

Abstract
Linear growth failure can be caused by many different genetic abnormalities. In many cases, the genetic defect affects not only the growth plate, causing short stature, but also other organs/tissues causing additional clinical abnormalities. The proband was evaluated at 10 years of age for impaired postnatal linear growth (height 113.3 cm, −4.6 SDS), a bone age that was delayed by 5 years, dysmorphic facies, cognitive impairment, and central nervous system anomalies. His younger brother, presented only with growth failure at 10 months of age.
Exome sequencing identified compound heterozygous variants in the gene encoding RNA polymerase III transcription initiation factor 90 kDa subunit (BRF1) in both affected siblings: a missense mutation (c.875 C>G:p.P292R) and a frameshift mutation (c.551delG:p.C184Sfs). The frameshift mutation is expected to lead to nonsense-mediated mRNA decay (NMD) and/or to protein truncation. Expression of BRF1 with the P292R missense mutation failed to rescue yeast lacking BRF1. The findings confirm a previous report showing that biallelic mutations in BRF1 cause cerebellar-facial-dental syndrome. Our findings also help define the growth phenotype, indicating that the linear growth failure can become clinically evident before the neurological abnormalities and that a severely delayed bone age may serve as a diagnostic clue.
Keywords: Exome sequencing, BRF1, severe short stature, delayed bone age, neurologic syndrome
Graphical Abstract
Introduction
Short stature is a heterogeneous condition which is caused by decreased chondrogenesis in the growth plate (1, 2). The diminished rate of chondrogenesis can be caused either by a primary abnormality intrinsic to the growth plate chondrocytes themselves or can be secondary to a systemic abnormality, including nutritional, inflammatory, or endocrine disorders. Some genetic conditions affect both the growth plate, causing short stature, and other tissues, causing additional developmental abnormalities. In many children with short stature, either isolated or syndromic, the etiology remains unknown despite careful clinical and laboratory evaluation (3).
Exome sequencing has been successfully applied to identify the genetic causes of various disorders (4). However, this approach generates large numbers of sequence variants and it is often difficult to identify the pathogenic variant(s), particularly when the disorder is rare, limiting study to a small number of affected individuals. To overcome this challenge, exome sequencing can be performed on unaffected and affected family members of the proband, and most non-pathogenic variants can often be excluded based on the inheritance pattern and minor allele frequency, leaving only a small number of candidate variants (5). This family approach has elucidated the etiology of numerous genetic diseases (6, 7), including childhood linear growth disorders (8, 9).
Here, we describe a child who was referred to our pediatric endocrinology clinic at the National Institutes of Health for evaluation of severe idiopathic short stature, markedly delayed bone age, dysmorphic facies, cognitive dysfunction, and CNS anomalies. His younger brother manifested only linear growth failure at 10 months of age. Exome sequencing of the family identified compound heterozygous mutations in BRF1 in the affected individuals, which is the genetic cause of a recently identified syndrome called cerebellar-facial-dental syndrome (10). The findings confirm the causative connection between BRF1 mutations and this syndrome and also help elucidate the clinical growth phenotype.
Subjects and Methods
Clinical presentation
A 10-year old boy was referred from St. Christopher’s Hospital for Children in Philadelphia to the pediatric endocrinology clinic at the NICHD, National Institutes of Health (NIH) for evaluation of severe short stature, dysmorphic facies and developmental delay (Figure 1A, Figure 2A & B). He was the product of a full term gestation with a birth weight of 2.9 kilograms (−1.5 SDS). He was found to have hydronephrosis in utero and postnatally, he was diagnosed with ureteropelvic junction obstruction and underwent surgical correction at three months of life. He was also noted to have dysmorphic features including microcephaly, micrognathia, and midface hypoplasia. During early infancy, he began to decline in growth percentiles for both height and weight (Fig 2A) and was diagnosed with failure to thrive which was managed with supplementation in formula. It was thought that tracheomalacia also contributed to his poor weight gain at the time. Through childhood, the height and weight remained well below the 3rd centile for age. His bone age has been delayed 5 years (Supplemental Figure 1). He has a significant global developmental delay with particularly delayed speech. He underwent inguinal hernia repair with bilateral orchiopexy and surgical correction for foot contractures. He has been evaluated and followed by pediatric geneticists and endocrinologists without identifying a unifying diagnosis despite extensive metabolic, genetic and endocrine evaluations, which included a sweat chloride test, a karyotype (46,XY), FISH for DiGeorge and Williams syndrome, aCGH array, and growth hormone stimulation testing. An MRI of the brain revealed a small brainstem with flattening of the inferior vermis, cerebellar hemisphere volume loss, periventricular leukomalacia with marked white matter volume loss posteriorly, and thinning of the corpus callosum (Figure 3) but normal pituitary gland. Echocardiogram was normal.
Figure 1.


A and B. Photograph of proband and affected brother. A) The 10-year old boy showed dysmorphic facial features including anteverted nares, wide nasal bridge, mid-face hypoplasia, long philtrum, down-sloping upper lip, micrognathia and prominent enlarged superior central incisors. B) 10-month old affected brother did not show dysmorphic facial features.
Figure 2.



A, B and C. Growth charts of proband and his affected brother. The proband’s growth charts showed impaired linear growth causing progressive deviation of the height from the normal range through infancy and childhood. 2A: infantile growth chart (0–2 years). 2B: childhood growth chart (2–20 years). 2C: Affected brother’s growth chart shows pre- and post-natal growth deceleration. Both growth charts are based on normative data from the Centers for Disease Control and Prevention, National Center for Health Statistics, United States, http://www.cdc.gov/growthcharts/.
Figure 3.

Brain MRI of proband. MRI sagittal section at 1 year 9 months in the proband reveals a small brainstem with flattening of the inferior vermis (short white arrow), cerebellar hemisphere volume loss (long white arrow), periventricular leukomalacia with marked white matter volume loss posteriorly, and thinning of the corpus callosum (long black arrow).
On physical exam, at 10 years of age, his height was 113.3 cm (−4.6 SDS) and weight was 17.3 kg (−5.7 SDS). His sitting/standing height ratio was 0.489, which was at the 5th percentile for age. His arm span was 104.5 cm, 9 cm less than his standing height. These measurements suggest that growth of the vertebrae and upper extremities were affected more than growth of the lower extremities. His head circumference was 46.4 cm (−5.0 SDS). His face was dysmorphic with anteverted nares, wide nasal bridge, mid-face hypoplasia, down-sloping upper lip and micrognathia. He had prominent enlarged superior central incisors and a long philtrum (Figure 1). He had prepubertal testicular volume with no pubic hair. He had a single transverse crease noted bilaterally and increased lower extremity tone. His hand and middle finger length were 12 cm and 5 cm, respectively which are average length for a 4-year old child. Despite his extreme short stature, his skeletal survey was normal (Supplemental figure 2A–D).
The proband’s younger brother was 10 months of age at the time of the NIH evaluation. He was born at 36 weeks gestation with a weight of 2.51 kilograms (−0.5 SDS) and length of 46 cm (−0.48 SDS). His medical history included neonatal jaundice, pertussis, and multiple other respiratory tract infections. He showed normal developmental milestones. He had noisy breathing which was thought to be due to laryngomalacia by clinical evaluation. At 10 months of age, his length was 63 cm (−4.4 SDS) and weight was 6.55 kg (−3.8 SDS)(Figure 2C). The head circumference was 42.5 cm (−2.7 SDS). Physical examination was otherwise unremarkable (Figure 1B). An evaluation for failure to thrive was normal, including standard screening tests of electrolytes, mineral metabolism, hepatic function, thyroid function and blood cell counts. IGF-1 was 24 ng/mL (normal range for 1 year old, 55–327). An MRI of the brain has not yet been performed because of the need for sedation at the patient’s young age and the lack of clinical urgency.
The rest of the nuclear family was in good health. The parents are not related. The proband’s father is German and Italian in descent and mother is Polish in descent. The mother’s height was 167 cm with a normal sitting/standing height ratio. Father’s height was 175 cm with normal sitting/standing height ratio. The height of the proband’s 7-year-old unaffected sister was 126.56 cm (0.20 SDS) with a normal sitting/standing height ratio.
Study approval and DNA samples
This study was approved by the NICHD IRB. Consent and assent, as appropriate, were obtained from all subjects studied or from the parents.
Genomic DNA was extracted from peripheral blood leukocytes of the proband and his affected brother, unaffected sister and parents.
SNP array
DNA samples from the proband, affected brother, parents and unaffected sister were genotyped by the Cancer Genetics and Comparative Genomics Branch at the National Human Genome Research Institute (NHGRI) with the Illumina Infinium HumanOmniExpress Exome BeadChip platform, using the Illumina “Infinium assay” protocol (11). This assay includes whole-genome amplification and fragmentation of DNA, hybridization to the BeadChip with specific oligonucleotide probe array (50-mers), enzymatic extension of the 3’ terminal base for incorporation of the allele specific nucleotide, detection with fluorescently tagged reagent and signal amplification. HumanOmniExpressExome BeadChip contains nearly 1 million SNPs. The allele type and its intensity were assessed using iScan, and visualized with the GenomeStudio v2011.1 genotyping module (www.Illumina.com). All samples had a call rate >0.99.
Exome sequencing
Exome sequencing was performed at the National Institute of Sequencing Center. A whole-genome library with ~325 base inserts was prepared for each sample from 1 µg of DNA using the Kapa DNA Library Preparation Kit (high throughput, with bead) (Kapa Biosystems, Wilmington, MA) on a Beckman Coulter FX robot. Exome capture was performed with the SeqCap EZ Human Exome + UTR Kit v3.0 according to the SeqCap EZ Library SR User’s Guide (Roche Nimblegen). Each captured exome pool was sequenced on a HiSeq2500 using version 4 chemistry. Batches of 24 or 48 uniquely barcoded libraries were pooled using equal volumes of input and run on a MiSeq with version 2 chemistry at a loading concentration of 6 pM. The run consisted of 25 cycles followed by an index read. The demultiplexed read counts were used to normalize the DNA input for exome capture where 300 ng of each of 8 libraries were pooled together. At least 38 million paired-end 125 base reads were obtained for each sample. Data was processed using RTA ver. 1.18.64 and CASAVA 1.8.2. Reads were mapped to NCBI build 37 (hg19). Further methods for alignment, genotype calling, coverage and annotation are described in the Supplemental Method.
Analysis of Variants using VarSifter
The candidate variants were identified by applying Boolean logic filters using the open source program VarSifter (12, 13). Candidate variants were selected by Mendelian consistency and population frequency < 2% in Clinseq, an institutional database. Then, the variants were further narrowed down by population frequency found in Exome Aggregation Consortium (ExAc) and CADD to prioritize candidate variants. The predicted impact on protein function was evaluated using PolyPhen2, SIFT and MutationTaster.
Sanger sequencing
The mutations detected by exome sequencing were confirmed by Sanger sequencing in all the members of this family. PCR amplicons were prepared with the following primer sets: 1) BRF1 frameshift deletion; forward 5’ AAATGGGACATCTTTCACCTCAA 3’ and reverse 5’ GTCA GGAAAGTGTGAGGCCAG 3’, 2) BRF1 missense mutation; forward 5’ CTCAAGCAATCCACCCACCT 3’ and reverse 5’ GCTCTGGCTGGTCATAGCACT 3’. BRF1 mutation nomenclature is based on transcript NM_001519.3.
Yeast proliferation test
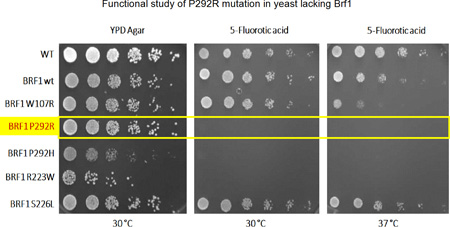
The yeast growth assay was performed as described by Borck et al. (10). In brief, the mutation corresponding to BRF1 P292R was introduced into the yeast BRF1 cDNA by site-directed mutagenesis and confirmed by Sanger sequencing. The plasmid harboring the BRF1 P292R mutation was transformed into a haploid yeast strain lacking the genomic BRF1 but carrying a plasmid with wild-type BRF1 cDNA as well as a selectable marker, URA3. To assess yeast viability, spot assays were performed. The cell density of overnight cultures was normalized to equal densities (107 cells/200 uL), serially diluted, and spotted on complete media (YPD) and medium supplemented with 5-fluoro-orotic acid (5-FOA), respectively. Subsequently, the agar plates were incubated for 2 days at 30 °C (the optimal temperature for yeast growth) and 37 °C. 5-FOA is converted into the toxic compound 5-fluorouracil by the URA3 gene product. This negative selection leads to loss of the rescue plasmid with wild-type BRF1 leaving only the extra-chromosomal BRF1 P292R, which thus allows evaluation of the missense mutation.
Results
SNP array
SNP array did not reveal significant copy number variation, uniparental isodisomy, or regions of homozygosity in the proband and in the affected brother. Paternity was confirmed.
Exome sequencing
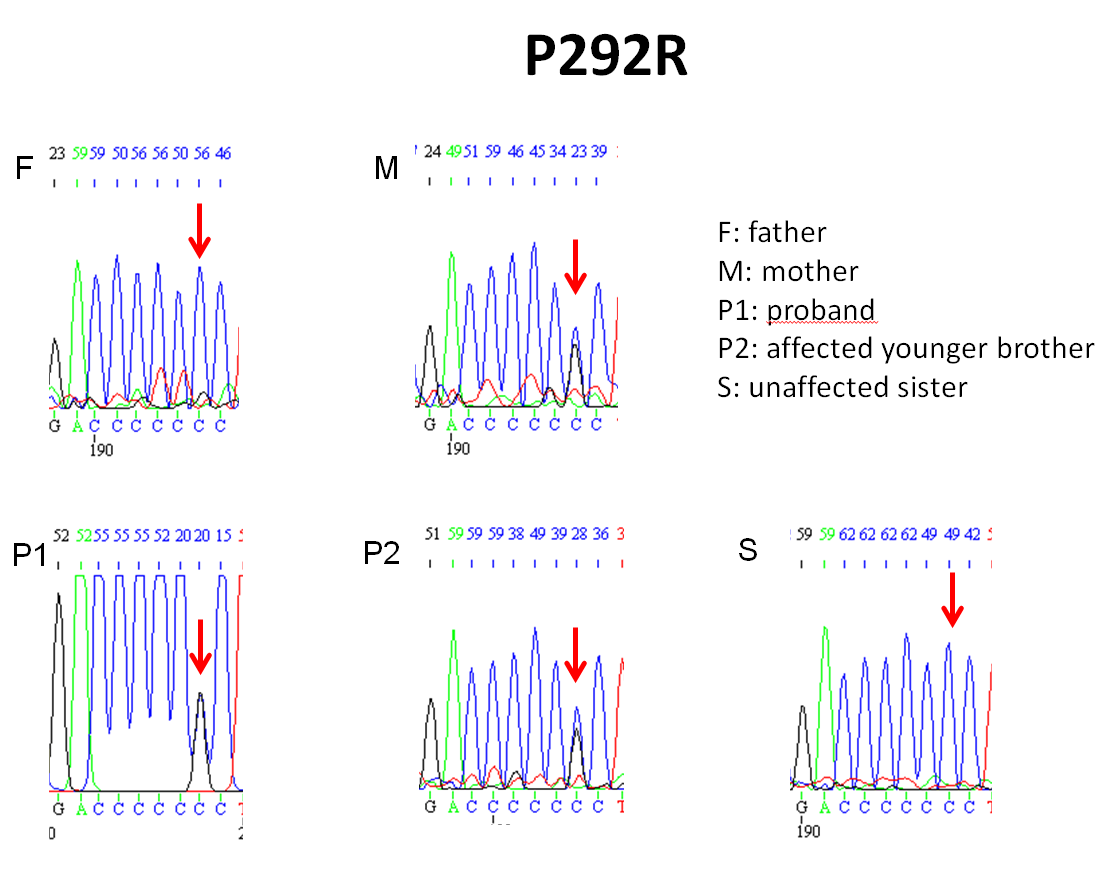
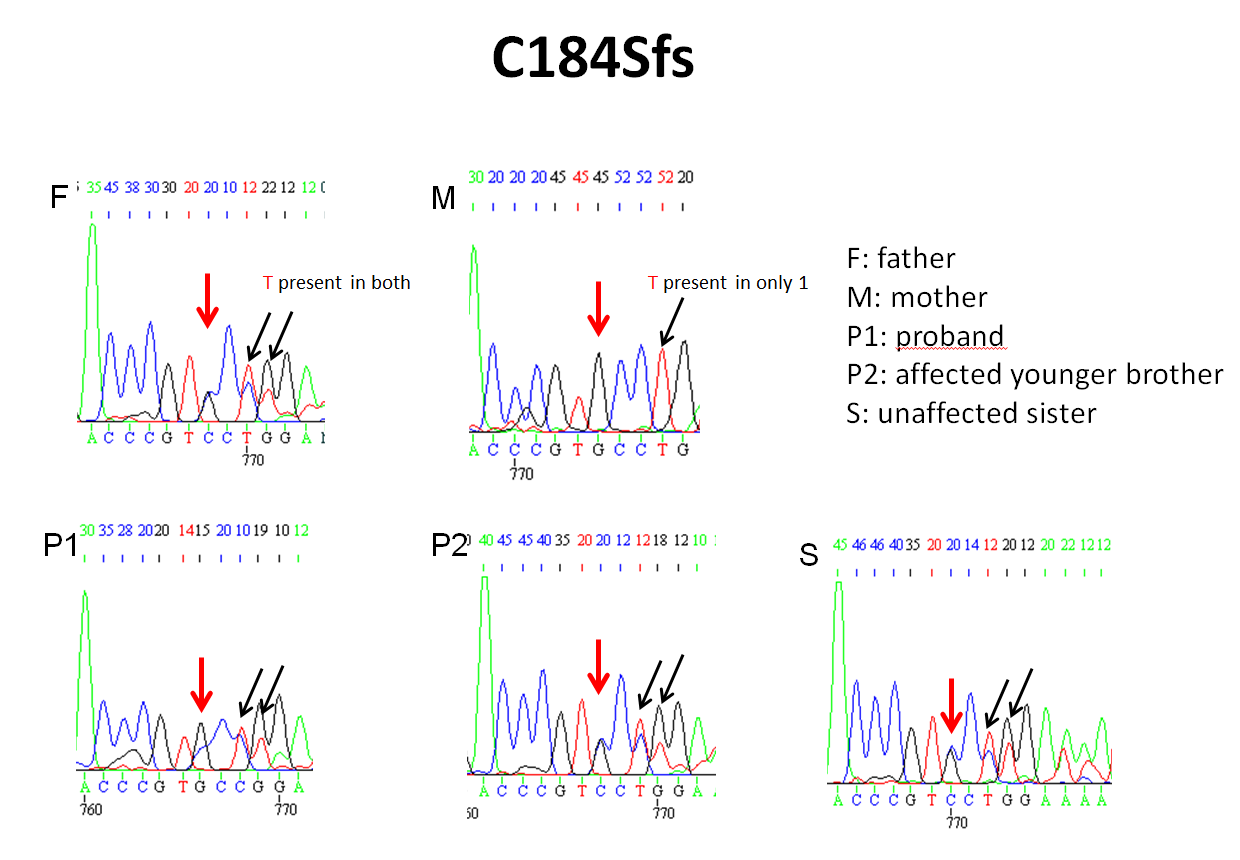
Exome sequencing yielded 494,079 variants genome-wide. The only variants that were rare (population frequency < 2% in Clinseq and ExAC) and that showed Mendelian consistency in the family (considering homozygous, compound heterozygous, X-linked, or heterozygous de novo variants) were compound heterozygous variants in RNA polymerase III transcription initiation factor 90 kDa subunit (BRF1); the proband and his affected brother shared a missense mutation (c.875 C>G:p.P292R; NM_001519.3) and a frameshift mutation (c.551delG: p.C184Sfs). p.P292R was inherited maternally and p.C184Sfs was inherited paternally (Figure 4). The unaffected sister carried only p.C184Sfs. Both mutations were not present in the ExAC database. The p.P292R missense mutation is located in a domain of BRF1 that is highly conserved and was predicted to be deleterious by CADD, Polyphen2, SIFT and MutationTaster. Both mutations were confirmed by Sanger sequencing to be inherited in a pattern that is consistent with the phenotype of all family members tested (Figure 4, Supplemental Figure 3A & B).
Figure 4.

Pedigree of the family and compound heterozygous mutations identified in two affected children. The proband and his affected younger brother shared a missense mutation (c.875 C>G:p.P292R) and a frameshift mutation (c.551delG:p.C184Sfs) in BRF1. P292R was inherited from their mother and p.C184Sfs was inherited from their father. The unaffected sister carried only C184Sfs. Filled symbols indicate an affected family member.
Yeast proliferation test
Yeast lacking the chromosomal BRF1 gene but expressing wild-type BRF1 from a plasmid construct survived on culture medium containing 5-fluoro-orotic acid in spot dilution test. In contrast, the same yeast strain lacking the chromosomal BRF1 gene but expressing BRF1 with the mutation corresponding to P292R did not grow under the same conditions (Figure 5) at either 30 °C or 37 °C, confirming a deleterious effect of the variant. Thus, P292R is deleterious, as is P292H, a BRF1 mutation that affects the same amino acid and that causes the cerebellar-facial-dental syndrome (10). The other mutation found in the proband was a deletion, which creates a frameshift starting at codon Cys184, very likely leading to loss of function. Therefore, no further functional study was performed on this mutant.
Figure 5.

Functional study of P292R mutation in yeast lacking Brf1. Yellow box indicates the missense mutation identified in the current study. Yeast carrying this mutation (P292R) failed to grow in YPD agar at either 30 °C or 37 °C in the presence of 5-fluoro-orotic acid (FOA), which negatively selects against an additional plasmid expressing wild-type BRF1. The P292R mutation behaves as the P292H (10) mutation in this assay. R223W and S226L are BRF1 mutations that we previously identified in patients with the cerebellar facial dental syndrome (10) and that lead to no growth and normal growth, respectively, in this assay. W107R is a previously described artificial temperature-sensitive mutation (26).
Discussion
We report a boy with severe short stature, extremely delayed bone age, facial dysmorphism and cognitive dysfunction. His younger brother, age 10 months, presented only with poor linear growth although other clinical abnormalities may become manifest at an older age. Exome sequencing, confirmed by Sanger sequencing, identified compound heterozygous variants in RNA polymerase III transcription initiation factor 90 kDa subunit (BRF1) in both affected brothers.
Several lines of evidence support a causal link between the identified sequence variants in BRF1 and the phenotypic findings: 1) Both variants are extremely rare, not found in large databases; 2) The variants were inherited in a pattern that would explain the phenotypic findings; 3) No other rare variants identified by exome sequencing showed Mendelian consistency. 4) One of the variants is a frameshift mutation that leads to an early stop codon and is likely to induce nonsense-mediated mRNA decay and/or protein truncation; 5) The other variant is a missense mutation which is located in a highly conserved domain of BRF1 and is strongly predicted to affect protein function. The effect of the missense mutation on protein function was confirmed experimentally based on its inability to rescue growth of yeast lacking BRF1. 6) Mutations in BRF1 have recently been reported to cause a new syndrome called cerebellar-facial-dental syndrome, and our proband shares multiple specific phenotypic characteristics with the patients previously described (10). Together, these findings strongly support the conclusion that the observed sequence variants in BRF1 caused the genetic disorder in the family reported here.
The proband in our study had a clinical presentation very similar to the patients previously reported with cerebellar-facial-dental syndrome and mutations in BRF1 (10). These common features include short stature, delayed bone age, upper airway abnormalities, speech delay, microcephaly, facial dysmorphism (especially prominent upper central incisors), cognitive dysfunction and hypoplastic cerebellum. Our proband also had a history of hydronephrosis in utero but without kidney problems after birth, which could either be a coincidental finding or an associated finding of this syndrome. The brother of our proband presented at 10 months of age with linear growth failure only, suggesting that this disorder should be considered in the differential diagnosis of a child who is evaluated for short stature at a young age, when other features may not yet be clinically evident.
One of the two mutations in our patients was a frameshift mutation whereas the other mutation in our patients and all previously reported mutations have been missense mutations. The phenotypic severity of our subjects appears similar to previously reported patients, showing similar growth parameters (height SDS −2.3 to −4.4 in previously reported patients vs. −4.4 to −4.6 in our patients). Also, cognitive function in the current proband is mild and does not appear to be more severe than in previously described subjects. Whether truncating mutations would tend to cause more severe forms of cerebellar-facial-dental syndrome may require larger case series.
BRF1 is a subunit of transcription initiation factor IIIB (TFIIIB) which participates in the recruitment of RNA polymerase (Pol) III, the polymerase that transcribes small RNAs including 5S rRNA, precursor tRNAs, some microRNAs, small nuclear RNAs, and mammalian-wide interspersed repeat and Alu elements (14–17). TFIIIB is required for initiation of Pol III transcription along with TATA box binding protein (TBP) and B double prime 1 (BDP1), which cooperatively bind to the promoter region upstream of the transcription start site (14) and then, recruit Pol III. In fibroblasts, Brf1 is phosphorylated by extracellular signal-regulated kinase (ERK). As a result, the ERK MAP kinase cascade, which promotes cell growth, is able to induce tRNA synthesis (18). In cardiomyocytes, it has also been noted that ERK-mediated induction of Brf1 as a subunit of TFIIIB is one of mechanisms to increase Pol III transcription in terminally differentiated cells (19) whereas low Brf1 expression limits Pol III transcription in resting cardiomyocytes.
The pathogenic mechanisms by which BRF1 mutations cause the decreased linear growth, the CNS developmental problems, and the dental and facial anomalies remain unknown. One possibility is that decreased Pol III transcription of tRNAs and/or 5S rRNA restricts the protein translational capacity of some cells. The apparent preferential involvement of certain tissues and organs, such as CNS, growth plate and teeth, in this syndrome might reflect the observation that levels of different tRNA transcripts vary greatly among different tissues (10, 20). Another possibility is that decreased Pol III transcription of non-coding regulatory RNAs might secondarily affect expression of protein-coding genes in the CNS, growth plate, and other tissues in which such small RNAs play important developmental roles (21–22).
Mutations in genes involved in tRNA transcription or processing impair brain development. The requirement of BRF1 for normal brain development is evident not only in humans but also in zebrafish, in which suppression or deletion of brf1b results in a small head and cerebellar hypoplasia, consistent with the human phenotype (10). Similarly, mutations in genes encoding subunits of Pol III (POLR3A, POLR3B, POLR1C) have been reported to cause various forms of leukodystrophies (23) or 4H syndrome (hypomyelination, hypodontia, hypogonadotropic hypogonadism)(24). In addition, mutations in genes that are required for tRNA processing cause pontocerebellar hypoplasia, which is a rare, progressive severe neurodegenerative disorder that includes hypoplasia of the cerebellum, pons and, to a lesser extent, the cerebral cortex causing microcephaly (25). Interestingly, mutations in subunits of Pol III or tRNA processing proteins do not seem to cause severe growth disorders or a skeletal phenotype, suggesting that BRF1 may have a distinctive role in growth plate chondrogenesis. To date, BRF1 is the only gene known to underlie cerebellar-facial-dental syndrome. However, our findings do not exclude the possibility of locus heterogeneity or modifier genes.
In conclusion, our findings confirm that mutations in BRF1 cause severe short stature, remarkably delayed bone age, dysmorphic features, cerebellar hypoplasia and cognitive dysfunction inherited in an autosomal recessive pattern. Our findings also suggest that the growth abnormality occurs in early postnatal life and that young patients may present clinically with linear growth failure before other manifestations of the disorder are apparent. Thus, cerebellar-facial-dental syndrome caused by biallelic mutations in BRF1 should be considered in the differential diagnosis of children with early unexplained linear growth failure.
Supplementary Material
Supplemental figure 1. Bone age was obtained at age 10.
Supplemental figure 2A–D. Skeletal x-rays were obtained at age 10.
Supplemental figure 3A–B. Both mutations were confirmed by Sanger sequencing.
{kind=link}
{kind=link}
Acknowledgments
This work was supported by the Intramural Research Programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), the Intramural Research Program of the National Human Genome Research Institute and the Common Fund of the National Institutes of Health.
Footnotes
Declaration of interest
All authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Reference
- 1.Jee YH, Baron J. The Biology of Stature. J Pediatr. 2016 doi: 10.1016/j.jpeds.2016.02.068. pii: S0022-3476(16)00291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baron J, Sävendahl L, De Luca F, et al. Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol. 2015;11(12):735–746. doi: 10.1038/nrendo.2015.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wit JM, Clayton PE, Rogol AD, et al. Idiopathic short stature: definition, epidemiology, and diagnostic evaluation. Growth Horm IGF Res. 2008;18(2):89–110. doi: 10.1016/j.ghir.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42(9):790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams DR, Sincan M, Fuentes Fajardo K, et al. Analysis of DNA sequence variants detected by high-throughput sequencing. Hum Mutat. 2012;33(4):599–608. doi: 10.1002/humu.22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gahl WA, Markello TC, Toro C, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14(1):51–59. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatton-Brown K, Seal S, Ruark E, Harmer J, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014;46(4):385–388. doi: 10.1038/ng.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glazov EA, Zankl A, Donskoi M, et al. Whole-exome re-sequencing in a family quartet identifies POP1 mutations as the cause of a novel skeletal dysplasia. PLoS Genet. 2011;7(3):e1002027. doi: 10.1371/journal.pgen.1002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarig O, Nahum S, Rapaport D, et al. Short stature, onychodysplasia, facial dysmorphism, and hypotrichosis syndrome is caused by a POC1A mutation. Am J Hum Genet. 2012;10;91(2):337–342. doi: 10.1016/j.ajhg.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borck G, Hög F, Dentici ML, et al. BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies. Genome Res. 2015;25(2):155–166. doi: 10.1101/gr.176925.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gunderson KL, Steemers FJ, Lee G, et al. A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet. 2005;37(5):549–554. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
- 12.Teer JK, Green ED, Mullikin JC, et al. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;15;28(4):599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gahl WA, Adams DR, Markello TC, et al. In: Genetic Approaches to Rare and Undiagnosed Diseases, chapter 83, Nelson's Textbook of Pediatrics. Twentieht. Kliegman, Stanton, St Gene, Shore, Behrman, editors. Philadelphia, PA: Elsevier; pp. 629–633. [Google Scholar]
- 14.Khoo SK, Wu CC, Lin YC, et al. Mapping the protein interaction network for TFIIB-related factor Brf1 in the RNA polymerase III preinitiation complex. Mol Cell Biol. 2014;34(3):551–559. doi: 10.1128/MCB.00910-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oler AJ, Alla RK, Roberts DN, Wong A, Hollenhorst PC, Chandler KJ, Cassiday PA, Nelson CA, Hagedorn CH, Graves BJ, Cairns BR. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat Struct Mol Biol. 2010;17(5):620–628. doi: 10.1038/nsmb.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13(12):1097–1101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 17.Dieci G, Fiorino G, Castelnuovo M, Teichmann M, Pagano A. The expanding RNA polymerase III transcriptome. Trends Genet. 2007;23(12):614–622. doi: 10.1016/j.tig.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Felton-Edkins ZA, Fairley JA, Graham EL, Johnston IM, White RJ, Scott PH. The mitogen-activated protein (MAP) kinase ERK induces tRNA synthesis by phosphorylating TFIIIB. EMBO J. 2003;15;22(10):2422–2432. doi: 10.1093/emboj/cdg240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodfellow SJ, White RJ. Regulation of RNA polymerase III transcription during mammalian cell growth. Cell Cycle. 2007;1;6(19):2323–2326. doi: 10.4161/cc.6.19.4767. [DOI] [PubMed] [Google Scholar]
- 20.Barski A, Chepelev I, Liko D, Cuddapah S, Fleming AB, Birch J, Cui K, White RJ, Zhao K. Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes. Nat Struct Mol Biol. 2010;17(5):629–634. doi: 10.1038/nsmb.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Y, Zhao X, Hsieh J, Wichterle H, Impey S, Banerjee S, Neveu P, Kosik KS. MicroRNA regulation of neural stem cells and neurogenesis. J Neurosci. 2010;30(45):14931–14936. doi: 10.1523/JNEUROSCI.4280-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi T, Lu J, Cobb BS, Rodda SJ, McMahon AP, Schipani E, Merkenschlager M, Kronenberg HM. Dicer-dependent pathways regulate chondrocyte proliferation and differentiation. Proc Natl Acad Sci U S A. 2008;105(6):1949–1954. doi: 10.1073/pnas.0707900105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thiffault I, Wolf NI, Forget D, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun. 2015;7;6:7623. doi: 10.1038/ncomms8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terao Y, Saitsu H, Segawa M, Kondo Y, Sakamoto K, Matsumoto N, Tsuji S, Nomura Y. Diffuse central hypomyelination presenting as 4H syndrome caused by compound heterozygous mutations in POLR3A encoding the catalytic subunit of polymerase III. J Neurol Sci. 2012;320(1–2):102–105. doi: 10.1016/j.jns.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 25.Namavar Y, Barth PG, Poll-The BT, Baas F. Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia. Orphanet J Rare Dis. 2011;12;6:50. doi: 10.1186/1750-1172-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hahn S, Roberts S. The zinc ribbon domains of the general transcription factors TFIIB and Brf: conserved functional surfaces but different roles in transcription initiation. Genes Dev. 2000;15;14(6):719–730. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1. Bone age was obtained at age 10.
Supplemental figure 2A–D. Skeletal x-rays were obtained at age 10.
Supplemental figure 3A–B. Both mutations were confirmed by Sanger sequencing.