


Abstract
Transposable element (TE) derived sequences are known to contribute to the regulation of the human genome. The majority of known TE-derived regulatory sequences correspond to relatively ancient insertions, which are fixed across human populations. The extent to which human genetic variation caused by recent TE activity leads to regulatory polymorphisms among populations has yet to be thoroughly explored. In this study, we searched for associations between polymorphic TE (polyTE) loci and human gene expression levels using an expression quantitative trait loci (eQTL) approach. We compared locus-specific polyTE insertion genotypes to B cell gene expression levels among 445 individuals from 5 human populations. Numerous human polyTE loci correspond to both cis and trans eQTL, and their regulatory effects are directly related to cell type-specific function in the immune system. PolyTE loci are associated with differences in expression between European and African population groups, and a single polyTE loci is indirectly associated with the expression of numerous genes via the regulation of the B cell-specific transcription factor PAX5. The polyTE-gene expression associations we found indicate that human TE genetic variation can have important phenotypic consequences. Our results reveal that TE-eQTL are involved in population-specific gene regulation as well as transcriptional network modification.
INTRODUCTION
Transposable elements (TEs) are mobile DNA sequences that create copies of themselves when they move among chromosomal locations. TE activity has had a major impact on the evolution and structure of the human genome; millions of TE sequence copies have accumulated over the last ∼100my. The initial sequencing and subsequent analysis of the human genome revealed that >50% of the genome sequence is derived from past TE sequence insertions (1,2).
TEs can also shape the function of the human genome, particularly with respect to the regulation of gene expression (3). Human TE-derived sequences have been shown to provide a wide variety of gene regulatory sequences including promoters (4–6), enhancers (7–11), transcription terminators (12) and several classes of small RNAs (13–15). Human TEs also influence various aspects of chromatin structure throughout the genome (1,16–20).
The vast majority of human TE sequences are remnants of ancient transposition events that occurred many millions of years ago (1). Accordingly, studies that have uncovered the regulatory properties of TE-derived sequences have dealt with fixed TE insertions that are present at the same locations in the genome sequences of all human individuals. Such fixed TE-derived regulatory sequences are not expected to provide for gene regulatory variation based on insertional polymorphisms between individuals.
It has only recently become possible to systematically evaluate the effects of TE genetic variation within and between human populations, i.e. TE polymorphisms. Human TE polymorphisms are primarily generated via the activity of three families of retrotransposons: Alu (21,22), L1 (23,24) and SVAs (25,26). Transposition events by members of these polymorphic TE (polyTE) families yield numerous differences in the presence/absence of insertions at specific loci among individual human genome sequences. The recent phase 3 variant release of the 1000 Genomes Project included a catalog of presence/absence genotypes for >16 000 polyTE loci among 2504 individuals from 26 human populations (27,28). This genome-wide collection of polyTE genotypes provides an opportunity to explore the phenotypic consequences of TE activity at the level of human populations.
Considering the known regulatory properties of human TEs, together with the fact that TE insertional activity is known to be highly disruptive (29,30), we hypothesized that polyTE activity can lead to gene expression differences between human individuals. We used an integrated analysis of polyTE genotypes and genome-wide expression profiles, for the same set of 1000 Genome Project samples, in order to test this hypothesis (Figure 1). Gene expression levels were regressed against polyTE genotypes to search for polyTE-gene expression associations. This approach revealed numerous human polyTE loci that correspond to expression quantitative trait loci (eQTL). The TE-eQTL uncovered here are involved in the establishment of population-specific expression profiles as well as transcriptional regulatory network modification.
Figure 1.

Scheme for the polymorphic transposable element (polyTE) expression quantitative trait loci (eQTL) analysis conducted here. Data were taken from 87 African and 358 European individuals from the 1000 Genomes Project. Genome (DNA-seq) and transcriptome (RNA-seq) data were used to characterize polyTE genotypes and gene expression levels for all individuals in the study. Individual gene expression levels were regressed against polyTE insertion genotypes in an effort to reveal associations between polyTE loci and human gene expression, i.e. TE-eQTL.
MATERIALS AND METHODS
Polymoprhic transposable element (polyTE) analysis
Genotype calls for three families of human polyTEs—Alu, L1 and SVA—in 445 individuals from five populations were taken from the phase 3 variant release of the 1000 Genomes Project (28). The phase 3 variant release corresponds to the human genome reference sequence build GRCh37/hg19. The five human populations are CEU: Utah Residents (CEPH) with Northern and Western Ancestry, FIN: Finnish in Finland, GBR: British in England and Scotland and TSI: Toscani in Italia from Europe along with YRI: Yoruba in Ibadan, Nigeria from Africa (Figure 1). These populations were chosen because they have matching RNA-seq data for the same individuals (see RNA-seq analysis section). PolyTE genotypes were characterized by the 1000 Genomes Project Structural Variation Group using the program MELT as previously described (27). The polyTE genotype data were accessed from the 1000 Genomes Project ftp server maintained at the NCBI: http://ftp-trace.ncbi.nlm.nih.gov/1000genomes/ftp/release/20130502/.
For any given polyTE insertion site, there are three possible presence/absence genotype values for an individual genome: 0-no polyTE insertion (homozygote absent), 1-a single polyTE insertion (heterozygote) and 2-two polyTE insertions (homozygote present). PolyTE genotypes were used for eQTL analysis as described below. PolyTE genotypes were also used to compute pairwise genetic distances between individuals as: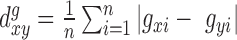 , where
, where 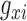 and
and 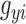 are the polyTE genotype value for individual
are the polyTE genotype value for individual 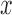 and individual
and individual 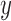 at insertion site
at insertion site 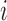 , for a total of
, for a total of 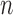 sites. The resulting pairwise polyTE genotype distance matrix was subject to dimension reduction using multidimensional scaling (MDS) (31), using the cmdscale function from the R statistical package version 3.2.2 (32), in order to visualize the genetic relationships between individuals based on their polyTEs (Figure 2C).
sites. The resulting pairwise polyTE genotype distance matrix was subject to dimension reduction using multidimensional scaling (MDS) (31), using the cmdscale function from the R statistical package version 3.2.2 (32), in order to visualize the genetic relationships between individuals based on their polyTEs (Figure 2C).
Figure 2.
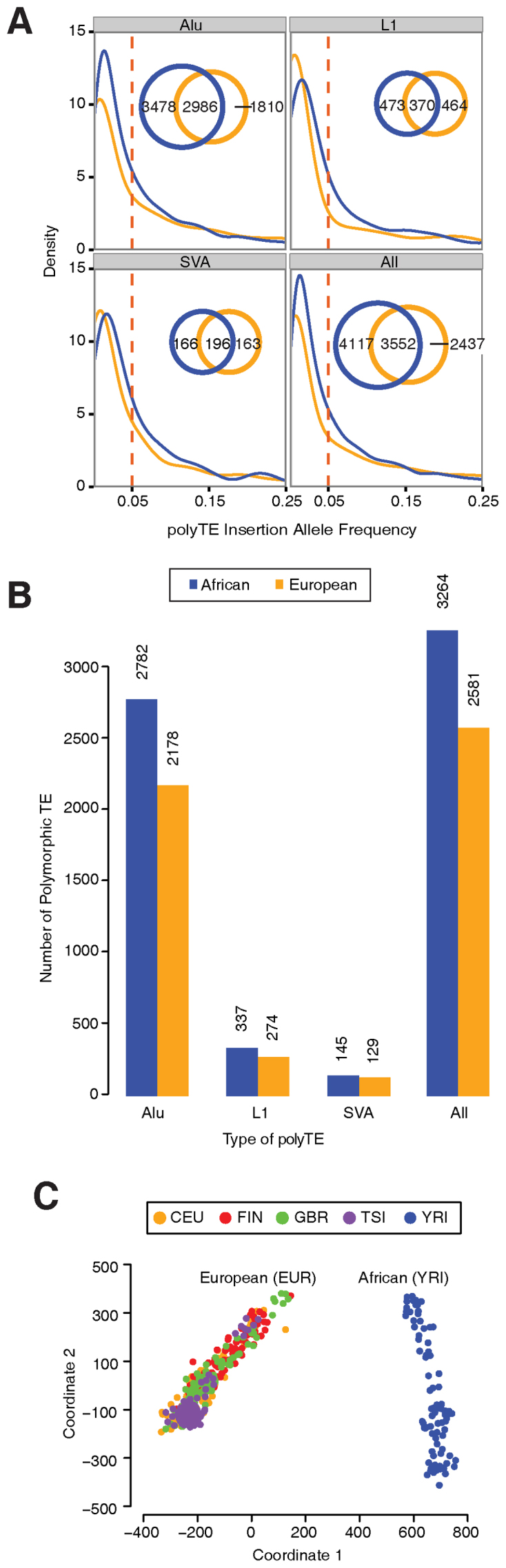
Distribution of polyTEs among the African and European population groups analyzed here. Data are broken down into Alu, L1 and SVA polyTE families. (A) PolyTE insertion allele frequency distributions for African and European populations are shown along with the numbers of shared and population-specific polyTE loci. (B) The numbers of African and European polyTE insertions with allele frequencies >5%. (C) Genetic relationships among the individuals analyzed here based on their polyTE genotypes. Individual's population origins are color coded as shown in the key.
RNA sequencing (RNA-seq) analysis
RNA-seq expression data, for the same 445 individuals from 5 human populations with polyTE genotypes characterized as described in the previous section, were taken from the GUEVADIS RNA sequencing project for 1000 Genomes samples (33). These RNA-seq data characterize genome-wide expression levels from the same lymphoblastoid cell lines, i.e. Epstein–Barr virus (EBV) transformed B-lymphocytes, used for DNA-seq analysis in the 1000 Genomes project. RNA isolation, library preparation, sequencing and read-to-genome mapping was performed as previously described (33). As with the polyTE data, the RNA-seq read mapping corresponds to the human genome build GRCh37/hg19. Mapped reads were used to quantify gene expression levels for ENSEMBL gene models (34) and normalization of gene expression levels was done using a combination of a modified RPKM approach followed by the probabilistic estimation of expression residuals (PEER) method (35) as previously described (36). The PEER normalized RNA-seq gene expression levels were accessed from the GUEVADIS project ftp server maintained at EBI: ftp://ftp.ebi.ac.uk/pub/databases/microarray/data/experiment/GEUV/E-GEUV-1/analysis_results/.
Genome-wide expression profiles were used to compute pairwise phenotypic distances between individuals as: 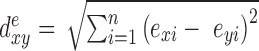 where
where 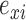 and
and 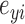 are the normalized gene expression level for individual
are the normalized gene expression level for individual 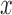 and individual
and individual 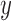 at gene
at gene 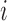 , for a total of
, for a total of 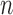 genes. The resulting pairwise expression distance matrix was subject to dimension reduction using MDS (31), using the cmdscale function from the R statistical package version 3.2.2 (32), in order to visualize the relationships between individuals based on their genome-wide expression profiles (Figure 3A). Differential gene expression between African and European populations was evaluated using a paired t-test implemented with the genefilter package from Bioconductor (37) (Figure 3B).
genes. The resulting pairwise expression distance matrix was subject to dimension reduction using MDS (31), using the cmdscale function from the R statistical package version 3.2.2 (32), in order to visualize the relationships between individuals based on their genome-wide expression profiles (Figure 3A). Differential gene expression between African and European populations was evaluated using a paired t-test implemented with the genefilter package from Bioconductor (37) (Figure 3B).
Figure 3.

Gene expression profiles within and between populations analyzed here. (A) Individuals from different populations are related based on their genome-wide expression profiles. Individual's population origins are color coded as shown in the key. (B) Heatmap showing genes that have expression profiles that are significantly different between the African and European population groups. Gene expression levels are color coded as shown in the key.
Expression quantitative trait loci (eQTL) analysis
PolyTE genotypes from the individuals analyzed here were related to their gene expression levels to identify eQTLs that correspond to polyTE insertion sites (TE-eQTLs) using the program Matrix eQTL (38) (Figure 1). Only polyTE insertion sites with >5% TE-present allele frequency were used for this purpose (Figure 2A and B). Matrix eQTL was run using the additive linear (least squares model) option with covariates for gender and population. This was done for all possible pairs of polyTE insertion sites and genes. Cis versus trans eQTLs were defined later as polyTE insertion sites that fall inside (cis) or outside (trans) 1Mb from gene boundaries. P-values were calculated for all pairs of polyTE-gene expression comparisons, and fale discovery rate (FDR) q-values were then calculated to correct for multiple statistical tests. The genome-wide significant polyTE-gene expression eQTL association threshold was set at FDR q < 0.05 (P < 4.7e-7).
A series of three additional control analyses were implemented in an effort to control for potentially confounding effects, for regulatory single nucleotide polymorphisms (SNPs) in particular, on the TE-eQTL associations that passed the genome-wide significance threshold (Supplementary Figure S1). [Control 1] TE-eQTL versus SNP-eQTL comparisons: for all of the genes found to be associated with TE-eQTLs, we searched the results of the GEUVADIS RNA-seq project (33) to identify the number of SNPs that were previously implicated as eQTLs for the same genes (Supplementary Figure S1A). [Control 2] Conditional association analysis: For the genes that were found to be associated with both TE-eQTLs and SNP-eQTLs, we performed conditional association analysis whereby multiple regression of expression against genotype is done using both TE and SNP genotype information used as explanatory variables in the same multiple regression model (Supplementary Figure S1B). The conditional association analysis was performed using the same multiple regression approach as implemented in the GCTA program (39). [Control 3] Regional association scans: Regional eQTL association scans were done by defining linked 1Mb regions that are centered on individual polyTE loci, and then all SNP and polyTE genotypes from the linked regions were further evaluated for association with gene expression using the same approach used for TE-eQTLs (Supplementary Figure S1A). Results of the regional eQTL association scans were visualized using the regional association plot R script from the Broad Institute of MIT and Harvard (40).
Functional enrichment analysis
Genes that correspond to best TE-eQTLs were used for gene set enrichment analysis using the KEGG, BIOCARTA and REACTOME data sets from the Molecular Signatures Database web server (version 5.1) (41) in order to identify functionally enriched gene categories. A FDR q-value threshold of 0.05 was used for this purpose.
Transcription factor (TF) target identification
TF (PAX5) target genes were taken from annotations of experimentally characterized TF binding sites from the 2015.1 version of GENOME TRAX™ (www.biobase-international.com/genome-trax) from BIOBASE corporation (42). TF-target gene interactions were visualized using the program Circos (version 0.69) (43).
RESULTS
The landscape of human TE polymorphisms
Computational analysis of next-generation (re)sequencing data can be used to identify the locations of polyTE insertions genome-wide (44). Recent applications of this approach to human genome sequences from the 1000 Genomes Project has resulted in a deep characterization of human genetic variation resulting from TE activity (27,28). We analyzed polyTE loci from the genome sequences of 445 individuals sampled from 5 human populations (4 European and 1 African) characterized as part of this project. There are a total of 10,106 polyTE insertions observed for these 445 individual genome sequences: 8,274 for Alu, 1,307 for L1 and 525 for SVA (Figure 2A). Most of the polyTE insertions that we observe (9,799) can be considered to be cis to human genes as they either fall within gene boundaries or within 1 Mb upstream or downstream of genes. Furthermore, consistent with previous results, the majority of polyTE loci for these five populations show low frequencies of TE insertions (i.e. low minor allele frequencies), suggesting that TE insertions are highly disruptive and subject to strong purifying selection (27,45). Nevertheless, there are 2617 polyTE loci that show >5% TE insertion frequency for these populations (Figure 2B); these common polyTE loci were used for the subsequent eQTL analysis. The vast majority of these are Alu polyTE loci with an order of magnitude fewer L1 and fewest SVA loci.
Despite the similar shapes of the TE insertion allele frequency distributions, many of the loci are specific to individual populations or continental population groups. Indeed, genetic distances between individuals calculated based on their polyTE genotypes clearly separates European from African populations (Figure 2C). Population-specific polyTE loci with higher insertion frequencies can be considered to be more likely to exert broad regulatory effects across individuals and populations. Accordingly, we focused our subsequent analysis on these (relatively) high frequency polyTE loci and searched for possible population-specific regulatory effects of such loci.
TE expression quantitative trait loci (TE-eQTL)
We analyzed genome-wide expression profiles for these same individuals in an effort to evaluate the relationship between TE genetic variation and human gene regulation. Genome-wide expression profiles were compared to compute a pairwise phenotypic (regulatory) distance matrix for the individuals analyzed here. Unlike what is seen for the polyTE genetic distances, genome-wide expression profiles do not separate individual humans among different population groups (Figure 3A). In other words, gene expression variation does not segregate globally in the same way that TE genetic variation does. Nevertheless, there are several hundred genes that do show statistically different levels of expression between the African and European populations analyzed here (Figure 3B).
We evaluated the relationship between TE genetic variation and human gene regulation by searching for expression quantitative trait loci (eQTL) that correspond to polyTE insertion sites. To do this, gene-specific expression levels were regressed against presence/absence genotypes—0, 1 or 2 TE insertions—for individual polyTE loci (Figure 1). We used an additive linear model as described in the ‘Materials and Methods’ section to search for statistically significant associations between the polyTE genotypes at any given locus and expression levels for individual genes. This was done separately for African and European population groups as well as for all individuals considered together. The total number of statistically significant (FDR q-value < 0.05, P < 4.7e-7) polyTE-gene expression associations (TE-eQTLs) for the different population cohorts, and different polyTE families, are shown in Figure 4A. Alu polyTE loci provide the greatest number of TE-eQTL by far, consistent with their substantially higher numbers in the genome. A quantile-quantile (Q-Q) plot for these data confirms a strong overall signal of statistically significant associations (Figure 4B), which are shown along individual chromosomes, and broken down by polyTE family, in the Manhattan plot in Figure 4C. A complete list of the TE-eQTL discovered here is provided as Supplementary Table S1.
Figure 4.

Polymorphic transposable element expression quantitative trait loci (TE-eQTL) detected here. (A) Numbers of statistically significant TE-eQTL are shown for different population group cohorts and different polyTE families. The numbers of individuals (n) are shown for each population cohort at the top of the table, and the total number of genes involved in TE-eQTL associations are shown at the bottom of the table. (B) Quantile-quantile (Q-Q) plot showing observed versus expected distributions of polyTE loci-gene expression association P-values (negative log transformed). (C) Manhattan plot showing the genomic distribution of polyTE-gene expression association values. The dashed yellow line indicates the FDR q-value cutoff of 0.05, which corresponds to a P-value of 4.7e-7. P-values are color coded according to polyTE families as shown in the key.
The set of genes that are associated with TE-eQTL is enriched for a number of immune-related functions including IgA production, antigen processing/presentation and several signalling pathways that lead to immune cell differentiation and activation (Figure 5 and Supplementary Table S2). This result is consistent with the fact that the expression data were taken from lymphoblastoid cell lines (i.e. transformed B-lymphocytes) and points to cell type-specific functional relevance of polyTE mediated gene regulation.
Figure 5.

Functional enrichment of polyTE loci associated genes. Enriched, immune-related gene sets are shown along with the FDR q-values indicating the significance of the enrichments.
We performed a series of additional analyses in an effort to control for the potential effects of other genomic variations, regulatory SNPs in particular, on the TE-eQTL associations uncovered by our initial screen (see ‘Materials and Methods’ section; Supplementary Figure S1). First, we assessed the extent of overlap between the genes that we observe to be associated with TE-eQTL here and genes previously found to be associated with SNPs using the same sequence and expression data. The overlap between the TE-eQTL genes identified here and the previously identified SNP-eQTL genes is extremely low (n = 50 or ∼1%), consistent with the fact that we are primarily identifying novel regulatory associations (Supplementary Figure S2). Second, for those genes that were found to be associated with both TE-eQTL and SNP-eQTL, we performed conditional association analyses that combine both TE and SNP genotypes. The majority of the TE-eQTL from the initial screen remain significant after conditioning on the SNP genotypes (Supplementary Table S3). Third, regional association scans were used to evaluate the regulatory effects of all genomic variants linked to the TE-eQTL discovered here (Supplementary Table S4). Examples of this analysis can be seen in the following section on population-specific TE-eQTLs.
Population-specific TE-eQTL
A number of factors suggested the possibility that TE-eQTL may exert population-specific effects on human gene regulation. The polyTE landscapes of human populations are very distinct, with polyTE genetic variation clearly delineating African from European populations. While gene expression does not show the same overall pattern of population divergence, there are hundreds of genes that do show population-specific expression. Finally, the numbers of TE-eQTL vary substantially for the African, European and merged population cohorts.
To evaluate the population-specific effects of TE-eQTL, we searched for gene-by-population interactions whereby specific polyTE loci are only associated with gene expression in the European or African populations (but not both). There are a total of 589 TE-eQTL that show such gene-by-population interactions: 407 for African and 182 for Europe (Supplementary Figure S3). These apparent population-specific effects of TE-eQTL can be attributed to cases where the polyTE genotypes are differentially distributed across population groups (Figure 6A and B) or where polyTE genotypes are shared across populations but their effects on gene expression are limited to one population group (Figure 6C).
Figure 6.

Examples of population-specific TE-eQTL detected here. Population-specific TE-eQTL where polyTE insertions are found primarily in only one population group are shown for the (A) REL and (B) PSD4 genes. Box-plots show the distributions of individual gene expression levels for each of the three possible polyTE insertion genotypes. Regional association plots show all associations with the gene expression centered on the polyTE locus. Association P-values are shown as indicated on the left y-axis along with the local recombination rate shown on the right y-axis. (C) A population-specific TE-eQTL is shown for PRDM2 gene where the associated polyTE locus has insertions found in both population groups but an association with gene expression is only seen in the African population. (D) A counter example of a polyTE locus with insertions shared among both population groups and similar associations with HSD17B12 are seen for both groups.
The polyTE locus Alu-5788 is strongly associated with REL expression levels when both population groups are considered together (Figure 6A). However, polyTE insertions at this locus are almost entirely African-specific and are associated with higher expression of the gene. Thus, consideration of the European population group alone would not turn up any association between this polyTE locus and the REL gene. REL encodes the c-Rel protein, which is part of the NF-κB family of transcription factors (46). REL is considered to be a proto-oncogene that influences the survival and proliferation of B-lymphocytes. The gene's function has clinical significance with somatic mutations that are associated with B-cell lymphomas (47) and SNPs that are associated with ulcerative colitis and rheumatoid arthritis (48,49).
A similar kind of a population-specific TE-eQTL is seen for the Alu-10841 locus, which is associated with PSD4 expression levels (Figure 6B). In this case, the presence of Alu insertions at the locus is associated with a reduction in gene expression levels. Alu insertions at this locus are far more common in European populations, and African individuals that lack the insertions tend to show higher expression levels for the gene. PSD4 encodes a guanine nucleotide exchange factor that works with the ARF6, ARL14/ARF7 protein complex to control the movement of major histocompatibility complex (MHC) class II containing vesicles along the actin cytoskeleton.
Gene-by-population interactions can also be seen for polyTE loci that are found in both the African and European population groups. While insertions at the Alu-1870 locus are commonly found in both population groups, polyTE insertion genotypes are only associated with decreased PRDM2 expression in the African population (Figure 6C). The population-specific effects of Alu insertions at this locus could be attributable to the distinct genetic background of each population, via interactions with population-enriched variants for instance. On the other hand, insertions at the Alu-8559 locus are similarly found in both African and European populations, but both populations show polyTE insertion associations with decreased expression levels of the HSD17B12 gene (Figure 6D). Interestingly, this particular example was detected with the FDR q-value cutoff employed here (0.05) for the both the European and merged population cohorts but not for the African population alone (P = 8.7e-5 and FDR q-value = 3.9e-1). This may be attributable to the relatively low number of human samples analyzed for the single African population and suggests the possibility that some bona fide African-specific associations may have been overlooked in this study.
Transcriptional network TE-eQTLs
We found a number of cases where polyTE loci corresponded to TE-eQTL for more than one human gene (Figure 4A and Supplementary Table S1). This suggested the possibility that individual polyTE loci may participate in coordinated gene regulatory networks. One possible mechanism by which this may occur is through indirect polyTE-expression associations that are mediated by transcription factors (TFs), which regulate the expression of multiple genes. In other words, if a polyTE loci affects the expression of a TF, it may also appear to affect the regulation of one or more gene targets of that TF (Figure 7A). The Alu-7481 locus exemplifies this phenomenon. Alu insertions at this locus are associated with increased expression of PAX5 (Figure 7B), which encodes a transcription factor crucial to the specific identity and function on B cells. In particular, PAX5 expression is critical for differentiation of lymphoid progenitor cells into B cells (Figure 7C). It achieves this by simultaneously activating B lineage-specific genes and repressing genes active in distinct lineages (50). There are 274 known Pax5 target genes that show the identical Alu-7481 insertion genotype expression pattern as seen for their cognate TF (Figure 7D and Supplementary Table S5). While the majority of these do not reach the FDR q-value cutoff used here, there are three immune related target genes—PIK3AP1, REL and ZSCAN23 – that all remain statistically significant after controlling for multiple tests (Figure 7E). These data suggest that polyTE insertions are also involved in establishing cell type-specific regulatory networks with phenotypically important consequences.
Figure 7.

TE-eQTL and a PAX5 transcriptional regulatory network. (A) Scheme for how a single polyTE loci can provide trans eQTL for multiple genes by modifying the expression of a transcription factor encoding gene (PAX5) and its downstream target genes. (B) The Alu-7481 PAX5 TE-eQTL. PAX5 expression levels are shown for individuals with different Alu-7481 insertion genotypes (0, 1 or 2 insertions); the association P-value is shown. (C) The role of PAX5 in B-cell development. (D) Average expression level of 274 PAX5 target genes for individuals with different Alu-7481 insertion genotypes. Normalized (z-score transformed) gene expression levels are color-coded as shown in the key. Target genes that correspond to the most significant Alu-7481 TE-eQTL (FDR q-value < 0.05, P < 4.7e-7) are indicated. (E) Circos plot showing the chromosomal locations of the Alu-7481 TE-eQTL, PAX5 and the downstream target genes. TE-gene associations are shown in red, and PAX5-target gene interactions are shown in blue. The association P-values are shown on the inner circle as indicated in the key.
DISCUSSION
Numerous previous studies have uncovered gene regulatory contributions of human TE sequences (3–15,17–20). However, these studies have dealt with TE sequences derived from relatively ancient insertion events, which now are fixed in human populations. In other words, these TE-derived sequences exist at the same genomic locations in all human genomes and thus may not contribute substantially to regulatory variation between individuals. Here, we present a systematic analysis of the regulatory contributions of polyTE loci that were generated by recent transpositional activity and thereby differ between individuals. The TE-eQTL that we discovered underscore the extent to which TE-generated human genetic variation can affect regulatory differences within and between populations.
Our results indicate that polyTE loci provide greater numbers of trans compared to cis eQTL (Figure 4A). This may be considered somewhat surprising given the fact that most human eQTL studies focus on cis eQTL (33,51). However, studies on eQTL are often limited to cis associations owing the large number of possible SNP-by-gene comparisons that whole genome (i.e. both cis and trans) analyses entail. Thus, it is not entirely clear whether cis eQTL are actually expected to be more common that trans eQTL. The relatively low number of polyTE loci studied here (∼16 000), combined with the introduction of a more computationally efficient eQTL detection algorithm (38), allowed us to evaluate all possible cis and trans TE-eQTL. There are several possible mechanisms by which polyTE loci could serve as trans eQTL. For example, they may exert trans eQTL effects indirectly by regulating transcription factors, which in turn regulate numerous target genes, as we have shown for PAX5 (Figure 7). In addition, TEs have been shown to influence three-dimensional genome architecture, via the formation of chromosome loops or association with the nuclear scaffold/matrix for instance (5). It is tempting to speculate that TEs can exert trans eQTL effects via similar mechanisms that bring distal, homologous TE sequences into close proximity.
It is worth noting that human TE activity has often been associated with disease (29,30). Indeed, transpositional activity of human TEs was confirmed via the discovery of de novo insertions with obvious effects on health (24). However, the samples analyzed here correspond to (presumably) healthy individuals from the 1000 Genomes Project and are thereby taken to represent the normal scope of human genetic variation. The fact that many of the polyTE loci analyzed here have accumulated to relatively high insertion allele frequencies (Figure 2) is consistent with the notion that they are not deleterious. Thus, the phenotypic impact of human TE activity is not limited to deleterious effects; it also includes the generation of regulatory differences that fall within the scope of naturally occurring human variation. These kinds of functionally relevant but subtle TE-genetic variations, which necessarily avoid elimination by purifying selection, may provide an important substrate for ongoing human evolution.
Supplementary Material
Footnotes
Present address: I. King Jordan, 950 Atlantic Drive, Atlanta, GA 30332-2000, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Georgia Institute of Technology Bioinformatics Graduate Program, IHRC-Georgia Tech Applied Bioinformatics Laboratory (ABiL); BIOS Centro de Bioinformática y Biología Computacional; Intramural Research Program of the National Institutes of Heatlh (NIH), National Library of Medicine (NLM), National Center for Biotechnology Information (NCBI). Funding for open access charge: Departmental funds.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. et al. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. [DOI] [PubMed] [Google Scholar]
- 2.de Koning A.P.J., Gu W.J., Castoe T.A., Batzer M.A., Pollock D.D.. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011; 7:e1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feschotte C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008; 9:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conley A.B., Piriyapongsa J., Jordan I.K.. Retroviral promoters in the human genome. Bioinformatics. 2008; 24:1563–1567. [DOI] [PubMed] [Google Scholar]
- 5.Jordan I.K., Rogozin I.B., Glazko G.V., Koonin E.V.. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 2003; 19:68–72. [DOI] [PubMed] [Google Scholar]
- 6.Marino-Ramirez L., Lewis K.C., Landsman D., Jordan I.K.. Transposable elements donate lineage-specific regulatory sequences to host genomes. Cytogenet. Genome Res. 2005; 110:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chuong E.B., Elde N.C., Feschotte C.. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016; 351:1083–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Notwell J.H., Chung T., Heavner W., Bejerano G.. A family of transposable elements co-opted into developmental enhancers in the mouse neocortex. Nat. Commun. 2015; 6:6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chuong E.B., Rumi M.A., Soares M.J., Baker J.C.. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat. Genet. 2013; 45:325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kunarso G., Chia N.Y., Jeyakani J., Hwang C., Lu X., Chan Y.S., Ng H.H., Bourque G.. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 2010; 42:631–634. [DOI] [PubMed] [Google Scholar]
- 11.Bejerano G., Lowe C.B., Ahituv N., King B., Siepel A., Salama S.R., Rubin E.M., Kent W.J., Haussler D.. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature. 2006; 441:87–90. [DOI] [PubMed] [Google Scholar]
- 12.Conley A.B., Jordan I.K.. Cell type-specific termination of transcription by transposable element sequences. Mobile DNA. 2012; 3:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapusta A., Kronenberg Z., Lynch V.J., Zhuo X.Y., Ramsay L., Bourque G., Yandell M., Feschotte C.. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013; 9:e1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piriyapongsa J., Marino-Ramirez L., Jordan I.K.. Origin and evolution of human microRNAs from transposable elements. Genetics. 2007; 176:1323–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weber M.J. Mammalian small nucleolar RNAs are mobile genetic elements. PLoS Genet. 2006; 2:1984–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavlicek A., Jabbari K., Paces J., Paces V., Hejnar J.V., Bernardi G.. Similar integration but different stability of Alus and LINEs in the human genome. Gene. 2001; 276:39–45. [DOI] [PubMed] [Google Scholar]
- 17.Sundaram V., Cheng Y., Ma Z., Li D., Xing X., Edge P., Snyder M.P., Wang T.. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014; 24:1963–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacques P.E., Jeyakani J., Bourque G.. The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet. 2013; 9:e1003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt D., Schwalie P.C., Wilson M.D., Ballester B., Goncalves A., Kutter C., Brown G.D., Marshall A., Flicek P., Odom D.T.. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell. 2012; 148:335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J., Vicente-Garcia C., Seruggia D., Molto E., Fernandez-Minan A., Neto A., Lee E., Gomez-Skarmeta J.L., Montoliu L., Lunyak V.V. et al. MIR retrotransposon sequences provide insulators to the human genome. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E4428–E4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batzer M.A., Deininger P.L.. A human-specific subfamily of Alu sequences. Genomics. 1991; 9:481–487. [DOI] [PubMed] [Google Scholar]
- 22.Batzer M.A., Gudi V.A., Mena J.C., Foltz D.W., Herrera R.J., Deininger P.L.. Amplification dynamics of human-specific (HS) Alu family members. Nucleic Acids Res. 1991; 19:3619–3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brouha B., Schustak J., Badge R.M., Lutz-Prigge S., Farley A.H., Moran J.V., Kazazian H.H. Jr. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:5280–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kazazian H.H. Jr, Wong C., Youssoufian H., Scott A.F., Phillips D.G., Antonarakis S.E.. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988; 332:164–166. [DOI] [PubMed] [Google Scholar]
- 25.Ostertag E.M., Goodier J.L., Zhang Y., Kazazian H.H. Jr. SVA elements are nonautonomous retrotransposons that cause disease in humans. Am. J. Hum. Genet. 2003; 73:1444–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H., Xing J., Grover D., Hedges D.J., Han K., Walker J.A., Batzer M.A.. SVA elements: a hominid-specific retroposon family. J. Mol. Biol. 2005; 354:994–1007. [DOI] [PubMed] [Google Scholar]
- 27.Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M.H.Y. et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015; 526:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altshuler D.M., Durbin R.M., Abecasis G.R., Bentley D.R., Chakravarti A., Clark A.G., Donnelly P., Eichler E.E., Flicek P., Gabriel S.B. et al. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hancks D.C., Kazazian H.H.. Active human retrotransposons: variation and disease. Curr. Opin. Genet. Dev. 2012; 22:191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Solyom S., Kazazian H.H.. Mobile elements in the human genome: implications for disease. Genome Med. 2012; 4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gower J.C. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika. 1966; 53:325–338. [Google Scholar]
- 32.Ihaka R., Gentleman R.. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 1996; 5:299–314. [Google Scholar]
- 33.Lappalainen T., Sammeth M., Friedlander M.R., 't Hoen P.A.C., Monlong J., Rivas M.A., Gonzalez-Porta M., Kurbatova N., Griebel T., Ferreira P.G. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013; 501:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flicek P., Ahmed I., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S. et al. Ensembl 2013. Nucleic Acids Res. 2013; 41:D48–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stegle O., Parts L., Piipari M., Winn J., Durbin R.. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 2012; 7:500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.t Hoen P.A., Friedlander M.R., Almlof J., Sammeth M., Pulyakhina I., Anvar S.Y., Laros J.F., Buermans H.P., Karlberg O., Brannvall M. et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat. Biotechnol. 2013; 31:1015–1022. [DOI] [PubMed] [Google Scholar]
- 37.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J.. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shabalin A.A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012; 28:1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J.A., Lee S.H., Goddard M.E., Visscher P.M.. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011; 88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diabetes Genetics Initiative of Broad Institute of, H., Mit, L.U., Novartis Institutes of BioMedical, R.. Saxena R., Voight B.F., Lyssenko V., Burtt N.P., de Bakker P.I., Chen H., Roix J.J. et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007; 316:1331–1336. [DOI] [PubMed] [Google Scholar]
- 41.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matys V., Kel-Margoulis O.V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K. et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006; 34:D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A.. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ewing A.D. Transposable element detection from whole genome sequence data. Mobile DNA. 2015; 6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rishishwar L., Tellez Villa C.E., Jordan I.K.. Transposable element polymorphisms recapitulate human evolution. Mobile DNA. 2015; 6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayden M.S., Ghosh S.. NF-kappa B, the first quarter-century: remarkable progress and outstanding questions. Gene Dev. 2012; 26:203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin-Subero J.I., Gesk S., Harder L., Sonoki T., Tucker P.W., Schlegelberger B., Grote W., Novo F.J., Calasanz M.J., Hansmann M.L. et al. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood. 2002; 99:1474–1477. [DOI] [PubMed] [Google Scholar]
- 48.McGovern D.P., Gardet A., Torkvist L., Goyette P., Essers J., Taylor K.D., Neale B.M., Ong R.T., Lagace C., Li C. et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010; 42:332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gregersen P.K., Amos C.I., Lee A.T., Lu Y., Remmers E.F., Kastner D.L., Seldin M.F., Criswell L.A., Plenge R.M., Holers V.M. et al. REL, encoding a member of the NF-kappa B family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat. Genet. 2009; 41:U820–U877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cobaleda C., Schebesta A., Delogu A., Busslinger M.. Pax5: the guardian of B cell identity and function. Nat. Immunol. 2007; 8:463–470. [DOI] [PubMed] [Google Scholar]
- 51.Consortium G.T. Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.