

Abstract
Sobetirome is one of the most studied TRβ-selective thyromimetics in the field due to its excellent selectivity and potency. A small structural change - replacing the 3,5-dimethyl groups of sobetirome with either chlorine or bromine - produces significantly more potent compounds, both in vitro and in vivo. These halogenated compounds induce transactivation of a TRβ-mediated cell-based reporter with an EC50 comparable to T3, access the CNS at levels comparable to their parent, and activate an endogenous TR-regulated gene in the brain with an EC50 roughly five-fold lower than sobetirome. Previous studies suggest that this apparent increase in affinity can be explained by halogen bonding between the ligand and a backbone carbonyl in the receptor. This makes the new analogs potential candidates for treating central nervous system disorders that may respond favorably to thyroid hormone stimulated pathways.
Keywords: thyroid, thyromimetic, brain, central nervous system
Graphical Abstract
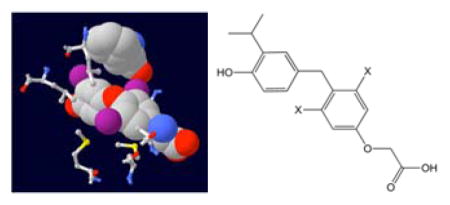
Modifying the structure of the potent TRβ-selective thyroid hormone analog sobetirome by replacing the 3,5-dimethyl groups with chlorine or bromine to potentially exploit halogen bonding interactions within the TR ligand-binding domain gives compounds that display significantly increased potency in vitro and in vivo while retaining distribution to the CNS. These improved characteristics make the new compounds attractive candidates for treating CNS demyelination diseases influenced by thyroid hormone stimulation.
Introduction
Thyroid hormone (TH) in the form of 3,3′,5-triiodothyronine (T3) derived from deiodination of the pro-hormone tetraiodothyronine (T4) (Fig 1) is a key signal for oligodendrocyte differentiation and myelin formation during development and also stimulates remyelination in adult models of multiple sclerosis (MS).1 However, TH is not an acceptable long-term therapy due to the lack of a therapeutic window in which remyelination can be induced while avoiding the cardiotoxicity and bone demineralization associated with chronic hyperthyroidism. A more promising approach comes from thyroid hormone analogs that can activate thyroid hormone-responsive genes while avoiding the associated downsides of TH by exploiting molecular and physiological properties of thyroid hormone receptors.2 These receptors are expressed in two major forms with heterogeneous tissue distribution and overlapping but distinct sets of target genes.3 TRα is enriched in the heart, brain, and bone while TRβ is enriched in the liver.4 Developing selective thyromimetics has been challenging due to the high sequence homology of thyroid hormone receptor subtypes - only one amino acid residue on the internal surface of the ligand-binding domain cavity varies between the α1 and β1 forms. Sobetirome (Fig 1) was one of the first potent analogs that demonstrated significant TRβ-selectivity in vitro,5,6 and in vivo.7–9 While sobetirome was designed as a cardiac-sparing drug for treating hypercholesterolemia by activating TRβ in the liver,10 it is unique within the thyromimetic field in possessing significant distribution in the brain.7,11 Recent studies highlight the potential advantage of this property by demonstrating its activity in in vitro models of human CNS demyelination diseases including multiple sclerosis (MS)12 and X-linked adrenoleukodystrophy (X-ALD).13
Figure 1.

Chemical structures of selected TR agonists.
Though sobetirome is a very promising drug candidate for CNS indications, its potency is lower than that of T3 (Fig 1). A more potent analog would evoke the same effects at a lower dose or greater stimulation of the target at the same dose. While many of the structural features of sobetirome are critical for its binding affinity and receptor selectivity6,12 the 3,5-dimethyl constituents have not been investigated. There is a large body of computational and biological structure-activity relationship data demonstrating that thyromimetics with 3,5-dimethyl substitutions have significantly reduced activity in comparison to structurally similar analogs with 3,5-position halogen substitutions. In the TRα-selective compounds CO-22 and CO-24 (Fig 1) replacement of 3,5-dimethyl groups with bromines improved binding affinity by 15-fold.14,15 A computational structure-activity relationship study of thyroid hormone analogs suggested a potential mechanism for these findings – halogens at the 3,5-position can form a dipole-dipole interaction with a backbone carbonyl in the TR ligand-binding domain, which influences binding affinity and selectivity.16 Halogen bonding interactions are strongly influenced by the geometry of the donor and halogen, as the dipoles must be close enough to interact and oriented so that the lone pairs of the donor and the partial positive charge on the halogen face each other.17 The interaction is generally stronger as the size of the halogen increases due to the increasing polarizability of larger halogens producing a greater partial positive charge on the atom. These data suggest that replacing the 3,5-dimethyl groups of sobetirome with halogens may result in thyromimetics with higher affinity and potency at the thyroid hormone receptor.
Results and Discussion
3,5-position halogen analogs of sobetirome required a new synthetic approach. Work by Dabrowski18 provided a template for producing the necessary 4-hydroxy-2,6-dihalobenzaldehyde intermediates by selectively deprotonating the 4-position of silyl protected 3,5-dihalophenols with lithium amide reagents. The method was improved by replacing the methyl ether and trimethylsilyl ether protecting groups used by Dabrowski with the more sterically bulky triethylsilyl ether protecting group, which significantly improved the selectivity of the deprotonation. These intermediates were used in a slightly altered version of the recently reported improved sobetirome synthesis.19 The 4-hydroxy-2,6-dihalobenzaldehyde intermediates could not be alkylated with tert-butyl chloroacetate using the standard cesium carbonate/DMF conditions due to the halogen substitutions reducing the nucleophilicity of the phenol. However, the reaction went to completion and in good yield after converting the chloroacetate into an iodoacetate via an in situ Finklestein reaction. After forming the tert-butyl oxyacetate intermediate, the carbon-carbon bond formation proceeded in the same fashion as with sobetirome by forming an arylmagnesium with 7 that attacked the benzaldehyde to form a carbinol intermediate. The arylmagnesium nucleophile was critical to the success of the synthesis as it will not exchange with aryl chlorides or bromides at cryogenic temperatures and is compatible with the tert-butyl ester protecting group. 20 Reduction of the carbinol and deprotection of the tert-butyl ester and methoxymethyl ether protecting groups proceeded simultaneously with TFA and triethylsilane in dichloromethane. The dibromo analog JD-20 was synthesized in 27% overall yield and the dichloro analog JD-21 was synthesized in 17% overall yield, both in five steps.
A electroporation-based transfection system with U2OS or HeLa cells was previously used for measuring the potency of thyroid hormone receptor-mediated transactivation.5,15 The electroporation method was limited by poor transfection efficiencies and reduced dynamic range that delivered inconsistent EC50 values for T3 at each receptor subtype. Previous reports suggested that lipofectamine reagents sequestered lipophilic compounds such as sobetirome.21 The electroporation-based assay was modified and optimized using Lipofectamine 2000 and HEK293 cells. In comparison to the previous method, the new version has significantly higher transfection efficiencies and a greater dynamic range while being operationally simpler and delivering similar EC50 values for T3 at both subtypes.
Transactivation assays showed that JD-20 and JD-21 have improved potency in comparison to their parent compound sobetirome. Increases in potency at TRα were significant, with more modest improvements at TRβ, boosting the analogs to match the EC50 of T3 (Fig 3, Table 1).
Figure 3.

TRE-driven dual luciferase transactivation assays with calculated sigmoidal dose-response curves against a) hTRα1 and b) hTRβ1 in transiently transfected HEK293 cells. Plots show data normalized to T3 response as means of triplicates with standard error.
Table 1.
Subtype selectivity measured by EC50 values from TRE-driven dual luciferase transactivation assays with standard error.
| Compound | EC50 TRα (nM) | EC50 TRβ (nM) | TRα/TRβ |
|---|---|---|---|
| T3 | 1.0 ±0.4 | 1.5 ±1.6 | 0.7 |
| Sobetirome | 74.7 ±28.9 | 2.8 ±1.8 | 26.5 |
| JD-20 | 8.0 ±7.0 | 0.9 ±1.1 | 9.0 |
| JD-21 | 7.8 ±3.6 | 1.2 ±1.3 | 6.3 |
A distribution study was carried out in C57BL/6J mice to determine the concentrations in brain and serum after systemic (ip) administration. Mice were given single 9.14 μmol/kg doses of sobetirome, JD-20, or JD-21. Tissue and blood were collected 1 hr post-injection and the concentration of the drugs was determined by LC-MS/MS analysis (Fig 4). JD-20 and JD-21 showed comparable brain uptake to sobetirome, while the serum levels of JD-20 and JD-21 were both significantly (p <0.05) lower than sobetirome. The brain:serum ratio of JD-21 trended higher than sobetirome (p = 0.055) while JD-20 had a brain:serum ratio comparable to sobetirome.
Figure 4.

in vivo concentrations of GC-1, JD-20, and JD-21 in C57/B mouse tissues 1 hr after systemic administration (ip) of GC-1, JD-20, and JD-21 9.14 μmol/kg doses measured by LC-MS/MS in brain and serum.
* P<0.05 compared to sobetirome.
Induction of the TR-regulated gene Hairless (Hr) in the brain was determined by qPCR and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA (Fig 5). Vehicle (1:1 saline/DMSO) was used as a negative control and saturating doses of T3 (0.305 μmol/kg) and sobetirome (9.14 μmol/kg) were used as positive controls. JD-20 and JD-21 at 0.914 μmol/kg had comparable Hr induction to a 10-fold higher dose of sobetirome..
Figure 5.

a) Expression of TR regulated gene Hairless (Hr) mRNA normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA measured by qPCR in C57/B mouse brain (3 mice/dose) 2 hr after systemic administration (ip) of saturating doses of T3 (0.305 μmol/kg) or sobetirome (9.15 μmol/kg) plus escalating doses of JD-20 and JD-21 (0.9 and 9.15 μmol/kg). b) Expression of TR regulated gene Hairless (Hr) mRNA normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA measured by qPCR in C57/B mouse brain (3 mice/dose) 2 hr after systemic administration (ip) of GC-1, JD-20, and JD-21.
The EC50 values of Hr mRNA induction in the brain normalized to GAPDH mRNA expression by sobetirome, JD-20, and JD-21 (Fig 5) were determined using the same experimental protocol. The EC50 for sobetirome was 8.20 ±12.65 μmol/kg, the EC50 for JD-20 was 1.49 ±1.08 μmol/kg, and the EC50 for JD-21 was 1.21 ±1.75 μmol/kg, making the halogenated analogs roughly 6-fold more potent than sobetirome at inducing Hr mRNA expression in the brain.
Conclusions
While sobetirome has become one of the standard TRβ-selective thyromimetics in the field, this study makes clear that a simple structural change - replacing the 3,5-dimethyl groups with bromine or chlorine - produces more potent compounds that largely maintain the TR-isoform selectivity and brain uptake properties of the parent. This improvement is potentially due to a halogen bonding interaction between the ligand and a backbone carbonyl in the TR ligand-binding domain. These results suggest that JD-20 and JD-21 may be useful molecules for studying and potentially treating CNS demyelination diseases such as X-ALD and MS.
The improved potency of JD-20 and JD-21 is consistent with numerous thyromimetic SAR studies that have found superior activity for analogs with halogens in the 3,5-position compared to similar analogs with methyl groups at those positions. It is surprising in this instance that by most measures JD-20 and JD-21 appear to have very similar properties. The previous thyromimetic SAR studies found that changing the halogen substitution frequently produced dramatic shifts in both potency and selectivity. 16 But the in vitro and in vivo potencies of JD-20 and JD-21 are roughly the same within error. This is different than most known thyromimetics and suggests something unique about the chemical context of the sobetirome scaffold. Additionally except for the improvements in potency, the only major difference between these novel compounds and sobetirome is the reduced serum concentrations. This hints that these compounds may be distributed by different mechanisms, such as being actively transported across the blood-brain barrier rather than by passive diffusion, where halogen bonding or sterics might alter the relative uptake rates through a putative transporter. It will be important to determine whether uptake of 0sobetirome and these analogs into the CNS occurs primarily through an active transport mechanism.
Experimental Section
Material and Methods
Transactivation Assay
Human epithelial kidney cells (HEK 293) were grown to 80% confluency in Dubelcco’s modified Eagles 4.5 g/L glucose medium (high glucose DMEM) containing 10% fetal bovine serum, 50 units/mL penicillin and 50 μg/mL streptomycin. The cells were trypsinized with 0.25% trypsin, then diluted to 5×105 cells/mL with high glucose DMEM. Cells were added to Costar 3917 96-well plates at 5×104 cells/well, then incubated at 37° C for 24 hours.
1.5 μg of TR expression vector (full length TRα-CMV or TRβ-CMV), 1.5 μg of a reporter plasmid containing a DR4 thyroid hormone response element (TRE) direct repeat spaced by four nucleotides (AGGTCAcaggAGGTCA) cloned upstream of a minimal thymidine kinase promoter linked to a firefly luciferase coding sequence, and 0.75 μg of a pRL-SV40 constitutive Renilla luciferase reporter plasmid were diluted into 540 μl of OptiMEM. 27 μL of Lipofectamine 2000 reagent was diluted into 540 μL of OptiMEM. The plasmid and lipofectamine dilutions were combined then incubated at RT for 10 min. The mixture was then diluted into 4.29 mL of OptiMEM. Plates were washed with 100 μL of phosphate buffered saline (PBS) at pH 7.2 without magnesium or calcium chloride per well. Transfection mixtures were added at 50 μL per well, then incubated at 37° C for 4 hours. Modified DME/F-12 Ham’s medium without phenol red containing 15 mM HEPES and bicarbonate, 5 mM L-glutamine, charcoal-stripped FBS, 50 units/mL penicillin and 50 μg/mL streptomycin was added at 50 μL per well, then the plates were incubated at 37° C for 20 hours.
Drug stocks were made at 10 mM in DMSO, then serially diluted to 1X concentrations in DME/F-12 Ham’s. Plates were washed with 100 μL of PBS (pH 7.2) per well. 100 μL of each drug stock was added to the wells in triplicate, then the plates were incubated at 37° C for 24 hours.
Cells were assayed for luciferase activity using the Promega DualGlo kit. 50 μl of Luciferase Reagent were added per well, the plate was rocked for 15 min at RT, then the plate was read of firefly luciferase activity. 50 μl of Stop & Glo Reagent were added per well, then the plate was read for Renilla luciferase activity. Data normalized to Renilla internal control were analyzed with GraphPad Prism v.4a using the sigmoid dose response model to generate EC50 values ±SEM.
Animal Studies
Experimental protocols were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Oregon Health & Science University Institutional Animal Care & Use Committee. Wild type male C57BL/6J mice, aged 8–10 weeks, were housed in a climate controlled room with a 12 hour light-dark cycle with ad libitum access to food and water.
Distribution Studies
Mice were injected once intraperitoneally (ip) with sobetirome at 9.14 μmol/kg, and analogs at 0.914, 9.14, and 30.5 μmol/kg. Euthanasia was performed on three mice per dose at 1 hr and the tissues and blood were harvested. Tissues were immediately frozen and blood was kept on ice for a minimum of 30 minutes and then spun down at 7,500 x G for 15 minutes. Serum (100uL) was collected and was stored with tissues at −80° C until samples were processed.
Serum Processing
The serum samples were warmed to RT and 10 uL of 2.99 μM internal standard (D6-Sobetirome) was added to them. Acetonitrile (500 uL) was added and the sample was vortexed for 20 seconds. The sample was then centrifuged at 10,000 x G for 15 minutes at 4 °C. Next, 90% of the upper supernatant was transferred to a glass test tube and concentrated using a speedvac for 1.5 hr at 45° C. The dried sample was then dissolved in 400 μL of 50:50 ACN:H2O and vortexed for 20 seconds. The resulting mixture was transferred to an eppendorf and centrifuged at 10,000 x G for 15 minutes. The supernatant was filtered with 0.22 μM centrifugal filters and submitted for LC-MS/MS analysis. The standard curve was made with 100 μL of serum from a 8–10 week old mouse not injected with T3, sobetirome, or analogs. The processing was performed exactly the same except after filtering the sample was split amongst 6 vials. To 5 out of the 6 vials was added sobetirome, JD-20, and JD-21 to make final concentrations of each compound in matrix of (0.1 pg/μL, 1 pg/μL, 10 pg/μL, 100 pg/μL, and 1000 pg/μL).
Brain Processing
The brain samples were warmed to RT and transferred to a homogenizer tube with 5 GoldSpec 1/8 chrome steel balls (Applied Industrial Technologies). The resulting tube was weighed and then 1 mL of H2O was added, followed by 10 uL of 2.99 μM internal standard (D6-Sobetirome). The tube was homogenized with a Bead Bug for 30 seconds and then transferred to a falcon tube containing 3 mL of ACN. ACN (1 mL) was used to wash homogenizer tube and the solution was transferred back to the falcon tube. The sample was then processed using the same method for the serum processing except the sample was concentrated in a glass tube using a speed vac for 4 hr at 45° C.
Gene activation
Mice were injected once intraperitoneally (ip) with vehicle (1:1 saline/DMSO), T3 at 0.305 μmol/kg, sobetirome at 9.14 μmol/kg, and analogs at 0.914, 9.14, and 30.5 μmol/kg. Euthanasia was performed on three mice per dose at 2 hr and the tissues were harvested. The brain tissues collected for qPCR analysis were processed according to a protocol for RNA extraction using Trizol reagent and the PureLink RNA mini kit, using Qiagen RNase-free DNase kit during the optional DNase treatment step. 1 μg of extracted RNA was used to synthesize cDNA via a reverse transcription (RT) reaction using the Qiagen QuantiTect Reverse Transcription kit. DNA contamination was controlled for by duplicating one sample without the addition of RT enzyme. Expression of the Hairless (Hr) gene was measured by QPCR using the QuantiTect SYBR green PCR kit from Qiagen. The primer sequences for hairless (Fwd: CCAAGTCTGGGCCAAGTTTG; Rev: TGTCCTTGGTCCGATTGGAA) were previously described by Barca-Mayo.22 The template cDNA was diluted 2-fold to minimize the interference of RT reagents in the qPCR reaction. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) was the housekeeping gene used for normalizing between samples. Data analysis for single dose experiment was done using the comparative CT method to look at the relative differences in Hr gene expression. Data analysis for dose-response experiment was done using GraphPad Prism v.4a with the sigmoid dose response model to generate EC50 values ±SEM.
Chemistry
Chemistry General
1H NMR were taken on a Bruker 400. All 1H and 13C NMR were calibrated to the NMR solvent reference peak (D6-acetone, CDCl3). High resolution mass spectrometry (HRMS) with electrospray ionization was performed by the Bioanalytical MS Facility at Portland State University. Anhydrous tetrahydrofuran (THF) and dimethylformamide (DMF) were obtained from a Seca Solvent System. All other solvents used were purchased from Sigma-Aldrich or Fisher. Purity analysis of final compounds was determined to be >95% by HPLC. HPLC analysis was performed on a Varian ProStar HPLC with an Agilent Eclipse Plus C18 5 μM column (4.6 × 250 mm) with a gradient of 10% to 95% acetonitrile (0.1% TFA) over 15 minutes.
Synthesis
(3,5-dibromophenoxy)triethylsilane (2a)
1a (5.04g, 20 mmol) and imidazole (4.09 g, 60 mmol) were dissolved in 80 mL of DCM. The solution was cooled to 0° C, then triethylsilyl chloride (5.03 mL, 30 mmol) was added, then the reaction was stirred at 0° C for 30 min. The reaction was diluted with 160 mL of Et2O, washed 2X with 50 mL of H2O and 2X with 50 mL of brine, then dried with MgSO4, filtered, and concentrated to give the 2a in quantitative yield, which was used without purification. 1H NMR (400 MHz, CDCl3): δ 7.37 (t, 1H, J=1.6 Hz), 7.10 (d, 2H, J=1.6 Hz), 1.02 (t, 9H, J=7.9 Hz), 0.82 (q, 6H, J=7.9 Hz).
4-hydroxy-2,6-dibromobenzaldehyde (3a)
A flask was loaded with molecular sieves, then flame-dried under vacuum. After cooling under argon, 2a (5.49 g, 15 mmol) was loaded, then the flask was sealed, evacuated, and flushed with argon. 30 mL of dry THF were added and degassed, then the solution was cooled to −78° C. A second flask was loaded with molecular sieves, then flame-dried under vacuum. After cooling under argon, diisopropylamine (4.6 mL, 33 mmol) was added, followed by 60 mL of dry THF, then the solution was degassed and cooled to −78° C. 2.5 M n-butyllithium solution in hexanes (12 mL, 30 mmol) was added, then the solution was stirred for 1 hr at −78° C. The lithium diisopropylamide solution was transferred dropwise via cannula to the 2a solution, then the deprotonation was stirred for 1 hr at −78° C. 5.8 mL of dry DMF (75 mmol) were added, then the reaction was stirred for 1 hr at −78° C. The reaction was decanted into 50 mL of 1 N aqueous HCl. The aqueous layer was extracted 3X with 90 mL of Et2O. The organic fractions were combined, washed 2X with 50 mL of brine, then dried with MgSO4, filtered, and concentrated to give the crude product, which was precipitated from hexanes at −78° C to give 2.8 g of 3a (67% yield). 1H NMR (400 MHz, d6-acetone) δ 10.16 (s, 1H), 7.27 (s, 2H).
tert-butyl 2-(3,5-dibromo-4-formylphenoxy)acetate (4a)
3a (2.8 g, 10 mmol), sodium iodide (3 g, 20 mmol), and cesium carbonate (3.24 g, 10 mmol) were dissolved in 40 mL of acetone. 2.86 mL tert-butyl chloroacetate (20 mmol) were added, then the reaction was refluxed at 65° C for 2 hr. The reaction was diluted with 80 mL of Et2O, washed 2X with 30 mL of water and 2X with 30 mL of brine, then dried with MgSO4, filtered, and concentrated. The product was precipitated from hexanes and collected by filtration, then dried under vacuum to give 3.49 g of 4a (88% yield). 1H NMR (400 MHz, CDCl3) δ 10.23 (s, 1H), 7.19 (s, 2H), 4.59 (s, 2H), 1.52 (s, 9H).
(3,5-dichlorophenoxy)triethylsilane (2b)
1b (6.54g, 40 mmol) and imidazole (8.18 g, 120 mmol) were dissolved in 160 mL of DCM. The solution was cooled to 0° C, then triethylsilyl chloride (10 mL, 60 mmol) was added, then the reaction was stirred at 0° C for 30 min. The reaction was diluted with 320 mL of Et2O, washed 2X with 100 mL of H2O and 2X with 75 mL of brine, then dried with MgSO4, filtered, and concentrated to give 2b, which was used without purification and weighed 10.58 g after drying (95% yield). 1H NMR (400 MHz, CDCl3) δ 6.98 (t, 1H, J=1.8 Hz), 6.76 (d, 2H, J=1.8 Hz), 1.02 (t, 9H, J=7.9 Hz), 0.77 (q, 6H, J=7.9 Hz).
4-hydroxy-2,6-dichlorobenzaldehyde (3b)
A flask was loaded with molecular sieves, then flame-dried under vacuum. After cooling under argon, 2b (3.6 g, 13 mmol) was loaded, then the flask was sealed, evacuated, and flushed with argon. 13 mL of dry THF were added and degassed, then the solution was cooled to −78° C. A second flask was loaded with molecular sieves, then flame-dried under vacuum. After cooling under argon, 2,2,6,6-tetramethylpiperdidine (1.84 g, 13 mmol) was added, followed by 13 mL of dry THF, then the solution was degassed and cooled to −78° C. 2.5 M n-butyllithium solution in hexanes (5.2 mL, 13 mmol) was added, then the solution was stirred for 20 min at 0° C. The lithium TMP solution was transferred dropwise via cannula to the 2b solution, then the deprotonation was stirred for 30 min at −78° C. 5 mL of dry DMF (65 mmol) were added, then the reaction was stirred for 30 min at −78° C. The reaction was decanted into 15 mL of 1 N aqueous HCl. The aqueous layer was extracted 3X with 15 mL of EtOAc. The organic fractions were combined, washed 2X with 15 mL of brine, then dried with MgSO4, filtered, and concentrated to give the crude product, which was recrystallized from hexanes at −20° C to give 1.39 g of 3b (56% yield). 1H NMR (400 MHz, d6-acetone) δ 10.37 (s, 1H), 7.01 (s, 2H).
tert-butyl 2-(3,5-dichloro-4-formylphenoxy)acetate (4b)
3b (1.15 g, 6 mmol), sodium iodide (1.8 g, 12 mmol), and cesium carbonate (1.94 g, 6 mmol) were dissolved in 24 mL of acetone. 1.72 mL tert-butyl chloroacetate (12 mmol) were added, then the reaction was refluxed at 60° C for 24 hr. The reaction was diluted with 30 mL of Et2O, washed 2X with 10 mL of water and 2X with 10 mL of brine, then dried with MgSO4, filtered, and concentrated. The crude oil was redissolved in a minimal amount of Et2O then added dropwise to 100 mL of vigorously stirring hexanes at −78°C. The precipitate was collected by filtration and dried under vacuum to give 1.545 g of 4b (84% yield). 1H NMR (400 MHz, CDCl3) δ 10.43 (s, 1H), 6.92 (s, 2H), 4.59 (s, 2H), 1.52 (s, 9H).
4-iodo-2-isopropylphenol (6)
5 (6.8g, 50 mmol), NaI (7.5 g, 50 mmol) were dissolved in 70 mL of MeOH. 10 M aqueous NaOH (5 mL, 50 mmol) was added, then the solution was cooled to 0° C. 6.25% w/v aqueous NaOCl (62.5 mL, 50 mmol) was added drop wise over 24 hr at 0° C. The reaction was acidified to pH 7 with 12 N aqueous HCl, then quenched with 10 mL of saturated aqueous Na2S2O3. The aqueous layer was extracted 3X with Et2O. The organic fractions were combined, washed 2X with brine, then dried with MgSO4, filtered, and concentrated to give the crude product, which was purified by flash chromatography (silica gel, hexane/ethyl acetate, 1–20%) to give 11.35 g of 6 (87% yield) as a reddish oil. 1H NMR (400 MHz, CDCl3) δ 7.47 (d, 1H, J=2.1 Hz), 7.36 (dd, 1H, J=8.4 Hz, 2.2 Hz), 6.54 (d, 1H, J=8.4 Hz), 3.16 (m, 1H, J=6.9 Hz), 1.25 (d, 6H, J=6.9 Hz).
4-iodo-2-isopropyl-1-(methoxymethoxy)benzene (7)
6 (2.62 g, 10 mmol) and tetrabutylammonium iodide (369 mg, 1 mmol) were dissolved in 100 mL of DCM. 10 mL of 10 M aqueous NaOH were added, followed by 5 mL of 6 M chloromethyl methyl ether in MeOAc. The reaction was stirred for 30 min at RT, then diluted with 200 mL of Et2O. The organic layer was washed 2X with 100 mL of H2O and 2X with 100 mL of brine, then dried with MgSO4, filtered, and concentrated to give the crude product, which was purified by flash chromatography (silica gel, hexane/ethyl acetate, 1–20%) to give 2.48 g of 7 (81% yield). 1H NMR (400 MHz, CDCl3) δ 7.47 (d, 1H, J=2.2 Hz), 7.42 (dd, 1H, J=8.6 Hz, 2.2 Hz), 6.83 (d, 1H, J=8.6 Hz), 5.18 (s, 2H), 3.47 (s, 3H), 3.27 (m, 1H, J=6.9 Hz), 1.20 (d, 6H, J=7 Hz).
tert-butyl 2-(3,5-dibromo-4-(hydroxy(3-isopropyl-4-(methoxymethoxy)phenyl)methyl)phenoxy)acetate (8a)
A flask was loaded with 4 Å molecular sieves and flame-dried under vacuum. 7 (1.47 g, 4.8 mmol) was loaded and the flask was sealed, evacuated, and flushed with argon. 24 mL of dry THF were added and degassed, then the solution was cooled to 0° C. Isopropylmagnesium chloride (2 M THF, 5.5 mL, 7.2 mmol) was added, then the reaction was stirred for 2 hours at RT. A second flask was loaded with 4 Å molecular sieves and and flame-dried under vacuum. 4a (946 mg, 2.4 mmol) was loaded and the flask was sealed, evacuated, and flushed with argon. 12 mL of dry THF were added and degassed. The arylmagnesium solution was cooled to −78° C, then the 4a solution was added drop wise via cannula and the reaction was stirred for 1 hour at −78° C. The reaction was quenched with 10 mL of 1 N aqueous HCl. The aqueous layer was extracted 3X with 10 mL of EtOAc. The organic fractions were combined and washed 2X with 10 mL of brine. The organic layer was dried with MgSO4, filtered, and concentrated to give the crude product, which was purified by flash chromatography (silica gel, hexanes/EtOAc 4–40%) to give 1.089 g of 8a (79% yield). 1H NMR (400 MHz, CDCl3) δ 7.24 (d, 1H, J=1.9 Hz), 7.17 (s, 2H), 7.00 (d, 1H, J=8.5 Hz), 6.90 (dd, 1H, J=8.7 Hz, 1.9 Hz), 6.51 (d, 1H, J=10.8 Hz), 5.21 (s, 2H), 4.53 (s, 2H), 3.49 (s, 3H), 3.36 (d, 1H, J=10.8 Hz) 3.34 (m, 1H, J=6.9 Hz), 1.52 (s, 9H), 1.21 (t, 6H, J=6.5 Hz).
2-(3,5-dibromo-4-((3-isopropyl-4-hydroxyphenyl)methyl)phenoxy)acetic acid (9a)
8a (1.089 g, 1.9 mmol) was dissolved in 19 mL of DCM with 1.21 mL of triethylsilane (7.58 mmol). The solution was cooled to 0°C, then 4.35 mL of trifluoroacetic acid (56.9 mmol) were added and the reaction was stirred for 30 min at 0° C, then 2 hr at RT. Solvent was removed under vacuum, then the product was precipitated by the addition of hexanes and collected by filtration. The solid was dried under vacuum to give 505 mg of JD-20 (9a) (58% yield). 1H NMR (400 MHz, CDCl3) δ 7.19 (s, 2H), 7.10 (d, 1H, J=1.9 Hz), 6.82 (dd, 1H, J=8.7 Hz, 1.9 Hz), 6.64 (d, 1H, J=10.8 Hz), 4.68 (s, 2H), 4.28 (s, 2H), 3.18 (m, 1H, J=6.9 Hz), 1.24 (d, 6H, J=6.9 Hz). 13C NMR (400 MHz, CDCl3) δ 173.12, 156.17, 151.03, 134.18, 133.67, 130.33, 126.90, 126.08, 126.02, 118.98, 115.13, 64.87, 28.04, 27.04, 22.57. HRMS exact mass calculated for C18H17Br2O4 [M+H]+: 459.94687. Found 459.94647.
tert-butyl 2-(3,5-dichloro-4-(hydroxy(3-isopropyl-4-(methoxymethoxy)phenyl)methyl)phenoxy)acetate (8b)
A flask was loaded with 4 Å molecular sieves and flame-dried under vacuum. 7 (459 mg, 1.5 mmol) was loaded and the flask was sealed, evacuated, and flushed with argon. 6 mL of dry THF were added and degassed, then the solution was cooled to 0° C. Isopropylmagnesium chloride (2 M THF, 1.125 mL, 2.25 mmol) was added, then the reaction was stirred for 2 hours at RT. A second flask was loaded with 4 Å molecular sieves and and flame-dried under vacuum. 4b (305 mg, 1 mmol) was loaded and the flask was sealed, evacuated, and flushed with argon. 4 mL of dry THF were added and degassed. The arylmagnesium solution was cooled to −78° C, then the 4b solution was added drop wise via cannula and the reaction was stirred for 1 hour at −78° C. The reaction was quenched with 5 mL of 1 N aqueous HCl. The aqueous layer was extracted 3X with 5 mL of EtOAc. The organic fractions were combined and washed 2X with 5 mL of brine. The organic layer was dried with MgSO4, filtered, and concentrated to give the crude product, which was purified by flash chromatography (silica gel, hexanes/EtOAc 2–20%) to give 260 mg of 8b (54% yield). 1H NMR (400 MHZ, CDCl3) δ 7.26 (d, 1H, J=2.1 Hz), 6.99 (d, 1H, J=8.5 Hz), 6.93 (dd, 1H, J=8.2 Hz, 2.2 Hz), 6.92 (s, 2H), 6.50 (d, 1H, J=10.8 Hz), 5.21 (s, 2H), 4.53 (s, 2H), 3.50 (s, 3H), 3.33 (m, 1H, J=6.9 Hz), 3.23 (d, 1H, J=10.8 Hz), 1.52 (s, 9H), 1.21 (t, 6H, J=6.8 Hz).
2-(3,5-dichloro-4-((3-isopropyl-4-hydroxyphenyl)methyl)phenoxy)acetic acid (9b)
8b (260 mg, 0.54 mmol) was dissolved in 5.4 mL of DCM with 0.345 mL of triethylsilane (2.16 mmol). The solution was cooled to 0°C, then 1.24 mL of trifluoroacetic acid (16.2 mmol) were added and the reaction was stirred for 30 min at 0° C, then 2 hr at RT. Solvent was removed under vacuum, then the product was precipitated by the addition of hexanes and collected by filtration. The solid was dried under vacuum to give 137 mg of JD-21 (9b) (69% yield). 1H NMR (400 MHz, CDCl3) δ 7.12 (d, 1H, J=2 Hz), 6.95 (s, 2H), 6.86 (dd, 1H, J=8.2 Hz, 2.2 Hz), 6.64 (d, 1H, J=8.2 Hz), 4.68 (s, 2H), 4.18 (s, 2H), 3.17 (m, 1H, J=6.9 Hz), 1.24 (d, 6H, J=6.9 Hz). 13C NMR (400 MHz, CDCl3) δ 172.18, 155.92, 151.09, 136.36, 134.15, 130.97, 130.67, 126.83, 121.46, 115.11, 115.04, 64.84, 27.06, 24.35, 22.53. HRMS exact mass calculated for C18H17Cl2O4 [M+H]+: 367.04984. Found 367.05053.
Figure 2.

Synthesis of JD-20 9a and JD-21 9b. Reagents and Conditions: (a) triethylsilyl chloride, imidazole, DCM, 0°C, 95%; (b) (i) nBuLi, DIA or TMP, THF, −78° C (ii) DMF, 56–67%; (c) tert-butylchloroacetate, NaI, Cs2CO3, acetone, 60–65° C, 84–88%; (d) NaI, NaOH, NaOCl, MeOH, H2O, 87%; (e) MOMCl, TBAI, NaOH, DCM, H2O, 81%; (f) (i) iPrMgCl, THF, 0° C to RT (ii) 4, −78° C, 54–79%; (g) TFA, triethylsilane, DCM, 0° C to RT, 58–69%.
Acknowledgments
This research was supported by a grant from the National Institutes of Health (T.S.S.). The authors would like to thank D. Koop and L. Bleyle in the OHSU Pharmacokinetics Core for their technical expertise and guidance.
References
- 1.Calzà L, Fernandez M, Giuliani A, D’Intino G, Pirondi S, Sivilia S, Paradisi M, Desordi N, Giardino L. Brain Research Reviews. 2005;48:339–346. doi: 10.1016/j.brainresrev.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 2.Malm J, Grover GJ, Färnegårdh M. Mini Rev Med Chem. 2007;7:79–86. doi: 10.2174/138955707779317885. [DOI] [PubMed] [Google Scholar]
- 3.Yen P. Physiol Rev. 2001 doi: 10.1152/physrev.2001.81.3.1097. [DOI] [PubMed] [Google Scholar]
- 4.O’Shea P, Bassett JHD, Cheng S. Nuclear Receptor …. 2006 doi: 10.1621/nrs.04011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiellini G, Apriletti JW, Yoshihara HA, Baxter JD, Ribeiro RCJ, Scanlan TS. Chem Biol. 1998;5:299–306. doi: 10.1016/s1074-5521(98)90168-5. [DOI] [PubMed] [Google Scholar]
- 6.Yoshihara HAI, Apriletti JW, Baxter JD, Scanlan TS. J Med Chem. 2003;46:3152–3161. doi: 10.1021/jm0301181. [DOI] [PubMed] [Google Scholar]
- 7.Trost SU, Swanson E, Gloss B, Wang-Iverson DB, Zhang H, Volodarsky T, Grover GJ, Baxter JD, Chiellini G, Scanlan TS, Dillmann WH. Endocrinology. 2000;141:3057–3064. doi: 10.1210/endo.141.9.7681. [DOI] [PubMed] [Google Scholar]
- 8.Grover GJ, Egan DM, Sleph PG, Beehler BC, Chiellini G, Nguyen NH, Baxter JD, Scanlan TS. Endocrinology. 2004;145:1656–1661. doi: 10.1210/en.2003-0973. [DOI] [PubMed] [Google Scholar]
- 9.Baxter JD, Webb P, Grover G, Scanlan TS. Trends Endocrinol Metab. 2004;15:154–157. doi: 10.1016/j.tem.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Scanlan TS. Heart Fail Rev. 2010 doi: 10.1007/s10741-008-9122-x. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi N, Asano Y, Maeda K, Watanabe N. Biol Pharm Bull. 2014;37:1103–1108. doi: 10.1248/bpb.b13-00915. [DOI] [PubMed] [Google Scholar]
- 12.Baxi EG, Schott JT, Fairchild AN, Kirby LA, Karani R, Uapinyoying P, Pardo-Villamizar C, Rothstein JR, Bergles DE, Calabresi PA. Glia. 2014;62:1513–1529. doi: 10.1002/glia.22697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genin EC, Gondcaille C, Trompier D, Savary S. The Journal of Steroid Biochemistry and Molecular Biology. 2009;116:37–43. doi: 10.1016/j.jsbmb.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Ocasio CA, Scanlan TS. ACS Chem Biol. 2006;1:585–593. doi: 10.1021/cb600311v. [DOI] [PubMed] [Google Scholar]
- 15.Ocasio CA, Scanlan TS. Bioorg Med Chem. 2008;16:762–770. doi: 10.1016/j.bmc.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valadares NF, Salum LB, Polikarpov I, Andricopulo AD, Garratt RC. J Chem Inf Model. 2009;49:2606–2616. doi: 10.1021/ci900316e. [DOI] [PubMed] [Google Scholar]
- 17.Auffinger P, Hays FA, Westhof E, Ho PS. Proceedings of the National Academy of Sciences. 2004;101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dabrowski M, Kubicka J, Luliński S, Serwatowski J. Tetrahedron Letters. 2005;46:4175–4178. [Google Scholar]
- 19.Placzek AT, Scanlan TS. Tetrahedron. 2015;71:5946–5951. doi: 10.1016/j.tet.2015.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krasovskiy A, Knochel P. Angew Chem Int Ed. 2004;43:3333–3336. doi: 10.1002/anie.200454084. [DOI] [PubMed] [Google Scholar]
- 21.Yoshihara H, Apriletti JW, Baxter JD, Scanlan TS. Bioorg Med Chem Lett. 2001;11:2821–2825. doi: 10.1016/s0960-894x(01)00521-2. [DOI] [PubMed] [Google Scholar]
- 22.Barca-Mayo O, Liao XH, Alonso M, Di Cosmo C, Hernandez A, Refetoff S, Weiss RE. Mol Endocrinol. 2011;25:575–583. doi: 10.1210/me.2010-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]