


Abstract
For reasons that remain unknown, the Plasmodium falciparum genome has an exceptionally high AT content compared to other Plasmodium species and eukaryotes in general - nearly 80% in coding regions and approaching 90% in non-coding regions. Here, we examine how this phenomenon relates to genome-wide patterns of de novo mutation. Mutation accumulation experiments were performed by sequential cloning of six P. falciparum isolates growing in human erythrocytes in vitro for 4 years, with 279 clones sampled for whole genome sequencing at different time points. Genome sequence analysis of these samples revealed a significant excess of G:C to A:T transitions compared to other types of nucleotide substitution, which would naturally cause AT content to equilibrate close to the level seen across the P. falciparum reference genome (80.6% AT). These data also uncover an extremely high rate of small indel mutation relative to other species, primarily associated with repetitive AT-rich sequences, in addition to larger-scale structural rearrangements focused in antigen-coding var genes. In conclusion, high AT content in P. falciparum is driven by a systematic mutational bias and ultimately leads to an unusual level of microstructural plasticity, raising the question of whether this contributes to adaptive evolution.
INTRODUCTION
The malaria parasite Plasmodium falciparum afflicts hundreds of millions of people worldwide and kills half a million African children each year. One of the major obstacles to controlling malaria is the parasite's prolific capacity for evolutionary adaptation. By a continual process of mutation and recombination, the parasite is able to generate very large numbers of new antigenic variants to evade the human immune system, thus complicating vaccine development, and to develop resistance to all drugs that are widely deployed as frontline antimalarial therapy. The most recent example of this phenomenon is the epidemic of artemisinin resistance that is spreading through Southeast Asia and threatening to destabilise malaria control worldwide (1).
Despite a large scientific literature on genetic diversity in P. falciparum, there remain important gaps in our understanding of the processes of mutation and recombination that underlie the parasite's ability to adapt rapidly to new evolutionary pressures. A question of fundamental interest is the role played by ongoing mutational processes in shaping the parasite's unusual genome architecture, particularly its exceptionally high AT content (80.6% AT in the 3D7 isolate), one of the most skewed base pair compositions of any eukaryote (2,3), and its expanses of repetitive, low-complexity regions, which are widely dispersed throughout the genome (4,5). For comparison, the AT contents of Homo sapiens, Drosophila melanogaster, Arabidopsis thaliana and the eukaryotic pathogens Toxoplasma gondii and Trypanosoma bruceii are 58.9%, 57.9%, 63.4%, 47.7% and 53.2%, respectively [NCBI Genome database: http://www.ncbi.nlm.nih.gov/genome/] (Supplementary Table S1). The Plasmodium genus as a whole exhibits high AT content, with the highest levels seen in P. falciparum and the related Laverania parasites P. reichenowi and P. gaboni, while P. vivax has a more even genome composition at 59.4% (Figure 1). The biological causes of these differences are currently unknown.
Figure 1.

AT content variation in Apicomplexan parasites. Phylogenetic relationships and AT:GC genome proportions are shown for a range of Apicomplexan parasite species. Phylogenetic lines are not to evolutionary scale. AT contents are derived from NCBI genome browser data (http://www.ncbi.nlm.nih.gov/genome/) and shown in Supplementary Table S1. Phylogeny of Plasmodium parasites is based on (42), Figure 1a. Phylogeny of the non-Plasmodium parasites Theileria parva, Babesia microti and Toxoplasma gondii is based on (44).
We investigated the mutational origins and consequences of high AT content using an experimental evolution approach in which P. falciparum parasites were cultured in vitro for hundreds of erythrocytic life cycles and de novo mutations were detected by whole genome sequencing. Mutation accumulation experiments, or ‘clone trees’, have previously been reported in P. falciparum using established long-term culture-adapted laboratory isolates (6,7). Here, we have enlarged the scale of these experiments by analysing six clone trees, including recently culture-adapted field isolates from Western Cambodia, producing the largest dataset of de novo mutation in P. falciparum published to date. This allowed us to calculate mutation rates for each type of base pair substitution (BPS, the mutation origin of single nucleotide polymorphisms (SNP) detected in population genetic data), and so quantify how ongoing mutational biases contribute to maintaining P. falciparum's skewed base pair composition. We then analysed what impact the parasite's AT-rich, repetitive DNA sequences has on the rate of spontaneous insertion/deletion (indel) mutations, demonstrating a prolifically high indel rate, over ten-fold higher than the BPS rate. We discuss the potential causes and biological significance of this BPS bias, AT-richness and high indel mutation rate.
MATERIALS AND METHODS
Parasite in vitro culture and clone tree generation
To analyse patterns of de novo mutation, we generated six mutation accumulation lines using the same approach previously reported (6). Briefly, we cultured parasites continuously in vitro with periodic subcloning to isolate single infected red blood cells, which were asexually expanded to provide DNA for whole genome Illumina sequencing. After 1–4 months of continuous culture, we selected one of these subclones for another round of cloning by limiting dilution. We refer to each of these cycles as clone tree ‘generations’ (Figure 2A). One advantage of this approach is that it reduces the impact of selection, thus approximating the molecular mutation rate (8).
Figure 2.

Detecting de novo mutations in mitotic ‘clone trees’. (A) Schematic of the KH1-01 clone tree. Each branch point represents a round of subcloning by limiting dilution, isolating single infected red blood cells, and asexually expanding them until we obtained sufficient amounts of DNA for whole-genome sequencing. The KH1-01 clone tree was cultured for ∼17 months and contained 59 subclone genomes derived from the parent isolate (obtained soon after venous sampling from a patient in Cambodia). Blue = de novo BPS detected; red = pairs of recombining var genes detected; yellow = both BPS and var gene recombination detected; asterisks = clonal dilutions performed. (B) Overall, in the clone trees of six wild-type P. falciparum isolates (3D7, HB3, Dd2, W2, KH1-01 and KH2-01), we identified 85 de novo BPS, 78 of which have known genomic coordinates (shown here). The 78 BPS with known genomic coordinates were distributed across the 14 nuclear chromosomes as expected by chance (P = 0.445, Fisher's exact test). Colors depict the isolate in which the BPS occurred. (C) We identified 164 de novo indels in the 3D7 clone tree, all of which have known genomic coordinates (shown here). Like BPS, indel distribution between the 14 chromosomes did not differ significantly from what is expected by chance (P = 0.194, Pearson's Chi-squared test). Insertions (n = 108) and deletions (n = 56) are colored blue and red, respectively.
All parasites were cultured in human O+ erythrocytes with heat-inactivated 10% pooled human serum as described in (9). Clone tree production in laboratory isolates (3D7, HB3, Dd2, W2) has been described previously, and the genomes from these clone trees were published in (6). We used the same method to produce clone trees from recently culture-adapted Cambodian field isolates, referred to as ‘KH1-01’ and ‘KH2-01’. Parasites were ultra-diluted into a 96-well plate, to reach a theoretical concentration of 0.2–0.5 parasites per well. We identified positive wells microscopically and further cultured parasites in a 10-ml flask, before extracting DNA and producing glycerolyte frozen stocks. One subclone was arbitrarily selected for the next round of clonal dilution to produce the next clone tree ‘generation’ (Figure 2 and Supplementary Figure S1). Median culture time between clonal dilution rounds was 49.5 days (range: 29–165; interquartile range (IQR): 28). We submitted ∼1–2 ug DNA for PCR-free library preparation by the Wellcome Trust Sanger Institute core sequencing teams for paired-end sequencing on the Illumina HiSeq 2000 platform. Conditions were as described in (10), except without the initial PCR amplification step. We selected DNA fragments with an insert size of ∼400 bp, producing paired-end reads of 100 bp each.
KH1-01 and KH2-01 parasites from Western Cambodia were frozen soon after patients’ sampling. KH2-01 had prolonged in vivo clearance following artesunate therapy (half-life: 10.08 hours), while KH1-01 had rapid clearance typical of artemisinin-sensitive parasites (half-life: 3.51 h). The isolates cluster within the KH1 and KH2 subpopulations defined by Miotto et al. (11), and KH2-01 carries the kelch13 C580Y mutation associated with artemisinin resistance, while KH1-01 is kelch13 wild-type. KH2-01 also possessed several of the highly differentiated SNPs in DNA repair genes identified by Miotto et al. (11), including P203S in mlh1, S506I in pms1, and C2010S in rad50.
Genome mapping
We processed all samples through the MalariaGEN pipeline, as described (6,12). Briefly, we mapped BAM files to the P. falciparum 3D7 reference genome (version 3) using BWA with default parameters. For the discovery of translocations within KH1-01 and KH2-01 var genes, we also mapped sequences to an assembled library of var gene sequences from KH1-01 and KH2-01 (ENA Accession numbers are in Supplementary Table S2).
Since we had no reference for the Cambodian isolates and we mapped their sequences to the 3D7 genome, we note that there may be a greater proportion of false negatives (i.e. missed mutations) in the Cambodian parasites. However, this is unlikely to dramatically affect the mutation rate, as the proportion of the 3D7 reference genome with ≥5× coverage was only slightly lower in the KH1-01 and KH2-01 subclone genomes (median 95.6%, IQR 4.7% versus 94.7%, IQR 2.1%, respectively) compared with the 3D7 subclone genomes (median 99.4%, IQR 2.5%) (difference between KH1-01 v 3D7 and KH2-01 v 3D7 both statistically significant: P = 8.96 × 10−9 and P = 1.94 × 10−6, respectively, two-sample Wilcoxon rank sum tests (discussed further in Supplementary Appendix S1)).
Base pair substitution calling
We used SAMtools mpileup followed by BCFtools (13) to detect BPS, exactly as described previously (6), and confirmed all variants by manual inspection on LookSeq (14). As expected, larger clone trees (with more subclone genomes analysed and longer in vitro culture times) yielded more de novo BPS than smaller ones (Supplementary Figure S2).
Indel calling
We used Genome Analysis Toolkit (GATK) software (version 3.3.0) (15) to call indels, as in (16). Using a known set of indels from the progeny of genetic crosses (3D7xHB3, Dd2xHB3, and 7G8xGB4) (16), we realigned each 3D7 BAM separately using options ‘RealignTargetCreator’ followed by ‘IndelRealigner.’ We then used the ‘UnifiedGenotyper,’ with ploidy = 1, to call indels in all re-aligned BAM files simultaneously. We used ‘VQSR’ to recalibrate indels and generate a VQSlod score (log odds ratio of being a true variant versus being false) for each indel. A list of indel hits found solely in a progeny sample and not its parent was generated and low-quality hits were filtered out if they failed to pass any of following parameters: Read counts ≥6, Genotype Quality ≥50, Phred-scale Likelihood ≥100, (Alternate reads/total reads) >0.6, VQSLOD >0. We visually inspected all 180 hits on LookSeq (14), and manually filtered out 16 hits (8.9%), as they could be sequencing/mapping errors (Supplementary Figure S3).
Structural variant calling
We called structural variants in var genes using DELLY (17) with default parameters as described (6). We used reads with one end mapped to a chromosome and the second end mapped to another to identify translocations, with a minimum cut-off of five reads. As with BPS, only structural variants found uniquely in progeny VCF files and not in the parent progressed to further analysis, thus identifying only de novo mutations. We manually inspected all resulting hits on LookSeq (14). We mapped reads from each sample with BWA to separate var gene sequences, i.e. each var sequence was considered its own ‘chromosome.’
Variant validation
The BPS validation process we used has been previously reported (6). Briefly, a 150–800 bp fragment containing the putative mutation is PCR amplified and capillary sequenced from wild-type controls and the sample containing the putative mutation, with sequences aligned and visualized using ClustalOmega and ClustalX. Primers were designed using Primer3 software (all primer sequences in Supplementary Table S11). We attempted to validate 17 indels from the 3D7 Illumina data using a similar approach, but only eight of these produced interpretable capillary sequence data (Supplementary Table S10) owing to the indels occurring in regions that were very difficult to PCR amplify (highly repetitive and AT rich). Eight out of eight indels called by Illumina were supported by the capillary sequence data.
Chimeric var genes identified by DELLY were also validated through PCR as described in (6), by designing primers that bridged the putative translocation sites and only amplified product in samples possessing that translocation, or amplified products of different sizes in wild-type and mutant parasites. In addition to the validations presented in (6), we used PCR amplification and capillary sequencing to validate the chimeric CARvar15-76 translocation in subclone KH2_2s from the KH2-01 clone tree.
Calculating the BPS, indel and var exon 1 recombination rates
We calculated mutation rates using the same methodology as in (6). We calculated separate mutation rates (μ1) per erythrocytic life cycle (ELC) for each clone tree generation using the number of de novo mutations (s) identified per subclone genome analysed within that generation (c), divided by the number of erythrocyte life cycles (L) since the previous clonal dilution (Equation 1). To derive mutation rates per ELC per base pair (μ2), we divided μ1 by the total genome size of P. falciparum (G), 2.33 × 107 bp (Equation 2).
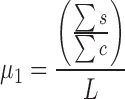 |
(1) |
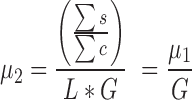 |
(2) |
The mutation rates for each isolate and for all isolates were then derived by taking the mean of the rates in each clone tree generation, with standard deviations and 95% confidence intervals. The mean was weighted by the size of each clone tree generation (w), described in Equation (3).
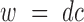 |
(3) |
where w is the weighting factor for that clone tree generation, d is total days of in vitro culture for that generation and c is number of subclone genomes analyzed in that generation. We chose this weighting because it had the best correlation with number of de novo BPS identified across the six clone trees (r2 = 0.908, P = 0.00211, linear regression), above d or c individually (Supplementary Figure S2).
To calculate BPS rates for each substitution type, the raw numbers of BPS in each type (A:T→G:C, G:C→A:T, A:T→C:G, A:T→T:A, G:C→T:A, and G:C→C:G) were divided by the total number of A/T or G/C nucleotides in the P. falciparum genome, taken as 18 779 800 bp and 4 520 200 bp, respectively (based on GC content of 19.36%), giving BPS/bp figures for each substitution type. These were then distributed between the mean overall BPS rate per ELC per bp for all clone trees (2.45 × 10−10 BPS/ELC/bp) to derive the BPS rates per ELC per bp for each substitution type, shown in Figure 3. (We use a colon to denote base pairing between complementary nucleobases on two DNA strands, and a slash to signify ‘either/or’ nucleotides on the same strand, e.g. A:T is a complementary base pairing whereas A/T can be either an adenine or a thymine.)
Figure 3.

Base pair substitution spectrum across all isolates. Boxplots show the median rate of each base pair substitution (BPS) type with first and third quartiles, while coloured dots indicate that value for each isolate. ELC = erythrocytic life cycle; bp = base pair.
Statistical methods
All statistical tests were performed using the programming language R. Datasets were assessed for normality by Shapiro–Wilk testing with α-level = 0.05. Normally distributed data were described with mean, standard deviation, and 95% Confidence Intervals, with two-sample Welch t-test for comparison between means with unequal variances and sample sizes. Bonferroni correction method was applied to P-values as stated in the text to account for multiple comparisons (18). Non-normal datasets were described with median and interquartile range, and compared via two-sample Wilcoxon rank sum test. Weighted Welch t-tests were performed using the R package weights.
To calculate statistical significance for BPS spectra, we took the raw BPS counts from the clone trees for the six different BPS types as the observed data. The ‘expected’ data assumed a null hypothesis in which all BPS types were equally likely, taking account of the skewed underlying A/T: G/C ratios in the P. falciparum genome. Observed vs expected data tables were compared using χ2 test. Indel size divisibility was assessed under the null hypothesis whereby indels would be of even length half the time, and divisible by three one third of the time. Further detail on contingency tables used for statistical tests is shown in Supplementary Appendix S2. Plots were created in R using ggplot2 and tidyr packages. 95% Clopper-Pearson confidence intervals for binomial data were generated using the package binom.
Var gene expression analysis
To analyse var gene expression, parasites in 10-ml flasks at 4% haematocrit were synchronized using 5% sorbitol and spun down at a ring-stage parasitaemia ≥4%. Parasites were stored in Ambion® RNAlater® Stabilization Solution (AM7024) at −4°C for a maximum of 48 h before RNA extraction with the Ambion® RiboPure™ RNA Purification Kit (AM1924) per the manufacturer's instructions. Stock RNA was stored at −80°C. cDNA was generated using the Applied Biosystems® High-Capacity cDNA Reverse Transcription Kit (4368814) per the manufacturer's instructions. Negative controls were included in the cDNA reaction step (with no reverse transcriptase enzyme added) to indicate DNA contamination. Reaction conditions were as follows: 10 min 25°C/120 min 37°C/5 min 85°C. cDNA test samples and negative controls were used as template for a PCR using DBLα ‘universal primers’ (19,20). The Platinum® Pfx DNA polymerase kit was used with reaction conditions per manufacturer's instructions and an annealing temperature of 42°C. Samples of the PCR reactions were run on a 1% agarose gel, and the remainder was used for blunt-end cloning into the Invitrogen pCR vector and plating on kanamycin–agar plates. The specialist and small faculty sequencing team at the Sanger Institute received the bacterial plates for each of the Dd2 and KH1-01 subclones tested. A median of 96 and 45 colonies were submitted for sequencing in Dd2 and KH1-01, respectively. Sequence tags were identified using BLAST and ClustalW, with the Dd2 and KH1-01 var genes.
RESULTS
Clone trees
We analysed data from six in vitro mutation accumulation lines, or ‘clone trees’: four laboratory isolates (3D7, HB3, Dd2 and W2, previously reported on for analysis of antigen-coding genetic recombination (6)) and the recently culture-adapted field isolates from Western Cambodia, KH1-01 and KH2-01. In total, we analysed 279 whole genomes from subclones cultured for 1404 days in vitro (∼700 asexual erythrocytic life cycles) (Table 1, Supplementary Figure S1). After mapping against the 3D7 reference genome, the median coverage with at least five reads of all 279 genomes was 94.7% (interquartile range, IQR, 2.30%) of the genome covered, and no individual subclone was <80% covered (Supplementary Table S2).
Table 1. Base pair substitution counts and mutation rates in the clone trees.
| Isolate | No. subclones sequenced | Days in culture | BPS | BPS/ELC mean (×10−3) | BPS/ELC 95% CI (×10−3) | BPS/ELC/bp mean (×10−10) | BPS/ELC/bp 95% CI (×10−10) |
|---|---|---|---|---|---|---|---|
| 3D7 | 37 | 203 | 15 | 4.91 | 3.48–6.33 | 2.10 | 1.49–2.72 |
| HB3 | 81 | 250 | 17 | 6.88 | 3.30–10.45 | 2.95 | 1.42–4.49 |
| Dd2 | 56 | 298 | 18 | 7.45 | 3.80–11.11 | 3.20 | 1.63–4.77 |
| W2 | 20 | 119 | 11 | N/A | N/A | N/A | N/A |
| KH1-01 | 59 | 447 | 20 | 5.29 | 1.29–9.28 | 2.27 | 0.550–3.98 |
| KH2-01 | 26 | 87 | 4 | 3.83 | 0.330–7.34 | 1.64 | 0.142–3.15 |
| Total | 279 | 1404 | 85 | 5.71 | 3.96–7.46 | 2.45 | 1.70–3.20 |
The mean base pair substitution (BPS) rate of all generations of each isolate's clone tree, along with 95% confidence interval (CI), are shown. All means were weighted by the size of the clone tree generated, defined as the product of the number of subclone genomes analysed and number of days in culture (see Materials and Methods and Supplementary Figure S2). ELC = erythrocytic life cycle. We excluded W2 data from mutation rate calculations due to an insufficient number of BPS observed after the first generation of the clone tree.
Biased nucleotide substitution rates drive AT-richness
We investigated whether biased BPS rates could explain the parasite's skewed genomic base pair composition. We identified 85 de novo nuclear BPS across all clone trees, plus one apicoplast BPS (Supplementary Table S3, Figure 2). Of the six possible BPS types, two are transitions (A:T→G:C and G:C→A:T) and four are transversions (A:T→C:G, A:T→T:A, G:C→T:A, and G:C→C:G). Hence, if all BPS were equally probable, the default transition-to-transversion ratio (Ts:Tv) would be 0.5. This differs significantly from the Ts:Tv ratio of 1.13 actually observed (Table 2) (P = 2.03 × 10−4, two-sided exact binomial test). This transition bias was mainly driven by an excess of G:C→A:T substitutions observed in all isolates (Figure 3). The mean G:C→A:T mutation rate per bp was higher than for any other substitution type (P < 0.01 for all pairwise comparisons, two-sample Welch t-test), while no other BPS type differed significantly from any other (Supplementary Table S4). The overall substitution spectrum across the six BPS types differed significantly from what would be expected if all substitutions occurred at an equal rate per bp (P = 5.13 × 10-4, Pearson's Chi-squared test).
Table 2. Raw base pair substitution counts in P. falciparum mutation-accumulation lines.
| Isolate | A:T→G:C | G:C→A:T | Ts | A:T→C:G | A:T→T:A | G:C→T:A | G:C→C:G | Tv | Ts:Tv |
|---|---|---|---|---|---|---|---|---|---|
| 3D7 | 4 | 2 | 6 | 0 | 5 | 1 | 3 | 9 | 0.67 |
| HB3 | 3 | 4 | 7 | 2 | 4 | 4 | 0 | 10 | 0.70 |
| Dd2 | 5 | 5 | 10 | 0 | 7 | 1 | 0 | 8 | 1.25 |
| W2 | 0 | 8 | 8 | 1 | 1 | 1 | 0 | 3 | 2.67 |
| KH1-01 | 7 | 4 | 11 | 2 | 3 | 2 | 2 | 9 | 1.22 |
| KH2-01 | 2 | 1 | 3 | 1 | 0 | 0 | 0 | 1 | 3.00 |
| Clone tree | 21 | 24 | 45 | 6 | 20 | 9 | 5 | 40 | 1.13 |
| Genetic crosses | 8 | 8 | 16 | 4 | 6 | 4 | 2 | 16 | 1.00 |
| Total | 29 | 32 | 61 | 10 | 26 | 13 | 7 | 56 | 1.09 |
The 3D7, HB3, Dd2, W2, KH1-01, and KH2-01 data are from clone tree experiments. ‘Clone tree’ shows the total for the six isolate clone trees. The ‘genetic crosses’ data are pooled de novo base pair substitutions from the progeny of three experimental genetic crosses: HB3xDd2, HB3×3D7, and 7G8xGB4. ‘Total’ shows combined counts for clone trees and genetic crosses. The overall Ts:Tv (Transition:Transversion) ratio for wild-type P. falciparum was 1.13, indicating transition bias. Every wild-type isolate had some degree of transition bias (i.e. Ts:Tv > 0.5), Ts = Transition; Tv = Transversion.
To increase the sample size, we analysed a set of 32 BPS identified in the progeny of three experimental genetic crosses: HB3xDd2, HB3×3D7 and 7G8xGB4 (16) (Table 2, Supplementary Table S5 and Figure S4). These isolates have been through many rounds of mitotic erythrocytic division before and after the crosses, and one round of meiotic division within mosquitoes. As with the clone trees, G:C→A:T mutations were the most common BPS per bp (Table 2, Supplementary Figure S4). The distribution of substitutions between the six BPS types did not differ significantly between the 85 clone tree BPS and 32 crosses BPS (P = 0.943, Pearson's Chi-squared test).
If the genome were at mutational equilibrium, the number of A/T conversions to G/C would equal G/C conversions to A/T, giving an expected ratio of 0.5. Our observed ratio (39:45, respectively) did not differ significantly from this (P = 0.586, two-sided exact binomial test). The total ratio of A/T→G/C versus G/C→A/T substitution rates per bp was 4.79, i.e. for every one A/T→G/C substitution there would be about five G/C→A/T substitutions, if the genome had an even base pair composition. We would expect this to equilibrate at a genomic base pair composition of ∼82.7% AT, close to the actual genomic average in P. falciparum (80.6% AT overall in the 3D7 reference genome). These data demonstrate that the parasite's high AT content is being maintained at or near mutational equilibrium by a skewed BPS spectrum.
We analyzed the pattern of nucleotides surrounding C→T transition sites for evidence of the underlying mechanism. C→T transitions commonly result from 5-methylcytosine deamination to thymine. Cytosine methylation is associated with CpG sites in mammalian genomes, though Ponts et al. have reported an enrichment for thymines surrounding conserved 5-methylcytosines in P. falciparum (21). We did not find any significant overrepresentation of any nucleotide in the +1 or −1 positions of our 24 G:C→A:T clone tree transitions compared with what would be expected by chance (Fisher's exact test). Another cause of G:C→A:T mutation is pyrimidine dimerisation, which can cause CpC→TpT transitions. We did not observe any instances of CpC→TpT mutations. However, with only 24 observations, our statistical power is low.
AT-rich repetitive sequences undergo prolific rates of indel mutation
The AT-rich P. falciparum genome contains stretches of highly repetitive, low complexity DNA. We investigated whether this predisposes to a raised rate of indel mutation. We limited this analysis to the nuclear 3D7 genome, as other isolates would have yielded a significant ‘false negative’ rate (i.e. missed indels) due to the lack of a near-perfect reference genome (unlike 3D7), particularly in telomeric and subtelomeric loci. We identified 164 de novo indels in 37 subclone genomes in 3D7, compared with 15 BPS (Figure 2C, Supplementary Table S6). There were 108 insertions and 56 deletions, with median insertion and deletion sizes of 2 and 8 bp, respectively (size difference between insertions and deletions was statistically significant, P = 6.01 × 10-9, two-sample Wilcoxon rank sum test). The 3D7 indel rate was extremely high, around 10-fold greater than the BPS rate, at 21.1 × 10−10 (95% confidence interval, CI, 13.2 × 10−10–28.9 × 10−10) indels/ELC/bp; i.e. about one in every 20 parasites (95%CI 15–33) would be expected to harbor a de novo indel per 48-h ELC, compared with one de novo BPS in every 175 parasites (95%CI 134–253) per ELC.
We analysed the genomic distribution and composition of de novo indels. Only 15/164 (9.1%) were observed in exons, a significantly different distribution from what would be expected by chance (P < 2.2 × 10−16, Pearson's Chi-squared test, Table 3). This contrasts with the distribution of BPS, which did not differ significantly from the background proportions of exons, introns and non-genic loci in the genome (Table 3, Figure 4A). 14/164 (8.5%) indels occurred outside of core genome coordinates (defined in (16)) close to chromosome ends. The AT content of combined inserted and deleted sequences was very high at 96.4% (83.7%, 99.3% and 99.4% in exons, introns and non-genic loci, respectively). This is high even by P. falciparum standards: in 3D7, the overall AT content is 76.3%, 86.5% and 86.4% in exons, introns and intergenic regions, respectively (2). Regardless of the coding or non-coding identity of the sequences, 100 bp regions containing an indel had a higher mean AT content (88.1%) than regions without identified indels (80.6%) (P < 2.2 × 10−16, Welch two-sample t-test, Figure 4C). All indels were flanked by at least one repeat of the same sequence. Collectively, these data show that indels occurred in highly AT-rich and repetitive sequences mainly in introns and outside of genes.
Table 3. Genomic distributions of indels and BPS.
| 3D7 clone tree | All wild-type clone trees | |||
|---|---|---|---|---|
| Observed indels (%) | Expected indels (%) | Observed BPS (%) | Expected BPS (%) | |
| Exonic | 15 (9.1) | 86.3 (52.6) | 48 (61.5) | 41.1 (52.6) |
| Intronic | 43 (26.2) | 9.50 (5.79) | 3 (3.8) | 4.52 (5.79) |
| Non-genic | 106 (64.6) | 68.2 (41.6) | 27 (34.6) | 32.4 (41.6) |
| Total | 164 (100) | 164 (100) | 78 (100) | 78 (100) |
The raw counts (and %) of observed indels from the 3D7 clone tree and BPS from all clone trees (3D7, HB3, Dd2, W2, KH1-01 and KH2-01) in exons, introns, and non-genic loci are shown, along with expected counts given a null hypothesis of the distribution being equal to the underlying genomic proportions (based on (2)). The distribution of indels between exons, introns and non-genic loci differed significantly from that expected by chance for indels (P < 2.2×10−16) but not for BPS (P = 0.511), Pearson's Chi-squared test.
Figure 4.

Indel distribution, size and AT content. (A) Relative distributions of indels and BPS in exons (Ex, red), introns (In, green) and non-genic loci (NG, blue). Indels (164 total from the 3D7 clone tree) were overrepresented in non-coding regions (non-genic and intronic), with only a small minority of indels (15/164, 9.1%) found in exons. In contrast, the distribution of BPS (78 pooled from all clone trees with known BPS coordinates) was similar to the underlying proportions of exonic, intronic, and non-genic sequence in the P. falciparum genome (raw data in Table 3). (B) Indel distribution in exonic, intronic, and non-genic loci, showing the number of indels whose nucleotide lengths are divisible (red) or indivisible (blue) by three (left) or two (right). Indels that are multiples of three preserve the reading frame when they occur in exons, and are overrepresented in exons compared with introns and non-genic loci (Table 4). In contrast, indels that are multiples of two bp are overrepresented from what is expected by chance in non-coding loci, reflecting the abundance of poly(AT) tracts in non-coding loci. (C) The 3D7 genome was divided into non-overlapping 100 bp bins, with the % AT content calculated for each bin and plotted as a histogram (blue bars, y-axis scale on left). The % AT content of the subset of all bins containing at least one indel was plotted separately (red bars, y-axis scale on right). The 100bp windows containing indels had a higher average AT content than those for the whole genome (88.1% and 80.6%, respectively) (P < 2.2 × 10−16, two-sample Welch t-test). Ex = Exon, In = Intron, NG = non-genic.
Poly(AT) dinucleotide repeats are more common in non-coding DNA than in exons, while around 24% of P. falciparum proteins contain low-complexity amino acid homorepeats (60% asparagines and 23% lysines) (5,22). Of the latter, the most common codons are AAT (asparagine) and AAA (lysine) (4,22). We tested whether these sequence properties were reflected in the types of indel mutation arising in exonic and non-coding loci. 86.7% (13/15) of exonic indels were divisible by three bp, significantly more than expected by chance (P = 3.14 × 10−5, two-sided exact binomial test), while 87.2% (130/149) of indels outside of exons were multiples of 2 bp (P < 2.2 × 10-16, two-sided exact binomial test) (Figure 4B, Table 4). 12/15 (80%) of exonic indels involved asparagine or lysine residues and 7/15 (47%) caused expansion or contraction of poly(N) homorepeats (Supplementary Table S7). Thus, the underlying sequences inside and outside of exons are associated with distinct forms of indel mutation. This may be due to the processes of DNA polymerase slippage and unequal crossing over, combined with the tendency for purifying selection to purge frameshift-causing indels from the gene pool. These data support the observation that short tandem repeats (STR) across diverse genomes are highly polymorphic and prone to high mutation rates, with dinucleotide STR tending to have higher mutation rates than trinucleotide repeats (23).
Table 4. Breakdown of indels divisible by two and three nucleotides by genomic context.
| Multiple of three bp | Multiple of two bp | |||
|---|---|---|---|---|
| % Observed indels (count) | P-value (expected ratio = one third) | % Observed indels (count) | P-value (expected ratio = one half) | |
| Exon | 86.7% (13/15) | 3.14 × 10−5 | 53.3% (8/15) | 1 |
| Non-exon | 6.71% (10/149) | 9.61 × 10−15 | 87.2% (130/149) | <2.2 × 10−16 |
% (and raw count) of observed indels from the 3D7 clone tree which were divisible by two and three nucleotides are shown, broken down into those occurring inside and outside of exons (outside of exons includes both introns and non-genic loci). By chance, the expected proportions of indels divisible by three and two base pairs are one third and one half, respectively. P-values are derived from binomial exact tests. Indels that were multiples of three bp were more likely to occur in exons, while introns and non-genic loci were more likely to contain indels that were multiples of 2 bp. Data also shown in Figure 4.
Mutation rate variation between isolates
It has been suggested that more drug resistant parasites have higher mutation rates, as this would increase the generation of novel genetic variants facilitating adaptive evolution (24–26). The KH2-01 parasite was artesunate slow-clearing in vivo and possessed several of the highly differentiated SNPs in DNA repair genes identified by Miotto et al. (11), including P203S in mlh1, S506I in pms1, and C2010S in rad50, whereas KH1-01 was artesunate fast-clearing and wild-type at these loci. The pooled mutation rate from all six P. falciparum isolates was 2.45 × 10−10 (95%CI, 1.70 × 10−10–3.20 × 10−10) BPS/ELC/bp, consistent with other estimates (6,7). The rates did not differ significantly between any isolate pairing (Table 1), including KH2-01 v KH1-01 (two-sample Welch t-test). Thus, we found no evidence of a BPS hypermutator phenotype in either the artesunate fast-clearing or slow-clearing Cambodian field isolates.
Unlike BPS, rates of non-allelic homologous recombination (NAHR) in and around var genes differ significantly between long-term laboratory adapted isolates (6). NAHR produces var gene sequence mosaicism during erythrocytic division, potentially adding to PfEMP1 antigenic diversity within infected people. We analysed the rate of var gene NAHR in our two Cambodian isolates, which are more biologically relevant as they are recently derived from their natural in vivo environment. We identified 27 pairs of recombining var genes in these isolates (Supplementary Table S8). Unlike BPS, the var gene exon 1 recombination rates were significantly higher in the Cambodian isolates than in the long-term culture-adapted laboratory isolates (P = 0.0306, two-sample Welch t-test with Bonferroni correction for multiple comparisons (18)) (Figure 5 and Supplementary Table S9). In KH1-01, there would be approximately one var gene recombination event, potentially producing an antigenically novel structure, for every 156 parasites (95%CI 117–233) per 48-h ELC. That is 1.21-fold higher than the BPS mutation rate in KH1-01 (Supplementary Table S9).
Figure 5.

Base pair substitution (BPS, red) and var exon 1 recombination rates (blue) shown per erythrocytic life cycle (ELC) in all isolates. BPS rates did not differ significantly between the different isolates, nor between the pooled long-term culture-adapted laboratory isolates (3D7, HB3 and Dd2) and the combined Cambodian isolates (KH1-01 and KH2-01) (all pairwise comparisons by two-sample Welch t-test). In contrast, the combined var gene exon 1 recombination rate in the Cambodian field isolates was significantly higher than for the combined laboratory isolates (P = 0.0306, two-sample Welch t-test after Bonferroni correction for multiple comparisons). The KH1-01 var exon 1 recombination rate was 6.40 × 10−3 (95%CI 4.30 × 10−3–8.51 × 10-3) recombining var pairs per ELC, or one parasite with recombined var genes for every 156 (95%CI 117–233) parasites following a single 48-h ELC. Error bars show standard error of the mean.
We investigated whether the higher var exon 1 recombination rate in the Cambodian isolates correlated with greater var gene transcriptional activity. Var genes are divided into three main families, A, B or C, based on phylogenetic analysis (27). Both the variety and quantity of var gene expression in wild parasites drop when they enter in vitro culture, with group B and C var genes predominating (28–30). Sampled Dd2 subclones from our clone tree expressed a very limited range of their var gene repertoire, with just two var genes dominating expression (Supplementary Figures S5 and S6). In contrast, no single var gene dominated consistently out of the five KH1-01 subclones analysed, and there were at least 37 var genes transcribed in the pooled KH1-01 dataset (Supplementary Figures S5 and S6). Thus, the more recently culture-adapted KH1-01 field isolate had both a higher var recombination rate and a greater variety of expressed var genes than the long-term culture-adapted Dd2 isolate. However, the var genes that underwent recombination were not those dominantly expressed in the preceding clone tree generation (Supplementary Figure S5), so a direct relationship between expression and recombination of specific var genes was not observed.
DISCUSSION
We have used mutation accumulation lines to study how spontaneous mutation across the P. falciparum genome sculpts the parasite's unusual genomic architecture. We found evidence of significant transition bias driven by an overrepresentation of G:C→A:T substitutions, expected to push the genome to its current skewed base pair composition of ∼80% AT. This is consistent with population genetic data suggesting an underlying mutation bias that favours G/C→A/T (31). This mutational bias may contribute to producing AT-rich, low-complexity sequences, particularly in non-coding loci where purifying selection is generally relaxed. Such AT-rich repetitive sequences can then cause DNA polymerase slippages and unequal crossing over events, as tandem repeats are prone to slipped-strand mispairing during DNA replication (32,33), and are associated with indel mutations (34). While previous studies have identified high indel variation between P. falciparum genomes (16,35), we have demonstrated the prolific rate at which these highly dynamic features occur during mitotic division: about 10-fold more frequently than de novo BPS. This is unusual amongst eukaryotes; in most other organisms studied BPS outnumber indel mutations (36–38), with substitution to indel ratios in sampled primate and bacterial genomes of 9.89 and 19.61, respectively (38). These data pose fundamental questions about P. falciparum genome biology. What is the evolutionary history of skewed genome composition in the Plasmodium family tree? What are the molecular mechanisms driving high rates of G:C→A:T transition? And what are the biological consequences of high AT content with high indel mutation rates; do they serve any adaptive roles for the parasite?
P. falciparum is not unique among Plasmodium species in possessing an AT-rich genome (Supplementary Table S1 and Figure 1). Closely related parasites in the Laverania subgenus from which P. falciparum emerged (39), such as P. reichenowi (40) and P. gaboni (41), also have markedly skewed genome compositions (80.7% and 79.4% AT, respectively). This suggests that systematic substitution bias driving high AT content predates the divergence of the Laverania group. How far back in evolutionary time does genome composition bias extend? The related primate parasites P. cynomolgi, P. knowlsei and P. vivax (42) have more even base pair compositions (59.4%, 60.8% and 59.4% AT, respectively), though this may be due to recent evolution towards higher GC, away from an AT-rich common Plasmodium ancestor (3). The bird parasite and evolutionary outgroup to Plasmodium, Haemoproteus tartakovskyi, also parasitizes erythrocytes and has high AT content, at 75% (43). The Apicomplexan parasites Babesia bovis, Theileria parva and Toxoplasma gondii, all outside of the Plasmodium/ Haemoproteus group (44), have more even AT contents (58.2%, 65.9% and 47.7%, respectively) (Figure 1) (45). Thus, a degree of AT bias may have evolved at least as far back as the common ancestor of Plasmodium and Haemoproteus. This model can be tested as more parasite genomes become available and phylogenetic trees with genome-wide AT contents (as in Figure 1) are more comprehensive.
Overrepresentation of G:C→A:T transitions and a mutation bias in favour of AT-richness is seen in diverse organisms (46–52), and transition mutations outnumber transversions in most organisms with intact DNA repair machinery (53–59). A major reason for transition bias and elevated G:C→A:T mutation rates is cytosine deamination to uracil, which is a common spontaneous biochemical change in DNA (60). We did not find a statistically significant enrichment of CpG or TpC dinucleotides at de novo C→T transitions, which would be expected if 5-methylcytosine deamination to thymine were playing a role. However, this finding should be cautioned by the low number of de novo C→T mutations identified.
In bacteria, obligate intracellular symbionts tend to evolve high AT-content (61,62) as guanine and cytosine nucleotides require higher energy for biosynthesis and are less readily available intracellularly. Relaxed selection pressure in intracellular parasites causes gene loss (63), and may facilitate the natural tendency for mutation to drive the genome towards high AT. Alterations in DNA repair and polymerase pathways also affect base pair composition (64–68), and DNA repair gene mutations or pathway degradation are often seen in obligate intracellular pathogens (61). Several features of P. falciparum DNA repair are unusual, such as the absence of a canonical non-homologous end joining pathway (69,70). The parasitized intraerythrocytic environment is also rich in reactive oxygen species, reactive nitrogen species, and NO radicals (71–74), which can cause cytidine deamination to uracil, driving C→T transitions (60,75). It may be that a combination of factors are responsible for P. falciparum G:C→A:T substitution bias, including cytosine deamination, exacerbated by exposure to intraerythrocytic oxidising agents, combined with underlying abnormalities in DNA repair. Further research is necessary to investigate these influences, for example, by culturing P. falciparum in oxidising conditions or generating lines with a range of genetically modified DNA repair activity and monitoring the effect on BPS rates and bias.
There are at least three potential biological consequences of widespread AT-rich, repetitive loci associated with high indel mutation: expansion and contraction of amino acid homorepeats, alterations to gene expression through indels in AT-rich regulatory DNA, and increased rates of copy number variation (CNV). Most of the exonic indels we observed contained asparagine and/or lysine residues in repetitive sequences, suggesting these low-complexity regions undergo a high rate of indel mutation. Various adaptive roles for these repeats in P. falciparum proteins have been suggested (76), such as tRNA sponges (77) and antigenic variation for immune evasion (78,79). They may provide a general platform for evolutionary adaptation, with the potential for mutation and selection to drive neofunctionalisation of the asparagine repeats in different contexts (76). AT-rich non-coding DNA surrounding genes is also likely to undergo a high indel rate. This could affect gene expression, as untranslated regions up- and down-stream of genes contain both enhancer and repressor elements (reviewed in (80)). Lastly, CNVs are correlated with repetitive sequences such as Alu repeats in human and mouse genomes, likely due to NAHR processes (81–84). P. falciparum CNVs contribute to the evolution of antimalarial drug resistance, for example amplification of pfmdr1 and gch1 genes driving resistance to mefloquine and antifolates, respectively (85,86). The amplicon breakpoints in pfmdr1 and gch1 occur in microsatellites and monomeric A/T tracts (87,88). It has been estimated that there are 37 177 monomeric A/T tracts in the P. falciparum genome, providing ample scope for the de novo emergence of CNV through NAHR-like processes (88). The mechanism first involves random duplication of DNA between two A/T tracts, followed by a more precise amplification of the sequence under selection (89). Thus, G:C→A:T transition bias may help generate widespread repetitive AT-rich low-complexity regions, both inside and outside of coding sequences, ultimately facilitating a form of microstructural genomic dynamism providing additional fuel for adaptive evolution.
We found no significant differences in BPS mutation rates between isolates, regardless of their geographic origins or the degree of drug resistance. This is consistent with a recent population genetic analysis which found no evidence of hypermutation in Southeast Asian parasites, including those possessing multiple highly differentiated SNPs in DNA repair genes found in the KH2 sub-population (90); though a mild mutator phenotype has been reported in a yeast model of the mlh1 203S variant (91). A more systematic evaluation of multiple isolates is needed to formally demonstrate that artemisinin resistant KH2 parasites, on average, have no difference in mutation rates to artemisinin sensitive parasites. In contrast, rates of var gene NAHR differed significantly between isolates, with the two Cambodian lines having higher rates than the long-term culture-adapted laboratory isolates. The Cambodian KH1-01 parasite also expressed a greater variety of its var gene repertoire than the Dd2 isolate. This is expected, as the Cambodian isolates were more recently taken from the in vivo setting where var gene transcriptional activity is higher (28–30). If transcription somehow opens up the chromosome to facilitate recombination, then higher levels of a more diversely expressed var gene repertoire could contribute to the higher recombination rates observed in KH1-01 and KH2-01. This could also explain why group A var genes have a lower recombination rate than group B or C var genes in the clone trees (discussed in Claessens et al. 2014 (6)) – group A var recombination may be more common in vivo where they are expressed more often, particularly in severe malaria (92,93). However, correlation does not demonstrate causation; further research is needed to investigate any mechanistic relationship between var gene transcription and mitotic recombination. Var gene recombination patterns in vivo are currently unexplored.
Even a relatively low parasitaemia of 0.01% (∼500 parasites/μl of blood) represents ∼1 billion parasites circulating in a single symptomatic human infection. Every two days, one would expect ∼6 million substitutions, 55 million indels and 4 million newly created mosaic var exon 1 sequences to emerge through de novo mutation inside an infected person. Thus, a continuous wellspring of genetic variation, particularly small indels, is generated across the ∼200 million symptomatic malaria cases each year. Effective population size, more relevant for adaptive evolution than total population, has been estimated at around 104–106 parasites in P. falciparum (31,94,95, http://dx.doi.org/10.1101/056291). These numbers highlight the need for effective artemisinin partner drug combinations (96), optimal patient adherence and high medicine quality to constrain resistance evolution. The C580Y mutation in kelch13 implicated in artemisinin resistance (97–100), for example, has arisen multiple times in Southeast Asia (100), and kelch13 mutations are detectable at low frequency in Africa (101). The great challenge facing malaria control efforts is to keep ahead of the curve, and eliminate these parasites before the evolutionary process has allowed particularly dangerous combinations of mutations, with high levels of resistance combined with strong biological fitness, to emerge and spread through parasite populations.
ACCESSION NUMBERS
Whole genome sequences can be found in the European Nucleotide Archive with accession numbers available in Supplementary Table S2.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Wellcome Trust [098051]; Medical Research Council [G0600718]. Funding for open access charge: Wellcome Trust.
Conflict of interest statement. None declared.
REFERENCES
- 1. Fairhurst R.M. Understanding artemisinin-resistant malaria: what a difference a year makes. Curr. Opin. Infect. Dis. 2015; 28:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gardner M.J., Hall N., Fung E., White O., Berriman M., Hyman R.W., Carlton J.M., Pain A., Nelson K.E., Bowman S. et al. . Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002; 419:498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nikbakht H., Xia X., Hickey D.A.. The evolution of genomic GC content undergoes a rapid reversal within the genus Plasmodium. Genome. 2015; 57:507–511. [DOI] [PubMed] [Google Scholar]
- 4. Pizzi E., Frontali C.. Low-complexity regions in Plasmodium falciparum proteins. Genome Res. 2001; 11:218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zilversmit M.M., Volkman S.K., Depristo M.a., Wirth D.F., Awadalla P., Hartl D.L.. Low-complexity regions in plasmodium falciparum: Missing links in the evolution of an extreme genome. Mol. Biol. Evol. 2010; 27:2198–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Claessens A., Hamilton W.L., Kekre M., Otto T.D., Faizullabhoy A., Rayner J.C., Kwiatkowski D.. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of var genes during mitosis. PLoS Genet. 2014; 10:e1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bopp S.E.R., Manary M.J., Bright a.T., Johnston G.L., Dharia N.V., Luna F.L., McCormack S., Plouffe D., McNamara C.W., Walker J.R. et al. . Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet. 2013; 9:e1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barrick J.E., Lenski R.E.. Genome dynamics during experimental evolution. Nat. Rev. Genet. 2013; 14:827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trager W., Jenson J.B.. Cultivation of malarial parasites. Nature. 1978; 273:621–622. [DOI] [PubMed] [Google Scholar]
- 10. Manske M., Miotto O., Campino S., Auburn S., Almagro-Garcia J., Maslen G., O'Brien J., Djimde A., Doumbo O., Zongo I. et al. . Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature. 2012; 487:375–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miotto O., Almagro-Garcia J., Manske M., Macinnis B., Campino S., Rockett K.a., Amaratunga C., Lim P., Suon S., Sreng S. et al. . Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat. Genet. 2013; 45:648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Manske M., Miotto O., Campino S., Auburn S., Almagro-garcia J., Maslen G., Brien J.O., Djimde A., Doumbo O., Zongo I. et al. . Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature. 2012; 487:375–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manske H.M., Kwiatkowski D.P.. LookSeq: a browser-based viewer for deep sequencing data. Genome Res. 2009; 19:2125–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. et al. . The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miles A., Iqbal Z., Vauterin P., Pearson R., Campino S., Theron M., Gould K., Mead D., Drury E., Brien J.O. et al. . Indels, structural variation, and recombination drive genomic diversity in Plasmodium falciparum. Genome Res. 2016; 26:1288–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rausch T., Zichner T., Schlattl A., Stütz A.M., Benes V., Korbel J.O.. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012; 28:i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bland J.M., Altman D.G.. Multiple significance tests: the Bonferroni method. BMJ. 1995; 310:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taylor H.M., Kyes S.A., Harris D., Kriek N., Newbold C.I.. A study of var gene transcription in vitro using universal var gene primers. Mol. Biochem. Parasitol. 2000; 105:13–23. [DOI] [PubMed] [Google Scholar]
- 20. Bull P.C., Berriman M., Kyes S., Quail M.A., Hall N., Kortok M.M., Marsh K., Newbold C.I.. Plasmodium falciparum variant surface antigen expression patterns during malaria. PLoS Pathog. 2005; 1:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ponts N., Fu L., Harris E.Y., Zhang J., Chung D.W.D., Cervantes M.C., Prudhomme J., Atanasova-Penichon V., Zehraoui E., Bunnik E.M. et al. . Genome-wide mapping of DNA methylation in the human malaria parasite Plasmodium falciparum. Cell Host Microbe. 2013; 14:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singh G.P., Chandra B.R., Bhattacharya A., Akhouri R.R., Singh S.K., Sharma A.. Hyper-expansion of asparagines correlates with an abundance of proteins with prion-like domains in Plasmodium falciparum. Mol. Biochem. Parasitol. 2004; 137:307–319. [DOI] [PubMed] [Google Scholar]
- 23. Fan H., Chu J.-Y.. A brief review of short tandem repeat mutation. Genomics. Proteomics Bioinformatics. 2007; 5:7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rathod P.K., McErlean T., Lee P.C.. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:9389–9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trotta R.F., Brown M.L., Terrell J.C., Geyer J.A.. Defective DNA repair as a potential mechanism for the rapid development of drug resistance in Plasmodium falciparum. Biochemistry. 2004; 43:4885–4891. [DOI] [PubMed] [Google Scholar]
- 26. Castellini M.a, Buguliskis J.S., Casta L.J., Butz C.E., Clark A.B., Kunkel T.a, Taraschi T.F.. Malaria drug resistance is associated with defective DNA mismatch repair. Mol. Biochem. Parasitol. 2011; 177:143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lavstsen T., Salanti A., Jensen A.T.R., Arnot D.E., Theander T.G.. Sub-grouping of Plasmodium falciparum 3D7 var genes based on sequence analysis of coding and non-coding regions. Malar. J. 2003; 2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peters J.M., Fowler E.V., Krause D.R., Cheng Q., Gatton M.L.. Differential changes in Plasmodium falciparum var transcription during adaptation to culture. J. Infect. Dis. 2007; 195:748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bachmann A., Petter M., Tilly A.K., Biller L., Uliczka K.a., Duffy M.F., Tannich E., Bruchhaus I.. Temporal expression and localization patterns of variant surface antigens in clinical Plasmodium falciparum isolates during erythrocyte Schizogony. PLoS One. 2012; 7:e49540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blomqvist K., Normark J., Nilsson D., Ribacke U., Orikiriza J., Trillkott P., Byarugaba J., Egwang T.G., Kironde F., Andersson B. et al. . var gene transcription dynamics in Plasmodium falciparum patient isolates. Mol. Biochem. Parasitol. 2010; 170:74–83. [DOI] [PubMed] [Google Scholar]
- 31. Chang H.-H., Park D.J., Galinsky K.J., Schaffner S.F., Ndiaye D., Ndir O., Mboup S., Wiegand R.C., Volkman S.K., Sabeti P.C. et al. . Genomic sequencing of Plasmodium falciparum malaria parasites from Senegal reveals the demographic history of the population. Mol. Biol. Evol. 2012; 29:3427–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Y.-C., Korol A.B., Fahima T., Beiles a, Nevo E.. Microsatellites: genomic distribution, putativa functions, and mutational mechanism: a review. Mol. Ecol. 2002; 11:253–256. [DOI] [PubMed] [Google Scholar]
- 33. Lovett S.T. Encoded errors: Mutations and rearrangements mediated by misalignment at repetitive DNA sequences. Mol. Microbiol. 2004; 52:1243–1253. [DOI] [PubMed] [Google Scholar]
- 34. Montgomery S.B., Goode D.L., Kvikstad E., Albers C.a, Zhang Z.D., Mu X.J., Ananda G., Howie B., Karczewski K.J., Smith K.S. et al. . The origin, evolution and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Res. 2013; 23:749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Volkman S.K., Sabeti P.C., DeCaprio D., Neafsey D.E., Schaffner S.F., Milner D.A., Daily J.P., Sarr O., Ndiaye D., Ndir O. et al. . A genome-wide map of diversity in Plasmodium falciparum. Nat. Genet. 2007; 39:113–119. [DOI] [PubMed] [Google Scholar]
- 36. Behringer M.G., Hall D.W.. The repeatability of genome-wide mutation rate and spectrum estimates. Curr. Genet. 2016; 62:507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sung W., Ackerman M.S., Dillon M.M., Platt T.G., Fuqua C., Cooper V.S., Lynch M.. Evolution of the insertion-deletion mutation rate across the tree of life. G3 Genes|Genomes|Genetics. 2016; 6:2583–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen J.Q., Wu Y., Yang H., Bergelson J., Kreitman M., Tian D.. Variation in the ratio of nucleotide substitution and indel rates across genomes in mammals and bacteria. Mol. Biol. Evol. 2009; 26:1523–1531. [DOI] [PubMed] [Google Scholar]
- 39. Liu W., Li Y., Learn G.H., Rudicell R.S., Robertson J.D., Keele B.F., Ndjango J.-B.N., Sanz C.M., Morgan D.B., Locatelli S. et al. . Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature. 2010; 467:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Otto T.D., Rayner J.C., Böhme U., Pain A., Spottiswoode N., Sanders M., Quail M., Ollomo B., Renaud F., Thomas A.W. et al. . Genome sequencing of chimpanzee malaria parasites reveals possible pathways of adaptation to human hosts. Nat. Commun. 2014; 5:4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sundararaman S.A., Plenderleith L.J., Liu W., Loy D.E., Learn G.H., Li Y., Shaw K.S., Ayouba A., Peeters M., Speede S. et al. . Genomes of cryptic chimpanzee Plasmodium species reveal key evolutionary events leading to human malaria. Nat. Commun. 2016; 7:11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Larremore D.B., Sundararaman S.A., Liu W., Proto W.R., Clauset A., Loy D.E., Speede S., Plenderleith L.J., Sharp P.M., Hahn B.H. et al. . Ape parasite origins of human malaria virulence genes. Nat. Commun. 2015; 6:8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bensch S., Canbäck B., DeBarry J.D., Johansson T., Hellgren O., Kissinger J.C., Palinauskas V., Videvall E., Valkiūnas G.. The Genome of Haemoproteus tartakovskyi and Its Relationship to Human Malaria Parasites. Genome Biol. Evol. 2016; 8:1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wasmuth J., Daub J., Peregrin-Alvarez J.M., Finney C.A.M., Parkinson J.. The origins of apicomplexan sequence innovation. Genome Res. 2009; 19:1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brayton K.A., Lau A.O.T., Herndon D.R., Hannick L., Kappmeyer L.S., Berens S.J., Bidwell S.L., Brown W.C., Crabtree J., Fadrosh D. et al. . Genome sequence of Babesia bovis and comparative analysis of apicomplexan hemoprotozoa. PLoS Pathog. 2007; 3:e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ossowski S., Schneeberger K., Lucas-Lledó J.I., Warthmann N., Clark R.M., Shaw R.G., Weigel D., Lynch M.. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010; 327:92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Artl D., Petrov D.. Patterns of nucleotide substitution in Drosophila and mammalian genomes. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:1475–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Keightley P.D., Trivedi U., Thomson M., Oliver F., Kumar S., Blaxter M.L.. Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genome Res. 2009; 19:1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Denver D.R., Dolan P.C., Wilhelm L.J., Sung W., Lucas-Lledó J.I., Howe D.K., Lewis S.C., Okamoto K., Thomas W.K., Lynch M. et al. . A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:16310–16314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lynch M., Sung W., Morris K., Coffey N., Landry C.R., Dopman E.B., Dickinson W.J., Okamoto K., Kulkarni S., Hartl D.L. et al. . A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hershberg R., Petrov D.A.. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet. 2010; 6:e1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Van Leuven J.T., McCutcheon J.P.. An AT mutational bias in the tiny GC-rich endosymbiont genome of Hodgkinia. Genome Biol. Evol. 2012; 4:24–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wakeley J. The excess of transitions among nucleotide substitutions: new methods of estimating transition bias underscores its significance. Trends Ecol. Evol. 1996; 11:158–162. [DOI] [PubMed] [Google Scholar]
- 54. Brown W.M., Prager E.M., Wang A., Wilson A.C., I S.. Mitochondrian DNA sequences of primates: tempo and mode of evolution. J. Mol. Evol. 1982; 18:225–239. [DOI] [PubMed] [Google Scholar]
- 55. Zhang Z. Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 2003; 31:5338–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gojobori T., Li W.-H., Graur D.. Patterns of nucleotide substitution in pseudogenes and functional genes. J. Mol. Evol. 1982; 18:360–369. [DOI] [PubMed] [Google Scholar]
- 57. Wakeley J. Substitution-rate variation among sites and the estimation of transition bias. Mol. Biol. Evol. 1994; 11:436–442. [DOI] [PubMed] [Google Scholar]
- 58. Curtis S.E., Clegg M.T.. Molecular evolution of chloroplast DNA sequences. Mol. Biol. Evol. 1984; 1:291–301. [DOI] [PubMed] [Google Scholar]
- 59. Schaaper R.M., Dunn R.L.. Spontaneous mutation in the Escherichia coli lacI gene. Genetics. 1991; 129:317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kreutzer D.a, Essigmann J.M.. Oxidized, deaminated cytosines are a source of C –>T transitions in vivo. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:3578–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Moran N.A., McCutcheon J.P., Nakabachi A.. Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 2008; 42:165–190. [DOI] [PubMed] [Google Scholar]
- 62. Rocha C., Danchin A.. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002; 18:291–294. [DOI] [PubMed] [Google Scholar]
- 63. Hershberg R., Tang H., Petrov D.A.. Reduced selection leads to accelerated gene loss in Shigella. Genome Biol. 2007; 8:R164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wu H., Zhang Z., Hu S., Yu J.. On the molecular mechanism of GC content variation among eubacterial genomes. Biol. Direct. 2012; 7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lind P.A., Andersson D.I.. Whole-genome mutational biases in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:17878–17883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lee H., Popodi E., Tang H., Foster P.L.. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2774–E2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Watanabe-Akanuma M., Woodgate R., Ohta T.. Enhanced generation of A:T-T:A transversions in a recA730 lexA51(Def) mutant of Escherichia coli. Mutat. Res. 1997; 373:61–66. [DOI] [PubMed] [Google Scholar]
- 68. Lang G.I., Parsons L., Gammie A.E.. Mutation rates, spectra, and genome-wide distribution of spontaneous mutations in mismatch repair deficient yeast. G3 Genes|Genomes|Genetics. 2013; 3:1453–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kirkman L.a, Lawrence E.a, Deitsch K.W.. Malaria parasites utilize both homologous recombination and alternative end joining pathways to maintain genome integrity. Nucleic Acids Res. 2014; 42:370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kirkman L., Deitsch K.. Antigenic variation and the generation of diversity in malaria parasites. Curr. Opin. Microbiol. 2013; 15:456–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ostera G., Tokumasu F., Oliveira F., Sa J., Furuya T., Teixeira C., Dvorak J.. Plasmodium falciparum: food vacuole localization of nitric oxide-derived species in intraerythrocytic stages of the malaria parasite. Exp. Parasitol. 2008; 120:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ghigo D., Todde R., Ginsburg H., Costamagna C., Gautret P., Bussolino S.F., Ulliers D., Giribaldi G., Deharo E., Gabrielli G. et al. . Erythrocyte stages of Plasmodium falciparum exhibit a high Nitric Oxide Synthase (NOS) activity and release an NOS-inducing soluble factor. J. Exp. Med. 1995; 182:677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Becker K., Tilley L., Vennerstrom J.L., Roberts D., Rogerson S., Ginsburg H.. Oxidative stress in malaria parasite-infected erythrocytes: host – parasite interactions. Int. J. Parasitol. 2004; 34:163–189. [DOI] [PubMed] [Google Scholar]
- 74. Müller S. Redox and antioxidant systems of the malaria parasite Plasmodium falciparum. Mol. Microbiol. 2004; 53:1291–1305. [DOI] [PubMed] [Google Scholar]
- 75. Caulfield J.L., Wishnok J.S., Tannenbaum S.R.. Nitric oxide-induced deamination of cytosine and guanine in deoxynucleosides. J. Biol. Chem. 1998; 273:12689–12695. [DOI] [PubMed] [Google Scholar]
- 76. Muralidharan V., Goldberg D.E.. Asparagine repeats in Plasmodium falciparum proteins: good for nothing. PLoS Pathog. 2013; 9:e1003488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Frugier M., Bour T., Ayach M., Santos M.A.S., Rudinger-thirion J., Théobald-dietrich A., Pizzi E.. Low complexity regions behave as tRNA sponges to help co-translational folding of plasmodial proteins. FEBS Lett. 2010; 584:448–454. [DOI] [PubMed] [Google Scholar]
- 78. Hughes A.L. The evolution of amino acid repeat arrays in Plasmodium and other organisms. J. Mol. Evol. 2004; 59:528–535. [DOI] [PubMed] [Google Scholar]
- 79. Verra F., Hughes A.L.. Biased amino acid composition in repeat regions of Plasmodium antigens. Mol. Biol. Evol. 1999; 16:627–633. [DOI] [PubMed] [Google Scholar]
- 80. Horrocks P., Wong E., Russell K., Emes R.D.. Control of gene expression in Plasmodium falciparum – ten years on. Mol. Biochem. Parasitol. 2009; 164:9–25. [DOI] [PubMed] [Google Scholar]
- 81. Cheeseman I.H., Gomez-escobar N., Carret C.K., Ivens A., Stewart L.B., Tetteh K.K.A., Conway D.J.. Gene copy number variation throughout the Plasmodium falciparum genome. BMC Genomics. 2009; 10:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cooper G.M., Nickerson D.A., Eichler E.E.. Mutational and selective effects on copy-number variants in the human genome. Nat. Genet. 2007; 39:S22–S29. [DOI] [PubMed] [Google Scholar]
- 83. de Smith A.J., Walters R.G., Coin L.J.M., Steinfeld I., Yakhini Z., Froguel P., Blakemore A.I.F.. Small deletion variants have stable breakpoints commonly associated with Alu elements. PLoS One. 2008; 3:e3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P. et al. . Origins and functional impact of copy number variation in the human genome. Nature. 2010; 464:704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Heinberg A., Siu E., Stern C., Lawrence E.A., Ferdig M.T., Deitsch K.W., Kirkman L.A.. Direct evidence for the adaptive role of copy number variation on antifolate susceptibility in Plasmodium falciparum. Mol. Microbiol. 2013; 88:702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Woodrow C.J., Krishna S.. Antimalarial drugs: recent advances in molecular determinants of resistance and their clinical significance. Cell. Mol. Life Sci. 2006; 63:1586–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nair S., Miller B., Barends M., Jaidee A., Patel J., Mayxay M., Newton P., Nosten F., Ferdig M.T., Anderson T.J.C.. Adaptive copy number evolution in malaria parasites. PLOS Genet. 2008; 4:e1000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nair S., Nash D., Sudimack D., Jaidee A., Barends M., Uhlemann A.-C., Krishna S., Nosten F., Anderson T.J.C.. Recurrent gene amplification and soft selective sweeps during evolution of multidrug resistance in malaria parasites. Mol. Biol. Evol. 2007; 24:562–573. [DOI] [PubMed] [Google Scholar]
- 89. Guler J.L., Freeman D.L., Ahyong V., Patrapuvich R., White J., Gujjar R., Phillips M.a, DeRisi J., Rathod P.K.. Asexual populations of the human malaria parasite, Plasmodium falciparum, use a two-step genomic strategy to acquire accurate, beneficial DNA amplifications. PLoS Pathog. 2013; 9:e1003375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Brown T.S., Jacob C.G., Silva J.C., Takala-Harrison S., Djimdé A., Dondorp A.M., Fukuda M., Noedl H., Nyunt M.M., Kyaw M.P. et al. . Plasmodium falciparum field isolates from areas of repeated emergence of drug resistant malaria show no evidence of hypermutator phenotype. Infect. Genet. Evol. 2015; 30:318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lee A.H., Fidock D.A.. Evidence of a mild mutator phenotype in Cambodian Plasmodium falciparum malaria parasites. PLoS One. 2016; 11:e0154166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Avril M., Tripathi A.K., Brazier A.J., Andisi C., Janes J.H., Soma V.L., Sullivan D.J., Bull P.C., Stins M.F., Smith J.D.. A restricted subset of var genes mediates adherence of Plasmodium falciparum-infected erythrocytes to brain endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E1782–E1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jensen A.T.R., Magistrado P., Sharp S., Joergensen L., Lavstsen T., Chiucchiuini A., Salanti A., Vestergaard L.S., Lusingu J.P., Hermsen R. et al. . Plasmodium falciparum associated with severe childhood malaria preferentially expresses PfEMP1 encoded by group A var genes. J. Exp. Med. 2004; 199:1179–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mu J., Awadalla P., Duan J., McGee K.M., Joy D.a., McVean G.a T., Su X.Z.. Recombination hotspots and population structure in Plasmodium falciparum. PLoS Biol. 2005; 3:e335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Joy D.A., Feng X., Mu J., Furuya T., Chotivanich K., Krettli A.U., Ho M., Wang A., White N.J., Suh E. et al. . Early origin and recent expansion of Plasmodium falciparum. Science. 2003; 300:318–321. [DOI] [PubMed] [Google Scholar]
- 96. Nosten F., White N.J.. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 2007; 77:181–192. [PubMed] [Google Scholar]
- 97. Ariey F., Witkowski B., Amaratunga C., Beghain J., Langlois A.-C., Khim N., Kim S., Duru V., Bouchier C., Ma L. et al. . A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014; 505:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ashley E.A., Dhorda M., Fairhurst R.M., Amaratunga C., Lim P., Suon S., Sreng S., Anderson J.M., Mao S., Sam B. et al. . Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2014; 371:411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Takala-Harrison S., Jacob C.G., Arze C., Cummings M.P., Silva J.C., Dondorp A.M., Fukuda M.M., Hien T.T., Mayxay M., Noedl H. et al. . Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in southeast Asia. J. Infect. Dis. 2014; 211:670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Miotto O., Amato R., Ashley E. a, MacInnis B., Almagro-Garcia J., Amaratunga C., Lim P., Mead D., Oyola S.O., Dhorda M. et al. . Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 2015; 47:226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. MalariaGEN Plasmodium falciparum Community Project. Genomic epidemiology of artemisinin resistant malaria. Elife. 2016; 5:e08714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.