

Abstract
Nanoparticle (NP) based drug delivery systems are frequently employed to improve the intravenous administration of chemotherapy; however, few reports explore their application as an intraperitoneal therapy. We developed a pH-responsive expansile nanoparticle (eNP) specifically designed to leverage the intraperitoneal route of administration to treat intraperitoneal malignancies, such as mesothelioma, ovarian and pancreatic carcinomatoses. This review describes the design, evaluation and evolution of the eNP technology and, specifically, a Materials-Based Targeting paradigm that is unique among the many active and passive targeting strategies currently employed among NP delivery systems. pH-responsive eNP swelling is responsible for the extended residence at the target tumor site as well as the subsequent improvement in tumoral drug delivery and efficacy observed with paclitaxel-loaded eNPs (PTX-eNPs) compared to the standard clinical formulation of paclitaxel, Taxol®. Superior PTX-eNP efficacy is demonstrated in two different orthotopic models of peritoneal cancer—mesothelioma and ovarian cancer; in a third model—of pancreatic cancer—PTX-eNPs demonstrated comparable efficacy to Taxol with reduced toxicity. Furthermore, the unique structural and responsive characteristics of eNPs enable them to be used in three additional treatment paradigms, including: treatment of lymphatic metastases in breast cancer; use as a highly-fluorescent probe to visually-guide the resection of peritoneal implants; and, in a two-step delivery paradigm for concentrating separately administered NP and drug at a target site. This case study serves as an important example of using the targeted disease-state’s pathophysiology to inform the NP design as well as the method of use of the delivery system.
Keywords: Nanoparticle drug delivery, expansile nanoparticle, peritoneal cancer, intraperitoneal administration, tumor targeting, Materials-Based Targeting, lymphatic metastases, visually-guided resection, two-step delivery
Graphical abstract
Expansile nanoparticles (eNPs) use Materials-Based Targeting to treat peritoneal cancers (e.g., mesothelioma, ovarian, pancreatic cancer).

Introduction
In this review we describe the design, evaluation, and evolution of the expansile nanoparticle (eNP) technology—a drug delivery system specifically designed for the treatment of intraperitoneal malignancies. We highlight the challenges of treating intraperitoneal cancers and summarize the state-of-the art in both clinical treatments and novel therapies. We describe how this technology leverages the pathophysiology of peritoneal tumors to enable a new type of Materials-Based Targeting strategy along with an efficient high-dose delivery of chemotherapy, both of which involve pH-triggered particle swelling—a keystone of the eNPs’ function and mechanism of action. We highlight the rigorous nanoparticle characterization and testing required to validate particle function and describe the efficacy of paclitaxel-loaded eNPs (PTX-eNPs) in three different models of peritoneal cancer: mesothelioma, ovarian and pancreatic carcinomatosis (i.e., cancer spread throughout the peritoneal cavity). Lastly, we describe the structural and responsive characteristics of eNPs that enable the technology to be uniquely leveraged in three additional treatment paradigms: as a means of treating lymphatic spread of disease (i.e. regional metastases); as a highly-fluorescent probe to visually-guide surgical resection of peritoneal implants and small tumors; and, as a two-step drug delivery device for concentrating separately administered drug at a target site.
Current treatments for patients with peritoneal malignancies frequently involve a complex combination of cytoreductive surgery, to remove large bulky disease, and peri- or post-operative administration of chemotherapy to treat residual, un-resected, or occult disease. Even though residual disease is located within the peritoneal cavity in the vast majority of patients, most chemotherapeutic regimens are administered intravenously. Intravenous administration leads to poor tumoral delivery with, frequently, less than one percent of the injected dose accumulating in tumors due to systemic distribution and rapid first-pass clearance.1 In contrast, drugs that are administered via the intraperitoneal route of administration are maintained in close proximity to the target tumor tissues leading to more rapid and prolonged tumoral accumulation and increased efficacy compared to intravenous administration.2–5
(Note to the Editorial Staff: The following definitions, which are critical to understanding the review, are intended to be included as a second “side-bar” to make them readily viewable for a reader skimming the text.)
Definitions:
PTX = paclitaxel (the drug alone)
PTX-eNP = paclitaxel-loaded expansile nanoparticle
Taxol® = paclitaxel dissolved in Cremophor EL/ethanol (i.e., the clinical formulation of paclitaxel)
Recent clinical studies demonstrate the benefit of intraperitoneal delivery. The results of two Gynecologic Oncology Group studies (GOG-0114 and GOG-0172) conclusively demonstrated improved outcomes for ovarian cancer patients treated with intraperitoneal Taxol® + cisplatin as opposed to standard intravenous injections of the same combination (overall survival of 61.8 v. 51.4 months, 23% decreased risk of death).6 Intraperitoneal delivery is also being investigated in peritoneal carcinomatosis of pancreatic ductal adenocarcinoma origin (PDAC-PC) where several clinical trials are ongoing.7–9 In patients with malignant peritoneal mesothelioma, observational studies document a similar improvement in patient survival for those undergoing cytoreductive surgery in combination with intraperitoneal chemotherapy (e.g., Taxol + cisplatin).2, 5 While no Phase III randomized clinical trials have been performed due to the limited number of patients presenting with peritoneal mesothelioma in the U.S., there is discussion that these combined surgical / chemotherapeutic regimens lead to improved outcomes. These conclusions must be handled with caution since, in the absence of randomized clinical trials, evaluation may be skewed by confounding variables or from referral bias due to patient selection favoring those with earlier-stage disease and fewer co-morbidities who are likely to have better outcomes. Unfortunately, randomized clinical trials may become possible in the future as the worldwide incidence of mesothelioma continues to rise with continued use of asbestos as an insulating material in the developing world.10–11
Despite the benefits to be gained from intraperitoneal administration, significant drawbacks have hampered the adoption and implementation of this treatment strategy. Primary among these are the heightened risk of systemic toxicity, surgical wound healing and technical complexity associated with intraoperative circulation of chemotherapy.1, 12–13 While intraperitoneal administration improves the pharmacokinetic profile of higher molecular weight (MW) agents (e.g., paclitaxel, MW = 853.9 g/mol) compared to intravenous administration with a t½ of 3–7 hours,14–15 systemic clearance is nevertheless still rapid with plasma t½ on the order of 4–12 hours for intraperitoneal administration.4
In addition, the large surface area of the peritoneal cavity, widespread dissemination of peritoneal tumors, and the presence of microtumors resulting from dissemination and seeding of tumors throughout the peritoneal cavity,16–17 make complete surgical resection both a herculean effort and one that is rarely achieved. Lastly, peritoneal tumors can be inoperable due to proximity to vital organs and structures. As a consequence of incomplete surgical resection, intraoperative tumor-cell seeding, or growth of occult microtumors, the majority of patients eventually develop recurrent disease that is typically more aggressive, frequently inoperable, and less responsive to chemotherapy / radiation therapy.18 It is the local progression of peritoneal disease, rather than metastatic disease, that results in the poor 5-year survival rates associated with mesothelioma and ovarian cancer.19–20 In resectable pancreatic cancer, the stagnant, 5-year survival rate remains low due to recurrence or metastases, with 50% of patients presenting with peritoneal metastases.21
We identified the above challenges as a significant clinical opportunity that could be addressed by the development of a nanoparticle-based drug delivery system for administration via the intraperitoneal route. The intraperitoneal route of administration has been leveraged by others developing nanoparticle-based drug delivery systems22–26 and is in contrast to the majority of nanoparticle systems, which are currently designed for intravenous administration.27–32 Specifically, we identified five design requirements necessary for the success of such a system, including the ability to: 1) leverage the intraperitoneal route of administration for peritoneal malignancies; 2) deliver a clinically relevant chemotherapeutic agent; 3) preferentially localize to tumor tissue through a unique Materials-Based Targeting mechanism that does not rely on the use of passive [i.e., enhanced permeability and retention (EPR) effect] or active (i.e., antibody-based) targeting; 4) respond to a physiological cue to release its payload only at the target site thereby minimizing off-target side effects and toxicity; and, 5) provide prolonged (days to weeks) delivery of therapeutic concentrations of drug and thereby improve treatment efficacy as measured by quantitative clinical endpoints (e.g., tumor growth or overall survival). To fulfil these design requirements and address this challenge, we engineered the expansile nanoparticle (eNP) delivery system (Fig. 1).
Figure 1.
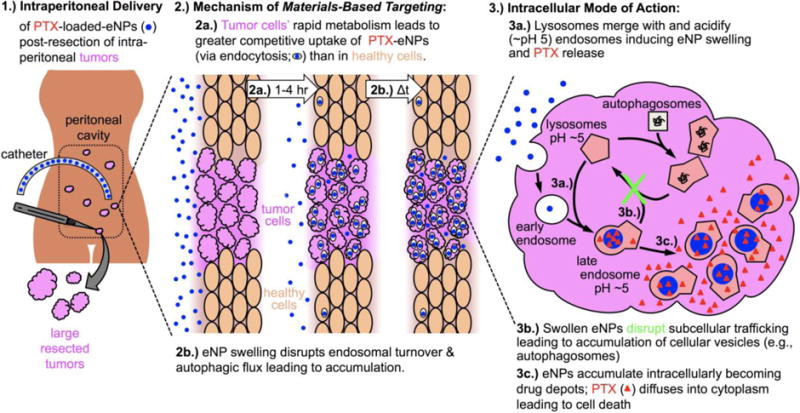
(Note to the editorial staff, this figure is intended to be a double column figure) Schematic of expansile nanoparticle (eNP)-mediated drug delivery to peritoneal tumors. eNPs are administered intraperitoneally and localize to tumors via Materials-Based Targeting. Disruption of intracellular trafficking results in eNP accumulation and delivery of high concentrations of paclitaxel (PTX) to the target cell.
Design and Characterization of the eNP System
The essential and unique material properties of the eNPs are derived from their polymer structure. Each eNP monomer unit is composed of three basic building blocks: (1) a hydrophobic, pH-responsive protecting group that masks (2) a hydrophilic triol-linker, and (3) a methyl methacrylate end-group that enables polymerization (Fig. 2A).33 Incorporation of a small percentage of a hydroquinone-based di-ester methacrylate leads to crosslinking of the polymer chains. The hydrophobicity imparted by the protecting group allows the monomer to be dissolved in organic solvents (e.g., dichloromethane, ethyl acetate). This, in turn, enables synthesis of nanoparticles via formation of an oil-in-water emulsion followed by in situ polymerization of the monomer and crosslinker. The oil-in-water synthetic scheme also enables loading of hydrophobic drug payloads, such as the small molecule chemotherapeutic paclitaxel (PTX), which partitions into the oil/eNP phase. Following polymerization, particles are stirred under air to allow the evaporation of the organic solvent and dialyzed in pH 7.4 phosphate buffer to remove excess salts and surfactants. The eNPs are stabilized with an anionic sodium dodecyl sulfate (SDS) surface coating that imparts a strong negative surface charge (−30 mV to −50 mV). The resultant particles possess a smooth morphology and a mean diameter of 30 nm to 50 nm as visualized by scanning electron microscopy (SEM).
Figure 2.

(Note to the editorial staff, this figure is intended to be a double column figure) A.) Schematic of the expansile nanoparticle (eNP) and non-expansile nanoparticle (neNP) chemical structures and function. B.) Hydrolysis of the eNP and neNP protecting groups as a function of time under pH 5 and pH 7.4 conditions. C.) Hydrophobicity of the eNP as a function of time in swelling particles (pH 5) and un-swollen particles (pH 7.4). D–F.) Transmission, freeze-fracture transmission, and scanning electron micrographs of un-swollen eNPs at pH 7.4 and swollen eNPs at pH 5. A decrease in eNP density (opacity in D) and increase in eNP size (E,F) are readily apparent. G.) eNP swelling and neNP lack-of swelling is characterized via dynamic light scattering at pH 5 and 7.4. H.) Particle-by-particle swelling of eNPs incubated at pH 5 is observed using scanning ion occlusion sensing (qNano) technology. Adapted with permission from Ref. 33, Copyright 2009 American Chemical Society; and, Ref. 36, Copyright 2013 They Royal Society of Chemistry
The eNP’s lightly cross-linked polymer structure affords a pH-triggered swelling functionality. Specifically, the pH-labile, 2,4,6-trimethoxybenzilidene acetal-protecting group is stable at pH 7.4 but hydrolyzes slowly at pH 6 and faster at mildly acidic pH ~5; this deprotection is readily monitored via high-performance liquid-chromatography (HPLC; Fig. 2B).33 As a consequence, when eNPs reach the extracellular environment of microscopic hypoxic tumors,34 large hypoxic tumors35 (pH ~6.5) or the late endosomal/lysosomal compartments of tumor cells (pH ~5), the protecting group hydrolyzes, exposing the more hydrophilic alcohol moieties. Hydrolysis of the ester linkage between the polymer backbone and linker is slower than the release of the protecting group given that the former is an ester and the latter is an activated acetal. If minimal hydrolysis of the ester linkage between the polymer backbone and linker occurs, it would result in exposure of a hydrophilic carboxylic acid moiety instead of two hydrophilic hydroxyls. A corresponding change in the hydrophobicity of the eNPs can be monitored via covalent incorporation of pyrene-methyl methacrylate—a solvochromatic dye whose fluorescence emission spectrum reflects the polarity of the surrounding environment (Fig. 2C).36 This compositional change from hydrophobic to hydrophilic leads to water infiltration into the polymer network and swelling of the particle. Particles swell, but do not immediately degrade as a result of the light (1% wt/wt) crosslinking. Particle swelling can be visualized via electron microscopy including, transmission-, freeze-fracture transmission-, and scanning electron microscopy (Fig. 2D–F).33, 36–38 To complement these techniques, dynamic light scattering (DLS) and tunable resistive pulse-sensing (i.e., qNano technology from iZon Ltd.) which provides the added benefit of measuring particle concentration, are also used (Fig. 2G–H).33, 36, 39 In these “aqueous” studies, the particles swell, and the density and mechanical modulus of the particles decreases with the terminal state of swelling being dissolution of the particle into individual polymer chains.36, 40 Furthermore, these studies, which examine swelling over various time courses and under multiple pH and buffering conditions, reveal that eNP swelling is a heterogeneous process that occurs over a time-scale of hours to days and with an ultimate increase in particle diameter on the order of 2–10X.
Particle swelling plays a critical role in the drug-release kinetics of the eNP system. Specifically, drug is packaged securely within the eNP as long as the pH of the local environment remains neutral and the particle unswollen. Upon acidification, the eNP swells and the encapsulated drug payload is released slowly over a period of time that closely mirrors the time-course of polymer deprotection and particle swelling (Fig. 3).33 Three different control particles were employed to confirm the pH-responsive drug-release functionality. First, substituting benzaldehyde for the protecting group (Fig. 2A) increased the stability of the structure, preventing hydrolysis at pH 5–6.5 and necessitating exposure to strongly acidic conditions (pH ≤2) to induce deprotection and swelling. Particles with this structure are termed “non-expansile nanoparticles” (neNPs) and serve as a chemically similar functional control under mildly acidic (pH 5–6.5) conditions. As a result, drug release from neNPs is independent of pH (Fig. 3).33 Second, eNPs were synthesized with “heavy” crosslinking (30% wt/wt v. the “standard” 1% wt/wt; termed “30X-eNPs”). Unlike the neNPs, the 30X-eNPs still undergo hydrolysis at pH 5, however the increased cross-linking prevents swelling and release of the drug payload thereby confirming that deprotection of the polymer alone is not enough to induce drug release (Fig. 3).38 Third, non-responsive, generic PTX-loaded poly(lactic-co-glycolic) acid nanoparticles (PTX-PLGA-NPs) demonstrated rapid “burst” release within the first 4 hours regardless of pH.
Figure 3.

(Note to the editorial staff, this figure is intended to be a double column figure) Paclitaxel (PTX) release from eNPs is characterized by high-performance liquid chromatography (HPLC) at pH 5 and pH 7.4. eNPs release PTX in a pH- and time-dependent manner. Control non-expansile nanoparticles (neNPs) and poly(lactic-co-glycolic) acid (PLGA)-NPs exhibit burst release regardless of pH. Control heavily crosslinked-eNPs (30X-eNPs) do not exhibit pH-dependent release. Adapted with permission from Ref. 33, Copyright 2009 American Chemical Society; and, Ref. 38, Copyright 2016 Future Medicine Ltd.
Particle swelling also plays an important role in the mechanistic interplay between the eNP and the subcellular trafficking that occurs following internalization within a cell. Prolonged intracellular accumulation of eNPs is essential to their efficacy because the drug payload, paclitaxel, must be present at therapeutic concentrations within the cell for at least one cell-cycle (i.e., >20 hours) to be cytotoxic.41 Based on the results generated with pharmacological inhibitors, temperature-sensitive metabolic reduction and fluid-phase marker co-localization of fluorescently-labeled eNPs (Fig. 4A), macropinocytosis is the primary pathway of particle uptake in multiple cancer cell lines, including: mesothelioma (MSTO-211H) and triple-negative (i.e., ER-, PR-, Her2/neu-) breast carcinoma (MDA-MB-231) (Fig. 4B).42–43 A similar uptake pathway is found with mesothelioma spheroids, which provide a measure of improved modeling of the tumoral microenvironment, including a collagen extracellular matrix.44 Following internalization, eNPs disrupt subcellular trafficking, leading to a block in autophagic flux and prolonged particle accumulation.43
Figure 4.

(Note to the editorial staff, this figure is intended to be a double column figure) Materials-Based Targeting A.) Confocal microscopy demonstrates that eNPs localize to late endosomes/lysosomes in vitro. B.) Pharmacologic inhibition of eNP uptake demonstrates that macropinocytosis is the primary pathway of particle internalization. C.) Schematic of proposed intracellular action of eNPs leading to prolonged tumoral accumulation. D.) Quantification of LC3-II in MSTO-211H mesothelioma tumor cells demonstrates dose-dependent increases when treated with eNPs. E.) Fluorescently-labelled eNPs (blue/white) accumulate specifically within regions of intraperitoneal tumor (white dashed circles) and remain for at least two weeks. F–G.) eNP internalization into malignant tumor cells in vitro is an order of magnitude faster than in healthy epithelial cells. Adapted with permission from Ref. 42, Copyright 2013 American Chemical Society; and, Ref. 43, Copyright 2016 Elsevier B.V.
The exact mechanism by which eNPs interfere with subcellular trafficking is still being investigated, but control experiments demonstrate that it is likely linked to the swelling functionality of eNPs (Fig. 4C). Specifically, while treatment with eNPs led to dose-dependent increases in LC3-II—a protein sequestered in the membranes of autophagosomes and whose accumulation serves as a marker of the impairment of autophagy—treatment with PLGA-NPs had no significant effect (Fig. 4D). Furthermore, treatment with eNPs also inhibited autophagosomal degradation and flux as measured by accumulation of p62—a protein normally degraded during autophagy. Treatment with PLGA-NPs or neNPs did not inhibit autophagosomal degradation/flux.43 Though these control studies are not exhaustive, they suggest the centrality of swelling in achieving prolonged intracellular accumulation via disruption of subcellular trafficking. These results provide impetus for the development of new delivery systems specifically designed to optimize the disruption of particle trafficking as a stand-alone therapeutic strategy or as part of a more complex treatment scheme.
eNP swelling, and the ensuing disruption of subcellular trafficking, is integral to the in vivo performance of eNPs. To visually assess the tumoral accumulation of eNPs, particles were covalently labeled with a fluorophore [either PolyFluor407 (PF-eNPs) or rhodamine-methyl methacrylate (Rho-eNPs)] and administered to mice bearing established intraperitoneal mesothelioma tumors. PF-eNPs localize rapidly, within 4 hours of injection, to regions of tumor of varying sizes from sub-mm to sub-cm and ≥1 cm. Furthermore, particles persist in the tumors for at least 14 days (Fig. 4E).43 In contrast, neither rhodamine-labeled PLGA-NPs, neNPs nor PEG-ylated eNPs are observable within tumors 24 hours after injection.43, 45 To investigate the mechanism behind this localization, we evaluated the in vitro uptake of Rho-eNPs in both healthy, non-immortalized, mesothelial cells (LP-3) and malignant mesothelioma cells (MSTO-211H) using flow assisted cell sorting (FACS). Tumor cells internalize Rho-eNPs an order of magnitude faster than healthy cells (98% uptake v. 2% uptake after 2 hours of incubation, respectively) (Fig. 4F) which is consistent with the well-documented hyper-function of endosomal processes in tumor cells that leads to increased rates of endocytosis.43, 46 Notably, eNPs demonstrate this rapid, highly-specific and prolonged tumoral accumulation without the use of targeting ligands, antibodies, or external triggers (e.g., light, sound, heat). With control experiments demonstrating that the material properties of the eNP polymer (i.e., pH-triggered swelling) are an essential design requirement to achieve tumoral accumulation, rather than surface functionalization, we have termed the “targeting” observed with eNPs: “Materials-Based Targeting”. This stands in contrast to both passive-targeting strategies that rely upon the enhanced permeability and retention (EPR) effect as well as active-targeting strategies that rely upon the use of targeting moieties such as antibodies, oligonucleotides or other complementary, high-affinity probes (e.g., biotin-avidin).
(Note to the Editorial Staff: The following paragraph is intended to be included as a “side-bar” and not part of the main text. It highlights information from the paragraph above that would be useful for someone skimming the article)
“Materials-Based Targeting” leverages the rapid metabolism of tumor cells compared to healthy cells as well as nanoparticle swelling that, following internalization, disrupts sub-cellular trafficking to achieve accumulation of particles within the target tumor cells. Materials-Based Targeting stands in contrast to both passive-targeting strategies that rely upon the enhanced permeability and retention (EPR) effect as well as active-targeting strategies that rely upon the use of targeting moieties such as antibodies, oligonucleotides, or other complementary high-affinity probes (e.g., biotin-avidin).
eNPs in Intraperitoneal Mesothelioma
With the materials-based targeting strategy providing tumor-specific accumulation of eNPs, we hypothesized that intraperitoneal administration of paclitaxel-loaded eNPs (PTX-eNPs) would provide prolonged tumoral and target-specific drug delivery. To determine the pharmacokinetic profile of PTX delivered via eNPs, animals with established intraperitoneal mesothelioma tumors were treated with a single intraperitoneal injection of PTX-eNPs. Animals were euthanized at various time points out to 7 days post-injection, and PTX concentration in the tumors, plasma, and a peritoneal lavage performed at the time of euthanasia was quantified by HPLC-mass spectrometry (HPLC-MS). As a control, a second group of animals received “free” PTX dissolved in Cremophor EL/ethanol (i.e., the clinically used formulation of paclitaxel; Taxol®). Both groups were given the equivalent of 10 mg/kg paclitaxel which, when scaled by body surface area, is somewhat lower than a standard 135 mg/m2 dose used clinically in humans. Treatment with PTX-eNPs increased the intratumoral delivery of PTX by 10-fold compared to Taxol over the first 24 hours and by over 100-fold compared to Taxol seven days following the injection (i.e., Fig. 5A).43 Maintenance of these high, super-therapeutic concentrations of PTX is significant because it is well documented that treatment with low doses of PTX at concentrations below the IC50 of ~10–50 ng/mL may drive the development of drug resistance.41, 47–48 While the comparison of PTX per mass of tissue in vivo and the in vitro IC50 is not exact, it is nevertheless clear that eNPs deliver PTX to the target tissue at concentrations in excess of 10,000-fold the IC50. Due to the rapid clearance of paclitaxel administered as Taxol within the first 4 hours, PTX concentrations in the plasma and peritoneal lavage are higher when delivered as PTX-eNPs than Taxol at all time points after the first 4 hours. This is reflective of two factors: first, the prolonged accumulation of PTX within the tumors establishes, in effect, a “drug depot” that continues to elute PTX into the surrounding tissues and, subsequently, the plasma for at least 7 days; second, the intraperitoneal administration of Taxol results in a bolus of drug which diffuses from the peritoneal cavity into the plasma and is rapidly cleared with greater than 99% of the drug removed within the first 24 hours. To compare the eNP system against a generic, non-responsive nanoparticle control, the experiment was also run with PTX-PLGA-NPs and the intratumoral concentration of PTX was demonstrated to be <20% of that achieved via the PTX-eNP.43
Figure 5.

(Note to the editorial staff, this figure is intended to be a double column figure) A.) Pharmacokinetics of PTX administered via eNPs or as Taxol. Paclitaxel-loaded eNPs (PTX-eNPs) deliver 10–100-fold more PTX to intraperitoneal mesothelioma tumors than is achieved with Taxol. B.) In a multi-dose established disease model, PTX-eNPs significantly improve survival compared to all controls and 33% of animals demonstrate a complete clinical response. C.) Doubling the weekly dose of PTX-eNPs or Taxol to 20 mg/kg/week does not improve survival compared to the 10 mg/kg/week dose. D.) Doubling the duration of dosing from four to eight weeks increases overall survival of PTX-eNP-treated animals while providing no benefit to Taxol-treated animals. Adapted with permission from Ref. 43, Copyright 2013 Elsevier B.V.
With the improvements in tumoral drug delivery observed using PTX-eNPs as compared to Taxol, we hypothesized that intraperitoneal administration of PTX-eNPs would significantly improve overall survival in intraperitoneal mesothelioma. In a model of established disease, animals received four weekly intraperitoneal treatments of PTX-eNPs or Taxol (10 mg/kg), which was designed to mimic the multi-dose regimens clinically employed to treat mesothelioma in patients. Additional control groups included: unloaded-eNPs (vehicle control); PTX-loaded PLGA-NPs (PTX-PLGA-NPs; non-swelling, generic NP control); unloaded-PLGA-NPs (PLGA vehicle control); and saline (tumor growth control). The PTX-eNP group demonstrated a significant improvement in survival v. all controls and a >50% increase in median survival compared to Taxol (72 v. 46 days, respectively). Remarkably, ~30% of animals demonstrated a complete clinical response during the study (Fig. 5B).43 The Taxol group experienced a modest improvement in median survival v. saline (46 v. 38 days) consistent with the improvements observed clinically in patients treated with intraperitoneal Taxol. PTX delivery via PLGA-NPs afforded no improvement over Taxol (median survival 43 days) and we interpreted this to be the result of: 1) lack of tumor-specific accumulation—PLGA-NPs do not accumulate significantly in tumors;43, 45, 49 and, subsequently, 2) non-tumor-specific delivery of PTX resulting in sub-therapeutic tumor-tissue concentrations.
To determine whether the improvement in animal survival afforded by multiple PTX-eNP treatments could be increased further, two additional studies were conducted. First, the treatment-of-established disease model was repeated with the exception that animals received double the weekly dose of PTX (i.e., 4-weekly injections of 20 mg/kg). Interestingly, doubling the dose resulted in no measureable improvement to animal survival compared to the 10 mg/kg treatment (Fig. 5C).43 This indicated that dose was not the limiting factor and that, rather, duration of drug exposure was the key element in improving mortality. Therefore, a second study was conducted to evaluate the impact of doubling the duration of treatment from the initial 4-week study (i.e., 8-weekly injections of 10 mg/kg). Median survival of the PTX-eNP treated group was prolonged to 103 days, an increase of over 43% compared to the 4-week PTX-eNP (72 days) and an increase of over 110% compared to the 8-week Taxol group (49 days) (Fig.5D).42 Importantly, all but one of the animals receiving Taxol died or were euthanized due to morbidity, according to IACUC guidelines, before completing their 8-week treatment regimen reflecting the underlying failure of standard Taxol alone to effectively deliver drug to the tumor despite a chronic dosing regimen. Together, these results demonstrate that, contrary to the clinical impression that mesothelioma is “PTX resistant”, which the Taxol data would support, PTX is an effective treatment for intraperitoneal mesothelioma if its pharmacokinetic profile is properly tailored via the use of a delivery system, such as the eNP.
eNPs in Ovarian Cancer
In addition to treating mesothelioma, intraperitoneal administration of PTX is employed clinically to treat ovarian cancer. A recent clinical trial demonstrated that ovarian cancer patients who underwent cytoreductive surgery and received multiple intraperitoneal boluses of Taxol demonstrated a 23% increase in survival over 10 years compared to patients who received traditional IV Taxol therapy.50 These results further validate the benefit gained by leveraging the proximity of drug to tumor inherent in the intraperitoneal route of administration, and it may be expected that further improvements to the current PTX formulation may afford greater tumor specific delivery and a wider therapeutic window resulting in improved patient outcomes. For example, Taxol’s toxicity and high complication rate lead to more than 58% of patients with advanced ovarian cancer being unable to complete a standard course of intraperitoneal therapy due to peritoneal adhesions, bowel obstructions, infections, and catheter-related complications.51–54 We therefore hypothesized that the PTX-eNP technology would provide a significant improvement over the standard of care in an ovarian cancer model by specifically targeting tumor tissue and locally delivering PTX.
To evaluate PTX-eNPs in ovarian cancer, a series of in vitro and in vivo assays were employed. In vitro cell cytotoxicity assays demonstrated comparable efficacy between PTX-eNPs and Taxol in an immortalized OVCAR-3 cell line (Fig. 6A). In contrast, PTX-eNPs were significantly more cytotoxic than Taxol in fresh human ovarian cancer cells derived from the malignant pleural effusion of a multidrug-resistance patient (Fig. 6B).55 We hypothesize that the increased efficacy of PTX-eNPs in drug resistant cells may be due to the establishment of a PTX “drug depot” within the tumor cells. Internalization of PTX-eNPs establishes an intracellular source of PTX that overcomes the PTX-efflux initiated by cell-membrane proteins, such as ABC-transporters, involved in multi-drug resistance.56 Additional studies are ongoing to investigate the ability of eNPs to overcome drug-resistance. In in vitro cell-uptake assays, eNPs demonstrated similarly rapid rates of internalization into malignant ovarian cancer cells as was previously observed in the mesothelioma model.43, 55 Again, similar to the mesothelioma model, tumor-specific accumulation of fluorescently-labeled eNPs was observed in vivo in established ovarian tumors (Fig. 6C).55 To simulate the clinical scenario of tumor-resection followed by intraperitoneal chemotherapy, we developed a resection-based ovarian cancer model in which animals were xenografted and bulky tumor was allowed to develop over a four-week period. Animals underwent intraoperative debulking to remove intraperitoneal tumors with >95% having <3 mm tumors remaining with an operative mortality of <10%. Following debulking, animals received intraperitoneal injections of either PTX-eNPs (experimental; 10 mg/kg), Taxol (clinically-used control; 10 mg/kg), or unloaded-eNPs (vehicle control). Animals were euthanized four weeks later, when the control group (unloaded-eNP) developed sufficient tumor to require euthanasia according to IACUC guidelines. Treatment with PTX-eNPs resulted in a profound decrease in the degree of tumor recurrence as compared to treatment with Taxol (0% v. 40%, respectively) (Fig. 6D).55 The results of this ovarian cancer study validated the efficacy of PTX-eNPs in a second intraperitoneal tumor model and demonstrated that the mechanistic driver for this efficacy—tumor-specific localization of eNPs—is active in multiple cancer models.
Figure 6.

(Note to the editorial staff, this figure is intended to be a double column figure) A.) PTX-eNPs display equivalent cytotoxicity to Taxol in vitro against OVCAR-3 breast cancer cells. B.) PTX-eNPs are significantly (* P<0.01) more cytotoxic than Taxol in multi-drug resistant human ovarian cancer cells at concentrations greater than 10 ng/mL. C.) Fluorescent, rhodamine-labelled-eNPs (Rho-eNPs) localize specifically to regions of intraperitoneal ovarian cancer. D.) PTX-eNPs reduce the incidence of significant tumor-recurrence (0%) in an orthotopic model of ovarian cancer compared to Taxol (40% significant recurrence). Adapted with permission from Ref. 55, Copyright 2012 Society of Surgical Oncology.
eNPs in Pancreatic Cancer
Besides ovarian cancer and mesothelioma, intraperitoneal administration of chemotherapy is also used to treat pancreatic carcinomatosis and, similarly, we hypothesized that the eNP delivery system would demonstrate tumor-specific behavior in this model. Recent reports57–58 have highlighted the importance of cancer stem cells (CSCs) to tumor growth, invasion and metastasis, and drug resistance. We therefore developed a pancreatic carcinomatosis rat model using Panc-1-derived CSCs (Panc-1-CSCs) with the aim of recapitulating the aggressive and drug-resistant phenotype frequently observed in pancreatic cancer.59 In vitro evaluation of fluorescently-labeled eNPs demonstrated similar behavior as previously observed in ovarian and mesothelioma models with eNPs rapidly internalized into both Panc-1 cells and Panc-1-CSCs within 1–4 hours in vitro.38 To investigate the kinetics and dose-dependence of eNP tumor localization in vivo, Rho-eNPs were injected into animals bearing established Panc-1-CSC tumors and imaged 1, 4 and 24 hours post injection. Dose-titration studies revealed that intraperitoneal injection of 1 mL of Rho-eNPs was the optimum dose to achieve maximum tumor-specific fluorescence and this was used in the following kinetics study.38 As with the ovarian and mesothelioma models, tumor-specific accumulation was observed within 4 hours of injection (Fig. 7A).38 Minimal tumoral localization was observed after 1 hour with most particles still in the ascites fluid; after 4 hours minimal eNP-fluorescence was observed in the ascites fluid with the preponderance of fluorescence occurring in the tumors. Complete penetration into and throughout some, though not all, large tumors was observed (Fig. 7B).
Figure 7.

(Note to the editorial staff, this figure is intended to be a double column figure) A.) Fluorescent rhodamine-labelled eNPs (Rho-eNPs; orange in UV light) localize to pancreatic tumors within 1–4 hours of intraperitoneal injection. B.) Rho-eNPs penetrate deep into and entirely throughout some tumors. C.) PTX-eNPs provide an equivalent survival benefit to Taxol in an orthotopic, cancer-stem-cell-derived model of pancreatic cancer. PTX-eNPs show a trend, though not significant, toward reduced tumor burden. D.) PTX-eNPs demonstrate reduced toxicity, as quantified by duodenal wall thickness and diameter, compared to Taxol. Adapted with permission from Ref. 38, Copyright 2016 Future Medicine Ltd.
This study validated the principle of Materials-Based Targeting in a third orthotopic model and was in concordance with the in vitro uptake models in malignant mesothelioma and ovarian cancer cell lines. It is important to note that the predictive power of in vitro assays must be handled with caution as not all particle formulations that demonstrate rapid in vitro uptake also demonstrate tumor-specific localization.43 This is a reflection of the often 1-dimensional nature of in vitro assays, which can be a poor substitute for the multi-dimensional, complex, and many-faceted interactions of an in vivo system.60
Based upon the highly-specific tumoral accumulation observed in vivo, we expected that PTX-eNPs would be as efficacious in vivo as was observed in the ovarian and mesothelioma models. Animals were xenografted with Panc-1-CSCs and allowed two weeks for bulky tumors to establish. Treatments were then administered weekly for 4 weeks with all PTX groups receiving PTX at 10 mg/kg. Treatments included: PTX-eNPs (experimental), Taxol (clinical formulation control), unloaded-eNPs (vehicle control) and saline (tumor growth control). While both PTX-eNP and Taxol improved survival compared to the saline and unloaded-eNP control groups, there was no significant difference in survival between PTX-eNP and Taxol treatment groups. All animals were euthanized at day 50 post-xenografting and quantification of tumor burden in the remaining animals revealed a trend, though not statistically significant, towards reduced tumor burden with the PTX-eNPs as compared to Taxol (Fig. 7C).38 Secondary endpoints, including in-life activity and degree of cachexia, as quantified by muscle mass and gut size at sacrifice, demonstrated that the PTX-eNP treatment resulted in significantly less toxicity and fewer side-effects than Taxol (Fig. 7D).38
The differences in efficacy observed between the pancreatic, ovarian, and mesothelioma models are likely due to three factors. First, by using CSCs to establish the pancreatic xenografts, these tumors were inherently more aggressive and likely drug resistant, though resistance was not experimentally verified. Second, the use of rats instead of mice meant the physical size of the tumors was several-fold larger than in the mouse (ovarian and mesothelioma) models increasing the challenge of achieving nanoparticle penetration and drug delivery to the entire tumor. Third, pancreatic cancer is known to be a particularly intractable disease where fibrosis can interfere with drug penetration; the results verify both this challenge as well as the need for continued development of new strategies and technologies for treating this challenging disease.
New eNP-Based Treatment Paradigms
The positive in vivo results described above along with the comprehensive characterization data for the eNP prompted us to investigate three new concepts using the eNP technology: 1) in the treatment of lymphatic metastatic disease (i.e. regional metastases); 2) as a highly-fluorescent probe to visually-guide surgical resection procedures; and, 3) as a two-step drug delivery device for concentrating separately administered drugs at a target site.
eNP-Mediated Treatment of Lymph Node Metastases in Breast Cancer
In addition to the applications described above, eNPs have also been leveraged to achieve lymphatic drug-delivery in orthotopic models of breast cancer. While breast cancer patients with localized disease typically have a favorable long-term prognosis, occult metastases in the axillary (i.e., draining) lymph-nodes of patients undergoing radical mastectomy result in a significant incidence (18%) of subsequent tumor growth within the lymph nodes following surgery.61 Due to the rapid clearance of systemically administered Taxol (half-life of <6 hours), PTX concentrations within the regional lymph nodes are sub-therapeutic and this results in ineffective treatment of occult disease. We hypothesized that the eNP-delivery system could improve the treatment of lymphatic disease by leveraging their small diameter (30 nm – 50 nm) to traffic within the lymphatic channels and deliver a “nano-bolus” of drug to microscopic disease within the lymph node.
To evaluate eNP trafficking to lymph nodes, fluorescently labeled particles were injected in the mammary fat pad of nude mice and the lymph nodes resected and imaged 4 days later. Rho-eNPs appear throughout the sinusoidal spaces where micrometastases are frequently described (Fig. 8A).62 In a large animal pig model, similar lymphatic trafficking was observed over a much larger and clinically relevant distance of >40 cm (Fig. 8B). To confirm that PTX is maintained within the eNPs during lymphatic trafficking and delivered to the lymph nodes in tumor-bearing animals, animals bearing 2-week old, orthotopic MDA-MB-231 breast cancer xenografts received peritumoral injections of Oregon Green-labeled paclitaxel (PTX-OG)-loaded Rho-eNPs. Histological analysis of the lymph nodes 10 days post-eNP injection revealed the presence of PTX-OG co-localized with Rho-eNPs within the lymph nodes demonstrating that both the drug and the eNP delivery system were present within the lymph node (Fig. 8C).62 The study was repeated with unlabeled PTX-eNPs and a control injection of Taxol and the PTX concentration in the tissue was quantified by HPLC. PTX-eNPs delivered ~10-fold greater concentrations of PTX to the lymph nodes than did Taxol (Fig. 8D). To evaluate whether increasing local concentrations of PTX within the lymphatic tissue would result in decreased lymph node metastases, animals bearing 1 week-old MDA-MB-231 xenografts were treated with injections of: peritumoral PTX-eNPs (experimental; 4 mg/kg), peritumoral Taxol (local clinical formulation control; 4 mg/kg), intraperitoneal Taxol (systemic clinical formulation control; 12 mg/kg) or local unloaded-eNPs (vehicle/no-PTX control). Five weeks later, animals were sacrificed, lymph nodes harvested, and the presence of disease evaluated via bioluminescence imaging of the lymph nodes. Treatment with PTX-eNPs significantly decreased the incidence of lymphatic metastases compared to the unloaded-eNP control (33% v. 100%, respectively) (Fig. 8E).62 In contrast, while local and systemic Taxol treatments showed a trend towards decreased lymphatic disease, neither decrease was significant compared to the control. These data demonstrate that PTX-eNPs migrate directly via the lymphatics to the regional draining lymph nodes, which are at greatest risk of metastasis, without the need for targeting ligands/antibodies and deliver a therapeutic dose of PTX that decreased the incidence of metastatic disease. This was the first study to establish the superiority of nanoparticle-mediated delivery compared to Taxol in the treatment of occult nodal disease and encourages further development of such technologies towards the clinic.
Figure 8.

(Note to the editorial staff, this figure is intended to be a double column figure) A.) Rhodamine-labelled eNPs (Rho-eNPs; red) localize to the sinusoidal spaces within lymph nodes (blue = nuclei). B.) eNPs loaded with a near infrared (NIR) dye (green) and subcutaneously injected into the mammary fat pat of a pig traffic over 40 cm to the draining lymph node. C.) Confocal microscopy of the sentinel (i.e., draining) lymph node from mice receiving injections of Rho-eNPs loaded with Oregon Green-labelled PTX (PTX-OG) demonstrate co-localization of PTX and eNP within the lymph node. D.) PTX-eNPs deliver 10-fold more PTX to the lymph nodes following a subcutaneous injection in the mammary fat pad than is achieved with Taxol. E.) PTX-eNPs significantly reduce the incidence of lymphatic disease in an orthotopic model of breast cancer. Taxol does not significantly decrease lymphatic disease compared to the control. Adapted with permission from Ref. 62, Copyright 2013 Elsevier B.V.
eNPs as Visual Assists for Cytoreductive Surgery
Given the in vivo results of eNP tumor localization, we investigated whether the eNP technology could be used as a visual assist for cytoreductive surgery. We therefore developed a highly-fluorescent rhodamine-labeled eNP (HFR-eNP) with the goal of increasing the visibility of small, sub-cm and sub-mm, tumors to enable complete resections. By using a UV-active fluorophore (rhodamine), we ensured the system would be compatible with inexpensive hand-held UV-lamps available for clinical use in operating suites thereby averting the need for expensive near-infrared (NIR), CT, or MR imaging technologies. By titering the quantity of rhodamine incorporated into the eNP, we achieved an optimum balance between relative fluorescence (i.e., photons/mg of rhodamine; which was maximized at lower rhodamine incorporations) and absolute fluorescence (i.e., photons/mL of eNPs; which was maximized at higher rhodamine incorporations) with a 0.2% wt/wt rhodamine/polymer eNP formulation (Fig. 9A–B).45 Of note, the fluorescence of the Rho-eNPs exceeded that of an equivalent concentration of “free” rhodamine in solution by 10–100-fold indicating that the hydrophobic, conformationally constrained environment of the eNP provided a favorable milieu for optimizing the fluorophore’s output.
Figure 9.

(Note to the editorial staff, this figure is intended to be a double column figure) A.) Rhodamine fluorescence as a function of incorporation into eNPs is optimized in the 0.02% and 0.2% formulations. B.) Rhodamine fluorescence as a function of polymer concentration is optimized in the 0.2% formulation. C.) Highly-fluorescent rhodamine-labelled eNPs (HFR-eNPs) visually identify tumors (orange in UV light) during cytoreductive surgery. D.) Large, small sub-cm and microscopic sub-mm tumors labelled with HFR-eNPs are visualized post-resection. Adapted with permission from Ref. 45, Copyright 2016 American Chemical Society.
To evaluate the accuracy of HFR-eNP localization to tumors, we employed a similar in vivo model of pancreatic carcinomatosis to that described previously.38 Specifically, animals bearing 3-week old xenografts received intraperitoneal injections of HFR-eNPs and were euthanized 24 hours later with all major organs and tumors harvested and evaluated for the presence of HFR-eNPs. Comparing HFR-eNP identification of tumors to the gold-standard, histological identification of tumors, it was found that HRF-eNPs possess a specificity of 90%, a sensitivity of 99%, and an overall accuracy of 95%.45 HFR-eNPs were neither grossly nor histologically evident in any major organs including the heart, lungs, brain, liver, spleen, pancreas, stomach, intestines, or kidneys. In a follow-up study, HFR-eNPs were employed as visual assists during cytoreductive surgery. Complete resection of the HRF-eNP-marked sub-cm and sub-mm tissues (Fig. 9C) was achieved in situ demonstrating the feasibility of using this technology as a visual-guide for cytoreductive surgery (Fig. 9D).45
Two-Step Delivery Using eNPs
Finally, we leveraged the unique structural and responsive characteristics of the eNP to develop it as a “two-step” drug delivery device. The idea of a two-step strategy was first developed by Press et al., who employed high-affinity antibodies (i.e., biotin-avidin) to achieve in vivo localization of a targeting moiety (e.g., tumor-specific Ab conjugated to avidin) with a subsequently administered therapeutic (e.g., radiolabeled-biotin).63–64 Unlike the traditional drug-loaded, PTX-eNP approach described above, where PTX and eNP are administered as a single unit, the two-step eNP delivery paradigm “pre-treats” the tumor with unloaded-eNPs and allows sufficient time for the eNPs to localize to the target-tissue (i.e., tumor). Subsequently, free PTX is administered via a separate injection and accumulates both within the eNP and the nearby tumor tissue (i.e., eNP + PTX; Fig. 10A). While several variations on the two-step theme involving nanoparticles or other antibody-based systems have been developed,65–69 the eNP + PTX two-step delivery method was the first to leverage entirely non-specific hydrophobic-hydrophobic interactions to achieve the in vivo co-localization of the two components of the two-step system.
Figure 10.

(Note to the editorial staff, this figure is intended to be a double column figure) A.) Schematic of the Two-Step paradigm. B.) PTX partitions into swollen eNPs from an aqueous sink; partitioning is significantly reduced when the hydrophobicity of the polymer is reduced. C.) The core of swollen eNPs affords a similarly hydrophobic environment compared to ethyl acetate. D.) A pre-treatment of eNPs significantly increases the intracellular concentration of PTX compared to pre-treatment with media or PLGA-NPs. E.) Pre-treatment of established intraperitoneal mesothelioma tumors prior to administration of Taxol results in significantly increased (~10-fold) intratumoral concentrations of PTX compared to pre-treatment with media (i.e., Taxol alone) or pre-treatment with PLGA-NPs. Adapted with permission from Ref. 70, Copyright 2016 Nature Publishing Group.
To characterize the two-step delivery strategy, several in vitro and in vivo assays were employed. First, a partitioning study was conducted in which swollen eNPs were exposed to an aqueous sink of PTX for 24 hours and PTX quantified in both the aqueous and eNP phases. PTX partitioned from the aqueous sink and into the swollen eNPs in a >4:1 ratio (Fig. 10B).70 Partitioning is thought to occur because the polymer chains of the swollen eNP provide a more hydrophobic environment, of similar polarity index to ethyl acetate, than the surrounding aqueous sink (Fig. 10C). As a control, the experiment was also conducted with eNPs that possess more hydrophilic succinic acid moieties upon deprotection. These succinic acid-based eNPs resulted in reduced PTX partitioning from the aqueous sink (2:1 ratio) confirming that the hydrophobicity of the swollen eNP is one of the major drivers of PTX partitioning.
Second, MSTO-211H tumor cells were cultured in vitro and treated with either media or media containing unloaded-eNPs. Two days later, the media was exchanged with media containing PTX-labeled with Oregon Green (PTX-OG). After 4 hours, the cells were washed, lysed, and the concentration of PTX-OG compared between treatments. Similar to the partitioning study, PTX-OG partitioned into the cells pre-treated with eNPs at a ratio of >4:1 compared to media pre-treated cells (Fig. 10D).70 PLGA-NPs were also evaluated in this assay to provide a non-swelling generic nanoparticle control. PTX-OG partitioning into the PLGA-NP-treated cells was equivalent to media pre-treated cells indicating that the swelling functionality and relative hydrophobicity of the swollen eNP polymer hydrogel are essential to creating a favorable environment into which PTX will partition. Third, the in vitro results were confirmed in vivo by treating animals bearing established intraperitoneal mesothelioma tumors with either unloaded-eNPs (experimental), PLGA-NPs (non-swelling, generic nanoparticle control) or saline (no nanoparticle control), followed two days later by an intraperitoneal injection of Taxol. Tumors were harvested and PTX quantified by HPLC and, as with both in vitro studies, pre-treatment with eNPs resulted in a >4:1 increase in intratumoral PTX concentrations compared to the saline control while PLGA-NPs afforded no increase in tumoral accumulation of PTX (Fig. 10E).70 Comparison of the tumoral PTX concentrations achieved with the eNP + PTX two-step strategy vs. the “traditional” PTX-eNP drug-loaded strategy reveals that the in vivo loading of eNPs with systemically administered PTX is possible, albeit the intratumoral PTX concentration is ~100-fold lower than with the “pre-loaded” PTX-eNP formulation. These results indicate that the eNP + PTX two-step method will require further development in order to maximize the impact of this approach. Nevertheless, one of the potential advantages of the current two-step system may be clarification and simplification of the FDA regulatory process (i.e., two-step delivery—a ‘device’ with the potential for 510k approval used in concert with standard systemic delivery of a chemotherapeutic, versus single component delivery—‘drug-device’ combination product) and, therefore, shortened time to the Clinic while still providing higher concentrations of drug to the target tissue compared to administration of drug alone. In sum, these studies demonstrated a proof-of-principle approach to using a partition-coefficient driven strategy to achieve two-step delivery with an otherwise “untargeted” nanoparticle and drug.
Future Directions for the eNP Technology
Future development of the eNP technology will involve continuation of some of the above efforts as well as engagement in new directions. One area that presents particular promise is in the development of eNPs as a “platform” technology for the delivery of poorly water-soluble agents and, in particular, novel natural products. PTX itself stands as the archetypal natural product—developed over nearly a half-century—once its supply and formulation were solved, it became the most economically and medically successful chemotherapeutic ever used.71–72 Unfortunately, the success story of PTX is not widely shared. The inability to overcome poor water solubility is a challenge rampant in the literature of natural products. In fact, challenges in formulating PTX early in clinical trials nearly halted development before its promising therapeutic activity could be uncovered. The superiority of the PTX-eNP formulations compared to Taxol (i.e., PTX solubilized in Cremophor ELethanol) in the studies described herein is a testament to the value of eNP-mediated delivery of natural products. While cytotoxic natural products have experienced a resurgence due to the development of antibody-conjugated therapies73, chemical conjugation is not always possible and, where possible, can be challenging, ill-defined, costly and time consuming. In contrast, encapsulation within the eNP delivery system requires only that the agent be hydrophobic. Thus, eNPs may provide a means to overcome the most challenging, traits of natural products—hydrophobicity and poor water solubility—and leverage them to our advantage. Several natural products have shown compatibility with the eNP-delivery system including verticillin A74–75—a molecule with poor water solubility but potent anticancer activity as a single-agent, the ability to overcome drug-resistance to traditional chemotherapeutics, and the ability to act as an immune checkpoint inhibitor. With these and many other potentially therapeutic compounds at hand, the eNP technology is ripe for continued development as a drug delivery platform, especially for peritoneal oncologic applications.
Conclusion
We describe the eNP drug delivery technology from its origin and mechanistic characterization to its in vivo application as a therapy in peritoneal carcinomatoses, a treatment for lymphatic metastases, a fluorescent guide for surgical resections, and a two-step delivery system. The observation that one particle performs so many different roles is a testament to the importance of engineering systems with the end, physiological application, in mind. It is also scientifically satisfying, as there is nothing more disappointing than a nanoparticle system that only performs in one specific, well-defined scenario. Importantly, the Materials-Based Targeting affords tumor-specific localization without the need for targeting moieties or antibodies, and this may provide a new paradigm by which nanotechnology experts can design future drug-delivery systems. Nanoparticle complexity can be an enticingly elegant academic solution but an unwelcome curse for subsequent clinical translation. The lessons learned in this nanoparticle case study serve as an example and an inspiration for other researchers to pursue novel nanotechnology advances for the treatment of cancer. It has been 11 years since the FDA-approved Abraxane,76–81 yet significant opportunities remain and much work has yet to be done to fulfill the promise of nanotechnology to provide innovative platform solutions for the unmet cancer needs in human health care.
Acknowledgments
The authors would like to acknowledge funding support for this project in part from: The Boston University Nanotechnology Innovation Center (BUnano), the Cross-Disciplinary Training in Nanotechnology for Cancer (XTNC NIH R25 CA153955) Training Fellowship, the Biomaterials Training Fellowship (NIH T32 EB006359), the National Science Foundation (DMR-1006601), Brigham and Women’s Hospital, Boston University, and the Nanotheranostics ARC, Evans Center for Interdisciplinary Biomedical Research at the Boston University School of Medicine.
Contributor Information
Aaron H. Colby, Departments of Biomedical Engineering and Chemistry, Boston University, Boston, MA 02215 Division of Thoracic Surgery, Department of Surgery, Brigham and Women’s Hospital, Boston, MA 02115.
Nicholas H. Oberlies, Department of Chemistry and Biochemistry, University of North Carolina at Greensboro, Greensboro, NC 27402
Cedric J. Pearce, Mycosynthetix, Inc., Hillsborough, NC 27278
Victoria L.M. Herrera, Department of Medicine and Whitaker Cardiovascular Institute, Boston University School of Medicine, Boston, MA 02118
Yolonda L. Colson, Division of Thoracic Surgery, Department of Surgery, Brigham and Women’s Hospital, Boston, MA 02115
Mark W. Grinstaff, Departments of Biomedical Engineering, Chemistry, and Medicine, Boston University, Boston, MA 02215
References
- 1.Lu Z, Wang J, Wientjes MG, Au JLS. Intraperitoneal therapy for peritoneal cancer. Future oncology (London, England) 2010;6:1625–1641. doi: 10.2217/fon.10.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roviello F, Caruso S, Marrelli D, Pedrazzani C, Neri A, De Stefano A, Pinto E. Treatment of peritoneal carcinomatosis with cytoreductive surgery and hyperthermic intraperitoneal chemotherapy: State of the art and future developments. Surgical Oncology. 2011;20:e38–e54. doi: 10.1016/j.suronc.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Imano M, Peng YF, Itoh T, Nishikawa M, Satou T, Yasuda A, Inoue K, Kato H, Shinkai M, Tsubaki M, Yasuda T, Imamoto H, Nishida S, Furukawa H, Takeyama Y, Okuno K, Shiozaki H. A preliminary study of single intraperitoneal administration of paclitaxel followed by sequential systemic chemotherapy with S-1 plus paclitaxel for advanced gastric cancer with peritoneal metastasis. Anticancer Res. 2012;32:4071–5. [PubMed] [Google Scholar]
- 4.Innocenti F, Danesi R, Di Paolo A, Agen C, Nardini D, Bocci G, Del Tacca M. Plasma and tissue disposition of paclitaxel (taxol) after intraperitoneal administration in mice. Drug Metab Dispos. 1995;23:713–7. [PubMed] [Google Scholar]
- 5.Tristan D Yan, Stine Munkholm-Larsen CQC. A pharmacological review on intraperitoneal chemotherapy for peritoneal malignancy. World J Gastrointest Oncol. 2010;2:109–116. doi: 10.4251/wjgo.v2.i2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tewari D, Java JJ, Salani R, Armstrong DK, Markman M, Herzog T, Monk BJ, Chan JK. Long-term survival advantage and prognostic factors associated with intraperitoneal chemotherapy treatment in advanced ovarian cancer: a gynecologic oncology group study. J Clin Oncol. 2015;33:1460–6. doi: 10.1200/JCO.2014.55.9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tentes A-AK, Kyziridis D, Kakolyris S, Pallas N, Zorbas G, Korakianitis O, Mavroudis C, Courcoutsakis N, Prasopoulos P. Preliminary Results of Hyperthermic Intraperitoneal Intraoperative Chemotherapy as an Adjuvant in Resectable Pancreatic Cancer. Gastroenterology Research and Practice. 2012;2012:506571. doi: 10.1155/2012/506571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vrochides D. NCT02850874 HIPEC as Neoadjuvant Treatment for Resectable Pancreatic Adenocarcinoma. 2016 [Google Scholar]
- 9.Open-label Pilot Phase I / II Study on Hyperthermic Intraperitoneal Chemotherapy (HIPEC) After Macroscopically Complete Resection (R0 / R1) of Adenocarcinomas of the Pancreas (PanHIPEC) University Hospital Tuebingen; 2016. [Google Scholar]
- 10.Kazan-Allen L. Asbestos and mesothelioma: worldwide trends. Lung Cancer. 2005;49(Suppl 1):S3–8. doi: 10.1016/j.lungcan.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Mesothelioma Cancer Trends. https://www.asbestos.com/mesothelioma/mesothelioma-trends/
- 12.Rowan K. Intraperitoneal Therapy for Ovarian Cancer: Why Has It Not Become Standard? Journal of the National Cancer Institute. 2009;101:775–777. doi: 10.1093/jnci/djp151. [DOI] [PubMed] [Google Scholar]
- 13.Goodman MD, McPartland S, Detelich D, Saif MW. Chemotherapy for intraperitoneal use: a review of hyperthermic intraperitoneal chemotherapy and early post-operative intraperitoneal chemotherapy. Journal of Gastrointestinal Oncology. 2016;7:45–57. doi: 10.3978/j.issn.2078-6891.2015.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Tang J, Li Y, Yu J, Zhang B, Yu C. Pharmacokinetic profile of paclitaxel in the plasma, lung, and diaphragm following intravenous or intrapleural administration in rats. Thorac Cancer. 2015;6:43–8. doi: 10.1111/1759-7714.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vasantha J, Kannan G, Goud T, Palani T, Vanitha R, Anitha R, Priya JMM. Pharmacokinetic Evaluation of Paclitaxel in South Indian Cancer Patients: A Prospective Study. Journal of Young Pharmacists : JYP. 2011;3:322–328. doi: 10.4103/0975-1483.90245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sodek KL, Murphy KJ, Brown TJ, Ringuette MJ. Cell-cell and cell-matrix dynamics in intraperitoneal cancer metastasis. Cancer Metastasis Rev. 2012;31:397–414. doi: 10.1007/s10555-012-9351-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarin D, Price JE, Kettlewell MG, Souter RG, Vass AC, Crossley B. Mechanisms of human tumor metastasis studied in patients with peritoneovenous shunts. Cancer Res. 1984;44:3584–92. [PubMed] [Google Scholar]
- 18.Coccolini F, Gheza F, Lotti M, Virzì S, Iusco D, Ghermandi C, Melotti R, Baiocchi G, Giulini SM, Ansaloni L, Catena F. Peritoneal carcinomatosis. World Journal of Gastroenterology : WJG. 2013;19:6979–6994. doi: 10.3748/wjg.v19.i41.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Metintas M, Ak G, Parspour S, Yildirim H, Erginel S, Alatas F, Batirel HF, Sivrikoz C, Metintas S, Dundar E. Local recurrence of tumor at sites of intervention in malignant pleural mesothelioma. Lung Cancer. 2008;61:255–61. doi: 10.1016/j.lungcan.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong DK. Relapsed Ovarian Cancer: Challenges and Management Strategies for a Chronic Disease. The Oncologist. 2002;7:20–28. doi: 10.1634/theoncologist.7-suppl_5-20. [DOI] [PubMed] [Google Scholar]
- 21.Kim HJ, Lee WJ, Kang CM, Hwang HK, Bang SM, Song SY, Seong J. Risk Factors Associated with Loco-Regional Failure after Surgical Resection in Patients with Resectable Pancreatic Cancer. PLoS One. 2016;11:e0157196. doi: 10.1371/journal.pone.0157196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Smet L, Ceelen W, Remon JP, Vervaet C. Optimization of drug delivery systems for intraperitoneal therapy to extend the residence time of the chemotherapeutic agent. Scientific World Journal. 2013;2013:720858. doi: 10.1155/2013/720858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armstrong DK, Fleming GF, Markman M, Bailey HH. A phase I trial of intraperitoneal sustained-release paclitaxel microspheres (Paclimer) in recurrent ovarian cancer: a Gynecologic Oncology Group study. Gynecol Oncol. 2006;103:391–6. doi: 10.1016/j.ygyno.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 24.Delgado G, Potkul RK, Treat JA, Lewandowski GS, Barter JF, Forst D, Rahman A. A phase I/II study of intraperitoneally administered doxorubicin entrapped in cardiolipin liposomes in patients with ovarian cancer. Am J Obstet Gynecol. 1989;160:812–7. doi: 10.1016/0002-9378(89)90296-2. discussion 817–9. [DOI] [PubMed] [Google Scholar]
- 25.Sugiyama T, Kumagai S, Nishida T, Ushijima K, Matsuo T, Yakushiji M, Hyon SH, Ikada Y. Experimental and clinical evaluation of cisplatin-containing microspheres as intraperitoneal chemotherapy for ovarian cancer. Anticancer Res. 1998;18:2837–42. [PubMed] [Google Scholar]
- 26.Verschraegen CF, Kumagai S, Davidson R, Feig B, Mansfield P, Lee SJ, Maclean DS, Hu W, Khokhar AR, Siddik ZH. Phase I clinical and pharmacological study of intraperitoneal cis-bis-neodecanoato(trans- R, R-1, 2-diaminocyclohexane)-platinum II entrapped in multilamellar liposome vesicles. J Cancer Res Clin Oncol. 2003;129:549–55. doi: 10.1007/s00432-003-0481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 28.Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 29.Hofmann-Amtenbrink M, Grainger DW, Hofmann H. Nanoparticles in medicine: Current challenges facing inorganic nanoparticle toxicity assessments and standardizations. Nanomedicine: Nanotechnology, Biology and Medicine. 11:1689–1694. doi: 10.1016/j.nano.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 30.Oberoi HS, Nukolova NV, Kabanov AV, Bronich TK. Nanocarriers for delivery of platinum anticancer drugs. Advanced Drug Delivery Reviews. 2013;65:1667–1685. doi: 10.1016/j.addr.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Svenson S. What nanomedicine in the clinic right now really forms nanoparticles? Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2014;6:125–35. doi: 10.1002/wnan.1257. [DOI] [PubMed] [Google Scholar]
- 32.He C, Liu D, Lin W. Nanomedicine Applications of Hybrid Nanomaterials Built from Metal–Ligand Coordination Bonds: Nanoscale Metal–Organic Frameworks and Nanoscale Coordination Polymers. Chemical Reviews. 2015;115:11079–11108. doi: 10.1021/acs.chemrev.5b00125. [DOI] [PubMed] [Google Scholar]
- 33.Griset AP, Walpole J, Liu R, Gaffey A, Colson YL, Grinstaff MW. Expansile Nanoparticles: Synthesis, Characterization, and In vivo Efficacy of an Acid-Responsive Polymeric Drug Delivery System. J Am Chem Soc. 2009;131:2469–2471. doi: 10.1021/ja807416t. [DOI] [PubMed] [Google Scholar]
- 34.Li X-F, O’Donoghue JA. Hypoxia in Microscopic Tumors. Cancer letters. 2008;264:172–180. doi: 10.1016/j.canlet.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown JM. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007;435:297–321. doi: 10.1016/S0076-6879(07)35015-5. [DOI] [PubMed] [Google Scholar]
- 36.Colby AH, Colson YL, Grinstaff MW. Microscopy and tunable resistive pulse sensing characterization of the swelling of pH-responsive, polymeric expansile nanoparticles. Nanoscale. 2013;5:3496–504. doi: 10.1039/c3nr00114h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zubris KAV, Colson YL, Grinstaff MW. Hydrogels as Intracellular Depots for Drug Delivery. Mol Pharmaceut. 2012;9:196–200. doi: 10.1021/mp200367s. [DOI] [PubMed] [Google Scholar]
- 38.Herrera VL, Colby AH, Tan GA, Moran AM, O’Brien MJ, Colson YL, Ruiz-Opazo N, Grinstaff MW. Evaluation of expansile nanoparticle tumor localization and efficacy in a cancer stem cell-derived model of pancreatic peritoneal carcinomatosis. Nanomedicine (Lond) 2016;11:1001–15. doi: 10.2217/nnm-2015-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colson YL, Liu R, Southard EB, Schulz MD, Wade JE, Griset AP, Zubris KA, Padera RF, Grinstaff MW. The Performance of Expansile Nanoparticles in a Murine Model of Peritoneal Carcinomatosis. Biomaterials. 2011;32:832–40. doi: 10.1016/j.biomaterials.2010.09.059. [DOI] [PubMed] [Google Scholar]
- 40.Weatherall E, Thomos L, Dickinson M, Colby AH, Grinstaff MW, Willmott GR. Tunable Resistive Pulse Sensing and Nanoindentation of pH-Responsive Expansile Nanoparticles. International Journal of Nanotechnology. 2016 In Press. [Google Scholar]
- 41.Au JL-S, Li D, Gan Y, Gao X, Johnson AL, Johnston J, Millenbaugh NJ, Jang SH, Kuh H-J, Chen C-T, Wientjes MG. Pharmacodynamics of Immediate and Delayed Effects of Paclitaxel: Role of Slow Apoptosis and Intracellular Drug Retention. Cancer Research. 1998;58:2141–2148. [PubMed] [Google Scholar]
- 42.Zubris KA, Liu R, Colby A, Schulz MD, Colson YL, Grinstaff MW. In vitro activity of Paclitaxel-loaded polymeric expansile nanoparticles in breast cancer cells. Biomacromolecules. 2013;14:2074–82. doi: 10.1021/bm400434h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu R, Colby AH, Gilmore D, Schulz M, Zeng J, Padera RF, Shirihai O, Grinstaff MW, Colson YL. Nanoparticle tumor localization, disruption of autophagosomal trafficking, and prolonged drug delivery improve survival in peritoneal mesothelioma. Biomaterials. 2016;102:175–86. doi: 10.1016/j.biomaterials.2016.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lei H, Hofferberth SC, Liu R, Colby A, Tevis KM, Catalano P, Grinstaff MW, Colson YL. Paclitaxel-loaded expansile nanoparticles enhance chemotherapeutic drug delivery in mesothelioma 3-dimensional multicellular spheroids. J Thorac Cardiovasc Surg. 2015;149:1417–1425. doi: 10.1016/j.jtcvs.2015.02.020. [DOI] [PubMed] [Google Scholar]
- 45.Colby AH, Berry S, Paison K, Liu R, Colson YL, Ruiz-Opazo N, Grinstaff MW, Herrera V. Highly Specific and Sensitive Fluorescent Nanoprobes for Image-Guided Resection of Sub-millimeter Peritoneal Tumors. 2016 doi: 10.1021/acsnano.6b06777. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer. 2008;8:835–850. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- 47.Vassileva V, Allen CJ, Piquette-Miller M. Effects of sustained and intermittent paclitaxel therapy on tumor repopulation in ovarian cancer. Mol Cancer Ther. 2008;7:630–7. doi: 10.1158/1535-7163.MCT-07-2117. [DOI] [PubMed] [Google Scholar]
- 48.Demidenko ZN, Kalurupalle S, Hanko C, Lim CU, Broude E, Blagosklonny MV. Mechanism of G1-like arrest by low concentrations of paclitaxel: next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis. Oncogene. 2008;27:4402–10. doi: 10.1038/onc.2008.82. [DOI] [PubMed] [Google Scholar]
- 49.Lu Z, Tsai M, Lu D, Wang J, Wientjes MG, Au JL. Tumor-penetrating microparticles for intraperitoneal therapy of ovarian cancer. J Pharmacol Exp Ther. 2008;327:673–82. doi: 10.1124/jpet.108.140095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wright AA, Cronin A, Milne DE, Bookman MA, Burger RA, Cohn DE, Cristea MC, Griggs JJ, Keating NL, Levenback CF, Mantia-Smaldone G, Matulonis UA, Meyer LA, Niland JC, Weeks JC, O’Malley DM. Use and Effectiveness of Intraperitoneal Chemotherapy for Treatment of Ovarian Cancer. Journal of Clinical Oncology. 2015 doi: 10.1200/JCO.2015.61.4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wenzel LB, Huang HQ, Armstrong DK, Walker JL, Cella D, Group GO Health-related quality of life during and after intraperitoneal versus intravenous chemotherapy for optimally debulked ovarian cancer: a Gynecologic Oncology Group Study. Journal of Clinical Oncology. 2007;25:437–43. doi: 10.1200/JCO.2006.07.3494. [DOI] [PubMed] [Google Scholar]
- 52.Walker JL, Armstrong DK, Huang HQ, Fowler J, Webster K, Burger RA, Clarke-Pearson D. Intraperitoneal catheter outcomes in a phase III trial of intravenous versus intraperitoneal chemotherapy in optimal stage III ovarian and primary peritoneal cancer: a Gynecologic Oncology Group Study. Gynecol Oncol. 2006;100:27–32. doi: 10.1016/j.ygyno.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 53.Verwaal VJ, van Tinterin H, Ruth SV, Zoetmulder FAN. Toxicity of Cytoreductive Surgery and Hyperthermic Intra-Peritoneal Chemotherapy. J Surg Oncol. 2004;85:61–67. doi: 10.1002/jso.20013. [DOI] [PubMed] [Google Scholar]
- 54.Verwaal VJ, Kusamura S, Baratti D, Deraco M. The Eligibility for Local-Regional Treatment of Peritoneal Surface Malignancy. Journal of Surgical Oncology. 2008;98:220–223. doi: 10.1002/jso.21060. [DOI] [PubMed] [Google Scholar]
- 55.Gilmore MD, Schulz MM, Liu MR, PhD, Zubris PKAV, Padera MRF, Catalano SPJ, Grinstaff PMW, Colson MYL., PhD Cytoreductive Surgery and Intraoperative Administration of Paclitaxel-loaded Expansile Nanoparticles Delay Tumor Recurrence in Ovarian Carcinoma. Annals of Surgical Oncology. 2013;20:1684–1693. doi: 10.1245/s10434-012-2696-5. [DOI] [PubMed] [Google Scholar]
- 56.Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv. 2004;1:27–42. doi: 10.2174/1567201043480036. [DOI] [PubMed] [Google Scholar]
- 57.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727–738. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 58.Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–143. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]
- 59.Chand S, O’Hayer K, Blanco FF, Winter JM, Brody JR. The Landscape of Pancreatic Cancer Therapeutic Resistance Mechanisms. International Journal of Biological Sciences. 2016;12:273–282. doi: 10.7150/ijbs.14951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Charoen KM, Fallica B, Colson YL, Zaman MH, Grinstaff MW. Embedded multicellular spheroids as a biomimetic 3D cancer model for evaluating drug and drug-device combinations. Biomaterials. 2014;35:2264–71. doi: 10.1016/j.biomaterials.2013.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fisher B, Jeong JH, Anderson S, Bryant J, Fisher ER, Wolmark N. Twenty-five-year follow-up of a randomized trial comparing radical mastectomy, total mastectomy, and total mastectomy followed by irradiation. N Engl J Med. 2002;347:567–75. doi: 10.1056/NEJMoa020128. [DOI] [PubMed] [Google Scholar]
- 62.Liu R, Gilmore DM, Zubris KA, Xu X, Catalano PJ, Padera RF, Grinstaff MW, Colson YL. Prevention of nodal metastases in breast cancer following the lymphatic migration of paclitaxel-loaded expansile nanoparticles. Biomaterials. 2013;34:1810–9. doi: 10.1016/j.biomaterials.2012.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Green DJ, Pagel JM, Nemecek ER, Lin Y, Kenoyer A, Pantelias A, Hamlin DK, Wilbur DS, Fisher DR, Rajendran JG, Gopal AK, Park SI, Press OW. Pretargeting CD45 enhances the selective delivery of radiation to hematolymphoid tissues in nonhuman primates. Blood. 2009;114:1226–35. doi: 10.1182/blood-2009-03-210344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park SI, Shenoi J, Frayo SM, Hamlin DK, Lin Y, Wilbur DS, Stayton PS, Orgun N, Hylarides M, Buchegger F, Kenoyer AL, Axtman A, Gopal AK, Green DJ, Pagel JM, Press OW. Pretargeted radioimmunotherapy using genetically engineered antibody-streptavidin fusion proteins for treatment of non-hodgkin lymphoma. Clin Cancer Res. 2011;17:7373–82. doi: 10.1158/1078-0432.CCR-11-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.von Maltzahn G, Park J-H, Lin KY, Singh N, Schwöppe C, Mesters R, Berdel WE, Ruoslahti E, Sailor MJ, Bhatia SN. Nanoparticles that communicate in vivo to amplify tumour targeting. Nat Mater. 2011;10:545–552. doi: 10.1038/nmat3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park JH, von Maltzahn G, Xu MJ, Fogal V, Kotamraju VR, Ruoslahti E, Bhatia SN, Sailor MJ. Cooperative nanomaterial system to sensitize, target, and treat tumors. Proc Natl Acad Sci U S A. 2010;107:981–6. doi: 10.1073/pnas.0909565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simberg D, Duza T, Park JH, Essler M, Pilch J, Zhang L, Derfus AM, Yang M, Hoffman RM, Bhatia S, Sailor MJ, Ruoslahti E. Biomimetic amplification of nanoparticle homing to tumors. Proc Natl Acad Sci U S A. 2007;104:932–6. doi: 10.1073/pnas.0610298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meng H, Zhao Y, Dong J, Xue M, Lin Y-S, Ji Z, Mai WX, Zhang H, Chang CH, Brinker CJ, Zink JI, Nel AE. Two-Wave Nanotherapy To Target the Stroma and Optimize Gemcitabine Delivery To a Human Pancreatic Cancer Model in Mice. ACS Nano. 2013;7:10048–10065. doi: 10.1021/nn404083m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brudno Y, Silva EA, Kearney CJ, Lewin SA, Miller A, Martinick KD, Aizenberg M, Mooney DJ. Refilling drug delivery depots through the blood. Proc Natl Acad Sci U S A. 2014;111:12722–7. doi: 10.1073/pnas.1413027111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Colby AH, Liu R, Schulz MD, Padera RF, Colson YL, Grinstaff MW. Two-Step Delivery: Exploiting the Partition Coefficient Concept to Increase Intratumoral Paclitaxel Concentrations In vivo Using Responsive Nanoparticles. Scientific Reports. 2016;6:18720. doi: 10.1038/srep18720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oberlies NH, Kroll DJ. Camptothecin and Taxol: Historic Achievements in Natural Products Research. J Nat Prod. 2004;67:129–135. doi: 10.1021/np030498t. [DOI] [PubMed] [Google Scholar]
- 72.Camidge R. The Story of Taxol: Nature and Politics in the Pursuit of an Anti-Cancer Drug. BMJ : British Medical Journal. 2001;323:115–115. [Google Scholar]
- 73.Oroudjev E, Lopus M, Wilson L, Audette C, Provenzano C, Erickson H, Kovtun Y, Chari R, Jordan MA. Maytansinoid-Antibody Conjugates Induce Mitotic Arrest by Suppressing Microtubule Dynamic Instability. Molecular cancer therapeutics. 2010;9:2700–2713. doi: 10.1158/1535-7163.MCT-10-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu F, Liu Q, Yang D, Bollag WB, Robertson K, Wu P, Liu K. Verticillin A overcomes apoptosis resistance in human colon carcinoma through DNA methylation-dependent upregulation of BNIP3. Cancer Res. 2011;71:6807–16. doi: 10.1158/0008-5472.CAN-11-1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paschall AV, Yang D, Lu C, Choi JH, Li X, Liu F, Figueroa M, Oberlies NH, Pearce C, Bollag WB, Nayak-Kapoor A, Liu K. H3K9 Trimethylation Silences Fas Expression To Confer Colon Carcinoma Immune Escape and 5-Fluorouracil Chemoresistance. J Immunol. 2015;195:1868–82. doi: 10.4049/jimmunol.1402243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grinstaff MW, Soon-Shiong P, Wong M, Sandford PA, Suslick KS, Desai NP. US5498421. Composition useful for in vivo delivery of biologics and methods employing same. 1996
- 77.Grinstaff MW, Soon-Shiong P, Wong M, Sandford PA, Suslick KS, Desai NP. US5665382. Methods for the preparation of pharmaceutically active agents for in vivo delivery. 1997
- 78.Desai NP, Soon-Shiong P, Sandford PA, Grinstaff MW, Suslick KS. US5439686. Methods for in vivo delivery of substantially water insoluble pharmacologically active agents and compositions useful therefor. 1995
- 79.Soon-Shiong P, Desai NP, Grinstaff MW, Sandford PA, Suslick KS. US5439686. Methods for in vivo delivery of substantially water insoluble pharmacologically active agents and compositions useful therefor. 1995
- 80.https://en.wikipedia.org/wiki/Protein-bound_paclitaxel.
- 81.FDA. FDA Approves New Drug for the Most Common Type of Lung Cancer Drug Shows Survival Benefit. 2004 [Google Scholar]