predicted to position DNA for cleavage.
DISCUSSION
Proper terminase function in viral genome packaging requires precise spatiotemporal coordination and regulation of ATP hydrolysis, DNA binding and nucleolytic cleavage. Each of these functions must be individually examined in order to piece together a packaging mechanism. We previously built a low-resolution structural model of a pentameric TerL ring that accurately predicted the position of the arginine finger (R139 in TerLP74-26), as well as a key DNA-binding residue (R101 in TerLP74-26) (46). We observed ATP-dependent conformational changes in the ATPase domain, suggesting that the Lid subdomain generates the force for DNA translocation through a lever-like motion. In this study we provide insights into terminase function that can then be applied to improve the existing models of genome packaging and further our understanding of one of nature's most powerful bio-motors.
The TerL ATPase domain tightly grips DNA
The TerL ATPase domain is indispensable for DNA binding. This conclusion is based on two major observations. First, we observe strong DNA binding with both full-length TerLP74-26 and the isolated ATPase domain. In contrast, the isolated nuclease domain of TerLP74-26 does not detectably bind or cleave DNA. Thus, the ATPase domain is both necessary and sufficient for DNA binding. Second, mutation of basic patch residues (R101, R102, R104 or R128) in full-length TerLP74-26 abrogates both DNA binding and cleavage. Conversely, none of the mutations located in the nuclease domain severely impact DNA binding, despite the fact that several residues are critical for DNA cleavage. These results suggest that the bulk of TerL affinity for DNA derives from the ATPase domain, as predicted by our previous model (46).
Several models for terminase:DNA binding predict a larger role of the nuclease domain in gripping DNA during translocation (18,35). Because we can separately measure DNA binding and cleavage with TerLP74-26, we are able to directly test these predictions. Surprisingly we find that residues within the nuclease domain only make a small contribution to DNA affinity (Figure 6A and Supplementary Figure S5) and that the entire domain is dispensable for tight binding (Figure 7A and Supplementary Figure S7). Moreover, two semi-conserved residues (K399 and R406) in a region predicted to bind DNA (35) show no role in binding or cleaving DNA (Figure 6A). Therefore, we favor a model in which the ATPase domain is the primary DNA grip during both translocation and cleavage modes, and that the nuclease domain only engages DNA during genome cleavage (Figure 8). Although unlikely, it is possible that free TerLP74-26 is locked into ‘cleavage mode’ and uses the nuclease domain for gripping DNA when in ‘translocation mode’. However, we do not favor this model because we observe no measurable affinity between the nuclease domain and DNA (Figure 7B), similar to results seen with T4-TerL (74). Moreover, the isolated ATPase domain, which is unlikely to be locked into ‘DNA cleavage mode’, displays tight DNA binding, as shown here (Figure 7A) and elsewhere for TerLT4 (74).
Figure 8.

Proposed model for nuclease regulation. During ‘translocation mode’ the nuclease domain active site is sequestered from DNA by interactions of the TerL with portal and capsid, preventing premature cleavage. The ATPase domain serves as the sole surface for gripping DNA during packaging. Upon completion of packaging TerL enters ‘cleavage mode’. TerL dissociates from the portal and capsid, releasing the inhibition of the nuclease domains. The ATPase domains remains tightly bound to DNA. The nuclease domains rearrange to cleave each of the antiparallel DNA strands. Although depicted as a blunt cut, cleavage could also leave overhangs depending on how both nuclease domains engage DNA.
By separating the primary DNA gripping region from the nuclease active site, terminases have evolved an efficient means for regulating nuclease activity. First, the nuclease domain's low intrinsic DNA-binding affinity appears to be important for proper nuclease regulation. Although the nuclease active site must bind DNA with at least weak affinity in order to cleave, this affinity must be carefully balanced; TerL would catalyze spurious cleavage if the affinity were too strong, but would cleave inefficiently if the affinity were too weak. There appears to be a spectrum of intrinsic DNA binding affinities for TerL nuclease domains. Isolated T4- (37,74), Sf6- (18,40), CMV- (7) and HSV-TerL (41) nuclease domains cleave DNA, implying a modest affinity. On the other hand, isolated SPP1- (38), P22- (39) and P74-26-TerL nuclease domains fail to cleave DNA. Secondly, the flexible nature between the ATPase and nuclease domains allows the allosteric regulation of the nuclease. The ATPase domain places the nuclease domain in high local concentration with DNA to overcome the nuclease domain's intrinsically weak affinity for DNA. Moreover, by altering the position of the nuclease active site relative to DNA, terminase enzymes can easily regulate DNA cleavage (see below).
Kinetic analysis reveals details of TerL nuclease activity
Kinetic analysis of the TerLP74-26 nuclease activity reveals a mechanism of TerL cleavage consistent across the terminase family. TerLP74-26 rapidly cleaves supercoiled plasmid, with concomitant increases in both relaxed and linearized plasmid, followed by complete fragmentation. This pattern of cleavage qualitatively resembles a mechanism involving dual strand cleavage observed in DNaseI (76). Regarding the terminase family, our results also mirror those observed for T4 phage and CMV TerL-catalyzed nuclease activity (7,42). Similarly, increasing concentrations of HSV-1 TerL nuclease domain results in an initial increase in relaxed and linearized plasmid, followed by near complete digestion at high nuclease concentrations (41). These results were interpreted as single strand nicking that eventually results in linearization and complete degradation (7,41). In contrast, our quantitative approach using Freifelder-Trumbo analysis and kinetic fitting of the nuclease reaction (Figure 4B and C) reveals significant dual strand cleavage by TerLP74-26. This result places large constraints on the arrangement of the TerL nuclease domains during DNA cleavage. Because the qualitative cleavage data is consistent across the family, we propose that these constraints are universal to all terminases.
From a mechanistic perspective, dual strand cleavage requires flexibility of the nuclease domain relative to the ATPase domain. Endonucleases require two active sites arranged in an anti-parallel fashion for simultaneous dual strand cleavage (88,89). As previously suggested (37,42), the TerL nuclease domains would assume conformations roughly 180° relative to one another for their active sites to align with each of the antiparallel DNA strands. Therefore, there is significant flexibility between the ATPase and nuclease domains of TerL to allow this rearrangement. Indeed, there is much previous data to support this assertion. Limited proteolysis of the TerL proteins from P74-26 (Supplementary Figure S8), T4 (32) and P22 (39) indicates that the linker connecting the ATPase Lid subdomain to the nuclease domain is highly flexible. Additionally, crystal structures of TerL proteins from T4 and Sf6 show very different orientations of the nuclease domain relative to the ATPase domain (18,35). Therefore, we propose that the TerL ATPase domain ring tightly grips DNA while the TerL nuclease domain is flexibly tethered to adopt the necessary orientation for DNA cleavage.
How does dual strand cleavage occur? We envision two possibilities for cleavage of both strands: (i) a monomer of TerL cleaves both strands in rapid succession, or (ii) two subunits within a TerL oligomer cleave each strand contemporaneously. In the first mechanism, after cleaving the Watson strand, the nuclease domain of the TerL monomer must rapidly reorient by ∼180° to cleave the Crick strand. In the second mechanism, two separate nuclease domains within a TerL oligomer can adopt orthogonal orientations to efficiently cleave both strands. Both mechanisms require a large degree of flexibility between the tightly bound ATPase domain and the nuclease domain. We do not observe a dimer oriented for dual strand cleavage within the nuclease domain crystal lattice (Supplementary Figure S9). Although we cannot decisively rule out dual strand cleavage by a monomer, we favor cleavage by a TerL oligomer for two reasons. First, the steep dependence on TerL concentration for both DNA binding and cleavage implies cooperative assembly of a TerL oligomer on DNA (Figure 3). Second, TerL requires ATP for both DNA binding and cleavage (Figure 2B), which implies that the interfacial contacts afforded by ATP binding (46) promote oligomerization on DNA. It is formally possible that a second TerL ring binds DNA in the opposite orientation (head to head) to the first ring during the cleavage reaction. This would require forming a large interface between rings as well as regulating the second ring binding to ensure it occurs only after capsid filling. We disfavor this model as there is no evidence for a head-to-head interaction or binding of a second ring at the end of packaging.
Regulation of TerL nuclease activity
During translocation, TerL nuclease activity must be inhibited to prevent premature cleavage. Two non-mutually exclusive possibilities could explain how TerL nuclease activity is regulated: (i) the ‘kinetic competition model’, wherein the rate of TerL ATP hydrolysis and DNA translocation significantly outpaces the rate of DNA cleavage until translocation slows upon maximal packaging, allowing cleavage to occur, and (ii) the ‘steric block model’, in which portal and/or TerS regulate the accessibility of the nuclease active site for DNA. We discuss these two possibilities below.
In the kinetic competition model the relative rates of ATP hydrolysis and DNA cleavage self-regulate TerL cleavage (37,43). If DNA translocation is much faster than the rate of cleavage, then the nuclease active site cannot stably engage DNA long enough for cleavage. Packaging would progress until the rate of DNA translocation sufficiently slows near the end of packaging. As the rates of translocation and cleavage become similar, the nuclease domain has enough time to engage a segment of DNA for successful cleavage. In the kinetic competition model, the rates of ATP hydrolysis and nuclease activity must be precisely balanced to prevent premature genome cleavage. However, two lines of evidence suggest that kinetic competition is not the regulatory mechanism. First, motor stalling events regularly occur during packaging in phages T4 (2) and Lambda (34,90) for periods of time up to ∼5 s with no reported cleavage of DNA. Second, long-term motor stalls can been artificially induced in phage T4 with no significant cleavage occurring over the time scale of hours (57,91).
In the steric block model, inhibition is achieved by restricting the accessibility of the nuclease active site for DNA. Multiple lines of evidence indicate that the TerL C-terminal tail binds to the portal (32,46,54–59). We propose that this interaction with portal locks the nuclease domain in an orientation that prevents the nuclease active site from accessing DNA, thereby inhibiting premature genome cleavage during packaging. Upon completion of genome packaging, portal transmits a ‘headful signal’ that is thought to trigger DNA cleavage (92). We hypothesize that the headful signal facilitates dissociation of TerL from portal, releasing the nuclease domain from this restricted conformation, allowing the flexibly tethered nuclease domains to reorient and doubly cut DNA. Similarly, TerS can inhibit TerL-nuclease activity (37,39,42,44); therefore, similar contacts with TerS could sterically block TerL nuclease activity.
How does DNA engage the nuclease active site? We use both the structure of RNaseH bound to an RNA/DNA hybrid (85) or RuvC bound to a Holliday junction (86) to model how DNA accesses the active site. DNA binds in orthogonal orientations in these structures, leading to distinct predictions for behavior of some of the variants tested here. We favor a model of DNA binding similar to RuvC. TerL is more similar to RuvC than RNaseH in terms of structure and the surface of TerL is a better steric fit to the RuvC DNA orientation rather than that of RNaseH. As noted here and in other studies (38,39), the β-hairpin would clash with DNA in the RNaseH-like configuration (Figure 5C). These results have raised the question of whether the β-hairpin adjusts its conformation to allow for productive access to the active site or if DNA is bound in a different orientation to the RNaseH model. In contrast, the model based on RuvC does not result in any substantial clash between the DNA and the protein (Figure 5D). Instead, the β-hairpin is positioned such that it can make favorable interactions along the DNA backbone. Importantly, our mutagenesis results are most consistent with the orientation of DNA predicted from the RuvC model. A β-hairpin residue predicted to directly form a salt bridge with the DNA backbone of the scissile strand (R421) is critical for nuclease activity, but two residues on the opposite face of the hairpin (Y410 and R412) are dispensable. In addition, three other residues in our panel of mutations are also predicted to interact with the scissile strand in the RuvC-like model (K297, K377 and R412); all three are critical for nuclease activity (Figure 6B). Furthermore, K372 is predicted to interact with the non-scissile strand, and the K372A variant shows a modest defect in nuclease activity. The phenotypes of the D294A, K297A and K377A (important for cleavage) and K399A and R406A (no role in cleavage) variants match both the RuvC and RNaseH orientations and do not effectively discriminate between the two models. Future studies will map the interactions with DNA in greater detail.
Our findings do not agree with previously proposed models of TerL DNA binding and cleavage. Translocation models for T4 (35) and Sf6 (18) both propose the nuclease domain as a primary site of DNA binding during packaging. Conversely, our TerLP74-26 variant and domain analyses clearly demonstrate that the ATPase domain is the primary site of DNA binding. First, the TerLT4 model posits that residues R517, and R524 bind the DNA backbone during translocation (35). However, the equivalent residues in TerLP74-26 (K399 and R406) are dispensable for binding. Second, the Sf6 model contends that the nuclease domain collaborates with the Lid subdomain (also referred to as the ‘Linker subdomain’) to grip DNA (18). However, key residues in the Sf6 structure that were predicted to bind DNA are either not conserved (R194, R305, R306 and K328 in Sf6) or show no binding defect when mutated in TerLP74-26 (K192/R193, and R200 in Sf6; R236 and R245 in P74-26). Other residues predicted by the Sf6 model only have a slight effect; K372 in TerLP74-26 (K323 in Sf6) is dispensable for DNA binding and only plays a minor role in DNA cleavage. A residue not predicted by the Sf6 model to bind DNA (K360 or R361 in Sf6; K377 in P74-26) is absolutely critical for DNA cleavage and has a moderate effect on DNA binding. Therefore, our data is not consistent with these two models, because we find that the ATPase domain is the primary site of DNA binding. However, it is possible that isolated TerLP74-26 is locked in ‘cleavage’ mode, whereas these residues would have a measurable role in ‘translocation’ mode.
DNA must bind to the nuclease domain for the DNA cleavage reaction to occur, even if this binding is weak and/or transient. Our data are consistent with two of the residues that were predicted to be important for DNA cleavage in the TerLT4 and TerLSf6 models. Residues flanking the active site are not necessary for full DNA binding affinity but are critical for DNA cleavage (R406 in T4 (35), which is K297 in P74-26; K416 in Sf6 (18), which is R412 in P74-26). The models therefore share some similarity for the TerL cleavage mechanism.
Terminases are conserved across many different families of dsDNA viruses, including human pathogens of Herpesviridae (11). There are several FDA-approved drugs on the market that target the terminase motor, and it is thought that these drugs’ mode-of-action is through inhibition of the terminase's nuclease activity (3–9). Our studies reveal important aspects of the TerL nuclease mechanism and regulation, and provide a blueprint for future mode-of-action studies of small molecule inhibitors of terminase enzymes from human pathogens.
ACCESSION NUMBER
PDB Code: 5TGE.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the Severinov laboratory (Rutgers University) for providing a sample of phage P23-45. We thank the Schiffer, Royer and Ryder labs for use of instrumentation and for helpful discussions. We thank members of the Antson Lab (University of York) for sharing data before publication and for thoughtful discussions. We thank beamline scientists at SIBYLS 12.3.1 (Lawrence Berkeley National Laboratory) and APS 23-ID-B (Argonne National Laboratory, award number GUP-39936) for technical support with x-ray diffraction data collection.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Pew Charitable Trusts Scholarship (to B.A.K.); Lawrence Berkeley; Argonne [GUP-39936]. Funding for open access charge: Pew Charitable Trust.
Conflict of interest statement. None declared.
REFERENCES
- 1. Smith D.E., Tans S.J., Smith S.B., Grimes S., Anderson D.L., Bustamante C.. The bacteriophage straight phi29 portal motor can package DNA against a large internal force. Nature. 2001; 413:748–752. [DOI] [PubMed] [Google Scholar]
- 2. Fuller D.N., Raymer D.M., Kottadiel V.I., Rao V.B., Smith D.E.. Single phage T4 DNA packaging motors exhibit large force generation, high velocity, and dynamic variability. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:16868–16873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buerger I., Reefschlaeger J., Bender W., Eckenberg P., Popp A., Weber O., Graeper S., Klenk H.D., Ruebsamen-Waigmann H., Hallenberger S.. A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J. Virol. 2001; 75:9077–9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bogner E. Human cytomegalovirus terminase as a target for antiviral chemotherapy. Rev. Med. Virol. 2002; 12:115–127. [DOI] [PubMed] [Google Scholar]
- 5. Dittmer A., Drach J.C., Townsend L.B., Fischer A., Bogner E.. Interaction of the putative human cytomegalovirus portal protein pUL104 with the large terminase subunit pUL56 and its inhibition by benzimidazole-D-ribonucleosides. J. Virol. 2005; 79:14660–14667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hwang J.S., Kregler O., Schilf R., Bannert N., Drach J.C., Townsend L.B., Bogner E.. Identification of acetylated, tetrahalogenated benzimidazole D-ribonucleosides with enhanced activity against human cytomegalovirus. J. Virol. 2007; 81:11604–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nadal M., Mas P.J., Blanco A.G., Arnan C., Sola M., Hart D.J., Coll M.. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:16078–16083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goldner T., Hewlett G., Ettischer N., Ruebsamen-Schaeff H., Zimmermann H., Lischka P.. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 2011; 85:10884–10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Melendez D.P., Razonable R.R.. Letermovir and inhibitors of the terminase complex: a promising new class of investigational antiviral drugs against human cytomegalovirus. Infect. Drug Resist. 2015; 8:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burroughs A.M., Iyer L.M., Aravind L.. Comparative genomics and evolutionary trajectories of viral ATP dependent DNA-packaging systems. Genome Dyn. 2007; 3:48–65. [DOI] [PubMed] [Google Scholar]
- 11. Baines J.D., Weller S.K.. Catalano CE. Cleavage and Packaging of Herpes Simplex Virus 1 DNA. Viral Genome Packaging: Genetics, Structure, and Mechanism. 2005; 1st edn., New York City, USA: Springer; 135–150. [Google Scholar]
- 12. Feiss M., Catalano C.E.. Catalano CE. Bacteriophage Lambda Terminase and the Mechanisms of Viral DNA Packaging. Viral Genome Packaging Machines: Genetics, Structure, and Mechanism. 2005; 1st edn., New York City, USA: Springer; 5–39. [Google Scholar]
- 13. Rao V.B., Black L.W.. Catalano CE. DNA Packaging in Bacteriophage T4. Viral Genome Packaging Machines: Genetics, Structure, and Mechanism. 2005; 1st edn., New York City, USA: Springer; 40–58. [Google Scholar]
- 14. Casjens S., Weigele P.. Catalano CE. DNA Packaging by Bacteriophage P22. Viral Genome Packaging Machines: Genetics, Structure, and Mechanism. 2005; 1st edn., New York City, USA: Springer; 80–88. [Google Scholar]
- 15. Serwer P. Catalano CE. T3/T7 DNA Packaging. Viral Genome Packaging Machines: Genetics, Structure, and Mechanism. 2005; 1st edn., New York City, USA: Springer; 59–79. [Google Scholar]
- 16. Droge A., Tavares P.. Catalano CE. Bacteriophage SPP1 DNA Packaging. Viral Genome Packaging Machines: Genetics, Structure, and Mechanism. 2005; 1st edn., New York City, USA: Springer; 89–101. [Google Scholar]
- 17. Zhao H., Finch C.J., Sequeira R.D., Johnson B.A., Johnson J.E., Casjens S.R., Tang L.. Crystal structure of the DNA-recognition component of the bacterial virus Sf6 genome-packaging machine. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:1971–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao H., Christensen T.E., Kamau Y.N., Tang L.. Structures of the phage Sf6 large terminase provide new insights into DNA translocation and cleavage. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:8075–8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rao V.B., Feiss M.. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 2008; 42:647–681. [DOI] [PubMed] [Google Scholar]
- 20. Feiss M., Rao V.B.. The bacteriophage DNA packaging machine. Adv. Exp. Med. Biol. 2012; 726:489–509. [DOI] [PubMed] [Google Scholar]
- 21. Catalano C.E. Viral Genome Packaging Machines: Genetics, Structure, and Mechanism. 2005; 1st edn., New York City, USA: Springer. [Google Scholar]
- 22. Shinder G., Gold M.. The Nul subunit of bacteriophage lambda terminase binds to specific sites in cos DNA. J. Virol. 1988; 62:387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Casjens S., Sampson L., Randall S., Eppler K., Wu H., Petri J.B., Schmieger H.. Molecular genetic analysis of bacteriophage P22 gene 3 product, a protein involved in the initiation of headful DNA packaging. J. Mol. Biol. 1992; 227:1086–1099. [DOI] [PubMed] [Google Scholar]
- 24. Chai S., Lurz R., Alonso J.C.. The small subunit of the terminase enzyme of Bacillus subtilis bacteriophage SPP1 forms a specialized nucleoprotein complex with the packaging initiation region. J. Mol. Biol. 1995; 252:386–398. [DOI] [PubMed] [Google Scholar]
- 25. Lin H., Black L.W.. DNA requirements in vivo for phage T4 packaging. Virology. 1998; 242:118–127. [DOI] [PubMed] [Google Scholar]
- 26. Wu H., Sampson L., Parr R., Casjens S.. The DNA site utilized by bacteriophage P22 for initiation of DNA packaging. Mol. Microbiol. 2002; 45:1631–1646. [DOI] [PubMed] [Google Scholar]
- 27. Morita M., Tasaka M., Fujisawa H.. DNA packaging ATPase of bacteriophage T3. Virology. 1993; 193:748–752. [DOI] [PubMed] [Google Scholar]
- 28. Rao V.B., Mitchell M.S.. The N-terminal ATPase site in the large terminase protein gp17 is critically required for DNA packaging in bacteriophage T4. J. Mol. Biol. 2001; 314:401–411. [DOI] [PubMed] [Google Scholar]
- 29. Goetzinger K.R., Rao V.B.. Defining the ATPase center of bacteriophage T4 DNA packaging machine: requirement for a catalytic glutamate residue in the large terminase protein gp17. J. Mol. Biol. 2003; 331:139–154. [DOI] [PubMed] [Google Scholar]
- 30. Valpuesta J.M., Carrascosa J.L.. Structure of viral connectors and their function in bacteriophage assembly and DNA packaging. Q. Rev. Biophys. 1994; 27:107–155. [DOI] [PubMed] [Google Scholar]
- 31. Bhattacharyya S.P., Rao V.B.. A novel terminase activity associated with the DNA packaging protein gp17 of bacteriophage T4. Virology. 1993; 196:34–44. [DOI] [PubMed] [Google Scholar]
- 32. Kanamaru S., Kondabagil K., Rossmann M.G., Rao V.B.. The functional domains of bacteriophage t4 terminase. J. Biol. Chem. 2004; 279:40795–40801. [DOI] [PubMed] [Google Scholar]
- 33. Duffy C., Feiss M.. The large subunit of bacteriophage lambda's terminase plays a role in DNA translocation and packaging termination. J. Mol. Biol. 2002; 316:547–561. [DOI] [PubMed] [Google Scholar]
- 34. Fuller D.N., Raymer D.M., Rickgauer J.P., Robertson R.M., Catalano C.E., Anderson D.L., Grimes S., Smith D.E.. Measurements of single DNA molecule packaging dynamics in bacteriophage lambda reveal high forces, high motor processivity, and capsid transformations. J. Mol. Biol. 2007; 373:1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun S., Kondabagil K., Draper B., Alam T.I., Bowman V.D., Zhang Z., Hegde S., Fokine A., Rossmann M.G., Rao V.B.. The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell. 2008; 135:1251–1262. [DOI] [PubMed] [Google Scholar]
- 36. Black L.W. Old, new, and widely true: the bacteriophage T4 DNA packaging mechanism. Virology. 2015; 479-480:650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alam T.I., Draper B., Kondabagil K., Rentas F.J., Ghosh-Kumar M., Sun S., Rossmann M.G., Rao V.B.. The headful packaging nuclease of bacteriophage T4. Mol. Microbiol. 2008; 69:1180–1190. [DOI] [PubMed] [Google Scholar]
- 38. Smits C., Chechik M., Kovalevskiy O.V., Shevtsov M.B., Foster A.W., Alonso J.C., Antson A.A.. Structural basis for the nuclease activity of a bacteriophage large terminase. EMBO Rep. 2009; 10:592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roy A., Cingolani G.. Structure of p22 headful packaging nuclease. J. Biol. Chem. 2012; 287:28196–28205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao H., Lin Z., Lynn A.Y., Varnado B., Beutler J.A., Murelli R.P., Le Grice S.F., Tang L.. Two distinct modes of metal ion binding in the nuclease active site of a viral DNA-packaging terminase: insight into the two-metal-ion catalytic mechanism. Nucleic Acids Res. 2015; 43:11003–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Selvarajan Sigamani S., Zhao H., Kamau Y.N., Baines J.D., Tang L.. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J. Virol. 2013; 87:7140–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ghosh-Kumar M., Alam T.I., Draper B., Stack J.D., Rao V.B.. Regulation by interdomain communication of a headful packaging nuclease from bacteriophage T4. Nucleic Acids Res. 2011; 39:2742–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cue D., Feiss M.. Genetic evidence that recognition of cosQ, the signal for termination of phage lambda DNA packaging, depends on the extent of head filling. Genetics. 1997; 147:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun S., Gao S., Kondabagil K., Xiang Y., Rossmann M.G., Rao V.B.. Structure and function of the small terminase component of the DNA packaging machine in T4-like bacteriophages. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leipe D.D., Koonin E.V., Aravind L.. Evolution and classification of P-loop kinases and related proteins. J. Mol. Biol. 2003; 333:781–815. [DOI] [PubMed] [Google Scholar]
- 46. Hilbert B.J., Hayes J.A., Stone N.P., Duffy C.M., Sankaran B., Kelch B.A.. Structure and mechanism of the ATPase that powers viral genome packaging. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E3792–E3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maluf N.K., Gaussier H., Bogner E., Feiss M., Catalano C.E.. Assembly of bacteriophage lambda terminase into a viral DNA maturation and packaging machine. Biochemistry. 2006; 45:15259–15268. [DOI] [PubMed] [Google Scholar]
- 48. Andrews B.T., Catalano C.E.. The enzymology of a viral genome packaging motor is influenced by the assembly state of the motor subunits. Biochemistry. 2012; 51:9342–9353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang T.C., Ortiz D., Nosaka L., Lander G.C., Catalano C.E.. Thermodynamic Interrogation of the Assembly of a Viral Genome Packaging Motor Complex. Biophys. J. 2015; 109:1663–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Morais M.C., Koti J.S., Bowman V.D., Reyes-Aldrete E., Anderson D.L., Rossmann M.G.. Defining molecular and domain boundaries in the bacteriophage phi29 DNA packaging motor. Structure. 2008; 16:1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mao H., Saha M., Reyes-Aldrete E., Sherman M.B., Woodson M., Atz R., Grimes S., Jardine P.J., Morais M.C.. Structural and molecular basis for coordination in a viral DNA packaging motor. Cell Rep. 2016; 14:2017–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cao S., Saha M., Zhao W., Jardine P.J., Zhang W., Grimes S., Morais M.C.. Insights into the structure and assembly of the bacteriophage 29 double-stranded DNA packaging motor. J. Virol. 2014; 88:3986–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao Z., De-Donatis G.M., Schwartz C., Fang H., Li J., Guo P.. An arginine finger regulates the sequential action of asymmetrical hexameric ATPase in the double-stranded DNA translocation motor. Mol. Cell. Biol. 2016; 36:2514–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yeo A., Feiss M.. Specific interaction of terminase, the DNA packaging enzyme of bacteriophage lambda, with the portal protein of the prohead. J. Mol. Biol. 1995; 245:141–150. [DOI] [PubMed] [Google Scholar]
- 55. Morita M., Tasaka M., Fujisawa H.. Analysis of the fine structure of the prohead binding domain of the packaging protein of bacteriophage T3 using a hexapeptide, an analog of a prohead binding site. Virology. 1995; 211:516–524. [DOI] [PubMed] [Google Scholar]
- 56. Lin H., Rao V.B., Black L.W.. Analysis of capsid portal protein and terminase functional domains: interaction sites required for DNA packaging in bacteriophage T4. J. Mol. Biol. 1999; 289:249–260. [DOI] [PubMed] [Google Scholar]
- 57. Dixit A., Ray K., Lakowicz J.R., Black L.W.. Dynamics of the T4 bacteriophage DNA packasome motor: endonuclease VII resolvase release of arrested Y-DNA substrates. J. Biol. Chem. 2011; 286:18878–18889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dixit A.B., Ray K., Black L.W.. Compression of the DNA substrate by a viral packaging motor is supported by removal of intercalating dye during translocation. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:20419–20424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dixit A.B., Ray K., Thomas J.A., Black L.W.. The C-terminal domain of the bacteriophage T4 terminase docks on the prohead portal clip region during DNA packaging. Virology. 2013; 446:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hegde S., Padilla-Sanchez V., Draper B., Rao V.B.. Portal-large terminase interactions of the bacteriophage T4 DNA packaging machine implicate a molecular lever mechanism for coupling ATPase to DNA translocation. J. Virol. 2012; 86:4046–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yu M.X., Slater M.R., Ackermann H.W.. Isolation and characterization of Thermus bacteriophages. Arch. Virol. 2006; 151:663–679. [DOI] [PubMed] [Google Scholar]
- 62. Otwinowski Z., Minor W.. Carter CW Jr, Sweet RM. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997; 276, New York, USA: Academic Press; 307–326. [DOI] [PubMed] [Google Scholar]
- 63. Hendrickson W.A., Teeter M.M.. Structure of the hydrophobic protein crambin determined directly from the anomalous scattering of sulphur. Nature. 1981; 290:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zwart P.H., Afonine P.V., Grosse-Kunstleve R.W., Hung L.W., Ioerger T.R., McCoy A.J., McKee E., Moriarty N.W., Read R.J., Sacchettini J.C. et al. . Automated structure solution with the PHENIX suite. Methods Mol. Biol. 2008; 426:419–435. [DOI] [PubMed] [Google Scholar]
- 65. Strong M., Sawaya M.R., Wang S., Phillips M., Cascio D., Eisenberg D.. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:8060–8065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 67. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. . PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schneider C.A., Rasband W.S., Eliceiri K.W.. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Freifelder D., Trumbo B.. Matching of single-strand breaks to form double-strand breaks in DNA. Biopolymers. 1969; 7:681–693. [Google Scholar]
- 70. Cowan R., Collis C.M., Grigg G.W.. Breakage of double-stranded DNA due to single-stranded nicking. J. Theor. Biol. 1987; 127:229–245. [DOI] [PubMed] [Google Scholar]
- 71. Minakhin L., Goel M., Berdygulova Z., Ramanculov E., Florens L., Glazko G., Karamychev V.N., Slesarev A.I., Kozyavkin S.A., Khromov I. et al. . Genome comparison and proteomic characterization of Thermus thermophilus bacteriophages P23-45 and P74-26: siphoviruses with triplex-forming sequences and the longest known tails. J. Mol. Biol. 2008; 378:468–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nowotny M., Yang W.. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006; 25:1924–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang W., Lee J.Y., Nowotny M.. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol. Cell. 2006; 22:5–13. [DOI] [PubMed] [Google Scholar]
- 74. Alam T.I., Rao V.B.. The ATPase domain of the large terminase protein, gp17, from bacteriophage T4 binds DNA: implications to the DNA packaging mechanism. J. Mol. Biol. 2008; 376:1272–1281. [DOI] [PubMed] [Google Scholar]
- 75. Gual A., Camacho A.G., Alonso J.C.. Functional analysis of the terminase large subunit, G2P, of Bacillus subtilis bacteriophage SPP1. J. Biol. Chem. 2000; 275:35311–35319. [DOI] [PubMed] [Google Scholar]
- 76. Campbell V.W., Jackson D.A.. The Effect of Divalent Cations on the Mode of Action of DNase I. J. Biol. Chem. 1980; 255:3726–3735. [PubMed] [Google Scholar]
- 77. Povirk L.F., Wubter W., Kohnlein W., Hutchinson F.. DNA double-strand breaks and alkali-labile bonds produced by bleomycin. Nucleic Acids Res. 1977; 4:3573–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Colis L.C., Woo C.M., Hegan D.C., Li Z., Glazer P.M., Herzon S.B.. The cytotoxicity of (-)-lomaiviticin A arises from induction of double-strand breaks in DNA. Nat. Chem. 2014; 6:504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jin Y., Cowan J.A.. DNA cleavage by copper-ATCUN complexes. Factors influencing cleavage mechanism and linearization of dsDNA. J. Am. Chem. Soc. 2005; 127:8408–8415. [DOI] [PubMed] [Google Scholar]
- 80. Zhang Q., Xiang Y., Liang D., Peng Y., Guo H.. Efficient double-strand scission of plasmid DNA by quaternized-chitosan zinc complex. Bioorg. Med. Chem. Lett. 2012; 22:1814–1817. [DOI] [PubMed] [Google Scholar]
- 81. Branum M.E., Tipton A.K., Zhu S., Que L. Jr. Double-strand hydrolysis of plasmid DNA by dicerium complexes at 37 degrees C. J. Am. Chem. Soc. 2001; 123:1898–1904. [DOI] [PubMed] [Google Scholar]
- 82. Mirabelli C.K., Huang C.H., Crooke S.T.. Comparison of DNA damage and single- and double-strand breakage activities on PM-2 DNA by talisomycin and bleomycin analogs. Cancer Res. 1980; 40:4173–4177. [PubMed] [Google Scholar]
- 83. Shubsda M.F., Goodisman J., Dabrowiak J.C.. Quantitation of ethidium-stained closed circular DNA in agarose gels. J. Biochem. Biophys. Methods. 1997; 34:73–79. [DOI] [PubMed] [Google Scholar]
- 84. Li S., Fan H., An X., Fan H., Jiang H., Chen Y., Tong Y.. Scrutinizing virus genome termini by high-throughput sequencing. PLoS One. 2014; 9:e85806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nowotny M., Gaidamakov S.A., Ghirlando R., Cerritelli S.M., Crouch R.J., Yang W.. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell. 2007; 28:264–276. [DOI] [PubMed] [Google Scholar]
- 86. Gorecka K.M., Komorowska W., Nowotny M.. Crystal structure of RuvC resolvase in complex with Holliday junction substrate. Nucleic Acids Res. 2013; 41:9945–9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 88. Newman M., Strzelecka T., Dorner L.F., Schildkraut I., Aggarwal A.K.. Structure of Bam HI endonuclease bound to DNA: partial folding and unfolding on DNA binding. Science. 1995; 269:656–663. [DOI] [PubMed] [Google Scholar]
- 89. Kim Y.C., Grable J.C., Love R., Greene P.J., Rosenberg J.M.. Refinement of Eco RI endonuclease crystal structure: a revised protein chain tracing. Science. 1990; 249:1307–1309. [DOI] [PubMed] [Google Scholar]
- 90. delToro D., Ortiz D., Ordyan M., Sippy J., Oh C.S., Keller N., Feiss M., Catalano C.E., Smith D.E.. Walker-A motif acts to coordinate ATP hydrolysis with motor output in viral DNA packaging. J. Mol. Biol. 2016; 428:2709–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ray K., Oram M., Ma J., Black L.W.. Portal control of viral prohead expansion and DNA packaging. Virology. 2009; 391:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Casjens S., Wyckoff E., Hayden M., Sampson L., Eppler K., Randall S., Moreno E.T., Serwer P.. Bacteriophage P22 portal protein is part of the gauge that regulates packing density of intravirion DNA. J. Mol. Biol. 1992; 224:1055–1074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.