

Summary
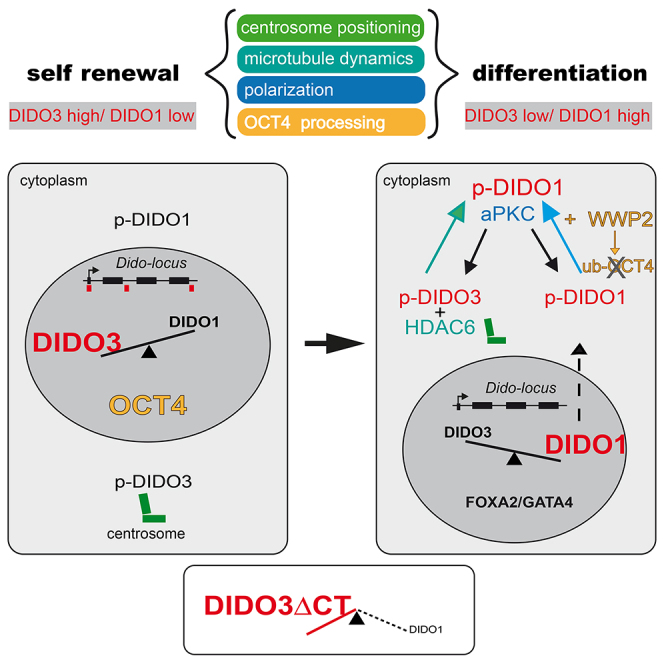
Transition from symmetric to asymmetric cell division requires precise coordination of differential gene expression. We show that embryonic stem cells (ESCs) mainly express DIDO3 and that their differentiation after leukemia inhibitory factor withdrawal requires DIDO1 expression. C-terminal truncation of DIDO3 (Dido3ΔCT) impedes ESC differentiation while retaining self-renewal; small hairpin RNA-Dido1 ESCs have the same phenotype. Dido3ΔCT ESC differentiation is rescued by ectopic expression of DIDO3, which binds the Dido locus via H3K4me3 and RNA POL II and induces DIDO1 expression. DIDO1, which is exported to cytoplasm, associates with, and is N-terminally phosphorylated by PKCiota. It binds the E3 ubiquitin ligase WWP2, which contributes to cell fate by OCT4 degradation, to allow expression of primitive endoderm (PE) markers. PE formation also depends on phosphorylated DIDO3 localization to centrosomes, which ensures their correct positioning for PE cell polarization. We propose that DIDO isoforms act as a switchboard that regulates genetic programs for ESC transition from pluripotency maintenance to promotion of differentiation.
Keywords: stem cell self-renewal, cell differentiation, cell polarization, primitive endoderm formation, transcriptional regulation
Graphical Abstract
Highlights
-
•
DIDO3 regulates DIDO1 expression
-
•
Cytoplasmic DIDO1 promotes cell fate
-
•
DIDO3 regulates centrosome position
-
•
DIDO1 and DIDO3 are phosphorylated by PKCiota
Fütterer et al. show that DIDO isoforms regulate symmetric and asymmetric cell division of ESCs. Whereas DIDO3 is involved in centrosome positioning to polarize PE cells, DIDO1 is determinant for their fate identity. Moreover, DIDO3 regulates DIDO1 expression at the onset of ESC differentiation.
Introduction
Mouse embryonic stem cells (ESCs) from early-stage embryos have indefinite self-renewal capacity and can differentiate into cell types derivative of all three germ layers through asymmetric cell division (Niwa, 2007). Asymmetric cell division is a complex process whereby transcription, cell differentiation, cell cycle, and cell polarity must be coordinated in time and space (Noatynska et al., 2013). In vitro embryoid bodies (EBs) are powerful tools with which to study and understand the molecular mechanisms that underlie this process, as they mimic the in vivo developmental stages of peri-implantation embryos from epiblast to egg cylinder stages. An early step in this process is formation of the primitive endoderm (PE), an outer layer of polarized cells, followed by epiblast and primitive ectoderm development (Niwa, 2010).
Genome-wide screens have identified many potential regulators of stem cell function. These include genes with established roles in transcription regulation, cell growth, and differentiation, as well as genes whose function is largely unknown or remains to be validated in the context of stem cell biology, including the Dido (death inducer obliterator) gene (Kim et al., 2008, Kidder et al., 2008, Chen et al., 2008, Brandenberger et al., 2004).
We identified Dido, a complex gene that encodes three proteins generated by alternative splicing. From smallest to largest, these isoforms are DIDO1, DIDO2, and DIDO3; they have a common N-terminal region and isoform-specific C-terminal parts (Futterer et al., 2005). DIDO3 composition, as determined by bioinformatics analysis, suggests that it helps maintain genomic stability (Rojas et al., 2005). DIDO3 comprises a plant homeodomain (PHD) finger, a transcription elongation factor S-II subunit M (TFSIIM) domain, a Spen paralog and ortholog (SPOC) module, and a long C-terminal region (CT) of unknown homology (Rojas et al., 2005, Futterer et al., 2005). The exact functions of DIDO3 TFSIIM and SPOC modules have not yet been defined. In the TFIIS transcription elongation factor, the TFSIIM domain binds to and facilitates RNA polymerase II activity (Kettenberger et al., 2003); the SPOC module is linked to Spen family proteins, which are involved in transcriptional repression (Sanchez-Pulido et al., 2004). The PHD, one of a large group of zinc-finger proteins, recognizes post-translationally modified histone tails, including methylated lysine residues (Musselman and Kutateladze, 2011).
We determined the crystal structure of the DIDO PHD finger in complex with histone 3 trimethylated on lysine 4 (H3K4me3), which showed an atypical aromatic cage-like binding site bearing a histidine residue. Biochemical, structural, and mutational analyses of this complex identified specificity and affinity determinants for DIDO PHD binding, which is disrupted by threonine 3 phosphorylation and triggers DIDO3 translocation from chromatin (Gatchalian et al., 2013).
DIDO1 localizes in the nucleus and cytosol, whereas DIDO2 is found only in the nucleus (Futterer et al., 2005). DIDO3, located primarily in the nucleus and centrosomes (Trachana et al., 2007), is expressed strongly in ESCs and decreases during development; expression is weaker in somatic cells, but recovers in induced pluripotent stem cells generated from somatic cells (Gatchalian et al., 2013).
All attempts to date to delete DIDO in cell lines or in mice show that deletion is incompatible with life. N-terminal truncation of DIDO (DidoΔNT) provokes aneuploidy, centrosome amplification, and DNA damage in the form of centromere-localized breaks (Guerrero et al., 2010), and leads to chromosomal instability (Martinez-A and van Wely, 2011, Trachana et al., 2007). Mice with the DidoΔNT mutation show hematological myeloid neoplasms, and alterations in DIDO are associated with the myelodysplastic syndrome in humans (Futterer et al., 2005). Characterization of DIDO isoform expression in myeloproliferative neoplasms showed no DIDO2 expression differences in CD34+ hematopoietic stem cells from polycythemia vera, essential thrombocythemia, and primary myelofibrosis patients. In contrast, DIDO2 expression is high in advanced phases of chronic myeloid leukemia (Berzoti-Coelho et al., 2016), suggesting that DIDO2 contributes to this disease.
DIDO3 has a role in delivery of actin-dependent histone deacetylase 6 (HDAC6) to the primary cell cilium (Sanchez de Diego et al., 2014). HDAC6 counteracts the activity of α-tubulin acetyltransferase (ATAT1) (d'Ydewalle et al., 2011), which acetylates TUBULIN and thus stabilizes primary cilium architecture.
In a second mutation (Dido3ΔCT), we replaced the Dido3-specific last exon, which encodes its C terminus and includes the centrosomal targeting domain (Sanchez de Diego et al., 2014). Deletion of this region leads to embryonic lethality; embryos die by gestation day 8.5 (Futterer et al., 2012). ESCs derived from Dido3ΔCT mutant embryos do not differentiate correctly after aggregation and leukemia inhibitory factor (LIF) withdrawal, but retain their capacity for self-renewal, as shown by sustained expression of OCT4 and other markers of undifferentiated stem cells. Differentiation can be recovered in vitro by reconstitution with full-length DIDO3, retinoic acid (RA) treatment, or in the teratoma assay (Futterer et al., 2012). Ectopic expression of the common DIDO NT region in Dido3ΔCT cells downregulates stemness genes in EBs only when the wild-type (WT) PHD is present (Gatchalian et al., 2013). In vivo differentiation can be rescued by crossing Dido3ΔCT with the DidoΔNT mutant. These double mutants overcome embryonic lethality, although the mice suffer high perinatal mortality and neurodevelopmental, morphogenetic, and metabolic alterations (Villares et al., 2015).
The DIDO1 isoform is upregulated when pro-B cells differentiate in vitro after growth factor starvation; its cytoplasmic localization requires phosphorylation and must be dephosphorylated for nuclear translocation (Garcia-Domingo et al., 1999, Garcia-Domingo et al., 2003). DIDO1 is also upregulated during WT ESC differentiation after LIF withdrawal and aggregation to EBs; this upregulation fails in mutant Dido3ΔCT ESCs (Futterer et al., 2012, Gatchalian et al., 2013).
Here we show an essential role for DIDO3 and DIDO1 isoforms at the onset of ESC differentiation. DIDO3 binds to its own promoter, associates with RNA polymerase II (RNA POL II), and regulates Dido1 transcriptional activation; after binding to the H3K4me3 domain, DIDO1 helps to downregulate stemness genes and upregulate genes associated with ESC differentiation. Atypical protein kinase C (aPKC) phosphorylates the NT domain; in DIDO1, this causes its translocation to the outer layer of PE cells, where it associates with WWP2, an E3 ubiquitin-protein ligase, to promote degradation of OCT4, the master regulator of stemness. In DIDO3, this phosphorylation triggers translocation to the centrosome and promotes correct centrosome positioning. We describe a model by which, in ESC, self-regulating Dido splice variants participate in the early stages of developmentally controlled gene expression patterns that determine cell self-renewal versus differentiation.
Results
Functional Effect of DIDO Domains on Regulation of Stemness Gene Expression
Dido3ΔCT ESCs are unable to differentiate correctly, and can serve as a model to study stem cell self-renewal and differentiation (Futterer et al., 2012). Here we used Agilent microarrays to compare transcriptomes of 10-day EBs (d10EBs) from WT and Dido3ΔCT cells. Gene ontology (GO) analysis showed persistent expression of stemness-related genes and genes involved in transcription regulation, and lack of genes associated with differentiation of all three germ layers (Figures 1 and S1; Tables S1 and S2).
Figure 1.

Functional Effect of DIDO Domain Expression in Day-10 Embryoid Bodies
(A) Group of genes, determined in Agilent microarrays, whose expression recovered to WT levels only after HA-DIDONT region expression in Dido3ΔCT ESCs, and GO analysis terms with example genes.
(B) Group of genes whose expression was fully recovered by HA-DIDONT and partially by the HA-DIDO3CT region, with GO analysis and example genes. All array data are from three biological replicates.
To test the influence of distinct DIDO regions (Figure S2A), we expressed the common N-terminal part of DIDO (HA-DIDONT) or the missing specific C-terminal part of DIDO3 (HA-DIDO3CT) in Dido3ΔCT ESC, and compared the d10EB gene expression profile with that of WT EB. Ectopic HA-DIDONT expression partially restored gene expression to the WT EB pattern. As predicted from previous results, HA-DIDONT EBs recovered downregulation of stemness genes (Gatchalian et al., 2013) and expression of a subset of other genes; GO analysis indicated that these genes are involved in endoderm and mesoderm differentiation (Figures 1A and 1B). Genes associated with ectoderm differentiation were not rescued (Figure S1; Tables S1 and S2). In contrast, stemness-related genes were not downregulated by ectopic HA-DIDO3CT expression, and endoderm- and mesoderm-related gene expression were only partially restored (Figures 1A and 1B). Although the WT DIDO PHD domain is necessary for the restoration effect by the NT region (Gatchalian et al., 2013), it is not sufficient, as the NT must localize to the nucleus. In a mutant lacking the nuclear localization domain (HA-DIDONTΔNLS), the protein remained in cytoplasm (Figure S2B) and d10EBs showed no decrease in stemness-related genes (Figure S2C). These findings show the distinct effect of DIDONT and DIDO3CT domains on ESC differentiation, which led us to analyze the implication of the DIDO1 and DIDO3 isoforms in differentiation.
DIDO1 and DIDO3 Expression in Early ESC Differentiation Stages
In floating culture conditions, ESC aggregation to form EBs accompanied by LIF withdrawal leads EBs to mimic the early steps of in vivo cell differentiation (Desbaillets et al., 2000, Doetschman et al., 1985). By day 3–4, the outer cell layer of WT EB forms the PE, characterized by expression of endoderm markers such as GATA4 and by downregulation of OCT4, a major component of the ESC circuit responsible for self-renewal (Hamazaki et al., 2004, Niwa, 2010) (Figure 2A). Affymetrix microarray analysis showed differential gene expression between WT and Dido3ΔCT ESC. Dido1 expression was 2.8-fold lower in Dido3ΔCT compared with WT ESCs (see Table S3 for full microarray data). At variance with WT EBs, Dido3ΔCT EBs did not correctly develop the PE cell layer (Figure 2A; Futterer et al., 2012) or induce increased Dido1 expression (Gatchalian et al., 2013). We monitored DIDO1 and DIDO3 levels in WT d4EBs using antibodies to peptides of the specific DIDO1 or DIDO3 CT domains (PAB-DIDO1 and PAB-DIDO3) (Figures S3A and S3B). Whereas DIDO3 was expressed mainly in the nucleus, DIDO1 was found at the apical site of the PE cells, with weak nuclear expression (Figure 2B). Staining with the monoclonal MAB-1C6 antibody (generated against amino acids 2–90 of the common NT region) (Figure S2A) also showed DIDO nuclear staining, with faint expression at the apical membrane and limited co-localization with PAB-DIDO1 (see below).
Figure 2.

DIDO1 and DIDO3 Expression in Early ESC Differentiation
(A) Left: immunostaining of WT day-4 embryoid bodies (d4EBs) for OCT4 (green) and GATA4 (red); arrowheads mark outer primitive endoderm (PE) cell layer. Right: lack of PE formation in Dido3ΔCT d4EBs.
(B) Left: localization of DIDO isoforms using anti-DIDO MAB-1C6 (green; mainly in nucleus) and DIDO3-specific PAB-DIDO3 (red). Right: localization of DIDO isoforms using MAB-1C6 (green) and DIDO1-specific PAB-DIDO1 (red; preferentially at the apical membrane of outer cell layer).
(C) Left: developing PE and apical localization of DIDO1 (arrowheads) in a time course of WT d1–d4EBs labeled for OCT4 (green) and DIDO1 (red) expression. Right: lack of PE formation in a time course of Dido3ΔCT d1–d4EBs labeled for OCT4 (green) and DIDO1 (red).
All panels are confocal images with nuclear counterstaining (DAPI, blue). Scale bars, 50 μm.
A time-course analysis of DIDO1 expression during EB differentiation showed that, in contrast to Dido3ΔCT, WT EBs showed prominent apical membrane localization in PE cells starting at day 3 and persisting to day 4 (Figure 2C).
DIDO1 Is Essential for PE Development during EB Differentiation
To clarify the role of DIDO1 in ESC differentiation, we generated various WT ESC clones in which DIDO1 expression was inhibited by RNAi. Specificity control by RT-PCR showed a decrease in Dido1, but not in Dido2 or Dido3 RNA expression compared with small hairpin RNA (shRNA) control-infected cells (Figure S4A). DIDO1 protein reduction was also observed in cells expressing HA-DIDO1, as shown by western blot analysis (Figure S4B). shRNA-Dido1 stable transfectants showed normal proliferation and self-renewal and were competent to form EBs. In contrast to shRNA-control, sh-Dido1 EBs did not generate PE at day 4, as determined by absence of the PE markers GATA4 and FOXA2, OCT4 persistence (Figure 3A), and no apparent signs of differentiation beyond day 7 (Figure S4C). Moreover, RA treatment, which rescues PE formation and differentiation of Dido3ΔCT EBs (Figure 3B), failed to do so in shRNA-Dido1 EBs (Figure 3C); FOXA2 staining was observed only inside the EBs and was independent of PE formation, as for WT EBs (Figure 3C).
Figure 3.

DIDO1 Is Essential for PE Formation
(A) (Left) shRNA-Control d4EBs with staining for PE formation (arrowheads). Top row: OCT4 (green; inside EBs), GATA4 (red; outer PE cells). Center: OCT4 (green; inside EBs) and DIDO1 (red; at apical membrane of outer PE cells). Bottom: DIDO1 (green; apical membrane) and FOXA4 (red; PE outer cell layer). (Right) All rows: lack of PE formation in shRNA-Dido1 d4EBs.
(B) (Left) Reconstitution of PE formation by ectopic HA-DIDO3 expression in Dido3ΔCT d4EBs; OCT4 (green; inside EBs) and DIDO1 (red; at apical membrane of PE cells). (Right) The same PE reconstitution pattern of Dido3ΔCT d4EBs after retinoic acid (RA) treatment.
(C) Comparison of solvent (DMSO) versus RA-treated shRNA-Co d4EBs. (Left) Top: OCT4 (green; reduction inside EBs) and DIDO1 (red; at apical membrane of PE cells). Bottom: DIDO1 (green; at apical membrane of PE cells) and FOXA2 (red; arrowheads indicate PE outer layer in DMSO-treated cells and inside EBs in RA-treated cells). (Right) shRNA-Dido1 d4EBs with lack of PE formation in DMSO- or RA-treated EBs, no DIDO1 labeling at apical membrane, and FOXA4 (red; only inside EBs).
(D) Ectopic HA-DIDO1 expression of reconstituted PE formation in Dido3ΔCT d4EBs. (Left) OCT4 (green; inside EBs) and DIDO1 (red; on apical membrane of PE cells). (Right) Co-localization of anti-HA (green) and DIDO1 (red) at apical membrane of outer PE cells.
Nuclei staining (DAPI, blue). Scale bars for EBs, 50 μm; Scale bar in boxed area of (D), 10 μm.
DIDO1 upregulation and its apical expression in PE outer layer cells with concomitant OCT4 downregulation appeared to be essential during Dido3ΔCT EB differentiation induced by RA treatment or ectopic expression of HA-DIDO3 (Figure 3B). We also generated stable Dido3ΔCT ESC transfectants expressing HA-DIDO1, which proliferated normally in self-renewal conditions and showed no signs of differentiation (not shown). In the absence of LIF, EBs derived from these cells were able to reconstitute PE formation completely. In control staining, anti-HA and anti-DIDO1 antibodies co-localized at the inner PE cell membrane, where DIDO1 is also located in WT EBs (Figure 3D).
Using qRT-PCR low-density arrays of representative genes related to undifferentiated stem cells or to differentiation to the three germ layers, we found that shRNA-Dido1 EBs do not differentiate in vitro (Figure S4D). The gene expression pattern resembled that for Dido3ΔCT EBs (Figure S4E).
These data indicate that EBs require DIDO1 to generate the PE outer cell layer and initiate ESC differentiation. Furthermore, DIDO1 must translocate from the nucleus to cytoplasm for correct PE formation.
DIDO3 Regulates Cell Polarization and DIDO1 Acts as a Cell Lineage Marker during Asymmetric ESC Division
During ESC differentiation, a hallmark of differentiated PE cells is their apico-basolateral polarity (Doughton et al., 2014). To identify the role of DIDO in establishing polarization of the nascent PE layer in EBs, we used various polarization markers such as MEGALIN (Moore et al., 2014), F-ACTIN, aPKC (Bedzhov and Zernicka-Goetz, 2014), and acetylated TUBULIN (Quinones et al., 2011). In WT and shRNA-control EB, we confirmed PE cell polarization as detected by positive MEGALIN, F-ACTIN, and aPKC staining. We detected overlapping DIDO1 co-expression with these markers in the PE cell apical membrane. This was not the case in Dido3ΔCT or shRNA-Dido1 EB, in which neither DIDO1 nor any of the polarization markers was found (Figure 4A). In the case of acetylated TUBULIN, expression was reduced only in Dido3ΔCT EB, whereas shRNA-Dido1 EBs showed no difference compared with shRNA-controls (Figure 4A). These lower acetylation levels in Dido3ΔCT coincide with reported DIDO3 and HDAC6 association (Sanchez de Diego et al., 2014).
Figure 4.

DIDO3 Regulates Polarization and DIDO1 Regulates Fate of PE Cells
(A) Left: comparison of WT and Dido3ΔCT d4EBs for apical polarization markers (MEGALIN, red; phalloidin-labeled F-ACTIN, red; aPKC, green; acetylated TUBULIN; green) and DIDO1 (green) at the PE layer (arrowheads). Right: comparison of shRNA-control and shRNA-Dido1 d4EBs (staining as in left panel). Scale bars, 50 μm.
(B) Left: WT rosettes stained for polarization markers as in (A) and DIDO1 for apical polarization at the central lumen (arrowheads). Right: examples of Dido3ΔCT rosettes with mislocalization of polarization markers; bottom row of rosettes stained with anti-αβ-TUBULIN (green). Nuclei (DAPI, blue). Scale bars, 20 μm.
(C) Quantification of the percentage of polarized rosettes of WT, Dido3ΔCT, shRNA-Co, shRNA-Dido1, HA-DIDO1-reconstituted Dido3ΔCT, and HA-DIDO3-reconstituted Dido3ΔCT. Data are shown as mean ± SD (n from more than three independent experiments).
(D) Left: acetylated TUBULIN (green) in Dido3ΔCT rosettes reconstituted with HA-DIDO1 or HA-DIDO3. Right: HA-DIDO3 (green) and DIDO1 (red) localization in HA-DIDO3-reconstituted Dido3ΔCT rosettes. Scale bars, 20 μm.
(E) Position of centrosomes labeled with anti γ-TUBULIN (green) in WT (left) and Dido3ΔCT 2-cell stage rosettes (right). Nuclei (DAPI; blue). Scale bars, 10 μm.
As PE outer layer formation is impaired in Dido3ΔCT and shRNA-Dido1 EBs, we could not determine whether DIDO1 was responsible for PE cell polarization or whether lack of PE cells defined the absence of DIDO1. To address this question, we used a culture model of ESC rosettes established in Matrigel, in which polarization in central lumen of rosettes is independent of PE formation (Bedzhov and Zernicka-Goetz, 2014, Martin-Belmonte and Perez-Moreno, 2012). WT ESCs growing in Matrigel without LIF expressed MEGALIN, F-ACTIN, aPKC, and acetylated TUBULIN in the rosette central lumen. In Dido3ΔCT ESC, rosette polarization was severely impaired, since >60% of rosettes were abnormal, with aberrant staining patterns for all polarization markers including acetylated TUBULIN (Figure 4B). None of the rosettes from WT or Dido3ΔCT ESCs showed DIDO1 associated with polarization, which indicates that DIDO1 is dispensable in cell polarization.
Since TUBULIN acetylation has a role in microtubule dynamics and stabilization during cell polarization (Matsuyama et al., 2002), we studied microtubule and cytoskeleton organization in the rosettes. Based on quantification of acetylated and non-acetylated αβ-TUBULIN, we concluded that lower acetylated TUBULIN levels in Dido3ΔCT compared with WT rosettes and EBs were not the result of low TUBULIN levels (Figure S5A). Rosettes derived from Dido3ΔCT, in the presence of the HDAC6 inhibitors BML-281 (Kozikowski et al., 2008) or trichostatin A (Quinones et al., 2011), showed normal or even hyperacetylated TUBULIN compared with rosettes from WT ESCs (Figure S5B). Ectopic expression of HA-DIDO3, but not HA-DIDO1, completely normalized rosette polarization and TUBULIN acetylation in Dido3ΔCT rosettes, which showed that rescue of rosette polarization required DIDO3 but was less dependent on DIDO1 (Figure 4C). This polarization did not depend on DIDO1 or DIDO3 co-localization at the central lumen of rosettes (Figure 4D).
DIDO3 localizes to centrosomes and centrosomal organization is severely altered in Dido mutants (Futterer et al., 2012, Trachana et al., 2007), especially in the Dido3ΔCT mutant, in which the centrosomal targeting domain is absent (Sanchez de Diego et al., 2014). Since correct centrosome positioning is needed for single and central lumen initiation of rosettes (Rodriguez-Fraticelli et al., 2012, Taniguchi et al., 2015), we analyzed centrosome organization during polarization in early stages of rosette formation. Whereas at the 2-cell stage the large majority of centrosomes is correctly located in WT cells (<6.7% ± 2.1% are incorrect), a substantial percentage of centrosomes was incorrectly located in Dido3ΔCT cells (28.3% ± 2.1%) (Figure 4E). This centrosome mislocation in Dido3ΔCT cells coincides with defective aPKC, αβ-TUBULIN, and F-ACTIN expression as well as of EZRIN, a marker of lumen formation (Taniguchi et al., 2015) (Figure S5C).
These findings clearly show the relevance during cell polarization of DIDO3 compared with DIDO1, probably due to its centrosomal location; centrosome misplacement in the Dido3ΔCT ESCs thus leads to formation of abnormal rosettes.
Translocation of DIDO1 to Cytoplasm and DIDO3 to the Centrosome Requires Association with and Phosphorylation by PKCiota
We found DIDO1 to be necessary for EBs to generate the PE outer cell layer, a process that requires DIDO1 translocation from the nucleus to cytoplasm. We identified DIDO1 at the PE cell apical membrane using the PAB-DIDO1 antibody, which recognizes phosphorylated and unphosphorylated DIDO1 forms. In EBs, the DIDONT-specific MAB-1C6 recognized DIDO1 and/or DIDO3 mainly in the nucleus, with faint expression at the PE inner membrane (Figure 2B). Epitope mapping and characterization of MAB-1C6 binding showed preferential reactivity with an unphosphorylated DIDO peptide (Figures S6A and S6B). We conclude that DIDO1 must be phosphorylated to localize at the PE apical membrane, which coincides with the finding that cytoplasmic DIDO1 is phosphorylated (Garcia-Domingo et al., 2003).
Tests for several markers on the polarized PE apical membrane showed localization of DIDO1 and aPKC (PKCiota and PKCzeta) (Figure 4A), both of which participate in cell polarization (Rosse et al., 2010). We analyzed the ability of both aPKCs to phosphorylate DIDO1 in an in vitro kinase assay with recombinant aPKC kinases that allow distinction between PKCiota and PKCzeta; as a substrate, we employed the same peptide used to characterize MAB-1C6 (Figure S6B). Mass spectrometry analysis (Navajas et al., 2011) showed that PKCiota phosphorylated the peptide at serine 8 (Figure 5A), as did PKCzeta to a lesser degree.
Figure 5.

DIDO Associates with and Is Phosphorylated by PKCiota
(A) PKCiota phosphorylation of DIDO peptide MDDKGHLSNEEAPK on residue Ser8. Top: electron transfer dissociation (ETD) tandem mass spectrometry (MS/MS) spectrum of the m/z 551.1 ion corresponding to the phosphorylated peptide; pS indicates a phosphorylated serine residue. Bottom: ETD MS/MS spectrum of the m/z 524.5 ion corresponding to the non-phosphorylated peptide. Peptide sequences are shown with the identified c-/z-type ions.
(B) Left: anti-HA immunoprecipitates of a negative control, WT HA-DIDONT, HA-DIDONTSer8Ala, shortened HA-DIDONT, and anti-aPKC as positive control were used as substrates in an in vitro kinase assay with recombinant PKCiota and zeta; autoradiograph shows [32P]ATP incorporation (arrowheads). Right: western blot controls of lysates and precipitates of corresponding proteins.
(C) Labeling of centrosomes (arrowheads) in WT 2-cell stage rosettes with γ-TUBULIN (red) and DIDO MAB-1C6 (green; nuclear only).
(D) Labeling of centrosomes (arrowheads) in WT 2-cell stage rosettes with PAB-DIDO3 (green) and γ-TUBULIN (red).
Scale bars: (C and D) 10 μm.
We tested whether recombinant PKCiota and/or PKCzeta phosphorylate WT HA-DIDONT, an HA-DIDONTSer8Ala mutant, or a shortened form of HA-DIDONT (cleaved at BglII) in an in vitro kinase assay with [γ-32P]ATP. PKCiota and PKCzeta both phosphorylated WT HA-DIDONT and HA-DIDONTSer8Ala; again, PKCiota phosphorylation was higher (Figure 5B). Only PKCiota phosphorylated the shortened HA-DIDONT(BglII), whereas PKCzeta did not (Figure 5B). The results show that PKCiota phosphorylates the DIDONT and that serine 8 is not the only phosphorylation site. A possible functional correlation of PKCiota and the DIDO protein is supported by the similarity between Dido3ΔCT mutant and PKCiota knockout mouse phenotypes, since both mutations result in early embryonic lethality (Soloff et al., 2004), and ESCs derived from either mutant embryo do not differentiate correctly in vitro.
Like DIDO3, aPKC localizes to centrosomes (Atwood et al., 2013, Rosse et al., 2010), and DIDO3 is involved in organizing microtubules and polarization; we thus tested whether the DIDO3 NT region is also phosphorylated. Confocal analysis of 2-cell stage rosettes using PAB-DIDO3 and anti-γ-TUBULIN antibodies detected co-localization in the centrosome (Figure 5C). In contrast, MAB-1C6 and anti-γ-TUBULIN antibodies did not co-localize (Figure 5D), which suggests that centrosomal DIDO3 is also phosphorylated.
We concluded that phosphorylation of the DIDO1 and DIDO3 NT domains is a shared mechanism for DIDO translocation from the nucleus to cytoplasm (DIDO1) or to the centrosome (DIDO3), and suggest that the kinase PKCiota is involved in this process.
DIDO1 Interacts with WWP2 and Regulates OCT4 Degradation in PE Cells
When cultured in Matrigel, blastocyst inner mass cells generate rosettes and a polarized outer endoderm layer (Bedzhov et al., 2014). In these conditions, ESC-derived rosettes form this PE layer, albeit at much lower efficiency, as defined by GATA4 and DIDO1 expression and OCT4 downregulation (Figure S7A). As described above for EBs, we found no PE cell that co-expressed DIDO1 at the inner membrane and OCT4. Whereas OCT4 protein is not detected in these cells, they express Oct4 RNA (Hamazaki et al., 2004), and OCT4 can be ubiquitinated by WWP2 (Liao and Jin, 2010, Xu et al., 2009). We thus tested for DIDO1 and WWP2 expression in PE from WT EBs, and found that they co-localized in PE cells (Figure 6A).
Figure 6.

DIDO1 Interacts with WWP2 in PE Formation
(A) Left: WT d4EBs stain positive with anti-WWP2 (red) at apical membrane of OCT4 (green)-negative PE cells. Right: overlapping staining of DIDO1 (cyan) with WWP2 (red) on PE cells of WT d4EBs. Scale bar, 50 μm.
(B) WWP2 co-immunoprecipitated with HA-DIDO1 or HA-DIDONT, but not with HA-DIDO3CT.
(C) Co-staining of d4EBs with OCT4 (green) and PE markers FOXA2 (red) and DIDO1 (cyan) in WT d4EBs infected with shRNA-Co (top row) or two clones of shRNA-Wwp2 A and B (center and bottom rows, respectively). Clone A completely lacked PE formation and clone B showed aberrant co-expression of OCT4 and FOXA2 in PE cells (zoomed images 1 and 2). Nuclei (DAPI, blue). Scale bars for EBs, 50 μm.
A western blot time-course analysis to monitor WWP2 expression during WT ESC differentiation showed the highest WWP2 expression in ESCs, which decreased during differentiation (Liao and Jin, 2010) (Figure S7B). WWP2 expression was lower in Dido3ΔCT than in WT ESCs, and its modification and downregulation were delayed during EB differentiation (Figure S7B). Using WWP2-specific antibody, we performed co-immunoprecipitation analyses in cells expressing HA-DIDO1, the HA-DIDONT domain, or HA-DIDO3CT. These studies confirmed interaction between HA-DIDO1 or HA-DIDONT and WWP2, whereas HA-DIDO3CT did not interact with WWP2 (Figure 6B). The results coincide with data in the BioGRID protein database that predict the DIDO and WWP2 interaction for Homo sapiens. Here we identify the DIDO NT domain as responsible for this interaction.
To define the cause of this WWP2/DIDO1 association, we used shRNA-Wwp2 to inhibit WWP2 expression. Two representative clones were characterized as expressing different WWP2 levels (Figure S7C). Whereas shRNA-control EBs showed the predicted PE cell formation pattern (expressing FOXA2 and DIDO1 but not OCT4), shRNA-Wwp2 clone A (trace WWP2 expression) did not develop PE (Figure 6C); this phenotype resembled shRNA-Dido1 ESC or Dido3ΔCT ESC differentiation to EBs. In shRNA-Wwp2 clone B, we observed PE formation in ∼50% of EBs, with abnormal OCT4, DIDO1, and FOXA2 co-expression in these cells (Figure 6C). The results suggest that both DIDO1 and WWP2 are needed for correct PE formation, and that they associate and thus contribute to OCT4 ubiquitination and degradation.
DIDO3 Recruitment to Its Promoter and 3′ UTR Regions Regulates DIDO1 Expression
Since ectopic DIDO3 expression rescues the ability of these EBs to differentiate by upregulating DIDO1, we analyzed the mechanism underlying this upregulation. From HA-DIDO3-expressing Dido3ΔCT ESC, we prepared chromatin immunoprecipitation sequencing (ChIP-seq) immunoprecipitates with anti-HA antibodies and sequenced bound DNA fragments. Bioinformatics analysis showed that HA-DIDO3 binds to its own genomic locus at least on three regions (Figure 7A and Table S4) including the Dido proximal promoter, a site after the Dido1 3′ UTR region, and the Dido3 3′ UTR. The chromatin status of the Dido locus in mouse stem cells (data from Mouse ENCODE project; ENCODE Project Consortium, 2012) confirmed Dido as an actively transcribed gene in ESCs, as indicated by H3K4me3 and H3K9ac at the distal and proximal Dido promoters. The active elongation histone mark H3K36me3 was positive, whereas the heterochromatic silencing markers H3K27me3 and H3K9me3 were absent (Figure 7A).
Figure 7.

DIDO3 Binds to the Dido Gene and Interacts with RNA POL II
(A) Image of the 55-kb genomic region containing the three alternatively spliced isoforms of the Dido gene. HA-DIDO3 ChIP-seq binding sites (red boxes); chromatin status is reflected by individual tracks of histone modifications H3K4me3, H3K9ac, H3K27ac, H3K4me1, H3K36me3, H3K27me3, and H3K9me3 (ChIP-seq data from mouse ENCODE project); individual tracks from ChIP-seq data for RNA POL II unphosphorylated, Ser5, Ser2, or Ser7 phosphorylated (Brookes et al., 2012).
(B) Top: DIDO3, HA-DIDO3, and DIDO3ΔCT expression in lysates of WT, mutant ESCs reconstituted with HA-DIDO3, and mutant ESCs (left) and in anti-RNA POL II co-immunoprecipitates (right), developed with anti-DIDO (MAB-1C6). Bottom: expression of total RNA POL II in the same lysates (left) and in anti-DIDO co-immunoprecipitates (right), developed with anti-RNA POL II.
(C) qPCR data demonstrating x-fold ChIP enrichment for anti-HA-DIDO3 and -RNA POL II on Dido proximal promoter (right) and Dido internal sequence after Dido1 3′ UTR (left) relative to negative control (Co; immunoglobulin G), normalized to input amounts. Data are shown as mean ± SD (n from at least three independent experiments).
Although no DNA binding motif has so far been identified for DIDO, the PHD of the common NT region binds to H3K4me3 (Gatchalian et al., 2013, Prieto et al., 2009). The peak detected in the proximal promoter is due to H3K4me3 binding, the only known binding site. There is no H3K4me3 at the second binding site (following the Dido1 3′ UTR), but rather H3K27ac, which is also present on both Dido promoters. This histone acetylation mark was described on poised enhancers that contribute to differentiation programs (Creyghton et al., 2010). H3K27ac was not detected as a DIDO binding site, but DIDO association to HDAC might participate in the H3K27 acetylation status.
As the DIDO TSFIIM domain is linked to RNA POL II (Rojas et al., 2005) and the yeast DIDO3 homolog BYE1 binds RNA POL II (Kinkelin et al., 2013, Pinskaya et al., 2014), we used co-immunoprecipitation experiments to test DIDO3 and DIDO3ΔCT association with RNA POL II. Both DIDO3 and DIDO3ΔCT associated with RNA POL II, which confirmed the link between DIDO and transcription (Figure 7B). When we included localization of RNA POL II, unphosphorylated or with various phosphorylation modifications (data from Brookes et al., 2012) in our representation of the Dido locus, we found RNA POL II peaks at DIDO3 ChIP-seq binding sites (Figure 7A).
To confirm DIDO3 and RNA POL II binding to the Dido locus, we performed further ChIP experiments followed by qPCR specific for the Dido proximal promoter and Dido internal binding site DNA, identified by ChIP-seq analysis. HA-DIDO3 (5.2-fold) and RNA POL II (6.5-fold) were enriched on the proximal promoter, as were HA-DIDO3 (3.8-fold) and RNA POL II (4.1-fold) on the internal Dido site (Figure 7C), which corroborates DIDO3 involvement in expression of Dido splice variants.
Discussion
Advances in the identification and generation of stem cells from numerous organisms have produced vast knowledge in many fields including embryology and development. ESCs are capable of prolonged self-renewal in an undifferentiated state, but can also differentiate into several cell types. The molecular mechanisms that underlie these alternative processes remain to be identified, and appear critical for stem cell potential in regenerative/reparative medicine. Here we combined characterization of a stemness-associated locus (Dido) with the EBs’ ability to mimic in vitro the early steps of in vivo ESC differentiation (Desbaillets et al., 2000, Doetschman et al., 1985).
ESC differentiation and formation of the PE cell layer require downregulation of stemness genes. In suspension culture, at 2–4 days after LIF deprivation, the polarized outer cell layer of EBs (the first cells to differentiate) forms the primitive endoderm, characterized by lack of stemness genes and expression of genes associated to the new PE cell fate. Dido is a gene complex that encodes three splice variants, Dido1 (the smallest), Dido2, and Dido3 (the largest) (Figure S3). We show that the DIDO1 isoform is a central player in regulating the EB switch from expansion to a differentiation/maintenance phase. During self-renewal, DIDO3 maintains stemness gene expression by binding to H3K4me3 and RNA POL II, and DIDO1 expression is repressed. Less is known of the role of DIDO2, the least-expressed isoform, which has only a few more amino acids than the DIDO3ΔCT mutant. We do not detect DIDO2 protein expression at differentiation onset, which is associated with changes in DIDO3/DIDO1.
Cell differentiation requires DIDO1 in cytoplasm and in the nucleus, where it probably competes with DIDO3 for binding to H3K4me3, promoting downregulation of stemness genes and DIDO3 degradation. Through its nuclear export signal, DIDO1 is exported to cytoplasm, which depends on its association to and phosphorylation by PKCiota (Figure 5). Once in the cytoplasm, DIDO1 associates to the E3 ubiquitin-protein ligase (WWP2), which leads to OCT4 ubiquitination and degradation (Figure 6). PKCiota also associates to and phosphorylates DIDO3 to form a complex that regulates correct centrosome positioning in daughter cells. DIDO3 interaction with HDAC6 (Sanchez de Diego et al., 2014) regulates TUBULIN acetylation, critical for its stabilization and dynamics (Figure 4). Both centrosome positioning and microtubule dynamics participate in the polarity pathway that controls stem cell self-renewal versus differentiation.
A model emerges in which DIDO acts as a switchboard that regulates a genetic program that drives stem cells between the transcription “modes” that operate to maintain pluripotency or promote differentiation. In these modes, control of the expression and cellular localization of different DIDO splice forms appears to be important. Whereas DIDO1 expression and phosphorylation commit cells to the first step in ESC differentiation, DIDO3 phosphorylation promotes its translocation to the centrosome, whose position it controls for the renewal or differentiation capacities of daughter cells.
Alternative mRNA isoforms are an important means of diversifying protein behavior, and have contributed to the complexity of vertebrate organisms (Roy et al., 2013). Here we show that two Dido isoforms generated by differential splicing are critical to determination of cell lineage early in ESC differentiation. Self-renewal and differentiation events apparently have distinct genetic requirements in the transcription modes for pluripotent ESCs and for differentiating cells, which must integrate all signals needed for each stage.
DIDO1 and DIDO3 have several mechanisms that might take part in the pluripotent and somatic cell transcription modes and in development. The composition of DIDO isoform domains might aid in establishing these modes. DIDO1 has the PHD, whereas DIDO3 encodes a much larger protein with three signature motifs, PHD, TFS2M, and SPOC, all typically found in proteins involved in transcription regulation. We found that DIDO3 binds RNA POL II and thus links DIDO to transcriptional control, as shown for the yeast DIDO homolog BYE1 (Kinkelin et al., 2013, Pinskaya et al., 2014). We show a direct DIDO3 role in regulating its own expression and that of DIDO1, as the Dido3ΔCT loss-of-function mutation confers a cell phenotype similar to that seen when DIDO1 is misregulated. ChIP mapping of DIDO3 recruitment to the Dido locus also indicated a binding pattern associated with a region that includes, but is not limited to, the Dido promoter (Figure 7). Results from chromatin binding indicated that DIDO3 binding to its own genomic DNA modulates Dido1 expression and might participate in regulating core stemness genes, a question currently under study. These findings coincide with the role of d(PPS), the DIDO homolog in Drosophila. d(PPS) protein also has four signature motifs typically found in proteins that act in transcriptional regulation; d(PPS) is needed to regulate Sxl (sex-lethal) splicing (Johnson et al., 2010) and DIDO3 is necessary for DIDO1 expression. Given the known d(PPS) splicing function, our data and the parallels with d(PPS) results suggest a role for DIDO in transcription control by splicing.
Experimental Procedures
Cell Culture and Transfection
All experiments using animal materials were designed in compliance with European Union legislation and approved by the Committee for Ethics in Animal Experimentation of the Centro Nacional de Biotecnología (CNB/CSIC) (Proex 322/15).
Mouse ESCs were maintained in KO-DMEM (Gibco, Invitrogen) with 20% fetal calf serum, 1× Glutamax (Invitrogen), non-essential amino acids, 0.05 mM 2-mercaptoethanol, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 1,000 U/mL mouse LIF supplement (Millipore) on a layer of mitomycin C-treated mouse embryonic fibroblasts or gelatinized tissue culture plates.
EBs were formed and cultured in suspension conditions in low-attachment dishes in the same medium without LIF. Rosettes were formed in the same medium without LIF and 5% Matrigel (Becton Dickinson) on 8-well chamber slides (iBidi) coated with 25 μL of Matrigel per well for 3 days, or 1 day for 2-cell stages.
Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocols. For overexpression experiments, Dido1, Dido3, or Dido fragments were cloned in mammalian expression vector pCAGG-HA-tagged Dido-IRES-puromycin resistance cassette.
For shRNA Dido1 experiments, we used Dido1-specific and negative control GIPZ lentiviral particles (Thermo Scientific). For Wwp2 shRNA, we used retroviral pGFP-V-RS vector with control or four different 29mer shRNAs to murine Wwp2 (Origene). Stable transfectants were obtained in all cases by puromycin selection (1 μg/mL). For target sequences, see Supplemental Experimental Procedures.
Immunofluorescence
ESC, EB, or rosettes were fixed in 4% formaldehyde, permeabilized with 0.2% NP-40, blocked with 1% BSA, and stained with primary (overnight, 4°C) and secondary antibodies (2 hr, room temperature) and DAPI (30 min, room temperature). For the list of antibodies, see Supplemental Experimental Procedures.
A Zeiss laser scanning confocal microscope was used for confocal microscopy; images were processed with ImageJ software (NIH).
Immunoprecipitation
For co-immunoprecipitation, cells were lysed in NETN buffer (20 mM HEPES [pH 7.5], 300 mM NaCl, 0.5 mM EDTA, 10% glycerol, 0.5% NP-40) with protease inhibitor cocktail (Roche) and 1 mM PMSF; cells for immunoprecipitation were lysed in RIPA buffer with the same inhibitors. Lysates were immunoprecipitated with specific antibody (overnight, 4°C), followed by addition of magnetic beads (Dynabeads, Invitrogen) coupled to Protein A or Protein G1, depending on the species and isotype of the first capture antibody. Immunoprecipitates were washed six times and released from beads in sample buffer (10 min, 70°C).
Western Blot
Samples were separated by 4%–10% SDS-PAGE depending on protein size, then transferred to nitrocellulose membrane (Bio-Rad), followed by western blot analysis with indicated antibodies. ECL solution (PerkinElmer) was used to visualize proteins.
Microarray and qRT-PCR
Total RNA was prepared with TRIzol (Invitrogen) and used for qRT-PCR in low-density arrays (TaqMan mouse stem cell, Applied Biosystems) and microarrays. Samples were labeled and hybridized on the mouse genome 430 2.0 array (Affymetrix) or Mouse GE 4x44K v2 microarrays (Agilent); three biological replicates were used for all conditions.
For detailed methods, see Supplemental Experimental Procedures.
Kinase Assay
Purified immunoprecipitates (magnetic beads with bound anti-HA + HA-DIDO1 or anti-HA + HA-DIDONT fragments) were washed once in kinase buffer (50 mM Tris [pH 7.5], 20 mM MgCl2, 10 mM MnCl2). Labeling reactions were performed with 10 μL of precipitated mixture +10 μL of recombinant PKC +5 μL of ATP-mix (cold ATP +10 μCi [γ-32P]ATP) (30 min, 30°C), washed twice in kinase buffer, released in sample buffer (10 min, 70°C) and resolved on a gel. The gel was fixed in methanol/acetic acid, dried, and exposed to X-ray film. Enzymes used were recombinant human PKCiota (14-505, Merck Millipore; 0.5 μg/reaction) and recombinant human PKCzeta (14-525, Merck Millipore; 0.2 μg/reaction).
Phosphopeptide Analysis
Phosphopeptides were analyzed by liquid chromatography-tandem mass spectrometry ion trap, alternating collision-induced dissociation, and electron transfer dissociation fragmentation techniques. For details, see Supplemental Experimental Procedures.
Chromatin Immunoprecipitation and qPCR
For ChIP, we used the MAGnify Chromatin Immunoprecipitation System (Invitrogen) according to the manufacturer’s protocol, with overnight immunoprecipitate incubation at 4°C. Inputs and eluates were used for qPCR with SYBR Green in an ABI PRISM 7900HT PCR (Applied Biosystems); x-fold probe enrichment was calculated relative to immunoglobulin G control precipitates, normalized to inputs. For primers, see Supplemental Experimental Procedures.
Chromatin Immunoprecipitation Sequencing
ChIP-seq was performed mainly as described by O'Geen et al. (2010). In brief, HA-DIDO3-expressing Dido3ΔCT ESCs were 1% formaldehyde-fixed and lysed in lysis buffer with proteinase inhibitor cocktail (Roche) and PMSF. Chromatin was sonicated in a Diagenode Bioruptor; fragments of ∼200–500 base pairs (verified by gel) were diluted with RIPA buffer and precipitated with anti-HA polyclonal antibody (ChIP-grade, ab9110; Abcam) coupled to magnetic beads coated with anti-rabbit immunoglobulin (Invitrogen; overnight, 4°C). Beads were washed extensively, and the precipitates were eluted and reverse-crosslinked; the DNA was purified on Qiagen columns. Further processing and massive sequencing was performed by the Genomics Service Unit, Parque Científico de Madrid (see Supplemental Experimental Procedures).
ChIP-Seq Analysis
Mapped reads were aligned with the Bowtie program on the mouse genome (NCBI Build 37/UCSC mm9) and significant peaks identified using MACS software (v. 1.4) with default thresholds. For detailed procedures, see Supplemental Experimental Procedures.
Statistical Analysis
Data are shown as mean ± SD. Statistical analyses were performed using Student’s t test.
Author Contributions
A.F. designed, performed, and analyzed most of the experiments and co-wrote the paper. J.d.C. analyzed the transciptome and ChIP-seq data and co-wrote the paper. R.N., L.A., and J.G. performed and analyzed some experiments. A.T.-G. and C.P.-B. provided reagents. I.B. and F.M.-B. provided rosette technology, expertise, and feedback. C.M.-A. conceptualized experiments, analyzed data, co-wrote the paper, and secured funding.
Acknowledgments
We thank the CNB Proteomics facility for peptide synthesis, the CNB Genomic Service Unit for performing microarray experiments, and C. Mark for editorial assistance. This work was supported by grants from the Spanish Ministerio de Economía y Competitividad (MINECO-FEDER SAF2013-42289-R and SAF2016-75456-R) and the Alfonso Martín Escudero Foundation.
Published: March 16, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.02.013.
Accession Numbers
The accession number for all genomic data (Affymetrix and Agilent microarrays, ChIP-seq) reported in this paper is GEO: GSE85029.
Supplemental Information
References
- Atwood S.X., Li M., Lee A., Tang J.Y., Oro A.E. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature. 2013;494:484–488. doi: 10.1038/nature11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedzhov I., Zernicka-Goetz M. Self-organizing properties of mouse pluripotent cells initiate morphogenesis upon implantation. Cell. 2014;156:1032–1044. doi: 10.1016/j.cell.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedzhov I., Leung C.Y., Bialecka M., Zernicka-Goetz M. In vitro culture of mouse blastocysts beyond the implantation stages. Nat. Protoc. 2014;9:2732–2739. doi: 10.1038/nprot.2014.186. [DOI] [PubMed] [Google Scholar]
- Berzoti-Coelho M.G., Ferreira A.F., de Souza Nunes N., Pinto M.T., Junior M.C., Simoes B.P., Martinez A.C., Souto E.X., Panepucci R.A., Covas D.T. The expression of Death Inducer-Obliterator (DIDO) variants in myeloproliferative neoplasms. Blood Cells Mol. Dis. 2016;59:25–30. doi: 10.1016/j.bcmd.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Brandenberger R., Wei H., Zhang S., Lei S., Murage J., Fisk G.J., Li Y., Xu C., Fang R., Guegler K. Transcriptome characterization elucidates signaling networks that control human ES cell growth and differentiation. Nat. Biotechnol. 2004;22:707–716. doi: 10.1038/nbt971. [DOI] [PubMed] [Google Scholar]
- Brookes E., de Santiago I., Hebenstreit D., Morris K.J., Carroll T., Xie S.Q., Stock J.K., Heidemann M., Eick D., Nozaki N. Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs. Cell Stem Cell. 2012;10:157–170. doi: 10.1016/j.stem.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V.B., Wong E., Orlov Y.L., Zhang W., Jiang J. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Ydewalle C., Krishnan J., Chiheb D.M., Van Damme P., Irobi J., Kozikowski A.P., Vanden Berghe P., Timmerman V., Robberecht W., Van Den Bosch L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011;17:968–974. doi: 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- Desbaillets I., Ziegler U., Groscurth P., Gassmann M. Embryoid bodies: an in vitro model of mouse embryogenesis. Exp. Physiol. 2000;85:645–651. [PubMed] [Google Scholar]
- Doetschman T.C., Eistetter H., Katz M., Schmidt W., Kemler R. The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J. Embryol. Exp. Morphol. 1985;87:27–45. [PubMed] [Google Scholar]
- Doughton G., Wei J., Tapon N., Welham M.J., Chalmers A.D. Formation of a polarised primitive endoderm layer in embryoid bodies requires fgfr/erk signalling. PLoS One. 2014;9:e95434. doi: 10.1371/journal.pone.0095434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futterer A., Campanero M.R., Leonardo E., Criado L.M., Flores J.M., Hernandez J.M., San Miguel J.F., Martinez A.C. Dido gene expression alterations are implicated in the induction of hematological myeloid neoplasms. J. Clin. Invest. 2005;115:2351–2362. doi: 10.1172/JCI24177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futterer A., Raya A., Llorente M., Izpisua-Belmonte J.C., de la Pompa J.L., Klatt P., Martinez A.C. Ablation of Dido3 compromises lineage commitment of stem cells in vitro and during early embryonic development. Cell Death Differ. 2012;19:132–143. doi: 10.1038/cdd.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Domingo D., Leonardo E., Grandien A., Martinez P., Albar J.P., Izpisua-Belmonte J.C., Martinez A.C. DIO-1 is a gene involved in onset of apoptosis in vitro, whose misexpression disrupts limb development. Proc. Natl. Acad. Sci. USA. 1999;96:7992–7997. doi: 10.1073/pnas.96.14.7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Domingo D., Ramirez D., Gonzalez de Buitrago G., Martinez A.C. Death inducer-obliterator 1 triggers apoptosis after nuclear translocation and caspase upregulation. Mol. Cell Biol. 2003;23:3216–3225. doi: 10.1128/MCB.23.9.3216-3225.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchalian J., Futterer A., Rothbart S.B., Tong Q., Rincon-Arano H., Sanchez de Diego A., Groudine M., Strahl B.D., Martinez A.C., van Wely K.H. Dido3 PHD modulates cell differentiation and division. Cell Rep. 2013;4:148–158. doi: 10.1016/j.celrep.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero A.A., Gamero M.C., Trachana V., Futterer A., Pacios-Bras C., Diaz-Concha N.P., Cigudosa J.C., Martinez A.C., van Wely K.H. Centromere-localized breaks indicate the generation of DNA damage by the mitotic spindle. Proc. Natl. Acad. Sci. USA. 2010;107:4159–4164. doi: 10.1073/pnas.0912143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki T., Oka M., Yamanaka S., Terada N. Aggregation of embryonic stem cells induces Nanog repression and primitive endoderm differentiation. J. Cell Sci. 2004;117:5681–5686. doi: 10.1242/jcs.01489. [DOI] [PubMed] [Google Scholar]
- Johnson M.L., Nagengast A.A., Salz H.K. PPS, a large multidomain protein, functions with sex-lethal to regulate alternative splicing in Drosophila. PLoS Genet. 2010;6:e1000872. doi: 10.1371/journal.pgen.1000872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenberger H., Armache K.J., Cramer P. Architecture of the RNA polymerase II-TFIIS complex and implications for mRNA cleavage. Cell. 2003;114:347–357. doi: 10.1016/s0092-8674(03)00598-1. [DOI] [PubMed] [Google Scholar]
- Kidder B.L., Yang J., Palmer S. Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS One. 2008;3:e3932. doi: 10.1371/journal.pone.0003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Chu J., Shen X., Wang J., Orkin S.H. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkelin K., Wozniak G.G., Rothbart S.B., Lidschreiber M., Strahl B.D., Cramer P. Structures of RNA polymerase II complexes with Bye1, a chromatin-binding PHF3/DIDO homologue. Proc. Natl. Acad. Sci. USA. 2013;110:15277–15282. doi: 10.1073/pnas.1311010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozikowski A.P., Tapadar S., Luchini D.N., Kim K.H., Billadeau D.D. Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: a new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J. Med. Chem. 2008;51:4370–4373. doi: 10.1021/jm8002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B., Jin Y. Wwp2 mediates Oct4 ubiquitination and its own auto-ubiquitination in a dosage-dependent manner. Cell Res. 2010;20:332–344. doi: 10.1038/cr.2009.136. [DOI] [PubMed] [Google Scholar]
- Martin-Belmonte F., Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer. 2012;12:23–38. doi: 10.1038/nrc3169. [DOI] [PubMed] [Google Scholar]
- Martinez-A C., van Wely K.H. Centromere fission, not telomere erosion, triggers chromosomal instability in human carcinomas. Carcinogenesis. 2011;32:796–803. doi: 10.1093/carcin/bgr069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama A., Shimazu T., Sumida Y., Saito A., Yoshimatsu Y., Seigneurin-Berny D., Osada H., Komatsu Y., Nishino N., Khochbin S. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002;21:6820–6831. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R., Tao W., Meng Y., Smith E.R., Xu X.X. Cell adhesion and sorting in embryoid bodies derived from N- or E-cadherin deficient murine embryonic stem cells. Biol. Open. 2014;3:121–128. doi: 10.1242/bio.20146254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman C.A., Kutateladze T.G. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011;39:9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navajas R., Paradela A., Albar J.P. Immobilized metal affinity chromatography/reversed-phase enrichment of phosphopeptides and analysis by CID/ETD tandem mass spectrometry. Methods Mol. Biol. 2011;681:337–348. doi: 10.1007/978-1-60761-913-0_18. [DOI] [PubMed] [Google Scholar]
- Niwa H. How is pluripotency determined and maintained? Development. 2007;134:635–646. doi: 10.1242/dev.02787. [DOI] [PubMed] [Google Scholar]
- Niwa H. Mouse ES cell culture system as a model of development. Dev. Growth Differ. 2010;52:275–283. doi: 10.1111/j.1440-169X.2009.01166.x. [DOI] [PubMed] [Google Scholar]
- Noatynska A., Tavernier N., Gotta M., Pintard L. Coordinating cell polarity and cell cycle progression: what can we learn from flies and worms? Open Biol. 2013;3:130083. doi: 10.1098/rsob.130083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Geen H., Frietze S., Farnham P.J. Using ChIP-seq technology to identify targets of zinc finger transcription factors. Methods Mol. Biol. 2010;649:437–455. doi: 10.1007/978-1-60761-753-2_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinskaya M., Ghavi-Helm Y., Mariotte-Labarre S., Morillon A., Soutourina J., Werner M. PHD and TFIIS-Like domains of the Bye1 transcription factor determine its multivalent genomic distribution. PLoS One. 2014;9:e102464. doi: 10.1371/journal.pone.0102464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto I., Kouznetsova A., Futterer A., Trachana V., Leonardo E., Alonso Guerrero A., Cano Gamero M., Pacios-Bras C., Leh H., Buckle M. Synaptonemal complex assembly and H3K4Me3 demethylation determine DIDO3 localization in meiosis. Chromosoma. 2009;118:617–632. doi: 10.1007/s00412-009-0223-7. [DOI] [PubMed] [Google Scholar]
- Quinones G.B., Danowski B.A., Devaraj A., Singh V., Ligon L.A. The posttranslational modification of tubulin undergoes a switch from detyrosination to acetylation as epithelial cells become polarized. Mol. Biol. Cell. 2011;22:1045–1057. doi: 10.1091/mbc.E10-06-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Fraticelli A.E., Auzan M., Alonso M.A., Bornens M., Martin-Belmonte F. Cell confinement controls centrosome positioning and lumen initiation during epithelial morphogenesis. J. Cell Biol. 2012;198:1011–1023. doi: 10.1083/jcb.201203075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A.M., Sanchez-Pulido L., Futterer A., van Wely K.H., Martinez A.C., Valencia A. Death inducer obliterator protein 1 in the context of DNA regulation. Sequence analyses of distant homologues point to a novel functional role. FEBS J. 2005;272:3505–3511. doi: 10.1111/j.1742-4658.2005.04759.x. [DOI] [PubMed] [Google Scholar]
- Rosse C., Linch M., Kermorgant S., Cameron A.J., Boeckeler K., Parker P.J. PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 2010;11:103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- Roy B., Haupt L.M., Griffiths L.R. Review: alternative splicing (AS) of genes as an approach for generating protein complexity. Curr. Genomics. 2013;14:182–194. doi: 10.2174/1389202911314030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez de Diego A., Alonso Guerrero A., Martinez-A C., van Wely K.H. Dido3-dependent HDAC6 targeting controls cilium size. Nat. Commun. 2014;5:3500. doi: 10.1038/ncomms4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Pulido L., Rojas A.M., van Wely K.H., Martinez A.C., Valencia A. SPOC: a widely distributed domain associated with cancer, apoptosis and transcription. BMC Bioinformatics. 2004;5:91. doi: 10.1186/1471-2105-5-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloff R.S., Katayama C., Lin M.Y., Feramisco J.R., Hedrick S.M. Targeted deletion of protein kinase C lambda reveals a distribution of functions between the two atypical protein kinase C isoforms. J. Immunol. 2004;173:3250–3260. doi: 10.4049/jimmunol.173.5.3250. [DOI] [PubMed] [Google Scholar]
- Taniguchi K., Shao Y., Townshend R.F., Tsai Y.H., DeLong C.J., Lopez S.A., Gayen S., Freddo A.M., Chue D.J., Thomas D.J. Lumen formation is an intrinsic property of isolated human pluripotent stem cells. Stem Cell Rep. 2015;5:954–962. doi: 10.1016/j.stemcr.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachana V., van Wely K.H., Guerrero A.A., Futterer A., Martinez-A C. Dido disruption leads to centrosome amplification and mitotic checkpoint defects compromising chromosome stability. Proc. Natl. Acad. Sci. USA. 2007;104:2691–2696. doi: 10.1073/pnas.0611132104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villares R., Gutierrez J., Futterer A., Trachana V., Gutierrez del Burgo F., Martinez A.C. Dido mutations trigger perinatal death and generate brain abnormalities and behavioral alterations in surviving adult mice. Proc. Natl. Acad. Sci. USA. 2015;112:4803–4808. doi: 10.1073/pnas.1419300112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Wang W., Li C., Yu H., Yang A., Wang B., Jin Y. WWP2 promotes degradation of transcription factor OCT4 in human embryonic stem cells. Cell Res. 2009;19:561–573. doi: 10.1038/cr.2009.31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.