


Abstract
Bacteria can become transiently tolerant to several classes of antibiotics. This phenomenon known as persistence is regulated by small genetic elements called toxin–antitoxin modules with intricate yet often poorly understood self-regulatory features. Here, we describe the structures of molecular complexes and interactions that drive the transcription regulation of the ccdAB toxin–antitoxin module. Low specificity and affinity of the antitoxin CcdA2 for individual binding sites on the operator are enhanced by the toxin CcdB2, which bridges the CcdA2 dimers. This results in a unique extended repressing complex that spirals around the operator and presents equally spaced DNA binding sites. The multivalency of binding sites induces a digital on-off switch for transcription, regulated by the toxin:antitoxin ratio. The ratio at which this switch occurs is modulated by non-specific interactions with the excess chromosomal DNA. Altogether, we present the molecular mechanisms underlying the ratio-dependent transcriptional regulation of the ccdAB operon.
INTRODUCTION
Persisters are non-growing, metabolically subdued cells within a bacterial population that are able to survive episodes of stress including antibiotic challenges (1,2). In contrast to resistance, which is an inherited trait that allows bacteria to grow in the presence of antibiotics, persistence is only temporary: when leaving the persistent state, bacterial cells again become sensitive and produce sensitive offspring (3). Persister cells are induced stochastically in a bacterial population, but their frequency depends on environmental conditions, and in particular the availability of nutrients. Persisters are rare in an exponentially growing culture and more abundant in the stationary phase (4). They are most important in biofilms and contribute to the difficulties in treating relapsing, chronic and biofilm-producing bacterial infections (5,6).
Toxin–antitoxin modules are found on the chromosomes and plasmids of most prokaryotes (7,8). The toxin generally targets the transcription or translation machinery of the cell, thereby inhibiting cell growth and potentially killing the cell. The antitoxin impairs the activity of the toxin, allowing cells to grow undisturbed. In type II toxin–antitoxin modules, this antitoxin is a protein that neutralizes the toxin by complex formation, but can be rapidly degraded by cellular proteases, releasing the active toxin. Different TA modules contribute cumulatively to persister frequency in Escherichia coli in non-stressed conditions during exponential growth (9). Activation of individual TA toxins has also been linked to specific stresses (10,11).
The ccdAB operon on the E. coli F plasmid was the first type II TA module to be discovered (12) and is well studied in terms of the gyrase poisoning activity of the toxin CcdB2 (13–15) and the rejuvenation action of CcdA2, i.e. its capacity to resolve poisoned CcdB2-gyrase complexes (16–18). Initially only seen as a locus that couples plasmid replication to cell division (12), it is now also known to contribute to persistence (19). The antitoxin CcdA2 can bind to DNA using its N-terminal ribbon–helix–helix domain (20) and is specifically degraded by Lon protease (21), while the interaction of CcdB2 with DNA gyrase results in DNA cleavage and inhibits transcription by forming a roadblock for the passage of the RNA polymerases (15). There are two equivalent but partially overlapping binding sites for the CcdA2 antitoxin on CcdB2. The first CcdA2 molecule binds the free CcdB2 with a high affinity, but its presence obstructs the binding of the second CcdA2, which subsequently binds with a low affinity. While only the high affinity binding is required for the rejuvenation of CcdB-poisoned gyrase, both binding sites play a role in the transcriptional regulation (18).
TA modules are precisely regulated, since their toxicity to the cell is determined by the molar toxin:antitoxin ratio. In many type II TA modules, this is accomplished by a negative autoregulation mechanism called conditional cooperativity. This mechanism has been investigated in detail at the molecular level for the phd/doc and the relBE TA modules (22–24). In general, the antitoxin binds the operator DNA, which overlaps with the promoter, and the toxin can either function as a co-repressor or a de-repressor for the antitoxin, depending on the amounts of toxin and antitoxin present in the cell (22,25).
Regulation of the F-plasmid ccdAB operon also involves conditional cooperativity (26), but the underlying mechanism is not understood at the molecular level. An intriguing open question is the role of the unusually long (113 bp) ccdAB promoter/operator region, containing eight putative antitoxin binding sites (27,28). It has been proposed that avidity plays a role in the transcriptional regulation of this operon (18,29). Avidity, also referred to as functional affinity, describes the cumulative strength of multiple non-covalent binding interactions (30). The concept of avidity was coined in the context of the polyvalency of antibodies, where multiple binding sites were observed to enhance antigen binding (31,32). Avidity effects have since been observed in many biochemical contexts including the interactions between carbohydrates and lectins (33,34) and pattern recognition by collectins in the innate immune system (35,36). Such avidity effects, induced by the bridging of antitoxins by toxins, cause an increase in the apparent affinity of the repressing complex for the operator DNA.
Here, we investigate the biochemical basis of the avidity in the transcriptional regulation of the ccdAB toxin–antitoxin module and present structures of complexes involved in this regulation. At low toxin:antitoxin ratios, the operator is repressed by a chain of alternating CcdA2 antitoxins and CcdB2 toxins spiraling around the DNA. As the toxin:antitoxin ratio increases, the repression is relieved because of the preferential formation of the V-shaped non-repressing CcdB2-CcdA2-CcdB2 heterohexamer. The forces that drive this ratio-dependent switching were identified by a global thermodynamic analysis. Together, these analyses put forward the molecular mechanisms underlying the avidity effects in the conditional cooperativity regulation of the ccdAB toxin–antitoxin module.
MATERIALS AND METHODS
Protein production
The GyrA fragments GyrA14 and GyrA59 were purified according to the protocol described for GyrA14 by Dao-Thi et al. (37).
The antitoxin CcdA2 was purified using a protocol modified from Van Melderen et al. (38). An overnight preculture of E. coli CSH50 lon::Tn10 (pULB2709) (16), grown at 37°C in Terrific Broth medium supplemented with ampicillin (100 μg/ml), was diluted 30 times in Terrific Broth and grown at 37°C. When the culture reached an OD600 of 0.6 to 0.8, it was induced by adding isopropyl β-D-thiogalactopyranoside at a final concentration of 1 mM. After overnight incubation at 28°C, the cells were harvested and resuspended in lysis buffer (50 mM Tris-HCl pH 8, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 2% glycerol, 0.1 mg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, 1 μg/ml leupeptine).
The cells were lysed by sonication and the cell debris was removed by centrifugation. Next, the protein was precipitated with 40% saturated ammonium sulfate. After centrifugation, the pellet was resuspended in and dialyzed against 20 mM Tris pH 8 (buffer A). It was then applied to a Source 30Q anion exchange column equilibrated with buffer A. Proteins were eluted using a linear gradient of 20 mM Tris pH 8, 1 M NaCl (buffer B). The fractions containing CcdA2 were pooled, concentrated and loaded on a Superdex 200 16/90 column (GE Healthcare), equilibrated with buffer B. After gel filtration, the relevant fractions were pooled, dialyzed against distilled water, flash-frozen in liquid nitrogen and lyophilized overnight. The protein was finally checked for purity using mass spectrometry and the quality of the preparation was verified using SAXS on an in-house Rigaku BioSAXS 2000.
The toxin CcdB2 was available from previous studies (18).
Small angle X-ray scattering
Data were collected at the SWING beamline, SOLEIL synchrotron (Gif-Sur-Yvette, France) in high pressure (or high performance) liquid chromatography mode (39). The CcdB2-CcdA2-CcdB2 complex was prepared at a concentration of 7 mg/ml in 10 mM Tris pH 7.3, 50 mM NaCl by slowly titrating CcdA2 into an excess of CcdB2. The sample was injected onto a Shodex KW402.5-4F column, which had been pre-equilibrated with running buffer (10 mM Tris pH7.3, 50 mM NaCl) for at least one column volume. The flow rate was 0.2 ml/min and data were collected with an exposure time of 750 ms and a dead time of 750 ms. Buffer data were collected at the beginning of the chromatogram and sample data were collected in the peak area. Data reduction was performed on-line using the FOXTROT software (SWING), while buffer subtraction and data averaging was performed using DATASW (40). Further analysis of the resulting scattering curve was performed using the ATSAS package (41). The indirect transform program GNOM was used to calculate the particle distance distribution function p(r) (42). Ten ab initio shape reconstructions were generated using DAMMIF (43) and averaged using DAMAVER (44). The shown ab initio envelope corresponds to the damfilt model.
Based on the N-terminal region of the solution structure of CcdA2 (20), encompassing residues 1 until 39 (PDB: 2ADL) and the crystal structure of the complex of CcdB2 with the C-terminal part of CcdA, CcdA37-72 (PDB: 3HPW), a model for the CcdB2-CcdA2-CcdB2 complex (model A) was built, assuming that the CcdA2 α-helices from the two models will extend into one continuous α-helix. The Allosmod-FOXS server (45,46) was then used to generate 100 structures similar to the input structure. The theoretical scattering curve of these models was then compared to the experimental data using CRYSOL (47).
Electron microscopy sample preparation
A 1000 bp long DNA fragment containing the ccdAB promoter/operator region was PCR amplified with primers EM1000centrFwd (5΄-AATTGTGATGCTTCTAAAATTACTA-3΄) and EM1000centrRev (5΄-GGTTAATGGCGTTTTTGATGT-3΄) from the F’ plasmid present in E. coli CSH100 (48). The resulting PCR fragment was then purified and concentrated using the Promega Wizard SV Gel and PCR clean-up system.
All binding reactions were performed in 30 mM Tris HCl pH 7.5, 200 mM NaCl, 0.5 mM EDTA. For the thin filaments, the binding reactions occurred at 20°C. DNA was first incubated with CcdA2 for 15 min. After adding CcdB2, the resulting mixture with final concentrations of 15 nM for the DNA duplex and 2.5 μM for CcdA2 and CcdB2 was incubated for another 15 min before preparing the EM sample. Thick filaments were prepared at 4°C. Again, the DNA was first incubated for 15 min with CcdA2 alone. After adding CcdB2, the mixture with final concentrations of 15 nM for the DNA duplex and 10 μM for CcdA2 and CcdB2 was incubated overnight at 4°C.
Negative stain samples were prepared by applying three microliters of CcdA2-CcdB2-DNA mixture supplemented with 5 mM MgAc2 on a freshly glow-discharged carbon-coated grid for 5 min after which the grid was washed with 5 mM MgAc2 and adsorbed protein stained with 1% uranyl formate.
Samples for cryogenic electron microscopy (cryo-EM) were prepared by applying 2 µl of CcdA2-CcdB2-DNA on a freshly glow-discharged holey carbon grid (Quantifoil, Germany) with additional 2 nm thick carbon layer. The sample was blotted manually for 2 s and vitrified by plunging in liquid ethane.
EM data collection and image processing
All EM images were collected on a JEOL JEM-1400 electron microscope equipped with LaB6 cathode and operated at 120 kV. Images were recorded on 4096 × 4096 pixels CMOS TemCam-F416 (TVIPS) camera. The negative stain data were collected at nominal microscope magnification of 50 000 and corresponding pixel size at the detector of 2.29 Å. The defocus was in the range between 1.2 and 3.0 µm. For the sample prepared at high and low CcdA-CcdB/DNA ratio 382 and 358 micrographs were collected, respectively. A total of 77 cryo-EM images were collected at nominal magnification of 40 000, corresponding pixel size of 2.87 Å and defocus range of 1.6 to 3.0 Å. For image processing, helices were boxed in e2helixboxer (49), 2D classes and 3D reconstruction by IHRSR procedure were performed in SPARX (50). The hand of the thick helices was determined by the tilt method (51) (see Supplementary Text: Electron Microscopy).
Model building
First, a model for the 113 bp ccdAB promoter/operator DNA was generated using the 3D-DART server (52). The helical axis of the model was aligned to the axis of the EM map. To correctly place the first CcdA2 dimer on the central 5΄-GTATAC-3΄ binding site, the solution structure of CcdA2 in complex with this binding site (PDB 2H3C) (20) was first aligned with the DNA. Then, the antitoxin CcdA2 from the CcdB2-CcdA2-CcdB2 heterohexamer refined to the experimental SAXS curve was aligned to this CcdA2 dimer. The helical symmetry of the 3D reconstruction was then applied to CcdA2 to generate the adjacent antitoxin on the DNA. To connect the two antitoxins, the crystal structure of CcdB2 in complex with two CcdA peptides was used (PDB 3G7Z) (18). The CcdA α helices in the latter structure were aligned with the CcdA2 α helices from the CcdB2-CcdA2-CcdB2 heterohexamer. The minimal helical unit for the protein chain finally consists of residues 1–40 of the two central CcdA antitoxin monomers from the CcdB2-CcdA2-CcdB2 heterohexamer, combined with residues 41–72 of the two CcdA peptides and the CcdB2 toxin from structure 3G7Z. The geometry of this minimal helical unit was then optimized using Coot (53). The helical symmetry of the 3D reconstruction was applied to this minimal helical unit to obtain a continuous model consisting of alternating CcdA2 and CcdB2 dimers. For the thick helices, two additional strands were generated by translating the minimal helical unit by 67.2 Å and 134.4 Å along the helical axis, and again applying the helical symmetry. Model building was performed in UCSF Chimera (54).
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSAs) were performed on 5΄-32P single-end-labeled oligonucleotide duplexes (Table 1). These were prepared by labeling one oligonucleotide of each duplex with γ-32P-ATP (Perkin Elmer) and T4 polynucleotide kinase (Fermentas). The duplexes were then annealed by incubating both oligonucleotides at 80°C during 10 min and cooling slowly overnight in a water bath. The labeled duplexes were purified by electrophoresis on a native 8% polyacrylamide gel.
Table 1. Oligonucleotides used in this work. Underlined sequences indicate the three central binding sites.
| Name | Sequence |
|---|---|
| Ccd1 | 5΄-ACGTACCTTCCTCTTTATGTATACCCGGCAGGACTGGAAATA-3΄ |
| 3΄-TGCATGGAAGGAGAAATACATATGGGCCGTCCTGACCTTTAT-5΄ | |
| Ccd2 | 5΄-ACGTACCTTCATATATACTGATATGTATACCCGCTGGAAATA-3΄ |
| 3΄-TGCATGGAAGTATATATGACTATACATATGGGCGACCTTTAT-5΄ | |
| Ccd3 | 5΄-ACGTGCGGTATAAGAATATATACTGATATGTATACCCGAATA-3΄ |
| 3΄-TGCACGCCATATTCTTATATATGACTATACATATGGGCTTAT-5΄ | |
| Random | 5΄-TGCCCAAGTGATGCTAAACAAGACTTAGCTGGTTCCTGTGTT-3΄ |
| 3΄-ACGGGTTCACTACGATTTGTTCTGAATCGACCAAGGACACAA-5΄ | |
| Full length | 5΄-CAGTATGCGTATTTGCGCGCTGATTTTTGCGGTATAAGAATATATACT |
| 3΄-GTCATACGCATAAACGCGCGACTAAAAACGCCATATTCTTATATATGA | |
| GATATGTATACCCGAAGTATGTCAAAAAGAGGTGTGCTATGAAGCAGCGTA | |
| CTATACATATGGGCTTCATACAGTTTTTCTCCACACGATACTTCGTCGCAT | |
| TTACAGTGACAGTT-3΄ | |
| AATGTCACTGTCAA-5΄ | |
| CcdITC | 5΄-GCGGTATAAGAATATATACTGATATGTATACCC-3΄ |
| 3΄-CGCCATATTCTTATATATGACTATACATATGGG-5΄ |
All binding reactions were performed at 20°C in 30 mM Tris-HCl pH 7.5, 200 mM NaCl, 0.5 mM EDTA, 150 μg/ml bovine serum albumin, in the presence of 0.25 μg/lane sonicated salmon sperm DNA. First, CcdA2 and labeled DNA (7500 cpm) were incubated together for 15 min in a total volume of 10 μl. After adding CcdB2, the resulting mixture of 15 μl was incubated again for 15 min. The samples were then mixed with 3 μl loading dye (25% ficoll, 0.1% xylene cyanol, 0.1% bromophenol blue) and loaded on a native 8% (for the 42 bp fragments) or 6% (for the full length operator) polyacrylamide gel prepared with TBE buffer (89 mM Tris-HCl, 89 mM boric acid, 2.5 mM EDTA). The electrophoresis was then performed at 12 V/cm until the sample had penetrated into the gel, and at 8 V/cm during 3 h, using TBE as the running buffer. An X-ray sensitive film was exposed to the gel overnight and then developed. The further data analysis is described in the supplementary text.
SwitchSENSE
All experiments were carried out on a switchSENSE analyzer DRX 2400 (Dynamic Biosensors GmbH, Munich, Germany). The switchSENSE technology evaluates the electrically actuated dynamic movement of DNA nanolevers that are immobilized on gold microelectrodes. Briefly, the DNA nanolevers which are modified with a fluorescent dye at the distal end are driven to oscillate on the surface of microelectrodes by alternating electric fields and their orientation-switching is analyzed by time-resolved single photon counting as described by Langer et al. (55). The nucleotide sequence of the DNA nanolevers was designed specifically to include the target six base-pair DNA sequence. Upon binding of an interaction partner to the DNA its movement is slowed down due to the increased hydrodynamic drag, which can be evaluated by a change in the Dynamic Response.
The on-chip exchange of DNA sequences was carried out by use of EXMAS system (exchangeable modular anchor sequences, Dynamic Biosensors GmbH, Munich, Germany), which is based on the specific hybridization of complementary overlapping strands.
Isothermal titration calorimetry (ITC)
Purified, lyophilized proteins were dissolved in water, then dialyzed against phosphate buffer (10 mM Na2HPO3, 10 mM NaH2PO3, 150 mM NaCl, 1 mM EDTA, pH 7.2). After dialysis, their concentrations (expressed as dimer equivalents for CcdA2, CcdB2 and the gyrase fragments) were determined by measuring absorbance of light at 280 nm (56). DNA oligonucleotides were similarly dissolved in water, complementary strands were mixed and annealed by heating to 90°C and cooling down to room temperature at 1°C/min. DNA concentration was determined by measuring absorbance at 260 nm (57) and correct 1:1 annealing was confirmed by a job plot (58). All solutions were filtered through a 0.45 μm membrane and degassed prior to titration. Titration was performed using the VP-ITC instrument (MicroCal, now part of Malvern Instruments, UK). Typical concentrations used were 1–3 μM protein in the cell and a 5- to 20-fold higher concentration of the titrant in the syringe. Typical injection volumes were 5–10 μl. The thermodynamic model of the ccdAB system (described below) was fitted to experimental data from all titrations simultaneously to yield the set of thermodynamic parameters that best describes the interactions between different binding partners at different temperatures.
Because of experimental limitations, not all binding reactions in the system could be measured directly. For instance, CcdA2 and CcdB2 tend to form insoluble aggregates at molar ratios around 1:1, presumably by forming long chains of alternating dimers (…CcdA2:CcdB2:CcdA2:CcdB2…) (28). To work around that, most titrations were performed using a truncated version of CcdA, CcdAC (Supplementary Figure S1A–D). CcdAC, also known as CcdA37–72, is comprised of the entire C-terminal domain that binds to CcdB2, but lacks the dimerization domain so it cannot form chains of alternating dimers (18,59). To study DNA binding, on the other hand, full-length CcdA2 was used; in this case DNA was always present in sufficient amounts to take up any CcdA2:CcdB2 1:1 complexes before they could aggregate. Another limitation is that affinity of the first CcdA2 dimer for CcdB2 cannot be accurately assessed from direct titrations because the very strong binding makes isothermal titration calorimetry (ITC) titration curves very sharp and compatible with a wide range of binding affinities during fitting (Supplementary Figure S1A). This was overcome by first titrating CcdB2 with a truncated gyrase dimer, GyrA142, (Supplementary Figure S1B) and then titrating the CcdB2:GyrA142 complex with CcdAC (Supplementary Figure S1C). In the second titration, CcdAC needs to displace GyrA142 before it can bind to CcdB2, so the free energy of this process gives us the difference between CcdAC:CcdB2 binding and CcdB2:GyrA142 binding, from which the CcdAC:CcdB2 affinity can be calculated. Similarly, the affinity of CcdB2 for the poorly soluble full-length gyrase (GyrA592) was obtained by titrating the CcdB2:GyrA592 complex with CcdAC and comparing that to the separately determined affinity of CcdAC for CcdB2.
The thermodynamic parameters of all these processes were determined by fitting a thermodynamic model to all data simultaneously. The global fitting methodology has already been described in detail (59). It is important to note that a global fit, i.e. one that optimizes every model parameter against all available data, was necessary to make sure all model parameters are consistent with each other and with all the data from different titrations (60).
Global thermodynamic model
The thermodynamics of the ccdAB system were described in terms of the following reaction equilibria:
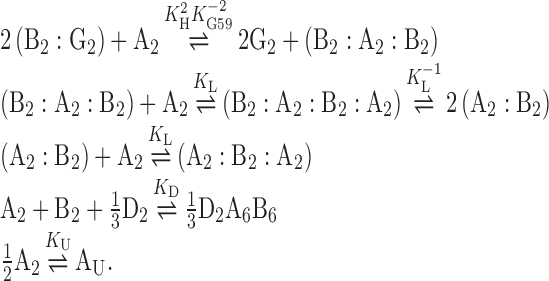 |
A, B, G and D represent CcdA, CcdB, GyrA and promoter DNA (central 3 binding sites), respectively. The different Ki are the equilibrium constants describing the high-affinity binding of the first CcdA2 antitoxin to CcdB2 (KH), the low-affinity binding of the second CcdA2 antitoxin to the same CcdB2 (KL), the binding of CcdB2 to GyrA2 (KG), the binding of each CcdA2:CcdB2 complex to operator DNA (KD) and the temperature-induced denaturation of CcdA2 (KU). The latter reaction was studied to verify that the unfolding of CcdA2 does not play a significant role in the regulation of ccdAB (see Supplementary Data for details). For each reaction in the above scheme, the equilibrium equation states that the equilibrium constant must equal the ratio between the equilibrium concentrations of products and reactants:
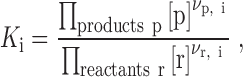 |
where [p] and [r] are the equilibrium concentrations of products and reactants, respectively, while νp,i and νr,i are their stoichiometric coefficients in reaction i. The six equilibrium reactions, combined with the conservation of mass for each of the four basic building blocks (CcdA, CcdB, GyrA and DNA), define a system of non-linear equations that can be solved to yield the equilibrium composition of the entire system. Thus the system composition is completely defined by the constants Ki and total concentrations of each building block cj.
c j were determined by measuring the concentrations of each solution going into an experiment, while Ki were calculated from the standard Gibbs free energy of the reaction, ΔG°i = -RT lnKi, where R is the gas constant and T is the absolute temperature. ΔG°i changes with temperature according to the Gibbs–Helmholtz relation,
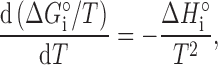 |
and the Kirchhoff relation,
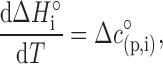 |
where ΔH°i and Δc°p,i are the standard enthalpy and heat capacity of reaction i. With Δc°p,i assumed to be constant across the range of experimental temperatures, these three equations allowed us to calculate Ki at any temperature using three parameters: ΔG°i(25°C), ΔH°i(25°C) and Δc°p,i. From Ki and cj we calculated the equilibrium concentrations of all molecular species using the Newton method with adaptive step size (61). By calculating the composition of the experimental system at each point during an experimental run (either ITC, DSC or CD temperature denaturation scans), we were able to calculate the model-based value of the experimental signal, dependent only on the parameters ΔG°i(25°C), ΔH°i(25°C) and Δc°p,i. These parameters were adjusted to produce the best fit of model-based signal to experimentally measured values by minimizing χ2 using the Nelder–Mead optimization algorithm with simulated annealing (61).
Simulations of the ccdAB regulation
All simulations are based on the experimentally determined equilibrium constants obtained from the global analysis of the ITC data (Supplementary Table S1). For a given model, a set of non-linear equations was defined by the equilibrium constants and total concentrations of the building blocks cj and solved using the trust-region dogleg algorithm (62) as implemented in the fsolve function from the scipy.optimize package (based on MINPACK1) (63). Total concentrations of toxin and antitoxin dimers were in 0–1 μM range, while fixed total concentrations of operator (1 nM), gyrase (100 nM) and non-specific DNA (42 μM) were assumed.
Two models were used in the simulations of the ccdAB system behavior: the simple and the extended model. In the absence of non-specific DNA, both models provide very similar results (Supplementary Figure S2A and B), therefore the computationally less demanding simple model was used in such cases. Simple model was introduced in the previous section (see Global Thermodynamic Model) and considers formation of only one kind of alternating (CcdA2-CcdB2)n complex, where n is the number of the binding sites on the operator. For three binding sites, such model is in complete agreement with the experimental data, therefore it was used to calculate the phase space. To study the effects of the multiplicity of antitoxin binding sites in the system, we varied the number of binding sites assuming the additivity of the corresponding free energies (Supplementary Figure S3A). Influence of affinity constants variations on the system behavior were studied with n = 8 using the simple model (Supplementary Figure S3B–E).
The extended model is superior in describing the system when non-specific DNA is included (Supplementary Figure S2C and D). The extended model considers all possible chain complexes (CcdA2)n:(CcdB2)n-1, (CcdA2)n:(CcdB2)n and (CcdA2)n:(CcdB2)n+1 where n runs from 1 to 8. These complexes can bind to the operator and to the non-specific DNA. Here, we assume that non-specific DNA is represented as a group of equivalent binding sites (64). Since the extended model assumes the presence of over 200 different species, the calculation of the whole phase diagram proved to be computationally too demanding. We performed calculations only on the 1D sections of the phase diagram.
RESULTS
The non-repressing CcdB2-CcdA2-CcdB2 heterohexamer is a V-shaped complex
Repression of the ccdAB operon is known to be relieved at high toxin:antitoxin ratios, when a soluble heterohexameric CcdB2-CcdA2-CcdB2 species is formed (26). To understand the molecular mechanism of derepression, we first investigated the structure of this non-repressing complex using HPLC-SAXS (Figure 1A, Supplementary Figure S4A). Molecular weight calculations based on the absolute I(0), DATMOW and the excluded volume in ab initio modeling correspond well with the expected molecular weight for a complex with a CcdA2-CcdB4 stoichiometry (Supplementary Table S2).
Figure 1.

The solution structure of the CcdB2-CcdA2-CcdB2 non-repressing complex is a V-shaped heterohexamer. (A) Experimental scattering curve for the CcdB2-CcdA2-CcdB2 heterohexamer (black) and fit of the theoretical scattering curve (blue) calculated based on the model shown in panel B. The Guinier plot is shown in the inset. (B) Structural model for the CcdB2-CcdA2-CcdB2 heterohexamer fitted in the ab initio envelope generated based on the experimental scattering curve shown in panel A. The CcdA2 antitoxin dimer is shown in green and the CcdB2 toxin dimers are shown in blue.
An initial model for this complex was built by combining the solution structure of the N-terminal DNA binding domain of CcdA2 (20) and the crystal structure of the complex of CcdB2 with the C-terminal peptide of CcdA (18). After refinement with AllosMod-FoXS (45), the theoretical scattering curve from this model fits the experimental scattering curve with a χ2 of 2.5 (Figure 1A and B). Furthermore, the refined model fits well into an ab initio envelope generated using DAMMIF (Figure 1B). Interestingly, the dimensionless Kratky plot suggests a limited degree of dynamics in the CcdB2-CcdA2-CcdB2 hexamer (Supplementary Figure S4B).
CcdA2-CcdB2 complexes assemble on the DNA into two types of helical structures
To elucidate the structure of the ccdAB repressor complex, we visualized CcdA2 and CcdB2 bound to a 1000 bp DNA fragment of the F plasmid encompassing the 113 bp ccdAB operator using negative stain transmission electron microscopy. We found that depending on the protein concentration, two types of structures are formed: short, relatively thin spirals at lower CcdA2 and CcdB2 concentrations, and longer, thick, continuous filaments at higher protein concentrations (Figure 2). These structures are not limited to the operator region, but extend over the length of the entire DNA fragments.
Figure 2.

Negative stain electron microscopy shows the different complexes CcdA2 and CcdB2 form with a 1000 bp DNA fragment containing the 113 bp ccdAB operator region. (A) DNA without proteins. (B) DNA with CcdA2 and CcdB2 at concentrations of 2.5 μM. (C) DNA with CcdA2 and CcdB2 at concentrations of 10 μM.
The thin filaments have a diameter of ∼90 Å and are typically 100–250 nm long, while the thick helices have a diameter of ∼130 Å and are often more than 1 μm long. This suggests that either the protein chain extends further than the length of the DNA (∼340 nm), or it concatenates several individual pieces of the DNA duplex. The high protein concentrations at which the thick filaments are observed (with an excess of protein over binding sites on the DNA), combined with DNase footprinting experiments showing only a limited protection of the operator in the presence of CcdA2 and CcdB2 (28), indicate that the thin filaments closer reflect the arrangement of proteins and DNA in the repressing complex. However, while both helical assemblies appear heterogeneous, the thick helices are more homogeneous and contain longer straight sections than the thin ones, making them more suitable for cryo-EM. Therefore, a pseudo-atomic model for the thick filaments was obtained first to aid the structure determination of the actual operator complex.
A triple helix of alternating CcdA2 and CcdB2 dimers saturates the operator
The 3D structure of the thick filaments was studied using negative stain and cryo-EM (Supplementary Figure S5A–F). The 3D helical reconstruction, calculated to a resolution between 20 and 25 Å, shows that the filaments are formed by right-handed triple helices (Supplementary Figure S6A and B). Each helical strand contains five helical units per turn. High density at the helical axis of the cryo-EM reconstruction is consistent with double stranded DNA being present in the center of this triple protein helix (Supplementary Figure S6A - Cryo).
We used the SAXS structure of the CcdB2-CcdA2-CcdB2 hexamer described above to fit a molecular model in the EM reconstruction. This EM reconstruction is consistent with three continuous chains of alternating CcdA2 and CcdB2 dimers spiraling around the DNA (Figure 3A). The build-up of this complex is shown in Supplementary Figure S7. In this model, the antitoxins CcdA2 are bound in the major groove of the DNA as in the previously published CcdA2-DNA NMR ensemble (20) while CcdB2 toxins line the outer surface of the triple helix. These CcdB2 dimers do not form direct contacts with the DNA or each other, neither within protein strands nor between protein strands. Therefore, the binding of CcdA2–CcdB2 complexes to DNA is likely independent for different strands. The spacing of the CcdA2 molecules on the DNA mirrors the ∼12 bp spacing of the imperfect palindromes on the 113 bp operator sequence. In the triple helix arrangement, CcdA2 antitoxins are stacked next to each other in the major groove of the B-form DNA, which they fill completely (Supplementary Figure S8).
Figure 3.

The structures of CcdA2-CcdB2-DNA helical assemblies consist of chains of DNA-bound antitoxins bridged by toxins. (A) Front view and top view of the cryo-EM density map and structural model for the thick filaments, corresponding to three strands of alternating CcdA2 and CcdB2 dimers spiraling around the DNA. The three-start right-handed helices have a pitch of 208 Å, a corresponding unit helical rise of 41.6 Å and a helical twist of 72°. (B and C) Front view and top view of the negative stain EM density map and structural model for the thin filaments. The panels show the EM density map for the data set with a pitch of 215 Å, a unit helical rise of 38.4 Å and a unit twist of 64.3°. (B) The DNA is bound by one strand of alternating CcdA2 and CcdB2 dimers. (C) The DNA is bound by two strands of alternating CcdA2 and CcdB2 dimers. The second strand of alternating dimers (shown in lighter colors) is only partially occupied in the negative stain-EM ensemble. The three main binding sites for CcdA2 on the operator are indicated in black.
It is also noteworthy that this model can explain the concatenation of several DNA fragments: each of the three protein strands surrounding the DNA will either end with a CcdA2 dimer or with a CcdB2 dimer. When two such protein-bound DNA fragments meet, these compatible ends will bridge the two DNA fragments resulting in the observed long assembly of thick filaments.
Structural properties of the repressing complex
Negative stain EM images of the thin filaments indicate a significant heterogeneity in the helical parameters. Reference-free 2D class averages display differences in the pitch of the helices ranging from ca. 190 Å to 340 Å (Supplementary Figure S5G and Supplementary text: Electron Microscopy), with the refined pitch of the most populated class (203–230 Å) close to the pitch of the thick helices.
Three-dimensional reconstructions from the three most populated classes, calculated to a resolution between 23 and 31 Å (Supplementary Figure S6C), show that these helices are formed by a main strand of density that is similar to the strands observed in the 3D reconstructions of the thick helices. The heterogeneity of the helical parameters originates from differences in the unit helical twist angle (∼15° for pitches between 180 and 258 Å) rather than from differences in the helical rise (Supplementary Table S3) and is consistent with the limited flexibility observed for the CcdB2-CcdA2-CcdB2 hexamer.
The CcdA2-CcdB2-DNA complexes that are observed as thin filaments thus constitute in a first approximation a substructure of the thick filaments (assuming DNA is in the center), with one strand of CcdA2-CcdB2 repeats spiraling along the DNA. Therefore, a single chain of alternating CcdA2-CcdB2 repeats was built into the 3D reconstructions for the thin helices, as described above for the thick helices (Figure 3B). Analysis of this helical structure shows that a single strand of alternating CcdA2 and CcdB2 dimers is sufficient to bind to all the eight imperfect palindromes (this is shown in Figure 3B for the three central binding sites) and is consistent with the DNase I footprinting results (27,28). Therefore, we expect that a single stranded assembly is sufficient to form the repressing complex assembling in vivo.
Nevertheless, additional density unexplained by the single strand structural model was present in the 3D reconstruction for the thin filaments (Figure 3B, Supplementary Figure S9). We interpret this additional density as a second strand of toxin and antitoxin dimers with partial occupancy. In some of the micrographs for the thin helices, a second strand of features can unambiguously be observed (e.g. Figure 2B, right). This second strand was included in the model for the most populated class of thin helices by translating and rotating the first strand, ensuring that the CcdA2 DNA binding domains are still located in the major groove of the DNA (Figure 3C).
CcdA2 has low affinity and specificity for single operator sites
In order to further understand how the observed structures confer operator specificity and affinity in a cellular context, we studied the interaction of CcdA2 and CcdB2 with the operator using EMSA and ITC. The F plasmid ccdAB operator consists of a rather long 113 bp stretch of DNA that contains at least eight binding sites for the antitoxin CcdA2 (28). At the center of this operator are three short GTATAC palindromes, one perfect and two imperfect ones (20) (Figure 4A). It has been proposed that these three palindromes serve as nucleation sites for the binding of CcdA2 and CcdB2 to the complete operator (28) and thus that CcdA2 specifically recognizes this palindrome. To test this hypothesis, we used the SwitchSENSE technology to measure the interaction of CcdA2 with all 64 possible 6 bp palindrome sequences. Surprisingly, the results indicate that CcdA2 does not discriminate significantly between the different sequences (Supplementary Figure S10). Moreover, several CcdA2 dimers appear to bind simultaneously to the nanolever DNA used in the experiment, indicating poor specificity.
Figure 4.

Antitoxin CcdA2 has a low affinity and sequence specificity for ccdAB operator fragments. (A) The promoter/operator region of the ccdAB operon on the F plasmid of E. coli. The promoter/operator is shown in pink, the ccdA gene in green and the ccdB gene in blue. The three central binding sites of the operator are indicated with a black line, the −10 and −35 promoter elements (12) with a dotted box, and the parts of the ccdAB promoter/operator embedded in the DNA duplexes for EMSA experiments are indicated with a colored line. (B) Autoradiographs of EMSA analyses of CcdA2 binding to 42 bp duplexes of random DNA, to 42 bp duplexes with fragments of the ccdAB operator (1–3 binding sites) embedded in random DNA and to the 113 bp full length operator. The concentration of CcdA2 is indicated above the lanes. The positions of free DNA (marked as F) and DNA bound by CcdA2 (marked as A) are indicated next to the lanes. (C) Binding profiles of the EMSAs shown in panel B.
EMSA experiments carried out in the presence of an excess of non-specific competitor DNA provide further indications that CcdA2 has a low affinity and specificity for DNA. The affinity for a 42 bp sequence containing a single GTATAC palindrome (Figure 4B and C, Supplementary Table S4) is only slightly higher than the one for the random sequence of the same length (Figure 4B and C, Supplementary Table S4). As more palindromes (binding sites) are inserted into the fragment, we observe an increase in the apparent affinity, which becomes highest for the full 113 bp operator sequence (Figure 4B and C, Supplementary Table S4). These experiments show that some sequence specificity is associated with the binding of CcdA2 to the operator.
CcdB2 increases both affinity and specificity of CcdA2 for its operator
The affinity of CcdA2 for DNA increases significantly in the presence of CcdB2 (Figure 5A). This increase in affinity is observed for both short and full-length operator fragments and depends on the ratio of CcdB2 to CcdA2, in agreement with the presence of conditional cooperativity (12). For both the 42 bp fragment containing three binding sites as well as the full length operator, a clear increase in the affinity of CcdA2 for the DNA is observed as long as the CcdB2:CcdA2 ratio remains below 1. For the full length operator, the switch from repression to non-repression occurs abruptly at the moment the CcdB2:CcdA2 ratio exceeds 1.
Figure 5.

The toxin CcdB2 increases the affinity and specificity of the antitoxin CcdA2 for the DNA at low toxin:antitoxin ratios. Autoradiographs of EMSA analyses of CcdA2 and CcdB2 binding to fragments of the ccdAB operator. The concentrations of CcdA2 and CcdB2 are indicated above the lanes. The positions of free DNA (marked as F), DNA bound by CcdA2 (marked as A) and DNA bound by CcdA2 and CcdB2 (marked as AB) are indicated next to the lanes. (A) The CcdB2 concentration is varied in the presence of a constant concentration of CcdA2. (B and C) The concentration of CcdA2 and CcdB2 is varied while maintaining a constant toxin:antitoxin ratio.
Moreover, CcdB2 enhances the specificity of CcdA2 for its cognate DNA. In the presence of the toxin, a clear band shift is observed for the DNA fragment with three central binding sites at 1.5 μM CcdA2 and 0.75 μM CcdB2 (Figure 5B). On the other hand, no binding is observed to a random DNA fragment of the same length even at three times higher concentrations (Figure 5C). Thus, CcdB2 increases the affinity of CcdA2 for a native promoter sequence (with specific 5΄-GTATAC-3΄ palindromes) more than it does for a non-specific random sequence.
Coordinated binding of multiple CcdA2 domains ensures a high affinity for the operator DNA
Next, we used ITC to investigate the thermodynamics governing the regulation of ccdAB expression. For practical reasons, the operator fragment containing three central binding sites for CcdA2 was used. A model that considers (CcdA2-CcdB2)3:DNA, a single chain of three CcdA2-CcdB2 units bound to the DNA, as the only repressing species was fitted globally to the ITC data (60,65) (see Materials and Methods). This simple model agrees well with the whole data set and suggests that the formation of the repressing complex is highly favorable (Figure 6, Supplementary Figure S1, Supplementary Table S1). Assuming that pairwise binding free energies are approximately additive (see the Supplementary Data for a more rigorous discussion), most of the stability of this complex (80%) comes from the formation of the (CcdA2-CcdB2)3 alternating chain. The remainder can be ascribed to the binding of three CcdA2 dimers to three binding sites on the DNA (Figure 6B, ΔG°). This gives a free energy contribution of only −5.4 kcal/mol per binding site, which corresponds to a Kd value in the high micromolar range (Kd = eΔG°/RT). However, by coupling the three CcdA2 dimers together and forcing them to bind to the DNA simultaneously, their binding free energies combine. As the Kd values multiply, this results in a high affinity of (CcdA2-CcdB2)3 for DNA. The entropic cost of constraining multiple CcdA2 units into the alternating CcdA2-CcdB2 chain (−TΔS° >> 0) is paid for by the otherwise highly favorable CcdA2-CcdB2 interaction energy (ΔH° << 0) and desolvation of hydrophobic surfaces (ΔCp° < 0).
Figure 6.

Coupling multiple CcdA2 antitoxins together is necessary to obtain a high affinity for the operator DNA. (A) Titrations of CcdA2 into a CcdB2:DNA 3:1 mixture. Note that each DNA molecule (D2) has three binding sites for CcdA2 (A2). Inset: titration of CcdB2 (B2) into a CcdA2:DNA 3:1 mixture. Symbols represent experimental data while lines of the same color represent the best fit of the global thermodynamic model (see Supplementary Figure S1). (B) Thermodynamic fingerprint of the CcdA2-CcdB2-DNA interactions: thermodynamic parameters for the assembly of the repressing complex on a DNA fragment containing three binding sites (blue and green) compared to the binding of CcdA2 alone to one binding site on the DNA fragment (red; see also Supplementary Table S1).
The transcriptional autoregulation in the ccdAB operon is strongly ratio-dependent
The experimentally obtained thermodynamic parameters allow us to rigorously calculate the concentrations of each molecular species in the ccdAB system under a wide range of conditions. A schematic overview of the interactions in the toxin–antitoxin module, depending on the relative amounts of toxin and antitoxin, is shown in Figure 7A. More quantitatively, we calculated the regulatory phase space accessible to the ccdAB system (with an operator containing three binding sites for CcdA2) at given total concentrations of the antitoxin CcdA2 and the toxin CcdB2. The state of the operator (free or repressed) strongly depends on the ratio of the total CcdA2 and CcdB2 concentrations, with a characteristic diagonal division of the phase space, which is a hallmark of ratio-dependent gene regulation (66) (Figure 7B). The diagonal separates the area with the free operator (at toxin:antitoxin ratios above two) from the area with the repressed operator (at toxin:antitoxin ratios below two). Additionally, below a threshold toxin concentration the operator is always in the unbound state, allowing new antitoxin and toxin to be produced.
Figure 7.

Multiple coupled low-affinity binding sites on the operator ensure a tight ratio-dependent transcriptional regulation of the ccdAB operon. (A) Schematic overview of the regulation of the ccdAB operon. The toxin:antitoxin ratio increases from left to right. (B) Heat plot showing the fraction of repressed operator DNA as a function of the dimer concentrations of CcdA2 and CcdB2. (C) Horizontal cross-section of the phase diagram as indicated by the dotted line on the heat plot. The curves represent the fraction of free CcdA2 relative to the total amount of CcdA2 (green), the fraction of free CcdB2 relative to the total amount of CcdB2 (blue), the fraction of bound DNA relative to the total amount of DNA (magenta), the fraction of non-repressing complex CcdB2-CcdA2-CcdB2 relative to the total amount of CcdA2 (black) and the fraction of bound gyrase relative to the total amount of gyrase (orange). (D–F) Simulations of the regulation of the ccdAB operon demonstrate the effect of multiple binding sites and the effects of non-specific DNA. Panels C–E show simulations performed with the simple model in the absence of non-specific DNA, panel F shows the simulation with the extended model in the presence of non-specific DNA. Model descriptions are given in Materials and Methods. All calculations are performed at 25°C.
The ratio-dependent switching can then be inferred from one-dimensional sections in the phase diagram, where at a fixed antitoxin concentration we increase the concentration of the toxin (this corresponds to the setup of the EMSA experiments in Figure 5A). When CcdA2 is in excess and at least some CcdB2 is present to bridge the antitoxin, repressor complexes will form on the ccdAB promoter (Figure 7C). Such complexes are stabilized by both high and low affinity interactions and the interaction with the operator (Figure 6B). However, upon increase of the CcdB2 concentration, the more stable, non-repressing heterohexameric complexes form, breaking up the CcdA2-CcdB2 alternating chains and lifting repression. Although the switch to the heterohexameric complex is accompanied by the loss of favorable interactions with the operator and low-affinity CcdA2-CcdB2 interactions in the alternating chain, this is compensated by the formation of high-affinity interactions between CcdA2 and the additional CcdB2. Thus, switching between the repressing and the non-repressing complex is defined by the relative difference between the high and low affinity CcdA2-CcdB2 binding constants. The observed difference is six orders of magnitude (Supplementary Table S1), which is consistent with the optimal response predicted by our simulations (Supplementary Figure S3C and D).
A high multiplicity of operator binding sites ensures efficient repression in the presence of non-specific DNA
Thermodynamic modeling of the ccdAB system sheds light on how multiple binding sites affect the regulatory mechanism. If only one binding site on the operator is considered, the system does not exhibit repression at physiological concentrations due to the low DNA binding affinity of CcdA2 (Figure 7D). An increasing number of binding sites leads to stronger repression, eventually resulting in a digital-like switch for a model with eight binding sites on the operator (Figure 7E, Supplementary Figure S3A). Importantly, simulated systems with higher affinity lack the ratio-sensitive switch, indicating that the low affinity of CcdA2 for an individual binding site is crucial for the functionality of the mechanism (Supplementary Figure S3B). Low affinity for the operator ensures that only multivalent chain complexes of (CcdA2:CcdB2)n are active repressors while heterohexamers and isolated CcdA2 bind too weakly to be effective.
Given the low specificity of a single CcdA2 antitoxin for a binding site on the operator, we asked how functional repression can be achieved in the presence of non-specific DNA. The affinity constant for the non-specific DNA was assumed to be 1/3 of that for the specific DNA. Several values of the affinity constant for the non-specific DNA were tested and good agreement with EMSA results was found when the estimated value for the non-specific DNA was 1/3 of that for the specific one. The model of the system based on the ITC experiments that takes into account only (CcdA2-CcdB2)8 and CcdB2-CcdA2-CcdB2 as repressing and non-repressing complexes reproduces the EMSA results on the full length operator in the presence of non-specific DNA to a certain extent, in particular the ratio-dependent on-off behavior (Supplementary Figure S2C). However, agreement with the EMSA is significantly improved using an extended model, where all possible (CcdA2-CcdB2)n chain complexes on the operator and on non-specific DNA are considered (Figure 7F). Particularly, this extended model correctly predicts that in the presence of non-specific DNA the de-repression occurs at lower CcdB2 to CcdA2 ratios (but still above one), as observed in EMSA (Figure 5A). This stems from the ratio-dependence of the stoichiometry of the (CcdA2-CcdB2)n repressor complex: at low CcdB2:CcdA2 ratios, shorter chains with lower specificity are favored, which gradually grow to the more specific (CcdA2-CcdB2)8 complex at equimolar ratios, in accordance with gradual supershifting of the repressor complex observed in EMSA (Supplementary Figure S2E, Figure 5A). At CcdB2:CcdA2 ratios above 1, the species distribution changes rapidly and repression is relieved due to lack of specific high stoichiometry (CcdA2-CcdB2)n complexes and concomitant formation of the non-repressing CcdB2-CcdA2-CcdB2 complex. Thus, in contrast to what intuitively may be expected, a relatively weak specificity may not be detrimental but can be functionally relevant.
DISCUSSION
Conditional cooperativity is a central mechanism in the transcriptional regulation of several type II toxin–antitoxin modules, involving repression at low toxin:antitoxin ratios and derepression at high toxin:antitoxin ratios. Although the ccdAB operon on the F plasmid of E. coli was the first TA module found to be regulated via conditional cooperativity (26), the underlying molecular mechanisms are not yet fully understood.
We elucidated the autoregulation of the ccdAB module by determining the structures of the full repressing complex and the non-repressing CcdB2-CcdA2-CcdB2 heterohexamer and by a thermodynamic characterization of all known toxin–antitoxin–DNA interactions within the system. Based on this analysis, we also propose a quantitative model that allows us to describe and simulate the functional behavior of the entire system. The main hallmark of the ccdAB autoregulation is its dependence on the ratio of two proteins, which is manifested as the diagonal division of the regulatory phase space (Figure 7B).
The CcdB2-CcdA2-CcdB2 heterohexamer has a V-shaped architecture, with the DNA-binding domain of the antitoxin located in the center, and two toxins at the extremities. This general lay-out has also been found in several other TA modules, such as E. coli relBE (67), bacteriophage P1 phd/doc (68), E. coli mqsRA (69) and E. coli and Bacillus subtilis mazEF (70,71). The limited dynamics observed in the CcdB2-CcdA2-CcdB2 heterohexamer may serve to allow the CcdA2-CcdB2 dimers to accommodate operator binding sites with slightly different angles and distances between them without interrupting the repressing complex. This repressing complex has an unusual and hitherto unobserved architecture, consisting of an elongated chain of alternating DNA-bound CcdA2 antitoxins bridged by CcdB2 toxins. The ratio-dependent regulation can be entirely explained based on the increase of the affinity induced by the multiplicity of binding sites on the operator, in contrast to the phd/doc operon where cooperative and allosteric effects play a key role (22,23). This difference might be related to the structure of the operators – the ccdAB promoter/operator region contains eight putative antitoxin binding sites (28), compared to only two in phd/doc (72).
We found that the 3D map for the thin filaments cannot be explained by the presence of only one strand of alternating toxin and antitoxin dimers. However, as we discuss below, the second spiral detected with EM is most likely a result of a concentration-dependent effect observed only in the absence of an excess of non-specific DNA that would act as a sink for CcdA2-CcdB2 chain complexes. To clarify this issue, we performed simulations in which the formation of the second spiral is allowed via sequential binding of the CcdA2-CcdB2 chain to the pre-existing single spiral CcdA2-CcdB2-operator complex, using the ‘simple model’ in which only the formation of CcdA2-CcdB2 chains that cover the whole operator fragment is allowed. We find that at the concentrations used in EM measurements, the second spiral is always bound to the DNA fragment (the fraction of the operator bound by two spirals is more than 99% relative to the total operator DNA), while at the conditions of the ITC measurements, the fraction of the operator fragment with two bound CcdA2-CcdB2 chains is negligibly small.
By contrast, when an excess of non-specific DNA is included in these simulations, the fraction of the complex with a second spiral is less than 1% even at the highest protein concentrations used in the EM measurements. Based on these results, we conclude that the formation of the second spiral is strongly concentration dependent, but more significantly, that the presence of an excess of non-specific competitor DNA abolishes the formation of the second spiral. Therefore, we believe that in vivo, where the concentration of non-specific DNA is significant, the formation of the second spiral on the operator is very unlikely. Generally, the binding of the second spiral does not affect the ratio-dependent regulation, which was found to be independent of the total CcdA2 and CcdB2 concentrations in the 0.1–10 μM range. Since the in vivo toxin and antitoxin concentrations are very likely in this concentration range (25,73), we believe that the regulatory properties predicted by the model (presented in the phase diagram) correspond well with the situation in the cellular environment.
The influence of the non-specific competitor DNA on the operator binding by CcdA2 and CcdB2 is also clearly illustrated by the DNase I footprinting experiment performed by Dao-Thi et al. (28). While the region of DNA bound by CcdA2 and CcdB2 is larger than the actual ccdAB operator in our EM measurements, the DNase I footprint, performed in the presence of a large excess of non-specific competitor DNA, only includes eight well-defined binding sites in the 113 bp region identified by Tam and Kline (27).
The most unusual and intriguing aspect of the ccdAB autoregulation is the low affinity and specificity of CcdA2 for its binding sites on the operator. The low operator affinity of CcdA2 prevents repression in the absence of CcdB2. In the presence of the toxin, CcdB2 dimers will link CcdA2 dimers together to form multivalent chains where the weak affinities of individual CcdA2 molecules are multiplied to provide a strong avidity. The small difference between affinities for cognate and non-cognate binding sites is similarly enlarged, which explains why DNA binding is more sequence-specific in the presence of CcdB2. This effect breaks down at higher CcdB2:CcdA2 ratios when CcdB2-CcdA2-CcdB2 complexes start to form. With only one DNA binding domain per complex, they again bind the DNA weakly, with the additional limitation that adjacent binding sites cannot be occupied due to steric clashes between CcdB2 dimers. This ensures that repression is lifted once the toxin:antitoxin ratio becomes too high. To our knowledge, this mechanism is unique in transcription regulation. Even in the mazEF family, which is related to ccdAB in terms of toxin structure and dynamics as well as in the way by which the antitoxin interacts with the toxin and regulates its activity (74–76), no extended operator or poor discrimination of DNA sequences has been reported. The well-studied mazEF homologue kis/kid on plasmid R1 was indeed shown in vitro to form extended alternating chains of toxin and antitoxin (77), but the operator complex is limited to a Kid2-Kis2-Kid2-Kis2 complex binding to a piece of operator DNA with two binding sites (78).
The ccdAB system behaves non-intuitively in terms of specificity requirements. One would expect that poor discrimination of the correct binding sites on the operator from non-specific chromosomal DNA should be detrimental for the cell, both due to poorly controlled repression of the ccdAB operon (leading to CcdB2-based toxicity) and to uncontrolled random repression of genes, disturbing cellular physiology. Yet our model clearly shows that a tight repression can be established by the coupled binding of several antitoxins bridged by CcdB2 toxins to the binding sites on the operator. Furthermore, the fact that specificity for the operator increases with the length of the (CcdA2-CcdB2)n chain prevents random patches of chromosomal DNA from being covered with these high-affinity complexes, and therefore CcdA2 and CcdB2 are unlikely to deregulate normal gene expression in E. coli. Altogether, the ccdAB system provides a unique and non-intuitive solution to a complex problem of gene regulation that depends on the multiplicity of antitoxin binding sites on the operator to generate both affinity and specificity.
ACCESSION NUMBERS
The SAXS Scattering data and model for CcdB2-CcdA2-CcdB2 has been deposited to SASBDB (ID SASDBA9). The 3D reconstructions based on the EM data have been deposited to EMDB (ID EMD-3568 for the negative stain data set of the thin filaments and ID EMD-3569 for the cryo-EM data set of the thick filaments).
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the beamline staff from synchrotron Soleil – Swing for the technical support. The authors further thank Sarah Haesaerts for protein purification and Mike Sleutel for fruitful discussions.
Footnotes
Present addresses:
Alexandra Vandervelde, Department of Cellular and Molecular Medicine, KU Leuven, Leuven, 3000, Belgium.
Igor Drobnak, Department of Synthetic Biology and Immunology, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Vlaams Interuniversitair Instituut voor Biotechnologie [VIB6; Q1-2011 to R.L.]; Fonds voor Wetenschappelijk Onderzoek Vlaanderen [G.0135.15N to R.L. and R.E., G0C1213N, G.0878.12N; G.0090.11N to R.L., FWOTM637 to A.V.]; Onderzoeksraad of the Vrije Universiteit Brussel [OZR2232 to S.H., SPR13 to R.L.]; European Community's Seventh Framework Program (FP7/2007-2013) under BioStruct-X [projects 1673 and 6131 to R.L.]; Hercules Foundation [UABR/11/012 to R.L.]; Slovenian Research Agency [P1–0201 and J1-5448 to J.L.]; Bundesministerium für Bildung und Forschung (BMBF) [GO-Bio program 031A240 to T.W.]. Funding for open access charge: Fonds voor Wetenschappelijk Onderzoek Vlaanderen [G.0135.15N].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lewis K. Persister cells. Annu. Rev. Microbiol. 2010; 64:357–372. [DOI] [PubMed] [Google Scholar]
- 2. Lewis K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 2007; 5:48–56. [DOI] [PubMed] [Google Scholar]
- 3. Bigger J.W. Treatment of staphylococcal infections with penicillin. Lancet. 1944; 244:497–500. [Google Scholar]
- 4. Keren I., Kaldalu N., Spoering A., Wang Y., Lewis K.. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004; 230:13–18. [DOI] [PubMed] [Google Scholar]
- 5. Lewis K. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 2008; 322:107–131. [DOI] [PubMed] [Google Scholar]
- 6. Fauvart M., De Groote V.N., Michiels J.. Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J. Med. Microbiol. 2011; 60:699–709. [DOI] [PubMed] [Google Scholar]
- 7. Hayes F., Van Melderen L.. Toxins-antitoxins: diversity, evolution and function. Crit. Rev. Biochem. Mol. Biol. 2011; 46:386–408. [DOI] [PubMed] [Google Scholar]
- 8. Gerdes K., Christensen S.K., Lobner-Olesen A.. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005; 3:371–382. [DOI] [PubMed] [Google Scholar]
- 9. Maisonneuve E., Shakespeare L., Jørgensen M.G., Gerdes K.. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:13206–13211. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10. Dörr T., Vulić M., Lewis K.. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010; 8:e1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Christensen-Dalsgaard M., Jørgensen M.G., Gerdes K.. Three new RelE-homologous mRNA interferases of Escherichia coli differentially induced by environmental stresses. Mol. Microbiol. 2010; 75:333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miki T., Yoshioka K., Horiuchi T.. Control of cell-division by sex factor F in Escherichia coli. 1. The 42.84-43.6 F segment couples cell-division of the host bacteria with replication of F plasmid DNA. J. Mol. Biol. 1984; 174:605–625. [DOI] [PubMed] [Google Scholar]
- 13. Bernard P., Couturier M.. Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J. Mol. Biol. 1992; 226:735–745. [DOI] [PubMed] [Google Scholar]
- 14. Dao-Thi M.-H., Van Melderen L., De Genst E., Afif H., Buts L., Wyns L., Loris R.. Molecular basis of gyrase poisoning by the addiction toxin CcdB. J. Mol. Biol. 2005; 348:1091–1102. [DOI] [PubMed] [Google Scholar]
- 15. Critchlow S.E., O'Dea M.H., Howells A.J., Couturier M., Gellert M., Maxwell A.. The interaction of the F plasmid killer protein, CcdB, with DNA gyrase: induction of DNA cleavage and blocking of transcription. J. Mol. Biol. 1997; 273:826–839. [DOI] [PubMed] [Google Scholar]
- 16. Bernard P., Kézdy K.E., Van Melderen L., Steyaert J., Wyns L., Pato M.L., Higgins P.N., Couturier M.. The F plasmid CcdB protein induced efficient ATP-dependent DNA cleavage by gyrase. J. Mol. Biol. 1993; 234:534–541. [DOI] [PubMed] [Google Scholar]
- 17. Maki S., Takiguchi S., Horiuchi T., Sekimizu K., Miki T.. Partner switching mechanisms in inactivation and rejuvenation of Escherichia coli DNA gyrase by F plasmid proteins LetD (CcdB) and LetA (CcdA). J. Mol. Biol. 1996; 256:473–482. [DOI] [PubMed] [Google Scholar]
- 18. De Jonge N., Garcia-Pino A., Buts L., Haesaerts S., Charlier D., Zangger K., Wyns L., De Greve H., Loris R.. Rejuvenation of CcdB-poisoned gyrase by an intrinsically disordered protein domain. Mol. Cell. 2009; 35:154–163. [DOI] [PubMed] [Google Scholar]
- 19. Tripathi A., Dewan P.C., Barua B., Varadarajan R.. Additional role for the ccd operon of F-plasmid as a transmissible persistence factor. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:12497–12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Madl T., Van Melderen L., Mine N., Respondek M., Oberer M., Keller W., Khatai L., Zangger K.. Structural basis for nucleic acid and toxin recognition of the bacterial antitoxin CcdA. J. Mol. Biol. 2006; 364:170–185. [DOI] [PubMed] [Google Scholar]
- 21. Van Melderen L., Bernard P., Couturier M.. Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in piasmid-free segregant bacteria. Mol. Microbiol. 1994; 11:1151–1157. [DOI] [PubMed] [Google Scholar]
- 22. Garcia-Pino A., Balasubramanian S., Wyns L., Gazit E., De Greve H., Magnuson R.D., Charlier D., van Nuland N.A., Loris R.. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell. 2010; 142:101–111. [DOI] [PubMed] [Google Scholar]
- 23. Garcia-Pino A., De Gieter S., Talavera A., De Greve H., Efremov R.G., Loris R.. An intrinsically disordered entropic switch determines allostery in Phd-Doc regulation. Nat. Chem. Biol. 2016; 12:490–496. [DOI] [PubMed] [Google Scholar]
- 24. Overgaard M., Borch J., Jorgensen M.G., Gerdes K.. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 2008; 69:841–857. [DOI] [PubMed] [Google Scholar]
- 25. Overgaard M., Borch J., Gerdes K.. RelB and RelE of Escherichia coli form a tight complex that represses transcription via the ribbon-helix-helix motif in RelB. J. Mol. Biol. 2009; 394:183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Afif H., Allali N., Couturier M., Van Melderen L.. The ratio between CcdA and CcdB modulates the transcriptional repression of the ccd poison-antidote system. Mol. Microbiol. 2001; 41:73–82. [DOI] [PubMed] [Google Scholar]
- 27. Tam J.E., Kline B.C.. Control of the ccd operon in plasmid F. J. Bacteriol. 1989; 171:2353–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dao-Thi M.-H., Charlier D., Loris R., Maes D., Messens J., Wyns L., Backmann J.. Intricate interactions within the ccd plasmid addiction system. J. Biol. Chem. 2002; 277:3733–3742. [DOI] [PubMed] [Google Scholar]
- 29. Loris R., Garcia-Pino A.. Disorder and dynamics-based regulatory mechanisms in toxin-antitoxin modules. Chem. Rev. 2014; 114:6933–6947. [DOI] [PubMed] [Google Scholar]
- 30. Flock T., Weatheritt R.J., Latysheva N.S., Babu M.M.. Controlling entropy to tune the functions of intrinsically disordered regions. Curr. Opin. Struct. Biol. 2014; 26C:62–72. [DOI] [PubMed] [Google Scholar]
- 31. Crothers D.M., Metzger H.. The influence of polyvalency on the binding properties of antibodies. Immunochemistry. 1972; 9:341–357. [DOI] [PubMed] [Google Scholar]
- 32. Hornickt C.L., Karush F.. Antibody affinity - III the role of multivalence. Immunochemistry. 1972; 9:325–330. [DOI] [PubMed] [Google Scholar]
- 33. Brewer C.F., Miceli M.C., Baum L.G.. Clusters, bundles, arrays and lattices: Novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr. Opin. Struct. Biol. 2002; 12:616–623. [DOI] [PubMed] [Google Scholar]
- 34. Lasky L.A. Selectin-carbohydrate interactions and the initiation of the inflammatory response. Annu. Rev. Biochem. 1995; 64:113–139. [DOI] [PubMed] [Google Scholar]
- 35. Holmskov U., Thiel S., Jensenius J.C.. Collectins and ficolins: Humoral lectins of the innate immune defense. Annu. Rev. Immunol. 2003; 21:547–578. [DOI] [PubMed] [Google Scholar]
- 36. Turner M.W. The role of mannose-binding lectin in health and disease. Mol. Immunol. 2004; 62:4–9. [DOI] [PubMed] [Google Scholar]
- 37. Dao-Thi M.-H., Van Melderen L., De Genst E., Buts L., Ranquin A., Wyns L., Loris R.. Crystallization of CcdB in complex with a GyrA fragment. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:1132–1134. [DOI] [PubMed] [Google Scholar]
- 38. Van Melderen L., Dao-Thi M.H., Lecchi P., Gottesman S., Couturier M., Maurizi M.R.. ATP-dependent degradation of CcdA by Lon protease - Effects of secondary structure and heterologous subunit interactions. J. Biol. Chem. 1996; 271:27730–27738. [DOI] [PubMed] [Google Scholar]
- 39. David G., Pérez J.. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 2009; 42:892–900. [Google Scholar]
- 40. Shkumatov A.V, Strelkov S.V. DATASW, a tool for HPLC-SAXS data analysis. Acta Crystallogr. D Biol. Crystallogr. 2015; 71:1347–1350. [DOI] [PubMed] [Google Scholar]
- 41. Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D.T., Konarev P.V., Svergun D.I.. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012; 45:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Svergun D.I. Determination of the regularization parameter in indirect-transform. J. Appl. Crystallogr. 1992; 25:495–503. [Google Scholar]
- 43. Franke D., Svergun D.I.. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009; 42:342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Volkov V.V., Svergun D.I.. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003; 36:860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schneidman-Duhovny D., Hammel M., Sali A.. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010; 38:W540–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weinkam P., Pons J., Sali A.. Structure-based model of allostery predicts coupling between distant sites. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:4875–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Svergun D.I., Barberato C., Koch M.H.J.. CRYSOL – a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995; 28:768–773. [Google Scholar]
- 48. Miller J.H. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. 1992; NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 49. Tang G., Peng L., Baldwin P.R., Mann D.S., Jiang W., Rees I., Ludtke S.J.. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007; 157:38–46. [DOI] [PubMed] [Google Scholar]
- 50. Behrmann E., Tao G., Stokes D.L., Egelman E.H., Raunser S., Penczek P.A. Real-space processing of helical filaments in SPARX. J. Struct. Biol. 2012; 177:302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Finch J.T. The hand of the helix of tobacco virus. J. Mol. Biol. 1972; 66:291–294. [DOI] [PubMed] [Google Scholar]
- 52. van Dijk M., Bonvin A.M.J.J.. 3D-DART: A DNA structure modelling server. Nucleic Acids Res. 2009; 37:W235–W239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera - a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 55. Langer A., Hampel P.A, Kaiser W., Knezevic J., Welte T., Villa V., Maruyama M., Svejda M., Jähner S., Fischer F. et al. . Protein analysis by time-resolved measurements with an electro-switchable DNA chip. Nat. Commun. 2013; 4:2099–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pace C.N., Vajdos F., Fee L., Grimsley G., Gray T.. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995; 4:2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cantor C.R., Warshaw M.M., Shapiro H.. Oligonucleotide interactions. III. Circular dichroism studies of the conformation of deoxyoligonucleolides. Biopolymers. 1970; 9:1059–1077. [DOI] [PubMed] [Google Scholar]
- 58. Huang C.Y. Determination of binding stoichiometry by the continuous variation method: the Job plot. Methods Enzymol. 1982; 87:509–525. [DOI] [PubMed] [Google Scholar]
- 59. Drobnak I., De Jonge N., Haesaerts S., Vesnaver G., Loris R., Lah J.. Energetic basis of uncoupling folding from binding for an intrinsically disordered protein. J. Am. Chem. Soc. 2013; 135:1288–1294. [DOI] [PubMed] [Google Scholar]
- 60. Drobnak I., Vesnaver G., Lah J.. Model-based thermodynamic analysis of reversible unfolding processes. J. Phys. Chem. B. 2010; 114:8713–8722. [DOI] [PubMed] [Google Scholar]
- 61. Press W.H., Teukolsky S.A., Vetterling W.T., Flannery B.P.. Numerical Recipes in C++: the art of scientific computing. 2002; Cambridge: Cambridge University Press. [Google Scholar]
- 62. Powell M.J.D. A hybrid method for nonlinear equations. Numerical methods for nonlinear equations. 1970; London: Gordon and Breach; 87–114. [Google Scholar]
- 63. Moré J.J., Garbow B.S., Hillstrom K.E.. User guide for MINPACK-1. Argonne National Laboratory Report ANL-80-74. 1980; Argonne. [Google Scholar]
- 64. von Hippel P.H., Revzin A., Gross C. A, Wang A.C.. Non-specific DNA binding of genome regulating proteins as a biological control mechanism: I. The lac operon: equilibrium aspects. Proc. Natl. Acad. Sci. U.S.A. 1974; 71:4808–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Simic M., De Jonge N., Loris R., Vesnaver G., Lah J.. Driving forces of gyrase recognition by the addiction toxin CcdB. J. Biol. Chem. 2009; 284:20002–20010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Setty Y., Mayo A.E., Surette M.G., Alon U.. Detailed map of a cis-regulatory input function. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:7702–7707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bøggild A., Sofos N., Andersen K.R., Feddersen A., Easter A.D., Passmore L.A., Brodersen D.E.. The crystal structure of the intact E. coli RelBE toxin-antitoxin complex provides the structural basis for conditional cooperativity. Structure. 2012; 20:1641–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Arbing M.A, Handelman S.K., Kuzin A.P., Verdon G., Wang C., Su M., Rothenbacher F.P., Abashidze M., Liu M., Hurley J.M. et al. . Crystal structures of Phd-Doc, HigA, and YeeU establish multiple evolutionary links between microbial growth-regulating toxin-antitoxin systems. Structure. 2010; 18:996–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Brown B.L., Grigoriu S., Kim Y., Arruda J.M., Davenport A., Wood T.K., Peti W., Page R.. Three dimensional structure of the MqsR:MqsA complex: a novel TA pair comprised of a toxin homologous to RelE and an antitoxin with unique properties. PLoS Pathog. 2009; 5:e1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kamada K., Hanaoka F., Burley S.K.. Crystal structure of the MazE/MazF complex: molecular bases of antidote-toxin recognition. Mol. Cell. 2003; 11:875–884. [DOI] [PubMed] [Google Scholar]
- 71. Simanshu D.K., Yamaguchi Y., Park J.H., Inouye M., Patel D.J.. Structural Basis of mRNA Recognition and Cleavage by Toxin MazF and Its Regulation by Antitoxin MazE in Bacillus subtilis. Mol. Cell. 2013; 52:447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gazit E., Sauer R.T.. Stability and DNA binding of the Phd protein of the phage P1 plasmid addiction system. J. Biol. Chem. 1999; 274:2652–2657. [DOI] [PubMed] [Google Scholar]
- 73. Li G.-W., Burkhardt D., Gross C., Weissman J.S.. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014; 157:624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hargreaves D., Santos-Sierra S., Giraldo R., Sabariegos-Jareño R., de la Cueva-Méndez G., Boelens R., Díaz-Orejas R., Rafferty J.B.. Structural and functional analysis of the kid toxin protein from E. coli plasmid R1. Structure. 2002; 10:1425–1433. [DOI] [PubMed] [Google Scholar]
- 75. Zorzini V., Buts L., Sleutel M., Garcia-Pino A., Talavera A., Haesaerts S., De Greve H., Cheung A., Van Nuland N.A.J., Loris R.. Structural and biophysical characterization of Staphylococcus aureus SaMazF shows conservation of functional dynamics. Nucleic Acids Res. 2014; 42:6709–6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zorzini V., Mernik A., Lah J., Sterckx Y.G.J., De Jonge N., Garcia-Pino A., De Greve H., Versees W., Loris R.. Substrate recognition and activity regulation of the Escherichia coli mRNA endonuclease MazF. J. Biol. Chem. 2016; 291:10950–10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kamphuis M.B., Monti M.C., van den Heuvel R.H.H., Santos-Sierra S., Folkers G.E., Lemonnier M., Díaz-Orejas R., Heck A.J.R., Boelens R.. Interactions between the toxin Kid of the bacterial parD system and the antitoxins Kis and MazE. Proteins. 2008; 70:311–319. [DOI] [PubMed] [Google Scholar]
- 78. Monti M.C., Hernández-Arriaga A.M., Kamphuis M.B., López-Villarejo J., Heck A.J.R., Boelens R., Díaz-Orejas R., van den Heuvel R.H.H.. Interactions of Kid-Kis toxin-antitoxin complexes with the parD operator-promoter region of plasmid R1 are piloted by the Kis antitoxin and tuned by the stoichiometry of Kid-Kis oligomers. Nucleic Acids Res. 2007; 35:1737–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.