

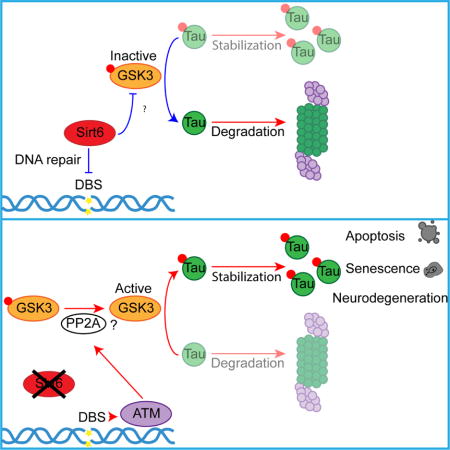
Summary
The histone deacetylase SIRT6 promotes DNA repair, but its activity declines with age, with a concomitant accumulation of DNA damage. Furthermore, SIRT6 knockout mice exhibit an accelerated aging phenotype and die prematurely. Here, we report that brain-specific SIRT6-deficient mice survive, but present behavioral defects with major learning impairments by 4 months of age. Moreover, the brains of these mice show increased signs of DNA damage, cell death and hyperphosphorylated Tau – a critical mark in several neurodegenerative diseases. Mechanistically, SIRT6 regulates Tau protein stability and phosphorylation through increased activation of the kinase GSK3α/β. Finally, SIRT6 mRNA and protein levels are reduced in patients with Alzheimer’s disease. Taken together, our results suggest that SIRT6 is critical to maintain genomic stability in the brain and its loss leads to toxic Tau stability and phosphorylation. Therefore, SIRT6 and its downstream signaling could be targeted in Alzheimer’s disease and age related neurodegeneration.
Keywords: SIRT6, DNA damage, Tau, Alzheimer’s disease, GSK3
Graphical abstract
Introduction
Neurodegeneration is a major health issue in developed countries, with more than 15 million people expected to be affected by 2050 in the US alone (Thies et al., 2013). The primary factor leading to neurodegenerative diseases is aging, which accepted theories associate with accumulation of unrepaired DNA damage (Madabhushi et al., 2014; Rulten and Caldecott, 2013). The accumulation of DNA damage can be particularly dangerous in the brain, where the high levels of metabolic activity create an environment with increased amounts of byproducts, such as reactive oxygen species. Since damaged post-mitotic neurons in the brain are hardly replaced, such damage leads to tissue degeneration.
Interestingly, DNA damage accumulation is even higher in brains affected by neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Amyotrophic lateral sclerosis (ALS) (Bajic et al., 2015; Lu et al., 2004; Madabhushi et al., 2014; Qiu et al., 2014). Two questions arise: (i) does physiological or pathological aging ultimately leads to decreased efficacy of the repair process? And (ii) is DNA damage accumulation the cause of neurodegeneration?
The Sirtuin protein family has been linked to aging and age-related diseases, and among the seven mammalian sirtuins, SIRT6 deficiency in mice causes the most striking phenotype, including genomic instability, premature aging, and ultimately death by approximately 4 weeks of age (Mostoslavsky et al., 2006). SIRT6 plays essential roles in metabolic homeostasis, inflammation, stress responses and genomic stability (Giblin et al., 2014; Kugel and Mostoslavsky, 2014). Indeed, cells lacking SIRT6 fail to repair various types of DNA damage, including Double Strand Breaks (DSB) and Base Excision Repair (BER). In this context, SIRT6 recruits the chromatin remodeler SNF2H to DNA breaks and deacetylates H3K56 on those sites, both critical for proper DNA repair (Toiber et al., 2013). Lack of SIRT6 or SNF2H affects recruitment of important repair factors, such as CtIP (Kaidi et al., 2010), Ku80, RPA, BRCA1 and 53BP1 (Toiber et al., 2013). Importantly, SIRT6-deficient mice present DNA damage in a tissue-specific manner, particularly in the brain (Toiber et al., 2013). Moreover, aging rats showed reduced levels of SIRT6 in all brain regions tested (Braidy et al., 2015). However, it remains unclear whether the observed reduced levels of SIRT6 in the aging brain contribute to aging-associated neurodegeneration.
Neurodegenerative diseases presenting hyperphosphorylated Tau (Tau-p) as a main pathological marker include AD, Frontotemporal Dementia with Parkinsonism-17, Pick’s disease and Huntington’s disease. Tau stabilizes neuronal microtubules, allowing axonal outgrowth, cargo transport, and cellular polarity (Wang and Mandelkow, 2016). Recent findings suggest Tau has additional roles in the nuclei, where it protects the DNA from genomic instability and chromatin relaxation (Sultan et al., 2011; Violet et al., 2014; Ziv et al., 2006). Tau protein, per se, is not toxic, however post-translational modifications such as phosphorylation and acetylation were shown to play important roles in its stability and toxicity (Min et al., 2010; 2015; Sohn et al., 2016; Tracy et al., 2016; Wang and Mandelkow, 2016). For example, Tau-p cannot bind microtubules with the same affinity, impairing neuronal function (Cook et al., 2014; 2015). In addition, phosphorylation stabilizes the protein, leading to the formation of toxic aggregates (Tenreiro et al., 2014; Wang and Mandelkow, 2016). Several kinases and phosphatases phosphorylate and dephosphorylate Tau. Of these, GSK3 and PP2A are more significant, relevant in AD and PD pathology (Cai et al., 2012; Gong and Iqbal, 2008; Lee et al., 2011; Lim, 2005; Ma, 2014; Voronkov et al., 2011). In addition, acetylation of Tau inhibits its degradation and increases its toxicity (Gorsky et al., 2016; Min et al., 2010; et al., 2015; Sohn et al., 2016; Tracy et al., 2016). These modifications are relevant in AD patients, who present increased acetylated and phosphorylated Tau (Min et al., 2015).
In this work, we developed a brain-specific SIRT6KO mouse model. SIRT6KO brains exhibit accelerated aging, present accumulated DNA damage, as well as increased apoptosis and hyperphosphorylated Tau levels. Functionally, SIRT6KO mice exhibit severe impairments in behavioral tasks as early as by 4 months of age. Accordingly, we found that SIRT6 depletion in cells led to increased Tau stability and phosphorylation through GSK3 activation and to increased apoptosis, which could be rescued by reducing GSK3 activity. Importantly, we show that Tau stability and phosphorylation are increased upon DNA damage. Finally, we analyzed samples from patients with Alzheimer’s disease and found a remarkable reduction in SIRT6 at both protein and mRNA levels, with further reduction with increased severity of Braak stages.
Together, our findings indicate that SIRT6 protects the brain from naturally accumulating DNA damage, in turn protecting against neurodegeneration.
Results
SIRT6 brain-specific KO mouse presents increased genomic instability and impaired learning phenotype
SIRT6 depletion is linked to both DNA damage and aging. We therefore hypothesized that brains missing the repair functionality of SIRT6 might experience accelerated degeneration. To this end, we generated a brain-specific SIRT6KO mice (brS6KO) using the Nestin-Cre promoter and measured DNA damage in brS6KO and WT brains.
Brains of brS6KO mice were significantly smaller, but otherwise structurally normal (Fig. S1A–C) (Schwer et al., 2010). In line with our hypothesis, brS6KO mice exhibited increased signs of DNA damage, marked by increased levels of ATM and H2AX phosphorylation, increased H3K56ac, and reduced SNF2H recruitment to chromatin (Fig. 1A, B). Moreover, we observed a significant increase in apoptotic cells in the cortex, as determined by TUNEL staining in young mice (3–4 month old) (Fig. 1C). Together, these results show that SIRT6-deficient brains have increased signs of DNA damage and cell death.
Figure 1. SIRT6 deletion in the brain leads to increase signs of DNA damage.
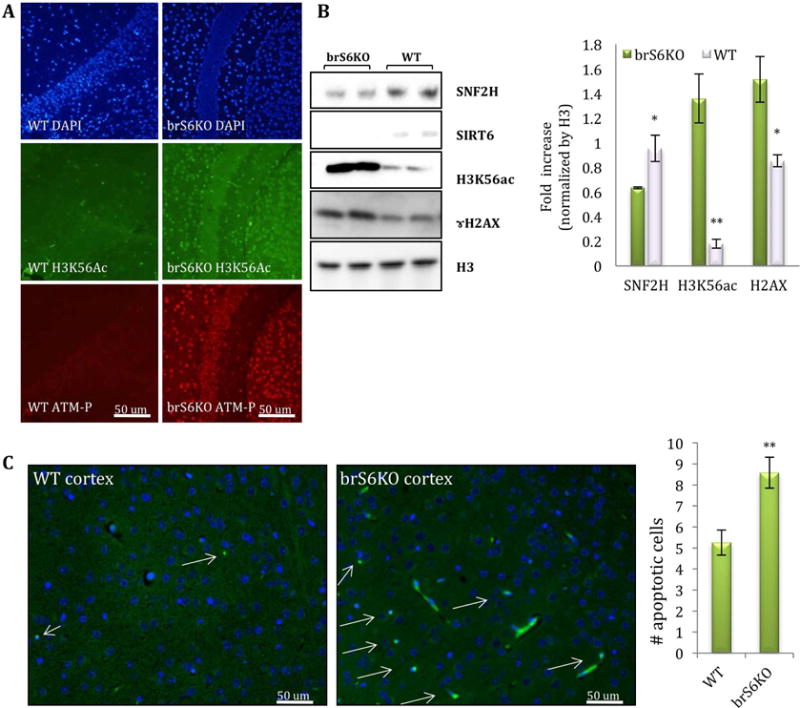
A–B. IF and protein blot of brain sections of 4-month-old brS6KO or WT mice (WT n=4, KO n=4) C. Apoptotic cells were labeled by TUNEL assay on brain slices. Positive cells per image were counted (in pictures with similar number of cells) (WT n=5, KO n=5).
Next, we examined behavior of brS6KO Mice. We first examined locomotor activity in the open field (OF) paradigm on two consecutive days. brS6KO mice displayed a striking increase in ambulation (Fig. 2A, S2A) along with a decrease in the number of rearing events (Fig. 2B, S2A), an index for exploratory behavior. Over time, control mice showed a time-dependent decrease of locomotor activity as well as increase in immobility, absent in brS6KO mice (Fig. S2B). brS6KO mice also showed persistent lack of habituation when re-exposed to the open field on subsequent day (Fig. 2A–C, S2A–B). The increased locomotor activity and failure to habituate indicate impaired hippocampus-dependent non-associative learning (Vianna et al., 2000) or striatal degeneration (Durieux et al., 2009). brS6KO mice showed no difference in time exploring the center of the OF on day 1 (Fig. 2D), an index of innate anxiety. Interestingly, this exploration phenotype habituated in control mice on day 2 but not in brS6KO mice (Fig. 2D), suggesting that SIRT6 deletion may impair contextual encoding and habituation.
Figure 2. SIRT6 brain-specific deletion elicits non-associative and associative learning deficits.
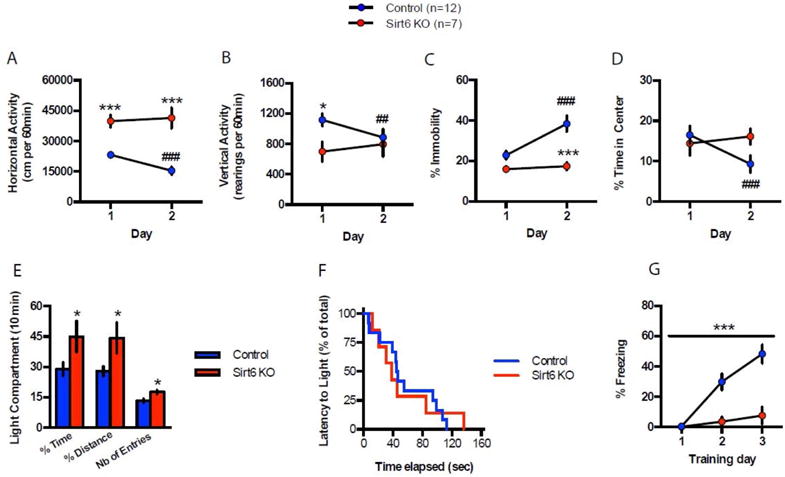
A. S6KO enhances spontaneous locomotor activity over two consecutive days in the OF (60 min/session). B. brS6KO decreases vertical activity in the OF test on day 1. Lack of difference on day 2 is due to decreased rearing events in Ctrl. C. S6KO decreases immobility in the OF test on both days. D. S6KO does not alter innate anxiety in the OF on day 1. Controls exhibit a significant decrease of time spent exploring the center but not S6KO. E–F. S6KO increases the time and distance percent and number of entries in the light-dark test (10 min session). G. S6KO markedly decreases freezing in the CFC paradigm. See statistical analyses in supplementary Fig. 2.
Lack of SIRT6 deletion effect on innate anxiety was further tested in the light-dark (LD) paradigm (Takao and Miyakawa, 2006), where brS6KO mice spent significantly more time exploring the lit compartment as compared to controls (Fig. 2E, S2C). Since the interpretation of these results can be encumbered by the aforementioned hyperlocomotor phenotype, we also measured the latency to first visit the lit compartment (Hovatta et al., 2005). Taking this parameter into account, we found no difference between brS6KO mice and WT control littermates, thus confirming that SIRT6 deletion in the brain does not alter innate anxiety (Fig. 2F).
Finally, we tested brS6KO mice in an associative learning task, namely the contextual fear-conditioning paradigm (CFC). Here, brS6KO mice showed a marked decrease in freezing behavior elicited by contextual cues explicitly associated to a foot shock (Fig. 2G).
Taken together, these results suggest that SIRT6 deletion markedly decreases non-associative (OF) and associative (CFC) learning. This profound effect on contextual encoding results in a striking deficit in behavioral habituation, translating to enhanced locomotor activity in rodents. These results are consistent with animal models for neurodegenerative diseases such as overexpression of human mutant Tau, different mouse models of neurodegeneration (Graham and Sidhu, 2010; Ikegami et al., 2000; Rao et al., 2011; Tanda et al., 2009) and irradiated mice phenotypes (Saxe et al., 2006).
Lack of SIRT6 leads to Tau hyperphosphorylation and GSK3 activation
Having established that SIRT6 deficiency results in increased DNA damage signaling, we set out to search for specific markers of neurodegeneration. We analyzed Tau-p levels, a common mark for several neurodegenerative diseases (Lee and Leugers, 2012; Spillantini and Goedert, 2013; Zhang et al., 2015).
Remarkably, brS6KO brain slices and protein extract showed increased levels of Tau-p in the cortex, but not in the hippocampus (Fig. 3A–B, S3A, D,E). To understand the molecular mechanism linking SIRT6 and Tau phosphorylation, we generated a cellular model where we deleted SIRT6 through CRISPR/Cas9 technology using two different guide RNA sequences in SH-SY5Y cells (S6KO), and an alternative model with shRNA in N2a cells. Both models exhibit similar phenotypes as brS6KO tissues, including increased levels of H3K56ac, ɤH2AX and PARP cleavage, as well as reduced SNF2H recruitment to chromatin (Fig. S3B–C). Remarkably, Tau-p was also seen in S6KO cells (Fig. S3C). Moreover, N2a cells were so severely impaired by SIRT6 reduction (Fig. S3C) that further experiments were done only in SH-SY5Y cell lines.
Figure 3. Lack of SIRT6 leads to increase in Tau stability and phosphorylation through GSK3 activation.
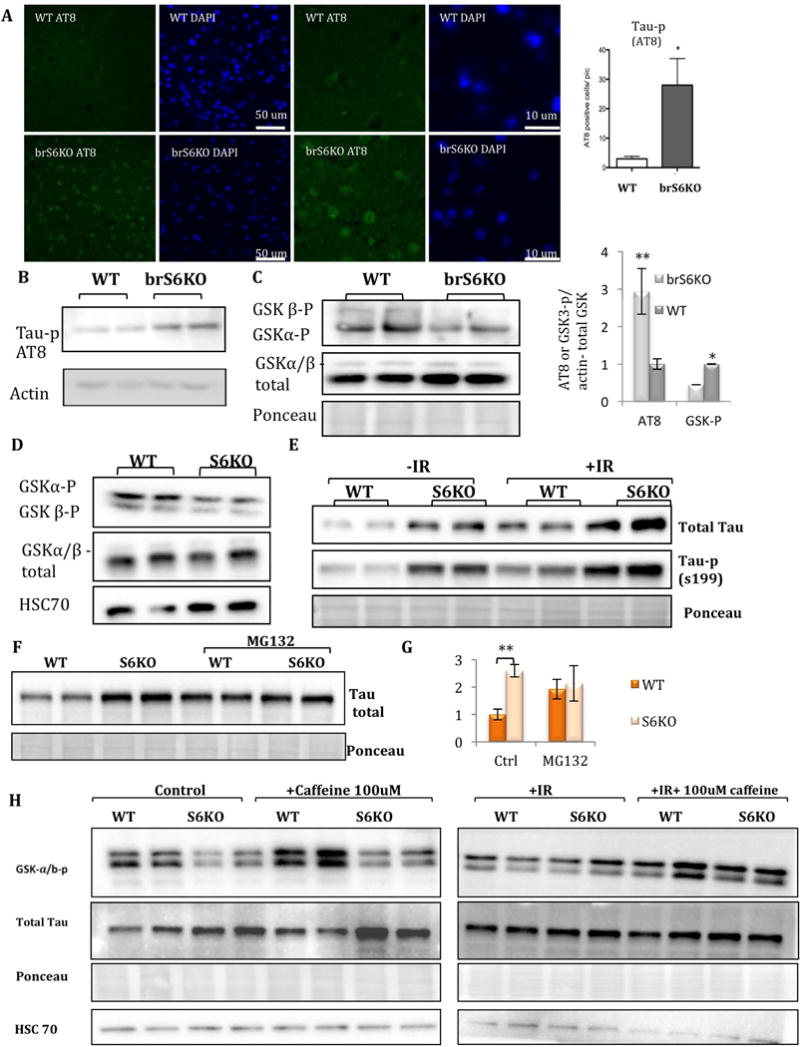
A–B. IF and protein blot of brain sections of 4-month-old S6KO or WT mice (IF, WB: WT n=4, KO n=4.) C–D. Protein blot of S6KO and WT brains or cells (SH-SY5Y). E–G. protein blots of SH-SY5Y S6KO and WT cells, transfected with Tau-emerald, before and after IR (400 rad), or cells treated with proteasome inhibitor (MG132). H. Protein blots of S6KO and WT cells treated with Caffeine.
Tau is phosphorylated by several kinases including CHK2 and GSK3 (Martin et al., 2013), known to be activated by DNA damage (Iijima-Ando et al., 2010; Ngok-Ngam et al., 2013; Watcharasit et al., 2002). While no changes were observed in CHK2 activation, we observed decreased levels of phospho-GSK3, a marker for increased GSK3 activity (Fig. 3C–D). Moreover, when SIRT6 was re-introduced to S6KO cells, GSK3 phosphorylation regained its normal levels (Fig. S3H–I).
Next, we asked how SIRT6 depletion may lead to Tau accumulation. One possibility is that SIRT6 directly represses Tau transcription through its histone deacetylation activity. To this end, we interrogated published ChIP-seq data (MAPT gene by ChIP-seq analysis (Ram et al., 2011)), and SIRT6 is not enriched in its genomic region. Indeed, Tau mRNA levels were not changed in mouse brains (Fig. S3J), ruling out possible Tau transcription repression by SIRT6.
Next, we asked if the increase in Tau phosphorylation observed in SIRT6-depleted brains and cell lines was a result of increased Tau stability. To this end, we ectopically expressed Tau-Emerald fusion in WT and S6KO cells, thus avoiding any transcriptional or splicing effect on Tau stability or phosphorylation. We found that under basal conditions S6KO cells have higher levels of Tau and Tau-p (Fig. 3E). Moreover, treating the cells with proteasome inhibitor MG132 results in equal amounts of Tau protein in WT and S6KO cells, suggesting that lack of SIRT6 stabilizes Tau from proteasomal degradation (Fig. 3F–G, S3M). Importantly, irradiation (IR) increased the levels of Tau and Tau-p (exogenous and endogenous) in both WT and KO cells (Fig. 3E, H, S3O). We therefore concluded that lack of SIRT6 resulted in the stabilization of Tau protein and its increased phosphorylation. To understand whether Tau stability and phosphorylation were ATM-dependent, we inhibited its activity by caffeine treatment. Tau stability was SIRT6- and IR-dependent, but ATM-independent, while GSK3 activation was ATM-dependent and IR-induced (Fig. 3H, S3N–O).
SIRT6KO cells are more sensitive to apoptosis, prevented by GSK3 or ATM inhibition
Since SIRT6-deficient brains and cells present increased signs of DNA damage and neurodegeneration, we predicted that S6KO cells are more sensitive to genotoxic damage. Indeed, an increase in apoptotic cell number was detected in S6KO cells under basal conditions and after IR (Fig. 4A, S4A–B), in line with our previous findings, however not all SIRT6-deficient tissues were more sensitive to genotoxic damage (Toiber et al., 2013). We therefore hypothesized that a specific protein within the brain proteome may be facilitating this toxicity, and speculated that Tau is the key. As Tau-p is toxic to the cells, reducing its phosphorylation could benefit S6KO cells. To achieve this, we treated the cells with lithium, a GSK3 inhibitor (Fig. 4E). Interestingly, lithium treatment and SIRT6 re-expression both rescued the sensitivity in S6KO cells, with concomitant reduction of Tau-p (Fig. 4D, F). Moreover, ATM inhibition, which inactivates GSK3, also rescued S6KO cells (Fig. 4C). Cells that were irradiated showed increased Tau levels as well as phosphorylation in WT cells, suggesting that DNA damage triggers Tau stability and phosphorylation. SIRT6 catalytic mutant HY showed a dominant negative effect on Tau stability and phosphorylation (in WT cells), leading to increased Tau levels and phosphorylation. On the other hand, overexpression of WT-SIRT6 could rescue Tau (GFP) levels even in irradiated cells, while phosphorylation was still higher after irradiation. Overall, this suggests that SIRT6 prevents DNA damage-induced Tau stability and to a lesser extent its phosphorylation, suggesting DNA damage has additional signaling on Tau-p (Fig. 4D). To confirm that GSK3 phosphorylation on Tau Ser199 was important for stability, we mutated Ser199 to Alanine or Glutamate. Our results show a striking decrease in Tau stability in the phospho-mutants S199A, but not in S199E, while S199E had an increase in the cleavage of Tau only in the S6KO cells (Fig. 4G, S4C–F). Irradiation increased the levels of Tau WT and S199E, but not a visible change in S199A. Treatment with proteasome inhibitor for a few hours increases protein in the Tau S199A, pointing to degradation of the mutant (Fig. S4D). Last, SIRT6 managed to interact with GSK3 through IP, suggesting it could control its activation through this interaction (Fig. S4G). Our studies indicate that lack of SIRT6 leads to DNA damage signaling, increasing activity of ATM and GSK3, phosphorylating Tau, leaving neurons more vulnerable to genotoxic insults.
Figure 4. GSK3 inhibition and Tau-p reduction rescue S6KO from apoptosis, as well as re-expression of WT SIRT6.
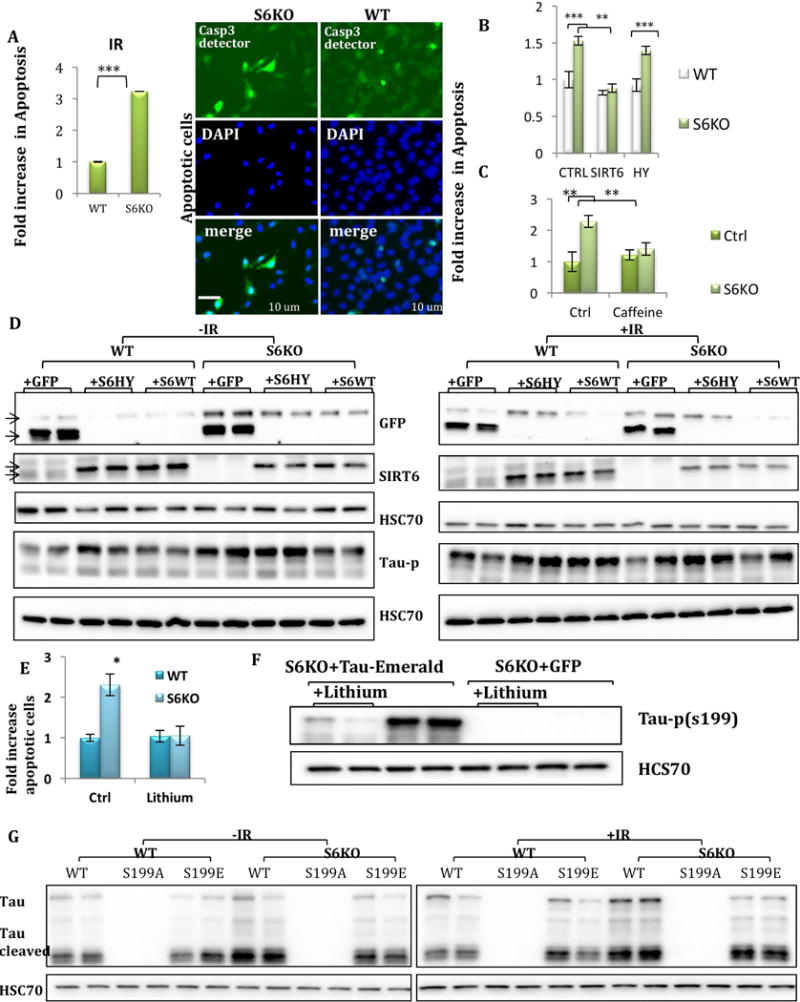
A–C. Apoptotic cells detected by ApoAlert. SH-SY5Y cells were transfected with pCasp3-sensor. A. treated with IR (400rad). Casp-3 activation lead to nuclear localization of GFP. B. Rescue with SIRT6 WT. C. Cells were treated with caffeine for 24hr D. Protein blots of cells that were transfected with GFP, SIRT6WT, SIRT6HY, with and without IR. E. Apoptosis was measured in cells treated by Lithium 1mM for 50hr. Blot showing reduction in Tau-p G. Tau Ser199 mutants were transfected in SH-SY5Y cells +/−IR and collected 2hr after treatment.
SIRT6 is highly expressed in human brains but reduced in AD patients
To understand if reduced levels of SIRT6 in the brain are relevant for human disease, we analyzed its levels in sporadic AD brain samples. We found a striking reduction in SIRT6 in the temporal cortex of AD patients (Fig. 5A). In addition, we analyzed mRNA expression in human datasets where we found SIRT6 reduction in temporal cortex and hippocampus (Fig. 5B), as well as incremented reduction with the increase in the severity of the disease stage (Braak stages iii–v, iii–vi) (Fig. 5C). Interestingly, SIRT6 and PP2A regulatory subunit (PPP2AR1A, the main Tau phosphatase) expression were positively correlated in normal brains, but this correlation was lost in AD brains (Fig. 5D). Our results indicate that reduced SIRT6 is indeed a feature of human AD.
Figure 5. SIRT6 reduction occurs in AD brains.
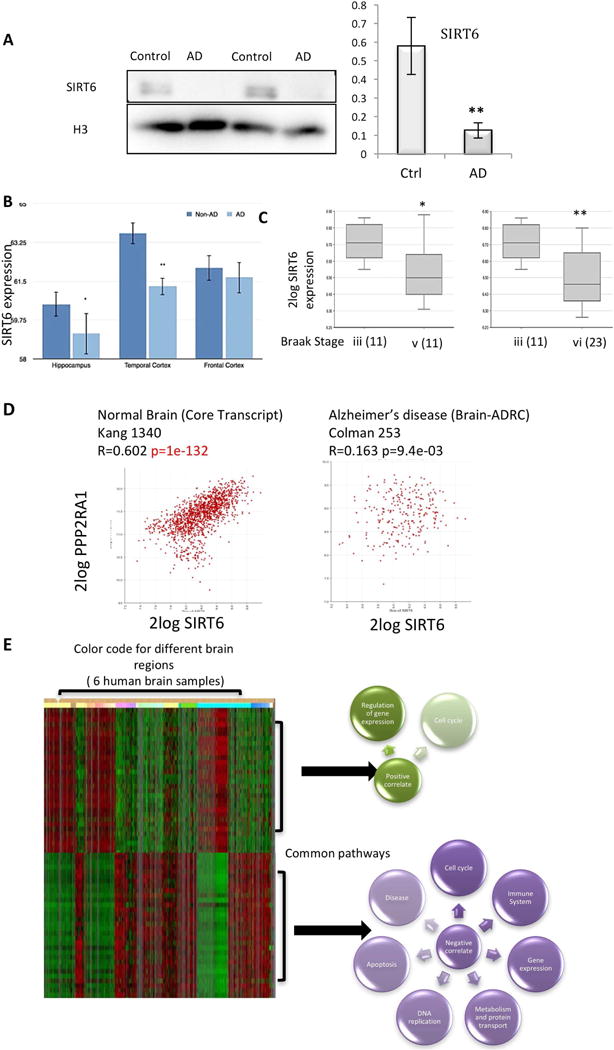
A. Protein blot for AD patients and controls (AD n=4, Ctrl n=4). B. Bioinformatic analysis of Geo profile GDS4758 samples. C–D. SIRT6 and PP2A analysis using R2: Genomics Analysis and Visualization. Datasets: Disease Alzheimer (Brain-ADRC) – Cotman – 253, Disease Alzheimer – Salomon – 74 and Normal Brain (Core Transcript) Kang 1340. E. Analysis of SIRT6 co-expression using the data in the Allen Human Brain Atlas, Allen Institute for Brain Science. Positive and negative genes were then analyzed for pathway enrichment in REACTOME database (http://www.reactome.org).
For all figures: Error bars represent ±SEM. * p<0.05, ** p<0.005, *** p<0.0005.
SIRT6 may have different roles in distinct tissues, but the DNA damage in the brain was striking, while other tissues were less affected (Mostoslavsky et al., 2006; Toiber et al., 2013). To better understand the brain-specific SIRT6 functions we used publicly available expression data and asked which genes in the brain are co-expressed with SIRT6, inferring that co-expression is associated with co-functions. We took advantage of available data from the ALLEN brain atlas (Hawrylycz et al., 2012) (Fig. 5E). We chose the top 2000 positively or 2000 negatively correlated genes for pathway enrichment and their common transcription factors. Not surprisingly, SIRT6 positively correlates with the regulation of gene expression and cell cycle control (two of its known roles). However, when SIRT6 is not expressed (negative correlation), categories related to apoptosis, disease and immune system were enriched. Interestingly, cell cycle regulation was also correlated, which may be relevant since several studies have shown aging and AD brains to re-enter the cell cycle – an interesting phenomenon in which SIRT6 involvement needs further investigation (Arendt, 2012; Bajić et al., 2008; Fischer et al., 2012) (Fig. 5E).
Discussion
SIRT6 is a critical protein in the DNA repair process (Ghosh et al., 2015; Toiber et al., 2013; Kaidi et al., 2010; Mao et al., 2011; McCord et al., 2009; Xu et al., 2015). Here we confirm that the brains of brS6KO mice exhibit pathological marks related to DNA damage and neurodegeneration, as well as behavioral defects. Moreover, SIRT6-deficient cells are vulnerable to further DNA damage accumulation, making it a good model for accelerated brain aging. Previous studies described a brain-specific SIRT6KO mouse (Schwer et al., 2010), yet those studies did not explore genome stability and behavior. In our research we focused on the role of SIRT6 in protecting the brain from increased DNA damage, appearance of toxic Tau-p, and vulnerability to cell death. Our results indicate that SIRT6 is critical for keeping the brain healthy by preventing DNA damage signaling.
Lack of SIRT6 leads to DNA damage and neurodegeneration
DNA damage accumulation is one of the most accepted theories of aging (López-Otín et al., 2013). In the brain, the presence of DNA damage correlates with age and with neurodegeneration. However, what is the cause and what are the consequences is still unclear. Our model strengthens the notion that DNA damage precedes neurodegeneration. Our studies suggest that SIRT6 deficiency leads to accelerated DNA damage accumulation and behavioral defects.
In our brS6KO model, SIRT6 is deleted starting at day 14–17 of embryonic development. Therefore, it is still possible that some of the behavioral effects could be due to deregulation of important pathways in brain formation. It is probable that SIRT6 has more than one role in the brain, however, keeping genomic stability seems to be critical in preventing neurodegeneration. These results are strengthened by our cellular model, where we show that even at the cellular level lack of SIRT6 will lead to increased DNA damage accumulation, Tau stabilization/phosphorylation, and increased sensitivity to genotoxic damage.
Lack of SIRT6 increases Tau phosphorylation and stability
brS6KO mice presented a behavioral phenotype similar to this of irradiated mice (where DNA damage is clearly induced) as well as the behavioral defects of dementia models such as AD. In addition, not all SIRT6-deficient tissues present the same sensitivity to DNA damage, suggesting a brain-specific cause of the hypersensitivity. Remarkably, Tau was hyperphosphorylated in SIRT6-deficient cells and brains, suggesting that this could be one of the proteins increasing the toxicity in the brain. Tau was not only hyperphosphorylated, but was also stabilized by lack of SIRT6 and DNA damage. It is possible that SIRT6 affects Tau by two different mechanisms.
First, SIRT6 could be increasing the activity of GSK3 by constant activation of DNA Damage repair signaling. We and others have shown that GSK3 can be activated upon DNA damage (as in SIRT6KO brains or aging). Such chronic activation would lead to the hypephosphorylation of various targets, such as Tau. Tau is phosphorylated at Ser199 by GSK3, and our and others’ results show that this residue is essential for Tau stability (S199A mutant is hardly expressed). In contrast, the phospho-mimic S199E is more stable, but cleaved in S6KO cells, probably through caspase activation in S6KO cells. Interestingly, SIRT6 is able to bind GSK3, and GSK3 and ATM inhibitors rescued the S6KO cells. GSK3 has been shown to have important roles in AD toxicity and its inhibition is being tested as potential therapeutic tool (Maqbool et al., 2016; McCubrey et al., 2014).
Recently Tau was shown to be acetylated in various residues. While some acetylations were important for Tau stability, others lead to its degradation. However, we did not see increased Tau280ac, but reduced levels in SIRT6KO and IR samples (Fig. S4H), indicating that SIRT6 is not directly regulating this residue.
SIRT6 may be regulating protein stability through other acetylations, or mechanisms that require further research. We see an increase in Tau protein, in S6KO cells as well as under DNA damage, strengthening our hypothesis that Tau’s effect on the SIRT6KO model is related to DNA damage. In our experiments, we separated the transcriptional regulation and splice variants choice from the effect on stability by transfecting Tau-Emerald plasmid. Therefore, using the same promoter, expressing the same cDNA show increased Tau in S6KO and WT irradiated cells. Moreover, proteasome inhibition resulted in the same amounts of Tau on both cell lines, suggesting it is being stabilized from degradation in the absence of SIRT6/DNA damage signaling.
SIRT6 axis in the brain and Alzheimer’s disease
Aging is a major risk for a variety of diseases, among them neurodegeneration. Therefore, common diseases such as AD, PD and other dementias may have a genetic cause or predisposition, but the main mechanism must be more universal. People of different genetic backgrounds have similar incidence of these diseases during aging, and DNA damage accumulation could be the key. Our results show that SIRT6 is drastically reduced in AD patients. Moreover, Jung et al. (2016) showed that the familiar AD model 5XFAD had reduced SIRT6 levels, showing also reduction in AD samples, suggesting that SIRT6 may be relevant for AD in familiar cases as well. SIRT6 is broadly expressed in the brain, therefore we do not assume DNA repair is its unique role. We analyzed normal brain gene expression, and found that SIRT6 is co-expressed with important pathways that regulate gene expression and cell cycle. Moreover, transcription factors important for learning and differentiation such as CREB1 and RORA may regulate SIRT6 expression. However, DNA damage accumulation plays a critical role in brS6KO phenotype, confirmed also by our cellular models, where development of differentiation could not be taken in account.
Overall, we found that SIRT6 depletion in the brain, which occurs during aging and in AD, results in increased GSK3 activity, Tau-p, DNA damage-induced neurodegeneration and behavioral defects, all of which might be linked to a brain-specific SIRT6-GSK3 axis. We believe that this axis holds promises as a therapeutic target in neurodegeneration.
Experimental Procedures
Generation of SIRT6 Conditional KO Mice
Mice were created as in Sebastian et al. (2012). Mice were backcrossed for 3 generations with C57BL6/J mice to obtain heterozygous mice that were 97% C57BL6/J background. These mice were bred with C57BL/Nestin-Cre/J (Jackson Laboratories). All animals were handled and experiments were conducted in accordance with procedures approved by the Institutional Animal Care and Use Committee at the Massachusetts General Hospital in accordance with NIH guidelines. 3–5 month old males and females were used in all experiments.
Generation of SIRT6KO cells
SH-SY5Y cells were infected with lentivirus GeCKO system. Using 2 sgRNAs targeting SIRT6: C2, GCTGTCGCCGTACGCGGACA; C3, GCTCCACGGGAACATGTTTG; and empty shRNA as a control. sgRNA was kindly donated by Aharoni’s lab. Cells were selected by 2ug/ml puromycin for a week, followed by serial dilutions to a single-cell colony.
Plasmids
mEm-MAPTau-N-10 was a gift from Michael Davidson (Addgene # 54155) (Em-Tau); ApoAlert™.
Em-Tau point mutations
Quick change site directed mutagenesis.
TauS199A-F ATCGCAGCGGCTACAGCGCCCCCGGCTCCCCAG
TauS199A-R CTGGGGAGCCGGGGGCGCTGTAGCCGCTGCGAT
TauS199E-F ATCGCAGCGGCTACAGCGAACCCGGCTCCCCAG
TauS199E-R CTGGGGAGCCGGGTTCGCTGTAGCCGCTGCGAT
Statistics
For Behavioral analysis we used Prism 6.0. Analysis include two-way ANOVA, followed by the post-hoc Bonferroni’s test. Latency analysis used the Log Rank Mantel-Cox test (See Supp. Table 1). Simple comparisons were assessed by unpaired two-tailed Student’s t-tests. In all cases, significance was set at p< 0.05.
Supplementary Material
Acknowledgments
This work was supported by the Israeli Ministry of Science and Space. A.B was supported by 2014 NARSAD Award, Bettencourt-Schueller Foundation, and Philippe Foundation. A.S is supported by Biobehavioral Research Awards for Innovative New Scientists (BRAINS)1-R01MH104175 and NIH-NIA 1R01AG048908-01A1. We thank Rafi Srebro for his assistance in the project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
S.K, M.P, D.S, M.E, L.Z performed the molecular Biology experiments. A.B performed the behavioral experiments. U.U and T.A extracted and classified the human tissues. D.T performed experiments and wrote the paper. T.A, A.S, R.M and D.T revised the paper and acquired funding.
See also supplementary data.
Conflict of interests:
The authors don’t have any conflict of interest.
References
- Arendt T. Cell cycle activation and aneuploid neurons in Alzheimer’s disease. Molecular Neurobiology. 2012;46:125–35. doi: 10.1007/s12035-012-8262-0. [DOI] [PubMed] [Google Scholar]
- Bajic V, Spremo-Potparevic B, Zivkovic L, Isenovic E, Arendt T. Cohesion and the aneuploid phenotype in Alzheimer’s disease: A tale of genome instability. Neuroscience and Biobehavioral Reviews. 2015;55:365–74. doi: 10.1016/j.neubiorev.2015.05.010. [DOI] [PubMed] [Google Scholar]
- Bajić V, Spremo-Potparević B, Zivković L, Djelić N, Smith M. Is the time dimension of the cell cycle re-entry in AD regulated by centromere cohesion dynamics? Bioscience Hypotheses. 2008;1:156–161. doi: 10.1016/j.bihy.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Poljak A, Grant R, Jayasena T, Mansour H, Chan-Ling T, Smythe G, Sachdev P, Guillemin G. Differential expression of sirtuins in the aging rat brain. Frontiers in Cellular Neuroscience. 2015;9:167. doi: 10.3389/fncel.2015.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Zhao Y, Zhao B. Roles of glycogen synthase kinase 3 in Alzheimer’s disease. Current Alzheimer Research. 2012;9:864–79. doi: 10.2174/156720512802455386. [DOI] [PubMed] [Google Scholar]
- Cook C, Carlomagno Y, Gendron T, Dunmore J, Scheffel K, Stetler C, Davis M, Dickson D, Jarpe M, DeTure M, et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Human Molecular Genetics. 2014;23:104–16. doi: 10.1093/hmg/ddt402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Murray M, Petrucelli L. Understanding biomarkers of neurodegeneration: Novel approaches to detecting tau pathology. Nature Medicine. 2015;21:219–20. doi: 10.1038/nm.3809. [DOI] [PubMed] [Google Scholar]
- Durieux P, Bearzatto B, Guiducci S, Buch T, Waisman A, Zoli M, Schiffmann S, Exaerde A de d’. D2R striatopallidal neurons inhibit both locomotor and drug reward processes. Nature Neuroscience. 2009;12:393–5. doi: 10.1038/nn.2286. [DOI] [PubMed] [Google Scholar]
- Fischer HG, Morawski M, Brückner M, Mittag A, Tarnok A, Arendt T. Changes in neuronal DNA content variation in the human brain during aging. Aging Cell. 2012;11:628–33. doi: 10.1111/j.1474-9726.2012.00826.x. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Liu B, Wang Y, Hao Q, Zhou Z. Lamin A Is an Endogenous SIRT6 Activator and Promotes SIRT6-Mediated DNA Repair. Cell Rep. 2015;13:1396–406. doi: 10.1016/j.celrep.2015.10.006. [DOI] [PubMed] [Google Scholar]
- Giblin W, Skinner ME, Lombard DB. Sirtuins: guardians of mammalian healthspan. Trends Genet. 2014;30:271–86. doi: 10.1016/j.tig.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong CX, Iqbal Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Current Medicinal Chemistry. 2008;15:2321–8. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsky M, Burnouf S, Dols J, Mandelkow E, Partridge L. Acetylation mimic of lysine 280 exacerbates human Tau neurotoxicity in vivo. Scientific Reports. 2016;6:22685. doi: 10.1038/srep22685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham D, Sidhu A. Mice expressing the A53T mutant form of human alpha-synuclein exhibit hyperactivity and reduced anxiety-like behavior. Journal of Neuroscience Research. 2010;88:1777–83. doi: 10.1002/jnr.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, Lagemaat LN van de, Smith KA, Ebbert A, Riley ZL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–9. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovatta I, Tennant R, Helton R, Marr R, Singer O, Redwine J, Ellison J, Schadt E, Verma I, Lockhart D, et al. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature. 2005;438:662–6. doi: 10.1038/nature04250. [DOI] [PubMed] [Google Scholar]
- Iijima-Ando K, Zhao L, Gatt A, Shenton C, Iijima K. A DNA damage-activated checkpoint kinase phosphorylates tau and enhances tau-induced neurodegeneration. Human Molecular Genetics. 2010;19:1930–8. doi: 10.1093/hmg/ddq068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami Harada, Hirokawa Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neuroscience Letters. 2000;279:129–32. doi: 10.1016/s0304-3940(99)00964-7. [DOI] [PubMed] [Google Scholar]
- Jung E, Choi H, Song H, Hwang Y, Kim A, Ryu H, Mook-Jung I. p53-dependent SIRT6 expression protects Aβ42-induced DNA damage. Scientific Reports. 2016;6:25628. doi: 10.1038/srep25628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010;329:1348–53. doi: 10.1126/science.1192049. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kugel S, Mostoslavsky R. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem Sci. 2014;39:72–81. doi: 10.1016/j.tibs.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Leugers C. Tau and tauopathies. Progress in Molecular Biology and Translational Science. 2012;107:263–93. doi: 10.1016/B978-0-12-385883-2.00004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee V, Brunden K, Hutton M, Trojanowski J. Developing therapeutic approaches to tau, selected kinases, and related neuronal protein targets. Cold Spring Harbor Perspectives in Medicine. 2011;1:a006437. doi: 10.1101/cshperspect.a006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Lu K. Pinning down phosphorylated tau and tauopathies. Biochimica et Biophysica Acta. 2005;1739:311–22. doi: 10.1016/j.bbadis.2004.10.003. [DOI] [PubMed] [Google Scholar]
- López-Otín C, Blasco M, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner B. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–91. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Ma T. GSK3 in Alzheimer’s disease: mind the isoforms. Journal of Alzheimer’s Disease : JAD. 2014;39:707–10. doi: 10.3233/JAD-131661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron. 2014;83:266–82. doi: 10.1016/j.neuron.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Hine C, Tian X, Meter M Van, Au M, Vaidya A, Seluanov A, Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443–6. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maqbool M, Mobashir M, Hoda N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. European Journal of Medicinal Chemistry. 2016;107:63–81. doi: 10.1016/j.ejmech.2015.10.018. [DOI] [PubMed] [Google Scholar]
- Martin L, Latypova X, Wilson C, Magnaudeix A, Perrin ML, Terro F. Tau protein phosphatases in Alzheimer’s disease: the leading role of PP2A. Ageing Research Reviews. 2013;12:39–49. doi: 10.1016/j.arr.2012.06.008. [DOI] [PubMed] [Google Scholar]
- McCord R, Michishita E, Hong T, Berber E, Boxer L, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame A, et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging. 2009;1:109–21. doi: 10.18632/aging.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubrey J, Steelman L, Bertrand F, Davis N, Sokolosky M, Abrams S, Montalto G, D’Assoro A, Libra M, Nicoletti F, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014;5:2881–911. doi: 10.18632/oncotarget.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SWW, Cho SHH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–66. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SWW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. 2015;21:1154–62. doi: 10.1038/nm.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua K, Lombard D, Pang W, Fischer M, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy M, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–29. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Ngok-Ngam P, Watcharasit P, Thiantanawat A, Satayavivad J. Pharmacological inhibition of GSK3 attenuates DNA damage-induced apoptosis via reduction of p53 mitochondrial translocation and Bax oligomerization in neuroblastoma SH-SY5Y cells. Cellular & Molecular Biology Letters. 2013;18:58–74. doi: 10.2478/s11658-012-0039-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Lee S, Shang Y, Wang WY, Au K, Kamiya S, Barmada S, Finkbeiner S, Lui H, Carlton C, et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. The Journal of Clinical Investigation. 2014;124:981–99. doi: 10.1172/JCI72723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, Kellis M, Gymrek M, Issner R, Coyne M, et al. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. 2011;147:1628–39. doi: 10.1016/j.cell.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Ye H, Decker P, Howe C, Wetmore C. Therapeutic doses of cranial irradiation induce hippocampus-dependent cognitive deficits in young mice. Journal of Neuro-Oncology. 2011;105:191–8. doi: 10.1007/s11060-011-0582-9. [DOI] [PubMed] [Google Scholar]
- Rulten S, Caldecott K. DNA strand break repair and neurodegeneration. DNA Repair. 2013;12:558–67. doi: 10.1016/j.dnarep.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Saxe MD, Battaglia F, Wang JWW, Malleret G, David DJ, Monckton JE, Garcia AD, Sofroniew MV, Kandel ER, Santarelli L, et al. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci USA. 2006;103:17501–6. doi: 10.1073/pnas.0607207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Schumacher B, Lombard DB, Xiao C, Kurtev MV, Gao J, Schneider JI, Chai H, Bronson RT, Tsai LHH, et al. Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc Natl Acad Sci USA. 2010;107:21790–4. doi: 10.1073/pnas.1016306107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn PD, Tracy TE, Son HII, Zhou Y, Leite RE, Miller BL, Seeley WW, Grinberg LT, Gan L. Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener. 2016;11:47. doi: 10.1186/s13024-016-0109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M, Goedert M. Tau pathology and neurodegeneration. The Lancet. Neurology. 2013;12:609–22. doi: 10.1016/S1474-4422(13)70090-5. [DOI] [PubMed] [Google Scholar]
- Sultan A, Nesslany F, Violet M, Bégard S, Loyens A, Talahari S, Mansuroglu Z, Marzin D, Sergeant N, Humez S, et al. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem. 2011;286:4566–75. doi: 10.1074/jbc.M110.199976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao K, Miyakawa T. Light/dark transition test for mice. Journal of Visualized Experiments : JoVE. 2006;104 doi: 10.3791/104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanda K, Nishi A, Matsuo N, Nakanishi K, Yamasaki N, Sugimoto T, Toyama K, Takao K, Miyakawa T. Abnormal social behavior, hyperactivity, impaired remote spatial memory, and increased D1-mediated dopaminergic signaling in neuronal nitric oxide synthase knockout mice. Molecular Brain. 2009;2:19. doi: 10.1186/1756-6606-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenreiro S, Eckermann K, Outeiro T. Protein phosphorylation in neurodegeneration: friend or foe? Frontiers in Molecular Neuroscience. 2014;7:42. doi: 10.3389/fnmol.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, Sebastian C, Cosentino C, Martinez-Pastor B, Giacosa S, et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell. 2013;51:454–68. doi: 10.1016/j.molcel.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy TE, Sohn PD, Minami SS, Wang C, Min SWW, Li Y, Zhou Y, Le D, Lo I, Ponnusamy R, et al. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron. 2016;90:245–60. doi: 10.1016/j.neuron.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vianna Alonso, Viola Quevedo, de Paris Furman, de Stein Medina, Izquierdo Role of hippocampal signaling pathways in long-term memory formation of a nonassociative learning task in the rat. Learning & Memory (Cold Spring Harbor, N.Y.) 2000;7:333–40. doi: 10.1101/lm.34600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R, Talahari S, Nesslany F, Lefebvre B, Bonnefoy E, et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci. 2014;8:84. doi: 10.3389/fncel.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronkov M, Braithwaite S, Stock J. Phosphoprotein phosphatase 2A: a novel druggable target for Alzheimer’s disease. Future Medicinal Chemistry. 2011;3:821–33. doi: 10.4155/fmc.11.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- Watcharasit P, Bijur G, Zmijewski J, Song L, Zmijewska A, Chen X, Johnson G, Jope R. Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7951–5. doi: 10.1073/pnas.122062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Zhang L, Zhang W, Meng D, Zhang H, Jiang Y, Xu X, Meter M, Seluanov A, Gorbunova V, et al. SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle (Georgetown, Tex.) 2015;14:269–76. doi: 10.4161/15384101.2014.980641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CCC, Xing A, Tan MSS, Tan L, Yu JTT. The Role of MAPT in Neurodegenerative Diseases: Genetics, Mechanisms and Therapy. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9415-8. [DOI] [PubMed] [Google Scholar]
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–6. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013;9:208–45. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.