


Abstract
Genome-wide enrichment of methylated DNA followed by sequencing (MeDIP-seq) offers a reasonable compromise between experimental costs and genomic coverage. However, the computational analysis of these experiments is complex, and quantification of the enrichment signals in terms of absolute levels of methylation requires specific transformation. In this work, we present QSEA, Quantitative Sequence Enrichment Analysis, a comprehensive workflow for the modelling and subsequent quantification of MeDIP-seq data. As the central part of the workflow we have developed a Bayesian statistical model that transforms the enrichment read counts to absolute levels of methylation and, thus, enhances interpretability and facilitates comparison with other methylation assays. We suggest several calibration strategies for the critical parameters of the model, either using additional data or fairly general assumptions. By comparing the results with bisulfite sequencing (BS) validation data, we show the improvement of QSEA over existing methods. Additionally, we generated a clinically relevant benchmark data set consisting of methylation enrichment experiments (MeDIP-seq), BS-based validation experiments (Methyl-seq) as well as gene expression experiments (RNA-seq) derived from non-small cell lung cancer patients, and show that the workflow retrieves well-known lung tumour methylation markers that are causative for gene expression changes, demonstrating the applicability of QSEA for clinical studies. QSEA is implemented in R and available from the Bioconductor repository 3.4 (www.bioconductor.org/packages/qsea).
INTRODUCTION
DNA methylation of CpG dinucleotides is a closely controlled epigenetic modification that impacts gene regulation and development (1). Aberrant DNA methylation has been identified as a hallmark of many diseases, in particular cancer (2). For example, down-regulation of tumour suppressor genes caused by focal hypermethylation of their promoters is a well described mechanism in the development of many cancer types (3). Thus, the systematic investigation of aberrant DNA methylation in cancer patients holds great potential in combatting cancer, since it not only contributes to the understanding of the functional role of epigenetic alterations in human disease, but also allows the identification of epigenetic biomarkers for noninvasive early cancer diagnosis (4) as well as targets for new molecular therapies (5).
The gold standard for measuring DNA methylation is bisulfite (BS) sequencing (6,7). DNA treated with sodium bisulfite converts unmethylated cytosines to uracil, but does not affect methylated cytosines (8). Subsequent sequencing of the BS-treated DNA reveals the fraction of unconverted (and thus methylated) cytosines. This approach measures DNA methylation at base resolution. However, it requires deep sequencing in order to generate sufficient read coverage, which remains a limiting cost factor when applying it at whole-genome scale (WGBS). Thus, bisulfite sequencing has been performed mainly as a targeted approach focusing on genomic regions of primary interest, for example with Methyl-seq (9,10) and reduced representation bisulfite sequencing (RRBS) (11). Other approaches, such as Illumina 450k arrays, use microarrays to measure methylation levels at genomic CpGs. All these approaches are limited to their respective target regions, and are not informative for discovery of epigenetic mechanisms outside the covered genome subset.
In contrast, assays based on enrichment of methylated DNA fragments target the entire genome and are, thus, not restricted to predefined sites: Methylated DNA Immuno-Precipitation (MeDIP) (12) and methyl-CpG binding domain (MBD) protein capture (13) are similar techniques, that enrich DNA fragments containing methylated cytosines. After sequencing, the measured read density can be related to the level of DNA methylation. This approach requires substantially less sequencing depth compared to WGBS, and is thus more cost effective. However, the resolution of enrichment-based methods is limited by the insert size of the sequencing library (typically 250 bp on average). With appropriate normalization, read density from these experiments provides a relative measurement for local methylation, and allows detecting relative differences between samples within a single region. However, due to dependence of the signal on CpG density, comparison of different genomic regions within and across samples, as well as derivation of absolute methylation levels requires further transformation. Many use cases presuppose absolute methylation levels, such as assessing, whether a specific region is methylated or unmethylated, comparing with bisulfite based assays, and charting whole genome methylation landscapes.
For processing enrichment-based methods, we have previously developed computational methods for the detection and annotation of aberrant DNA methylation, summarized in the MEDIPS software package (14). These methods have been applied to the analysis of MeDIP-seq data, for example, for identifying aberrant DNA methylation in colon cancer (15). Furthermore, they have been extended to other enrichment-based epigenetic sequencing data, for example, in order to profile hydroxymethylation changes during stem cell development (16) or to analyze cell type specific histone modification patterns from ChIP-seq experiments (17). Normalization of MeDIP-seq data implemented in the MEDIPS package corrects for local CpG densities and results in improved correlation of MeDIP signals to BS sequencing data. However, the current version of MEDIPS does not address transformation of these signals into absolute methylation estimates.
The task of estimating exact levels of methylation from enrichment experiments has recently been addressed by different methods: BayMeth is an approach that models read coverage with a Poisson distribution and quantifies methylation levels using Bayesian point estimators (18). The parameters of the model are calibrated with an additional fully methylated control enrichment experiment (DNA treated with SssI CpG methyltransferase). Another method, MeSiC, is based on a Random Forest Regression model, estimating methylation levels at base resolution from MeDIP-seq, without the need for additional calibration experiments (19). Both methods, however, have been developed for, and so far applied to, in vitro samples only, and their ability to conserve differences between pairs of in vivo tumour samples, and thus their applicability to clinical studies, has not yet been demonstrated.
Here, we present our novel workflow ‘Quantitative Sequencing Enrichment Analysis’ (QSEA). QSEA implements a statistical framework for modelling and transformation of MeDIP-seq enrichment data to absolute methylation levels similar to BS-sequencing read-outs. Furthermore, QSEA comprises functionality for data normalization that accounts for the effect of CNVs on the read-counts as well as for the detection and annotation of differentially methylated regions (DMRs). The transformation is based on a Bayesian model similar to BayMeth, but it extends this approach substantially by incorporating model parameters that explicitly take into account the signal-to-noise ratios of the experiments. Comparison of QSEA with BayMeth and MeSiC on different in vitro and in vivo benchmark data shows that QSEA outperforms both methods and that it is particularly suited for situations where no additional calibration experiments are available. We applied QSEA to the prediction of aberrant methylation on pairs of tumour and adjacent normal tissue from five non-small cell lung cancer (NSCLC) patients and validated the identified differentially methylated regions (DMRs) with BS sequencing on the same samples. Furthermore, we performed RNA-seq experiments on these tumour/normal pairs in order to monitor the effect of aberrant methylation on gene expression regulation and show that QSEA retrieves well-known lung cancer methylation markers that are causative for gene expression changes.
In summary, QSEA is a reliable workflow for detecting aberrant methylation in patient cohorts. Results are strongly correlated with BS-seq data and DMRs can be confirmed by the literature as well as experimental validation. QSEA is implemented as a user friendly R package, which is available at the Bioconductor repository (20).
MATERIALS AND METHODS
Patient-derived xenografts
Ethical approval (no. EA3/001/06) for the establishment of xenograft models from NSCLC patients was achieved from the local ethical review committee (Charité Berlin). All mice used in the study were handled in accordance with the Guidelines for the Welfare and Use of Animals in Cancer Research (21) and according to the German Animal Protection Law.Their use was approved by the local responsible authorities (approval no. G0030/15, H0023/09). Patient lung tumour samples were implanted subcutaneously into 1–3 nude or NOD/SCID mice (in-house breeding). Once tumours became palpable, tumour size was measured weekly with a caliper-like instrument. Individual tumour volume V was calculated with the formula 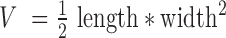 . Tumours of each model were further transplanted into 2–4 mice after a tumour volume of ∼1.2 cm³ was reached. Where possible, snap frozen tumour samples from each passage (up to 10 passages) were conserved and stored at −80°C for further analysis.
. Tumours of each model were further transplanted into 2–4 mice after a tumour volume of ∼1.2 cm³ was reached. Where possible, snap frozen tumour samples from each passage (up to 10 passages) were conserved and stored at −80°C for further analysis.
DNA library preparation and sequencing
DNA preparation
DNA from frozen tissue samples was isolated using a TissueLyser and the AllPrep DNA/RNA/Protein Mini Kit (Qiagen) according to the manufacturer΄s recommendations. Samples were quantified using the NanoDrop ND-2000 (Thermo Scientific).
MeDIP-Seq
1.3 μg of genomic DNA were randomly sheared using the Covaris S2 or M system to assess a size range of 100–300 bp. Illumina library preparation was performed by using the TruSeq DNA Sample Preparation Kit. Fragmented DNA was end repaired into dA-tailed fragments. The TruSeq indexed adaptor was then ligated to the fragmented DNA. Adaptor-ligated DNA was further cleaned up by AMPure XP beads (Beckman Coulter), denatured and then subjected to the methylated DNA immunoprecipitation (MeDIP) procedure. MeDIP was performed using 5 μg of a monoclonal antibody against 5-methylcytidine (Eurogentec) coupled to magnetic Dynabeads with M-280 sheep antibody against mouse IgG (Thermo Fisher Scientific). Sequencing libraries were denatured at 95°C for 10 min and incubation with the beads was carried out at 4°C for 4 h in the IP Buffer (10 mM sodium phosphate buffer (pH 7.0), 140 mM NaCl, 0.25% Triton X100). Beads were washed three times with the IP buffer and DNA was eluted in the elution buffer (50 mM Tris–HCl (pH 7.5), 10 mM EDTA, 1% SDS) at 65°C for 15 min. The beads were then treated with proteinase K for 2 h at 55°C, and methylated DNA was recovered using the QIAquick PCR Purification Kit from Qiagen. Assessment of the MeDIP efficiency was conducted with quantitative PCR (qPCR) targeting spiked-in controls as well as further methylated and unmethylated genomic regions. Following MeDIP enrichment, libraries were PCR amplified, size-selected and quantified using the Quant-iT dsDNA HS Assay Kit and a Qubit 1.0 Fluorometer from Invitrogen. Paired-end 2 × 50 bp libraries were sequenced using the HiSeq2500 platform (Illumina), yielding 57–111 million reads per sample.
Methyl-Seq
The Methyl-Seq experiments were conducted using the SureSelectXT Methyl-Seq Target Enrichment System by Agilent Technoligies. In brief, 3.0 μg of genomic DNA were fragmented to 100–200 bp using the Covaris S2 or M system followed by library preparation. Fragmented DNA was end repaired into dA-tailed fragments. The methylated adapter was then ligated to the fragmented DNA. Adapter-ligated DNA was further cleaned up by AMPure XP beads (Beckman Coulter), denatured and afterwards hybridized to the RNA capture library for 24 h at 65°C. Following the capturing of the RNA-DNA hybrids using streptavidine-coated magnetic beads, the DNA was separated from the beads, eluted and bisulfite converted using the EpiTECT Kit (Qiagen). The bisulfite-treated libraries were PCR amplified and purified. Finally, the DNA was again amplified and barcode sequences where attached to the sequences. The indexed DNA pool was analyzed with the 2100 Bioanalyzer High Sensitivity DNA assay (Agilent Technologies). Paired-end 2 × 50 bp libraries were sequenced using the HiSeq2500 platform (Illumina), yielding 41–101 million reads per sample.
RNA-seq
Sequencing libraries were prepared from 1μg of total RNA per sample following the TruSeq stranded RNA Low Sample protocol (single index; Illumina): Ribosomal RNA was depleted using the RiboZero Gold Kit (Epicentre) followed by chemical fragmentation of RNA, first and second strand cDNA synthesis, 3΄-end adenylation and adaptor ligation. Quality was tracked using the Bioanalyzer (Agilent). Libraries were amplified by PCR (15 cycles), quantified by qPCR and pooled for multiplex sequencing (3–4 libraries per lane). Paired-end 2 × 50 bp libraries were sequenced using the HiSeq2500 platform (Illumina), yielding 92–152 million reads per sample.
Demethylation experiment
Three lung cancer cell lines (H1299, H1650 and HCC827) were seeded at 3000–5000 cells per well in a 96-well plate and allowed to grow for 24 h. Subsequently, the cells were treated with one of four concentrations (2.5, 5, 10 and 20 μM) of decitabine (DMSO as negative control) for 120 h with growth medium change every 24 h. After isolation of the RNA (RNeasy Mini kit, Qiagen) and reverse transcription (RevertAid reverse transcriptase, Thermo Fisher Scientific), the expression values of the selected genes were measured with qRT-PCR (Universal Probe Library, Roche). ACTB was used as a houskeeping gene.
Computational analysis
Processing of MeDIP-seq
MeDIP paired-end reads were aligned using bwa Version 0.7.12-r1044 (22). In order to remove sequencing reads that originated from mouse DNA fragments, MeDIP reads from both PDX and human tissue samples were aligned to the mouse/mm10 reference sequence first. Only read pairs that, according to the aligner, did not align properly to the mouse reference were aligned to the human reference GrCh37/hg19, and processed in R 3.2.0 with ‘QSEA’. According to the average fragment length, the size of the genome-wide windows was set to 250 bases. CNVs were calculated from input and MeDIP reads based on 1 megabase windows. CpG enrichment function was calibrated in three different ways: (i) ‘BS calibration’, based on Methyl-Seq methylation values from regions with at least 70% methylation in at least half of the samples; (ii) ‘TCGA calibration’, based on mean Illumina 450k methylation values from TCGA LUSC and LUAD cohorts (n = 172) (28,29), for regions with mean methylation >90% and variance <0.05 and (iii) ‘Blind calibration’, based on the assumption, that average methylation level of CpG depleted regions is 80%, and decreases linearly with CpG density to 25% at regions with 15 CpGs per fragment. These averages match our observations for the analyzed samples (Supplementary Data). The two alternative methods, BayMeth and MeSiC were applied with default parameters, following the authors’ instructions on the project web pages. In order to minimize the effect of read counting, CNV inference and CpG density estimation, these steps have been conducted in QSEA for BayMeth as well. Parameters for BayMeth empirical Bayes function were method = ‘DBD’, mode = ‘fixedWeights’, weights = c(0.1,0.8,0.1). For MeSiC the preprocessing scripts provided by the authors were used for read counting. On the web-page, all available sequence features were selected, and the algorithm was set to Random Forest Regression. Resulting base-specific methylation estimates were averaged in genomic windows in order to compare the results with the window based methods. The sequence files of IMR-90 dataset were downloaded from SRA (accession numbers SRR513111 and SRR513112 for IMR-90 MBD seq; SRR068932 and SRR068933 for SssI treated MBD seq) and aligned to reference GrCh37/hg19 using bwa 0.7.12-r1044. Processed IMR-90 450k Illumina human methylation files were downloaded from GEO, accession GSM1314099, and filtered for detection P-values <0.01.
Processing of Methyl-Seq
Adapter sequences in paired-end Methyl-Seq reads were trimmed using trim_galore version 0.4.0 and then aligned using bismark v0.10.0 (23) based on bowtie2 version 2.2.1 (24) with default parameters. Corresponding to the MeDIP alignment strategy, Methyl-Seq reads were aligned to the mouse/mm10 reference first. Read pairs, not properly aligned to the mouse genome according to the aligner have been aligned to the human reference GrCh37/hg19. Using bismark_methylation_extractor, the methylation levels were called at all covered CpG sites. For subsequent analysis, only regions covered by 20 or more reads were considered. Methyl-Seq methylation levels were averaged in 250 base windows in order to compare Methyl-Seq to MeDIP.
Processing of RNA-seq
By analogy with MeDIP and Methyl-Seq analysis, RNA-seq paired-end reads were first aligned to the mouse/mm10, and remaining reads to the human reference GrCh37/hg19 using rna-star alignment tool version 2.4.1.d (25). For both references, we provided the RefSeq gene annotation file to facilitate mapping of reads spanning exon-exon junctions. Reads were counted per gene, using htseq-count version 0.6.1p1 in union mode (26). For normalization and detection of differentially expressed genes, we used the Bioconductor package DESeq2 (27). In general, genes with more than 1 FPKM were considered expressed. For the analysis of variable genes, we applied variance stabilizing transformation of the DESeq2 package in order to normalize for the dependency of the variance to the mean expression.
RESULTS
Workflow for Quantitative Sequencing Enrichment Analysis
The QSEA workflow comprises the following steps (Figure 1):
Import of alignment results and counting of fragments along genomic windows,
Normalization for copy number variation,
Normalization for sequencing depth and library composition,
Transformation and quantification of enrichment signals,
Computation of differentially methylated regions,
Annotation of DMRs.
Figure 1.

Overview of the QSEA workflow. Green boxes represent data input, functions implemented in QSEA are depicted in blue, and red boxes describe the respective analysis step performed within these functions.
Details for each step are given in Supplementary Data.
Modeling enrichment profiles enables transformation of MeDIP-seq read densities to absolute methylation levels
Comparing MeDIP-seq read densities with absolute methylation levels derived from BS sequencing on the same samples reveals particular characteristics. The MeDIP enrichment signal is dependent on the number of methylated cytosines within the fragment, which is limited by the number of CpGs. By extracting genomic windows with similar CpG densities, we observe a linear relation between absolute methylation (β) and mean normalized sequence read coverage (Figure 2A). On the other hand, for a fixed level of absolute methylation, we observe an increase of MeDIP enrichment from lower to medium CpG density that becomes saturated at higher levels of CpG density (Figure 2B). We further observe that regions lacking DNA methylation as well as regions lacking CpG dinucleotides are covered by an offset of reads. These ‘background reads’ represent the noise of the experiment. Especially at regions with low CpG density and regions with low methylation levels, these background reads lead to distortion of the signals.
Figure 2.

Modelling MeDIP enrichment. (A) MeDIP read density (depicted exemplary for normal sample from patient 3) has linear relation to methylation level, and (B) gets saturated for high CpG density. (C) CpG dependent enrichment profile plot shows heat-color coded density of MeDIP reads by CpG density, the mean MeDIP coverage (red line), the observed MeDIP coverage at fully methylated regions (black line), and the sigmoidal function fitted to the coverage of fully methylated regions (dashed green line). (D) Exemplary illustration of methylation estimates depending on MeDIP read density, assuming four background reads and 25 reads at fully methylated windows.
Based on these observations, we model the number of reads, y, with a Poisson distribution with mean parameter λ linear in the methylation level β:
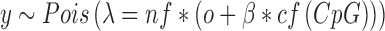 |
The offset o is the expected read density without enrichment (‘background reads’), which corresponds to the experimental noise. The enrichment signal is the absolute methylation level β multiplied by the CpG-dependent enrichment function cf(CpG). This function describes the sample specific dependency of the MeDIP enrichment and the CpG density, and can be interpreted as the expected enrichment if the region were fully methylated. Both the enrichment signal and the noise are scaled by the sample- and region-specific normalization factor (nf) which accounts for library size and composition as well as potential CNV influences (Supplementary Data).
To estimate the sample specific enrichment profiles cf(CpG), we rely on knowledge about the methylation status for a set of genomic regions, for example, derived from targeted BS sequencing. As highly methylated regions have the best signal to noise ratio, these regions are most suitable for calibration. Further selected regions should span a broad range of CpG densities, in order to cover the genomic spectrum. In the following, we use three strategies for conducting calibration of model parameters.
-
‘BS calibration’: This strategy works with additional calibration experiments. For the studied samples, we selected between 146 455 and 184 099 genomic windows that are at least 50% methylated in the corresponding Methyl-Seq experiment, and at least 70% methylated in at least half of the samples. To estimate the expected enrichment of fully methylated regions, the observed read densities of selected regions are scaled according to the observed BS methylation levels in these regions. These estimates are grouped into bins of similar CpG density and averaged. We deduce sample-wise enrichment profiles by scaling and shifting the sigmoidal function
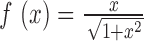 to these averages. This function is capable of describing the observed saturation of enrichment regarding CpG density levels, and fits the observed enrichment profile (Figure 2C).
to these averages. This function is capable of describing the observed saturation of enrichment regarding CpG density levels, and fits the observed enrichment profile (Figure 2C).In addition to ‘BS calibration’ we explored two approaches that do not require additional experiments and thus preserve the cost advantage of MeDIP-seq over whole-genome bisulfite experiments.
‘TCGA calibration’: Here, we rely on publicly available data for comparable samples. We used methylation values from microarray measurements of 54 adenocarcinoma samples and 32 adjacent normal tissue samples (28), as well as 49 squamous cell lung cancer samples and 37 adjacent normal tissue samples (29) published by the TCGA consortium. From these cohorts we identified 18 587 genomic windows with average methylation levels >0.9 over all samples, covering the full range of CpG density. These regions have low methylation variability over a large set of samples and are used to calibrate the MeDIP enrichment profiles.
‘Blind calibration’: This approach is based on the inverse relationship between methylation and CpG density in vertebrate methylomes. Commonly, regions with low CpG density are highly methylated whereas methylation decreases with higher CpG density levels (30). Accordingly, we assume that regions with low CpG density are 80% methylated on average and that with increasing CpG density, methylation decreases linearly to 25% for the mean CpG density of CpG islands (CGIs). This assumption provides a rough estimate for the average methylation levels of windows in this range of CpG density that is used analogously to the previous calibration strategies.
The Poisson model describes the distribution of the read coverage y in genomic regions where methylation levels β are known. In order to estimate methylation levels β given the read coverage y, we apply Bayes’ theorem using an uninformative, uniform prior and derive a Bayesian posterior distribution for the methylation level β, given the number of reads y (Supplementary Data).
Approximating the quantiles of the posterior distributions with binary search allows the calculation of credibility intervals for the estimates. Figure 2D shows an example of estimated methylation and credibility intervals dependent on MeDIP coverage at a hypothetical genomic window that, when completely unmethylated, would be covered by four reads, and when fully methylated, by 25 reads on average.
QSEA accurately quantifies methylation and improves over existing methods
We compared the accuracy of QSEA methylation estimates with BayMeth and MeSiC using two experimental data sets. The first dataset (in vitro) consists of an enrichment-based methylation assay data (MBD-seq) of IMR-90 cells, and Illumina 450k HumanMethylation array data from the same cell line (18). The samples of the second data set (in vivo) are derived from tumours from five human non-small cell lung cancer (NSCLC) patients, that had been transplanted after surgery onto xenograft mice (patient derived xenografts, PDXs), as well as normal lung tissue adjacent to the tumours. We generated genome-wide methylation experiments for all samples using MeDIP-seq, as well as targeted BS sequencing, using Methyl-Seq. Additionally, within the MeDIP protocol, parts of the sequencing libraries have been sequenced prior to MeDIP enrichment at low coverage (input sequencing) in order to estimate CNV levels of the PDX samples.
For the IMR-90 dataset, the enrichment profile was calibrated by QSEA following two of the strategies described in the previous section: first calibration was based on the ‘true’ methylation information obtained from 450k HumanMethylation arrays (‘BS calibration’), and second calibration was based on the inverse relation of CpG density and methylation (‘blind calibration’). BayMeth was run in two calibration modes as well: the first used the SssI treated IMR-90 control sample (‘SssI calibration’), the second did not use the additional experiment (‘blind calibration’). For the calibration of MeSiC, we selected all available sequence features. To compare MeSiC methylation estimates, which are reported at CpG resolution, with the two window-based approaches, BayMeth and QSEA, we averaged MeSiC methylation values within the windows. We quantified the accuracy of the different methylation estimates resulting from the three different methods by calculating the Spearman correlation coefficient with 450k HumanMethylation values.
Spearman correlations of QSEA methylation estimates with 450k are high for both ‘BS calibration’ (0.819) and ‘blind calibration’ (0.805). BayMeth results in a correlation of 0.786 with ‘blind calibration’ and 0.655 with ‘SssI calibration’. Methylation estimates of the MeSiC RFR model compared to 450k results in a correlation of 0.594 (Figure 3A).
Figure 3.

Benchmark and comparison to alternative methods. (A) Spearman correlation of QSEA, BayMeth and MeSiC estimates vs BS-seq for IMR-90 cell line. (B) High density scatterplots of MeDIP methylation estimates and 450k methylation levels for IMR 90 cell line. (C) Spearman correlation of QSEA, BayMeth and MeSiC estimates vs Methyl-Seq for chr1 of PDX samples. (D) Clustering of methylation differences between PDX and normal tissue for QSEA estimates.
Particularly at lower to medium methylation levels, QSEA benefits from explicitly modeling background reads: while BayMeth tends to overestimate methylation for these regions, QSEA shows less deviation from 450k values (Figure 3B). Taken together, we see that without SssI calibration, QSEA and BayMeth perform comparably well on the IMR-90 benchmark. With SssI calibration, Baymeth overestimates intermediate methylation levels.
Next, we compared the performance of QSEA with MeSiC and BayMeth using the lung cancer PDX dataset. In addition to allowing the assessment of the accuracy of the methylation estimates, this dataset provides the opportunity to analyze how well the methods correctly quantify methylation differences between pairs of samples which is essential for comparative methylation analysis and practical applicability to patient cohorts.
For QSEA, we used all three different strategies for enrichment estimation described in the previous section: (i) enrichment estimation based on targeted BS sequencing (‘BS calibration’), (ii) enrichment estimation based on invariable methylated regions in TCGA LUAD and LUSC cohorts (‘TCGA calibration’) and (iii) enrichment estimation based on the inverse correlation of CpG density and methylation (‘blind calibration’). Since no corresponding SssI experiments for these samples are available, we applied BayMeth in ‘blind calibration’ mode only. For the MeSiC RFR model, we used all available sequence features.
In line with the results from the IMR 90 dataset, QSEA performs comparably well for all three calibration configurations, as expected for the similar enrichment profiles, resulting in Spearman correlation coefficients between 0.75 and 0.84 for all patients. Correlation for BayMeth is 0.64 on average and 0.38 for MeSiC (Figure 3C). Again, especially for regions with low to medium levels of methylation QSEA estimates are less biased compared to BayMeth and MeSiC (Supplementary Data).
In order to analyze the ability of the different methods to capture individual differences between tumour and normal tissues, we calculated Spearman correlation coefficients between MeDIP-seq and BS-seq tumour-normal methylation differences for each patient. On average this correlation is 0.71 for ‘blind calibration’ and 0.73 for ‘TCGA calibration’ and ‘BS calibration’ modes, 0.44 for BayMeth ‘blind calibration’, and 0.02 for MeSiC. For comparison, the pairwise correlation between the BS tumour-normal differences of different patients is 0.51 on average. Based on this correlation analysis, we performed hierarchical clustering. For all calibration modes, QSEA estimates tightly cluster with the corresponding BS values (Figure 3D), while for the other methods the sample relationships can not be recovered (Supplementary Data). This implies that the differences between BS sequencing and the QSEA MeDIP estimates are minor compared to the differences between the tumour patients and, thus, that QSEA can be used to infer cross-sample methylation markers from patient cohorts.
Thus, the comparison shows that QSEA can reliably estimate methylation levels, without the need for additional experiments. For the following sections, we thus use QSEA with ‘TCGA calibration’ (option 2 above). Since this calibration mode is completely independent from the Methyl-Seq experiments, the Methyl-Seq β values can be used as a validation dataset in the following.
Differentially methylated regions computed with QSEA are supported by bisulfite sequencing and the literature
We further explored the performance of QSEA with respect to the detection of DMRs between PDX and normal tissues using the five patients as replicates. This comparison yields 105,426 genome-wide DMRs with an FDR <0.01, of which 11 098 are hypermethylated and 94 328 are hypomethylated in the tumours. Of these DMRs, 62.7% are located in intergenic regions, 33.4% in introns, 6.1% in promoter regions and 3.6% in exons.
QSEA found DMRs within CGI promoters of 1556 different genes, of which 1306 were hypermethylated, and 250 were hypomethylated (Supplementary Data). Confirming the results from the previous sections, for these regions we also observed a very good correlation (0.87) between QSEA methylation estimates and BS methylation values, emphasizing the high reliability of the method (Figure 4A). In total, 81 CGI promoter DMRs were not directly covered by Methyl-Seq probes, but could be approximated from probes in neighbouring regions. Another 63 CGI promoter DMRs were solely identified by the genome-wide MeDIP approach, without any neighbouring Methyl-Seq probes (visible as red points on the horizontal axis in Figure 4A). Even though probes for targeted approaches are designed to cover CGIs, the genome-wide MeDIP approach is more exhaustive for these regions: while 99.9% of regions overlapping CGIs are sufficiently enriched in all MeDIP experiments (>3 reads expected enrichment), these numbers drop significantly with targeted BS-methods where 74.2% of the CGIs are covered by Methyl-Seq probes and 43% with HumanMethylation 450k arrays (Supplementary Data).
Figure 4.

Validation and functional interpretation of differentially methylated Regions: (A) Scatterplot of methylation difference from QSEA methylation estimate and Methyl-Seq of CpG island promoter DMRs. Blue: directly covered by Methyl-Seq. Orange: neighbourhood covered by Methyl-Seq, red: not covered by Methyl-Seq. (B) Methylation difference of 20 most hypermethylated tumour suppressor genes form QSEA estimates (red) and Methyl-Seq(blue). (C) Histogram of correlation between CpG island promoter methylation and gene expression. (D) Mean methylation difference versus gene expression log2 ratio of differentially expressed genes with differentially methylated CpG island promoter. Red numbers are gene counts per quadrant. (E) Gene expression in four NSCLC cell lines after demethylation validation experiment.
Among genes with hypermethylated CGI promoters, we found 107 known tumour suppressor genes (TSGs), according to the TSGene database (31). Figure 4B shows the 20 TSGs with largest differences in methylation between tumour and normal samples. The literature supports the detected methylation differences: Differential promoter methylation for cysteine dioxygenase 1 (CDO1, mean QSEA methylation level of 4% in normal and 78% in PDX) has already been described as part of a DNA methylation signature to detect minimally and non-invasive lung cancer (32–34). Other prominent methylation markers of NSCLC are, for example, paired box 6 (PAX6, mean QSEA methylation level of 7% in normal and 79% in PDX) whose promoter hypermethylation has been found to be significantly associated with poor overall survival (35) but also genes like CDX2, CEBPA, HOXB13 and SOX11, that are well described epigenetically regulated genes involved in tumourigenesis of several cancers (36–40).
In summary, DMRs identified with QSEA could be validated with BS data and reveal important and well-known markers for NSCLC tumour progression.
QSEA reveals gene regulation by CGI promoter hypermethylation
In order to assess the effects of differential methylation on gene expression regulation, we additionally performed gene expression experiments using RNA-seq of the PDX and normal samples. Out of 1556 genes with promoter DMRs, 757 are expressed (>1 FPKM) in at least two of the analyzed samples. For 330 of these genes, expression and promoter methylation are anti-correlated (Spearman correlation < –0.5), corresponding to the expected regulatory effect of DNA methylation at CGI promoters (Figure 4C). Additionally, from the 757 expressed genes with promoter DMR, we identified 300 to be differentially expressed between PDX and normal tissue. According to the anticorrelation of promoter methylation and expression, 233 of these are either hypermethylated and down-regulated or hypomethylated and up-regulated (Figure 4D).
In order to confirm selectively the causative effect of promoter hypermethylation on repression of gene expression in vitro, three different NSCLC cell lines (H1299, H1650, HCC827) were demethylated by treatment with different concentrations of decitabine, an inhibitor of DNA methyltransferase. We selected seven genes with promoter hypermethylation and accordingly anti-correlated gene expression (<–0.5) and compared gene expression changes relative to untreated control samples. Among the selected genes were well-studied cancer-relevant genes like tumour suppressors (HLF, FOXA2, STAT5A, ALDH1A2), receptor tyrosine kinases (PDGFRA), and potential biomarkers (NKX2-1, CLECL14A), most of them with a distinct role in NSCLC. All down-regulated genes showed increased expression in at least one cell type after reversal of the promoter hypermethylation, suggesting that gene expression of those genes is indeed controlled by promoter methylation (Figure 4E).
QSEA detects functional mechanisms affected by differential methylation
In order to exploit the full potential of the genome-wide methylation information from the MeDIP-seq experiments, we interrogated QSEA for inference of functional mechanisms other than CGI promoter hypermethylation. As mentioned above only 6.1% of the detected DMRs were located in promoter regions and 3.6% in exons, respectively, whereas 33.4% were located in introns and 62.7% in intergenic regions. The function of DNA methylation at those regions is still elusive, and they are covered to a lesser extent by targeted methods such as 450k arrays or Methyl-Seq. To infer functional mechanisms we analyzed the enrichment of genomic features and annotations within the DMRs (Supplementary Data).
As expected, we found strong enrichment of hypermethylation at CGI promoters: from 11 098 hypermethylated regions genome-wide, 2,971 regions are overlapping CGIs at promoters, corresponding to 42.7-fold enrichment. On the other hand, this implies that 73% (i.e. the remaining 8127) of hypermethylated DMRs are outside those well studied regions. Interestingly, we found even stronger enrichment (48-fold) for CGIs that are not in proximity to promotor regions of known genes. These regions may act as enhancer sites, whose functions have been reported to depend on methylation and, in the case of cancer genes, influence tumour properties (41). We therefore analyzed the enrichment of DMRs in 161 transcription factor binding sites, obtained from ENCODE (42).
We found the enrichment of hypermethylated sites highly variable for the binding sites of individual transcription factors. In line with the functional impact of Polycomb Repressive Complex 2 (PRC2) in tumour development, regions targeted by components of this factor are highly enriched for gain of methylation: 54.5% of all hypermethylated regions overlap with PRC2 binding sites (EZH2 and SUZ12), which corresponds to a 102-fold enrichment compared to the genome (Figure 5A).
Figure 5.

Enrichment of differentially methylated regions. (A) Sixteen most enriched transcription factor binding sites for hypermethylated regions and (B) hypomethylated regions respectively. (C) Tumour-Normal methylation differences chr22 show large hypomethylated blocks (LHB). Black dots represent mean methylation difference between PDX and normal, dashed lines are smoothed methylation differences for individual patients. Violet bars indicate two LHB, abundant in most of the tumour samples.
Globally, hypomethylation is predominant in cancer compared to hypermethylation, but less enriched in annotated regions. We found 94 328 regions with loss of methylation, corresponding to 0.82% of the genome. These regions are agglomerated in large hypomethylated blocks (LHB) of 0.1–12 Mb in size (Figure 5C), which has been shown to be a characteristic feature for several types of cancer (43). Interestingly, within the hypomethylated regions, we found less hypomethylated regions in promoter CGIs than expected by chance (one third), but a 2-fold enrichment of CGIs distal to promoters. Again, many of these distal DMRs overlap with enhancers containing particular TFBS. For example, the binding sites of the histone modifiers SMARCC1,SMARCC2 and SMARCB1 show 2- to 3-fold enrichment in loss of methylation, suggesting a role of DNA methylation in chromatin remodeling by the SWI/SNF complex, a mechanism known to be involved in carcinogenesis (44). Further, TFBS enriched for loss of methylation belong to RNA polymerase III (RPC155, GTF3C2, BDP1) and activator proteins AP-1 (FOSL1, FOSL2, JUNB) and AP-2 (TFAP2A, TFAP2C) (Figure 5B).
In general, the fact that DMRs are enriched with specific transcription factor binding sites suggests that DNA methylation dependent mechanisms related to those factors are involved in tumourigenesis. In particular, it is frequently the case that regulatory sites with loss of methylation are not located near promoters of known genes, indicating a role of DNA methylation in the control of transcription of distal genes.
QSEA is fast, flexible and easy to use
The described methods are implemented as an R package, ‘QSEA’, which is available at the Bioconductor repository (20): www.bioconductor.org/packages/qsea.
The complete analysis of 10 human MeDIP-seq samples with low coverage input sequencing took 95 min on a single core computer, and allocated a maximum of 14 GB main memory. A large part of the runtime is required for processing the alignment files: Import of MeDIP-seq alignment files and counting of reads overlapping genome-wide 250 base windows took 37 minutes, and CNV analysis including the import of low coverage input alignment files took 11 min. The analysis of CpG density of the human genome took 21 min. Calculation of the remaining normalization parameters, including calculation of effective library size, estimation of offset reads and analysis of MeDIP enrichment took ∼2 min. The detection of differentially methylated regions took 13 min for fitting the null model and estimating the dispersion for genome-wide windows, and 12 min for fitting a reduced model and testing the contrast. QSEA supports parallel scanning of alignment files on multicore computers, which reduced runtime for this step to 5 min on 10 cores (Supplementary Data).
Importantly, once these computational steps have been performed, QSEA provides functions to retrieve all information for any regions of interest, for example, regions defined by genome annotations or by differential methylation. Normalized values and methylation estimates for those regions are computed on request, instead of storing different values for all genome-wide windows. This approach allows both efficient usage of memory, as well as fast and flexible access to results of interest. For example, it takes obout one minute to compile a table for all 105,426 genome-wide DMRs, containing the raw read counts, normalized coverage, and estimated methylation values including the credibility interval for the estimates and adding comprehensive annotation.
DISCUSSION
We developed a novel analysis workflow, QSEA, for Quantitative Sequencing Enrichment Analysis, in particular for MeDIP-seq experiments. The workflow contains a Bayesian model for estimating absolute levels of methylation from genome-wide enrichment of methylated DNA fragments. This approach is based on a Poisson model that accounts for experimental noise by explicitly modeling background reads. The parameters for the model can be calibrated using data from additional experiments, published data sets or general assumptions. Furthermore, QSEA provides functionality for the estimation of CNVs from sequencing data, and incorporates this information to normalize local read density. For detecting differentially methylated regions we implemented a method based on generalized linear models. A collection of methods and functions for descriptive analysis and depiction of the results complements the software package.
We tested the practical applicability of QSEA for patient studies on MeDIP-seq data from five pairs of lung cancer PDX models and adjacent normal tissue. Using targeted bisulfite sequencing, we showed that the QSEA methylation estimates are highly accurate and, in contrast to previous methods, the performance is not dependent on additional experiments on the same samples. Additionally, the identified differentially methylated regions (DMRs) were confirmed by the literature. By integrating CGI promoter methylation and gene expression, we quantified the functional impact of gene silencing by promoter methylation. We found CGIs and binding sites for members of PRC2 enriched for gain of methylation, and CGIs distant to promoters as well as different specific TFBS enriched for loss of methylation. Overall, we demonstrated a comprehensive methylome analysis of cancer samples from MeDIP-seq experiments.
We further evaluated the importance of additional experiments for the analysis of MeDIP-seq data. Besides bisulfite calibration data, which can be replaced by alternative calibration strategies, sequencing of input libraries is commonly required for normalization of enrichment based sequencing assays. Within the QSEA pipeline, this input sequencing is used for the estimation of CNVs, by comparing the read densities within broad genome-wide windows (typically between 100 kb and 2 Mb). In the absence of input sequencing, QSEA can apply this procedure on methylation enriched sequencing data by considering only read fragments without CpG dinucleotides (typically ∼10% of the reads) This approach enables the user to estimate and incorporate CNV without additional experimental efforts. However, the strategy is only suitable for samples where DNA methylation is occurring exclusively in CpG context.
Targeted approaches based on bisulfite conversion limit the analysis on selected regions, which usually correspond to known functions of the methylome. For our samples, only 39% of hypermethylated windows overlap with Illumina 450k probes, and 78% overlap Agilent Methyl-Seq target regions (Supplementary Data). Thus, only a fraction could have been discovered using these targeted approaches. The full extent of this advantage becomes clear when comparing the coverage of hypomethylated regions: only 2% of hypomethylated windows overlap with Illumina 450k HumanMethylation probes and 5% with Methyl-Seq probes. Thus, MeDIP-seq offers a far more complete picture of the methylome and allows the investigation of yet unexplored functions of DNA methylation.
For computing DMRs we applied generalized linear models, which are a very popular approach for the detection of differential sequencing read counts, originally for RNA-seq. They can account for complex experimental designs and for unwanted influences like batch effects. It would be desirable to be able to test the linear relation of enrichment and numerical factors (for example patient age, response to treatment or other clinical parameters). However, a direct application of numerical factors on the implemented GLM would detect exponential rather than linear relations, due to the logarithmic link function, which is required to match the domains of the linear predictor and the response variable (the read density). An adaption of the approach might provide such functionality.
We also compared the quantified DMRs from our study to published studies which used larger cohorts of patients and microarray technology. Strikingly, a moderate to strong correlation (0.62) of the methylation differences between tumour and normal tissue was observed when compared with the TCGA lung cancer study. This is remarkable since these DMRs seem stably detectable across multiple platforms (MeDIP-seq vs Illumina HumanMethylation 450k array), different cancer models (PDX vs primary tumours) and even different tumour subtypes. (Supplementary Data)
Furthermore, we observed a group of regions that show high beta levels with respect to BS-seq but rather low levels estimated from MeDIP-seq (Supplementary Data). This might in fact be explained by the differences to BS-technology since the antibody used for MeDIP enrichment is specific for 5mC, while bisulfite conversion based methods cannot distinguish 5mC and 5hmC. The observed differences might thus reflect the level of 5hmCs in the samples under analysis.
Although we focused on MeDIP-seq specific functionality for this report, QSEA offers useful functions for the analysis of ChIP-seq as well. The commonly applied peak based approaches are limited, especially for the detection of differentially enriched regions between groups of samples. In addition to the functionality of MEDIPS, which has been applied to H3K4me2 ChIP-seq of blood samples from asthma patients (17), the ability to normalize for the effect of CNV also allows the application of QSEA methods to cancer samples.
In summary, QSEA is a highly reliable, flexible and efficient method to quantify DNA methylation from enrichment based experiments, and to identify aberrant methylation.
ACCESSION NUMBERS
Data for MeDIP sequencing, RNA sequencing and Methyl-Seq have been deposited at the European Genome-phenome Archive (EGA, http://www.ebi.ac.uk/ega/), which is hosted by the EBI, under accession number EGAS00001001822.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jacob Taylor for proofreading the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Federal Ministry of Education and Research under its e:BIO program [0316190 to R.H., 0316065 E to M.R.S.]; European Commission under its 7th Framework program [602156 to R.H.]; Lichtenberg program of VolkswagenStiftung (M.R.S.); IMPRS-CBSC of the Max-Planck-Society (to M.L.). Funding for open access charge: European Commission.
Conflict of interest statement. Jana Rolff, Michael Becker and Iduna Fichtner are employees of ‘Experimental Pharmacology & Oncology Berlin-Buch GmbH’, a company that provides service in preclinical research and testing of new cancer drugs.
REFERENCES
- 1. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002; 16:6–21. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D., Weinberg R.A.. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 3. Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002; 21:5427–5440. [DOI] [PubMed] [Google Scholar]
- 4. deVos T., Tetzner R., Model F., Weiss G., Schuster M., Distler J., Steiger K.V., Grutzmann R., Pilarsky C., Habermann J.K. et al. . Circulating methylated SEPT9 DNA in plasma is a biomarker for colorectal cancer. Clin. Chem. 2009; 55:1337–1346. [DOI] [PubMed] [Google Scholar]
- 5. Heyn H., Esteller M.. DNA methylation profiling in the clinic: applications and challenges. Nat. Rev. Genet. 2012; 13:679–692. [DOI] [PubMed] [Google Scholar]
- 6. Lister R., O'Malley R.C., Tonti-Filippini J., Gregory B.D., Berry C.C., Millar A.H., Ecker J.R.. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008; 133:523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cokus S.J., Feng S., Zhang X., Chen Z., Merriman B., Haudenschild C.D., Pradhan S., Nelson S.F., Pellegrini M., Jacobsen S.E.. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008; 452:215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frommer M., McDonald L.E., Millar D.S., Collis C.M., Watt F., Grigg G.W., Molloy P.L., Paul C.L.. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deng J., Shoemaker R., Xie B., Gore A., LeProust E.M., Antosiewicz-Bourget J., Egli D., Maherali N., Park I.H., Yu J. et al. . Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009; 27:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ivanov M., Kals M., Kacevska M., Metspalu A., Ingelman-Sundberg M., Milani L.. In-solution hybrid capture of bisulfite-converted DNA for targeted bisulfite sequencing of 174 ADME genes. Nucleic Acids Res. 2013; 41:e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meissner A., Gnirke A., Bell G.W., Ramsahoye B., Lander E.S., Jaenisch R.. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005; 33:5868–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weber M., Davies J.J., Wittig D., Oakeley E.J., Haase M., Lam W.L., Schubeler D.. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005; 37:853–862. [DOI] [PubMed] [Google Scholar]
- 13. Serre D., Lee B.H., Ting A.H.. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010; 38:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chavez L., Jozefczuk J., Grimm C., Dietrich J., Timmermann B., Lehrach H., Herwig R., Adjaye J.. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res. 2010; 20:1441–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grimm C., Chavez L., Vilardell M., Farrall A.L., Tierling S., Bohm J.W., Grote P., Lienhard M., Dietrich J., Timmermann B. et al. . DNA-methylome analysis of mouse intestinal adenoma identifies a tumour-specific signature that is partly conserved in human colon cancer. PLoS Genet. 2013; 9:e1003250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Etchegaray J.P., Chavez L., Huang Y., Ross K.N., Choi J., Martinez-Pastor B., Walsh R.M., Sommer C.A., Lienhard M., Gladden A. et al. . The histone deacetylase SIRT6 controls embryonic stem cell fate via TET-mediated production of 5-hydroxymethylcytosine. Nat. Cell Biol. 2015; 17:545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seumois G., Chavez L., Gerasimova A., Lienhard M., Omran N., Kalinke L., Vedanayagam M., Ganesan A.P., Chawla A., Djukanovic R. et al. . Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nat. Immunol. 2014; 15:777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Riebler A., Menigatti M., Song J.Z., Statham A.L., Stirzaker C., Mahmud N., Mein C.A., Clark S.J., Robinson M.D.. BayMeth: improved DNA methylation quantification for affinity capture sequencing data using a flexible Bayesian approach. Genome Biol. 2014; 15:R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiao Y., Yu F., Pang L., Zhao H., Liu L., Zhang G., Liu T., Zhang H., Fan H., Zhang Y. et al. . MeSiC: a model-based method for estimating 5 mC levels at single-CpG resolution from MeDIP-seq. Sci. Rep. 2015; 5:14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huber W., Carey V.J., Gentleman R., Anders S.. Orchestrating high-throughput genomic analysis with bioconductor. 2015; 12:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Workman P., Aboagye E.O., Balkwill F., Balmain A., Bruder G., Chaplin D.J., Double J.A., Everitt J., Farningham D.A., Glennie M.J. et al. . Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer. 2010; 102:1555–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H., Durbin R.. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krueger F., Andrews S.R.. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011; 27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anders S., Pyl P.T., Huber W.. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deaton A.M., Bird A.. CpG islands and the regulation of transcription. Genes Dev. 2011; 25:1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao M., Kim P., Mitra R., Zhao J., Zhao Z.. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016; 44:D1023–D1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kwon Y.J., Lee S.J., Koh J.S., Kim S.H., Lee H.W., Kang M.C., Bae J.B., Kim Y.J., Park J.H.. Genome-wide analysis of DNA methylation and the gene expression change in lung cancer. J. Thorac. Oncol. 2012; 7:20–33. [DOI] [PubMed] [Google Scholar]
- 33. Wrangle J., Machida E.O., Danilova L., Hulbert A., Franco N., Zhang W., Glockner S.C., Tessema M., Van Neste L., Easwaran H. et al. . Functional identification of cancer-specific methylation of CDO1, HOXA9, and TAC1 for the diagnosis of lung cancer. Clin. Cancer Res. 2014; 20:1856–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Diaz-Lagares A., Mendez-Gonzalez J., Hervas D., Saigi M., Pajares M.J., Garcia D., Crujeiras A.B., Pio R., Montuenga L.M., Zulueta J. et al. . A novel epigenetic signature for early diagnosis in lung cancer. Clin. Cancer Res. 2016; 22:3361–3371. [DOI] [PubMed] [Google Scholar]
- 35. Zhang X., Yang X., Wang J., Liang T., Gu Y., Yang D.. Down-regulation of PAX6 by promoter methylation is associated with poor prognosis in non small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015; 8:11452–11457. [PMC free article] [PubMed] [Google Scholar]
- 36. Liu X., Zhang X., Zhan Q., Brock M.V., Herman J.G., Guo M.. CDX2 serves as a Wnt signaling inhibitor and is frequently methylated in lung cancer. Cancer Biol. Ther. 2012; 13:1152–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cantu L., Corti M., Sonnino S., Tettamanti G.. Evidence for spontaneous segregation phenomena in mixed micelles of gangliosides. Chem. Phys. Lipids. 1990; 55:223–229. [DOI] [PubMed] [Google Scholar]
- 38. Lin T.C., Hou H.A., Chou W.C., Ou D.L., Yu S.L., Tien H.F., Lin L.I.. CEBPA methylation as a prognostic biomarker in patients with de novo acute myeloid leukemia. Leukemia. 2011; 25:32–40. [DOI] [PubMed] [Google Scholar]
- 39. Ghoshal K., Motiwala T., Claus R., Yan P., Kutay H., Datta J., Majumder S., Bai S., Majumder A., Huang T. et al. . HOXB13, a target of DNMT3B, is methylated at an upstream CpG island, and functions as a tumor suppressor in primary colorectal tumors. PLoS One. 2010; 5:e10338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40. Sernbo S., Gustavsson E., Brennan D.J., Gallagher W.M., Rexhepaj E., Rydnert F., Jirstrom K., Borrebaeck C.A., Ek S.. The tumour suppressor SOX11 is associated with improved survival among high grade epithelial ovarian cancers and is regulated by reversible promoter methylation. BMC Cancer. 2011; 11:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aran D., Sabato S., Hellman A.. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013; 14:R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.K., Cheng C., Mu X.J., Khurana E., Rozowsky J., Alexander R. et al. . Architecture of the human regulatory network derived from ENCODE data. Nature. 2012; 489:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Timp W., Bravo H.C., McDonald O.G., Goggins M., Umbricht C., Zeiger M., Feinberg A.P., Irizarry R.A.. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014; 6:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roberts C.W., Orkin S.H.. The SWI/SNF complex–chromatin and cancer. Nat. Rev. Cancer. 2004; 4:133–142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.