

Abstract
Theories for the origin of sex traditionally start with an asexual mitosing cell and add recombination, thereby deriving meiosis from mitosis. Though sex was clearly present in the eukaryote common ancestor, the order of events linking the origin of sex and the origin of mitosis is unknown. Here, we present an evolutionary inference for the origin of sex starting with a bacterial ancestor of mitochondria in the cytosol of its archaeal host. We posit that symbiotic association led to the origin of mitochondria and gene transfer to host’s genome, generating a nucleus and a dedicated translational compartment, the eukaryotic cytosol, in which—by virtue of mitochondria—metabolic energy was not limiting. Spontaneous protein aggregation (monomer polymerization) and Adenosine Tri-phosphate (ATP)-dependent macromolecular movement in the cytosol thereby became selectable, giving rise to continuous microtubule-dependent chromosome separation (reduction division). We propose that eukaryotic chromosome division arose in a filamentous, syncytial, multinucleated ancestor, in which nuclei with insufficient chromosome numbers could complement each other through mRNA in the cytosol and generate new chromosome combinations through karyogamy. A syncytial (or coenocytic, a synonym) eukaryote ancestor, or Coeca, would account for the observation that the process of eukaryotic chromosome separation is more conserved than the process of eukaryotic cell division. The first progeny of such a syncytial ancestor were likely equivalent to meiospores, released into the environment by the host’s vesicle secretion machinery. The natural ability of archaea (the host) to fuse and recombine brought forth reciprocal recombination among fusing (syngamy and karyogamy) progeny—sex—in an ancestrally meiotic cell cycle, from which the simpler haploid and diploid mitotic cell cycles arose. The origin of eukaryotes was the origin of vertical lineage inheritance, and sex was required to keep vertically evolving lineages viable by rescuing the incipient eukaryotic lineage from Muller’s ratchet. The origin of mitochondria was, in this view, the decisive incident that precipitated symbiosis-specific cell biological problems, the solutions to which were the salient features that distinguish eukaryotes from prokaryotes: A nuclear membrane, energetically affordable ATP-dependent protein–protein interactions in the cytosol, and a cell cycle involving reduction division and reciprocal recombination (sex).
Keywords: origin, eukaryotes, endomembrane system, meiosis, mitosis, syngamy, karyogamy, coeocytic, reduction division, chromosome segregation, alternation of generations
Sex Is Essential In Eukaryotes
Few problems have baffled evolutionary biologists more thoroughly than the origin of meiotic sex. Eukaryotes do it, prokaryotes do not. The basic machinery of sexual recombination was present in the last common ancestor of extant eukaryotes, because 1) the genes underpinning sexual recombination are homologous across all eukaryotes studied so far (Ramesh et al. 2005) and 2) all eukaryotic clades either undergo sexual recombination or have the machinery in their genomes to do so (Speijer et al. 2015; Bloomfield 2016). Thus, sex was clearly present in the eukaryote common ancestor and was furthermore manifest in a form homologous to its modern-day incarnations. The classical questions have remained the same for decades: How did it arise, from what, what benefits bore its origin, and what benefits maintained its presence? Many papers and books have been written on the origin of sex (Cleveland 1947; Williams 1975; Maynard Smith 1978; Bell 1982; Bernstein et al. 1984; Uyenoyama and Bengtsson 1989; Hurst and Nurse 1991; Otto and Goldstein 1992; Maynard Smith and Szathmary 1997; Cavalier-Smith 2002; Solari 2002; Wilkins and Holliday 2008; Goodenough and Heitman 2014; Cavalier-Smith 2010; Hörandl and Hadacek 2013). Papers still continue to come in on the topic, a good indication that no one has solved the problems to everyone’s satisfaction (Havird et al. 2015; Radzvilavicius and Blackstone 2015; Speijer et al. 2015).
The issue of why eukaryotes as a lineage never lost sex is most readily attributed to genetic load, or the cumulative effects of sublethal mutations in clonally growing organisms, a population genetic process called Muller’s ratchet (Muller 1964; Felsenstein 1974). Without recombination, reproduction is strictly clonal, mutation being inevitable and leading to the steady accumulation of deleterious mutations (Muller 1964). In the absence of recombination, these mutations will ultimately lead to extinction (Muller 1964; Felsenstein 1974; Moran 1996; Crow 2005). Though occasional high ploidy can possibly delay the effects of Muller’s ratchet, it cannot alleviate the effects (Kondrashov 1994); ploidy is not a substitute for sex.
In this article, we will be arguing that recombination is essential for long-term lineage survival of both prokaryotes and eukaryotes. We will also argue that recombination rescues organisms from extinction at the hands of Muller’s ratchet. Because sex is the only means of recombination known in eukaryotes, it seems likely to us that the avoidance of Muller’s ratchet is the reason that eukaryotes have preserved meiotic sex throughout their history, which spans some 1.7 Gyr (Parfrey et al. 2011). However, for readers who doubt the power of Muller’s ratchet, we interject that is indeed possible that selective pressures other than the escape of Muller’s ratchet are responsible for eukaryotes having retained meiotic recombination. Yet for the purposes of this article, it is immaterial whether Muller’s ratchet or some other selective force is responsible for the retention of sex (meiotic recombination) throughout all of eukaryotic history up to the present. For such skeptics, we emphasize: It is an observation from biology (not a prediction from theoretical population genetics) that recombination and the proteins required have been strictly conserved during eukaryote evolution (Ramesh et al. 2005; Speijer et al. 2015). From that we can readily and robustly infer that recombination is essential to long-term eukaryote survival. We are also fully aware that various eukaryotes appear to have lost the ability to undergo sex in some terminal branches (Maynard Smith 1986; Welch and Meselson 2000; Rougier and Werb 2001; Halary et al. 2011; Hand and Koltunow 2014; Speijer et al. 2015). But we reaffirm: If homologous recombination was not essential over the long term in eukaryotes, it would have been lost long ago and in many independent lineages. We posit that sex was conserved throughout eukaryote evolution by purifying selection as a means to escape Muller’s ratchet. Only the conserved and clearly essential nature of sex (meiotic recombination) is vital to our inference for its origin, not the exact reason for why sex has been conserved. As Maynard Smith (1986) put it: “… it is clear that for one reason or another it is very difficult to give up sex once you have it.” Our article is not about the “… hard to give up …” part, it is about the “… once you have it …” part, which is more challenging, because it falls into the prokaryote–eukaryote transition.
It is also very important to note this: Eukaryotes and prokaryotes use conserved mechanisms and homologous enzymes to perform DNA recombination (Camerini-Otero and Hsieh 1995; Ramesh et al. 2005), given the presence of two different DNA molecules within the cell. But crucially, the way(s) in which DNA substrates for recombination enter the cell and come into contact for recombination differ fundamentally across the prokaryote–eukaryote divide, as explained in the following.
In prokaryotes, the mechanisms that bring DNA into the cell for recombination are the mechanisms of lateral gene transfer (LGT): Transformation, conjugation, transduction, and gene transfer agents (Jones and Sneath 1970; Doolittle 1999; Martin 1999; Ochman et al. 2000; Lang et al. 2012). These mechanisms operate unidirectionally, from donor to recipient. Except for some archaeal lineages that undergo cell fusion and recombination in the fused state (Naor and Gophna 2014) these mechanisms do not obey taxonomic boundaries, species or otherwise, and over time they generate the pangenomes typical of prokaryotic taxa (Rasko et al. 2008). What are pangenomes? Pangenomes are readily illustrated as follows: Although 61 different humans (or individuals of any eukaryotic species) possess essentially the same genes (some copy number variation notwithstanding), 61 strains of Escherichia coli, each harboring about 4,500 genes, possess in total about 18,000 genes (the pangenome), with only 1,000 genes being present in all strains (the core genome) (Lukjancenko et al. 2010). Thus, homologous recombination in prokaryotes entails the introduction of foreign DNA into the cell through the mechanisms that we typically associate with LGT (transformation, conjugation, transduction, and/or gene transfer agents), and in these cases recombination is never reciprocal. In fusing archaeal species, there is no clear evidence that recombination is homologous (Papke et al. 2004; Naor and Gophna 2014). In prokaryotes, recombination is not reciprocal, is always unidirectional from donor to recipient, and operates with LGT machinery: Conjugation, transformation, transduction, or gene transfer agents.
In eukaryotes, homologous recombination occurs during meiosis, is always reciprocal and occurs between individuals of the same species. The DNA substrates for eukaryotic recombination come into contact through gamete fusion (syngamy) followed either immediately or after a dikaryon or multinucleated stage by nuclear fusion (karyogamy). Because eukaryotes arose from prokaryotes (Williams et al. 2013), eukaryotes must have lost the prokaryotic LGT mechanisms present in their ancestors, while retaining the enzymatic machinery that performs homologous recombination (Ramesh et al. 2005; Speijer et al. 2015; Bloomfield 2016). Without sex (reciprocal recombination), eukaryotes would have succumbed to Muller’s ratchet long ago. Sex was an invention of the eukaryote common ancestor that has neither been replaced nor fundamentally improved upon in the approximately 1.7 Gyr since eukaryotes arose.
The Nature of the Cell That Evolved Sex
Traditionally, approaches to the origin of sex start with a mitotic cell (typically a hypothetical “primitive” eukaryote) and introduce factors and effects that lead to meiosis (Cleveland 1947; Williams 1975; Maynard Smith 1978; Bell 1982; Bernstein et al. 1984; Uyenoyama and Bengtsson 1989; Hurst and Nurse 1991; Otto and Goldstein 1992; Maynard Smith and Szathmary 1997; Cavalier-Smith 2002, 2010; Solari 2002; Wilkins and Holliday 2008; Hörandl and Hadacek 2013; Goodenough and Heitman 2014). That approach sounds reasonable enough at first encounter, but upon closer inspection, some fairly severe problems quickly become apparent. First, if the ancestral eukaryote (the cell that evolved sex) was mitotic, it was an asexual mitotic cell, obviously. In our view, an asexual mitotic cell is a very problematic intermediate, because it raises the question of how it escaped Muller’s ratchet both during the time 1) before it evolved sex and 2) while it was evolving mitosis. Yet more pressingly from our perspective, if meiosis arose from mitosis, how and from what did mitosis arise?
Recent progress in understanding eukaryote origins has changed the nature of the problem concerning the origin of sex in some salient respects. Current data on eukaryote origin have it that the host for the origin of mitochondria was an archaeon, not a eukaryote (Cox et al. 2008; Lane and Martin 2010; Williams et al. 2013; Spang et al. 2015). That would in turn suggest that the evolutionary inventions that distinguish eukaryotes from prokaryotes (including sex) arose in a prokaryotic (archaeal) host cell that possessed a mitochondrial (bacterial) symbiont, providing good reasons to doubt that the cell that acquired the mitochondrion was even mitotic at the time that the mitochondrion became established. There are a growing number of reports that implicate a role for mitochondria at the origin of sex (Lane and Martin 2010; Hadjivasiliou et al. 2013; Radzvilavicius and Blackstone 2015; Lane 2015). The basic idea that mitochondria came before sex (Radzvilavicius and Blackstone 2015) is worth exploring.
In fact, one can probably even exclude the possibility that a mitosing cell arose in the absence of mitochondria. How so? A short calculation is insightful. All eukaryotes separate their chromosomes with the help of microtubules. A tubulin dimer has 110 kDa (Oakley 2000), corresponding to about 1,000 amino acids, each of the peptide bonds requiring four ATP for polymer formation (Stouthamer 1978), or 4,000 ATP per dimer. A microtubule filament has about 13 dimers for one 360° turn in a 25-nm filament, the turn covering about 10 nm length, such that 10 nm of microtubule requires about 50,000 ATP for its synthesis (Nogales 2000). If a eukaryote cell is 10 µm long, and the microtubule has to go end to end, that corresponds to 50 million ATP to make one microtubule. If there is one microtubule per centromere (as in yeast), the cell can move one chromosome at a cost of 50 million ATP, the cost of microtubule depolymerization being 1/1,000th that of making the tubulin. If there are ten microtubules per centromere, as in many eukaryotes, we need 0.5 billion ATP to move a chromosome. If we have ten chromosomes per cell, we are at 5 billion ATP to move the chromosomes, but only if every single microtubule hits/attaches to a centromere, which does not happen—maybe 1–10% of the microtubules formed during mitotic cell division actually hit centromeres. That puts us at about 50–500 billion ATP to divide ten chromosomes in a 10-µm cell (or about 5–50 billion ATP to divide one chromosome)—in the modern, highly refined and regulated process. At the onset of microtubule-dependent chromosome segregation (MDCS), when the process was still primitive and improving via purifying selection, the cost of chromosome segregation was probably much higher. How much is 50–500 billion ATP? For comparison, E. coli needs a total of about 10–20 billion ATP per cell division (Neidhardt et al. 1990) to synthesize the daughter cell, to physically divide and to keep both cells alive during the process. Similarly, the amount of ATP that ancestral mitochondrial endosymbionts could make available to their host, the nascent eukaryote, simply by not synthesizing 5% unneeded proteins (such as for cell wall and the like) also comes in at about 50 billion ATP, but 50 billion per day (Lane 2014). Such calculations serve to highlight the amount of ATP required by eukaryotic cell biological processes and how mitochondria could contribute to these energetic needs.
Thus, the ancestral eukaryotic cell, the one that learned to divide its chromosomes using microtubules, expended as much ATP to merely segregate one prefabricated chromosome as normal prokaryotes expend to generate an entire daughter cell. This strongly suggests that the cell that learned to segregate chromosomes with microtubules had mitochondrial power (Lane and Martin 2010)—an inference that is consistent with, but independent of, data on the antiquity of mitochondria (McInerney et al. 2014; Ku et al. 2015) and the archaeal ancestry of the host (Cox et al. 2008; Lane and Martin 2010; Williams et al. 2013; Raymann et al. 2015; Spang et al. 2015). Although it should be mentioned that there are criticisms of the idea that mitochondrial power was important at eukaryote origin (Booth and Doolittle 2015; Lynch and Marinov 2015), it should also be mentioned that those criticisms have their own criticisms (Lane and Martin 2015, 2016). It should also be mentioned that mitochondria were not only a source of innovation, but they also caused problems (Blackstone 2013): Having a foreign cell in one’s cytosol is a great perturbation in the day-to-day life of any viable prokaryotic cell. Yet by virtually any measure, it is increasingly clear that mitochondria played an important role at eukaryote origin; indeed, even more genes in eukaryotes stem from the ancestral mitochondrion than stem from the archaeal host (Esser et al. 2004; Ku et al. 2015). Though views on eukaryote origin have changed radically in recent years (Martin and Müller 1998; Cox et al. 2008; Lane and Martin 2010; Katz 2012; Williams et al. 2013; McInerney et al. 2014; Raymann et al. 2015; Spang et al. 2015; Ku et al. 2015), views on the origin of sex have not at all kept pace with that development.
Sex: Embedded in the Cell Cycle and Dependent Upon Energy
As outlined in figure 1, meiosis and mitosis are just part of a more general process at the heart of eukaryotic cell growth and survival: The eukaryotic cell cycle. The cell cycle is, in turn, itself embedded in an even more general process: Carbon and energy metabolism, which run all processes of the cell to begin with. Without carbon and energy, no cell can survive, and no evolution can take place. Carbon and energy govern the immediate survival of the individual, hence its ability to evolve. Energy means ATP synthesis and is the first limiting factor for evolution. Without ATP synthesis, life and evolution come to an immediate halt. Population genetic effects operate within generations, bioenergetic effects operate within minutes.
Fig. 1.
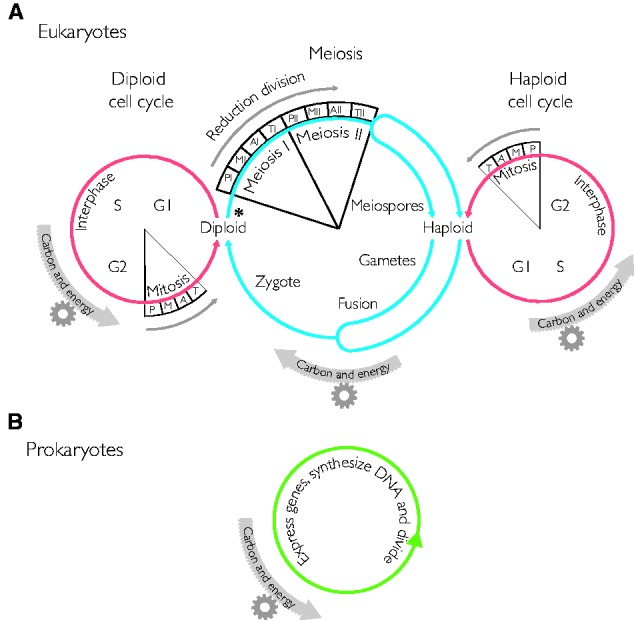
(A) Cell cycles and life cycles in eukaryotes. Meiosis (reduction division) connects haploid and diploid cell cycles. Each cell cycle is divided into distinct phases G (gap), S (synthesis) and the meiotic or mitotic phase, within which P (prophase), M (metaphase), A (anaphase), and T (telophase) phases are distinguished (see text). Carbon and energy are required to drive all cellular processes, including these cycles, upon which population genetic effects can subsequently operate. The asterisk indicates where, roughly, we would place the starting point in the origin of the process, that is, the symbiotic merger of host and symbiont. (B) Cell cycles and life cycles in prokaryotes, schematic. We recognize that there are spore-forming types, stalk-forming types, heterocyst-forming types, and other exceptions among prokaryotes. But in the main, we find it fair to generalize that the prokaryotic life cycle, if compared with the eukaryotic state, is a matter of continuously simultaneous cell and chromosome division.
The typical eukaryotic cell cycle comprises two major stages: The interphase and the mitotic phase or M-phase (fig. 1A) (Mitchison 1971). Interphase is further separated into the synthesis (S-) phase, during which the genome is replicated, and the gap (G-) phases, G1 and G2 (Norbury and Nurse 1992). During G1, the cell is metabolically active and prepares for genome replication, sensing the favorability of the environment; DNA replication during S phase follows. During G2, the cell carefully checks the integrity of its genetic material and prepares for mitosis (Norbury and Nurse 1992). M-phase or mitosis entails chromosome segregation followed by cell division or cytokinesis marking the end of the cell cycle. Many variations on this theme exist, for example, the presence or absence of the nuclear envelope in closed and open mitosis (Raikov 1994) or other variants such as cell senescence or specialization where the cells enter into a stage where they cease dividing (Blagosklonny 2011).
Meiosis, also called reduction division (because ploidy is reduced), can also be seen as a part of the cell cycle that is manifested in conjunction with sexual reproduction (Solari 2002). In this article, we use the term “sex” to designate a process in which the nucleus-bound genomes of two parents are brought together in a common cytoplasm (syngamy), whereupon the nuclei eventually fuse (karyogamy) to produce a cell with a double set of chromosomes, which will eventually undergo meiotic recombination and reduction, giving rise to progeny (meiospores) with a single (haploid) set of chromosomes that contain reassorted portions of the parental genomes (Bernstein et al. 1984) starting from a diploid cell. Meiosis typically results in four haploid cells (meiospores or gametes, depending upon how the organism undergoes alternation of generations) whose chromosomes have been reassorted and recombined relative to the mother cell, whereas mitosis yields two haploid or diploid cells, again depending upon alternation of generations, that have identical genomes.
Meiosis begins with duplicated sister chromatids paired with their homologous counterparts. Double-strand breaks (DSBs) initiate recombination. After crossovers are resolved, the sister chromatids are independently assorted. Following cytokinesis, a second round of chromosome segregation without DNA replication ensues, sister chromatids are distributed to the daughter cells, typically haploid gametes. Fusion of two such meiotically generated gametes, which may have a prolonged mitotic life cycle of their own (fig. 1A), eventually restores diploidy (Wilkins and Holliday 2008). Importantly, the transition from one cell cycle phase to another is a highly controlled process involving cyclin-dependent kinases (CDKs), which are temporally regulated by their respective cyclins (Morgan 1997). These act as checkpoints that ensure faithful chromosome replication and segregation are followed by cell division (Hartwell and Weinert 1989), initiating mitosis at its specific phase during the cell cycle.
Prokaryotes clearly have recombination (Camerini-Otero and Hsieh 1995; Papke et al. 2004; Naor and Gophna 2014). But meiosis, mitosis, and a eukaryotic type cell cycle are lacking in prokaryotes altogether (fig. 1B). Some might counter that prokaryotes have mitosis or that archaea have a cell cycle (Lindås and Bernander 2013) but these are misnomers: Prokaryotes do not present anything resembling bona fide mitosis (chromosome condensation and microtubule-dependent chromosome separation to nuclear poles or cell poles) that resides at the heart the eukaryotic cell division process, nor do prokaryotes have anything that could be viewed as faintly homologous to the eukaryotic cell cycle, of which mitosis is a part (fig. 1). Archaeal chromosomes typically have multiple origins of replication, requiring a bit more care to ensure proper chromosomal partitioning (Lindås and Bernander 2013), yes. But a eukaryote-like cell cycle? Hardly. The eukaryotic cell cycle has no homolog among prokaryotes.
Reinspecting Old Premises
Compared with literature on the origin of sex or the origin of eukaryotes, literature concerning the evolution of the cell cycle is fairly scarce, with Nasmyth (1995), Novak et al. (1998) and Cross et al. (2011) being notable exceptions, though they do not specifically address cell cycle origin. Literature covering all three topics in one place is scarcer still, Cavalier-Smith’s essays (2002, 2010) being exceptions. Yet, like de Duve (2007) did in his day, Cavalier-Smith (2002, 2010, 2014) still rejects the idea that archaea participated in any way in the origin of eukaryotic lineage, steadfastly maintaining that both eukaryotes and archaea arose from actinobacteria. That makes it virtually impossible to integrate his views into any kind of modern synthesis, because phylogenetic analyses indicate that the host for the origin of mitochondria was an archaeon (Cox et al. 2008; Williams et al. 2013; McInerney et al. 2014; Raymann et al. 2015; Spang et al. 2015) and that the ancestor of the mitochondrion was an alphaproteobacterium, with no evidence for other partners at eukaryote origin (Ku et al. 2015). Genomes harbor evidence neither for an actinobacterial origin of eukaryotes (Ku et al. 2015) nor for an actinobacterial origin of archaea (Nelson-Sathi et al. 2015). Hence we acknowledge Cavalier-Smith’s contributions to the topic, but address no specifics of his hypotheses regarding actinobacterial origins of archaea, phagotrophy, eukaryotes, or sex (Cavalier-smith 1975, 2014). Not surprisingly, the phylogeny of the proteins involved trace the cell cycle to the eukaryote common ancestor (Krylov et al. 2003).
If meiosis evolved from mitosis, as traditional theories for the origin of sex posit, it arose in some hypothetical lineage of asexual, mitosing eukaryotes that, like all lineages, had to escape Muller’s ratchet. Therefore, it utilized either 1) the well-characterized prokaryotic mechanisms of getting DNA into the cell for recombination (the typical prokaryotic LGT mechanisms), or 2) some mechanism of recombination that was compatible with mitosis but did not involve meiosis. In either case, the lineage was necessarily recombining (to avoid Muller’s ratchet, we contend), leaving neither selective pressure to evolve anything as complicated as meiosis and sex, nor benefit from it once it arose. This line of thought actually renders the origin of meiosis from mitosis altogether unlikely.
Ordering Events at the Prokaryote–Eukaryote Transition
The eukaryote ancestor had mitochondria, a nucleus, a cell cycle and sex. In what order did these traits arise? Traditional theories holding that meiosis evolved from mitosis, also entail the assumption that mitochondria had nothing to do with the origin of eukaryote complexity. Yet from the energetic standpoint, mitochondria had everything to do with the origin of eukaryote complexity (Lane and Martin 2010), and several recent publications even report how genetic effects emanating from mitochondria could have impacted the origin of sex (Lane 2009; Hörandl and Hadacek 2013; Lane 2014; Havird et al. 2015; Radzvilavicius and Blackstone 2015; Speijer 2015). Cleveland (1947) clearly considered meiosis in the context of the life cycle. Our approach is similar. In eukaryotic microbes, whose common ancestor possessed mitochondria (Embley and Martin 2006), the life cycle is an iteration of the cell cycle (fig. 1). In ordering the sequences of events surrounding the origin of six key characters (mitochondria, the nucleus, meiosis, mitosis, the cell cycle, and sex), we start with minimal premises: A mitochondrial endosymbiont in an archaeal host that lacked the other five traits. For further justification of why we embark from such a simple cell biological starting point, see Gould et al. (2016) with regard to the origin of the endomembrane system and Sousa et al. (2016) with regard to the archaeal nature of the host.
Starting with Carbon and Energy
We start with an endosymbiosis, an archaeon (hereafter called the host) that acquired a bacterial endosymbiont, the common ancestor of mitochondria and hydrogenosomes (hereafter called the symbiont). Various prokaryotes harbor prokaryotic endosymbionts (Wujek 1979; von Dohlen et al. 2001; McCutcheon et al. 2009; Husnik et al. 2013; Kobialka et al. 2016), in cases investigated so far, the symbiotic interactions are metabolic and the host is not phagocytotic. At eukaryote origin, metabolic interactions (Martin and Müller 1998; Searcy 2003; Müller et al. 2012; Degli Esposti 2014) likely facilitated interactions between the mitochondrial symbiont and its host (fig. 2A), for which we posit an archaeal cellular organization. The symbiosis must be stable, neither partner digesting the other, and with a suitable metabolic flux fueling both partners, for example, anaerobic syntrophy with a hydrogen-dependent host (Sousa et al. 2016).
Fig. 2.
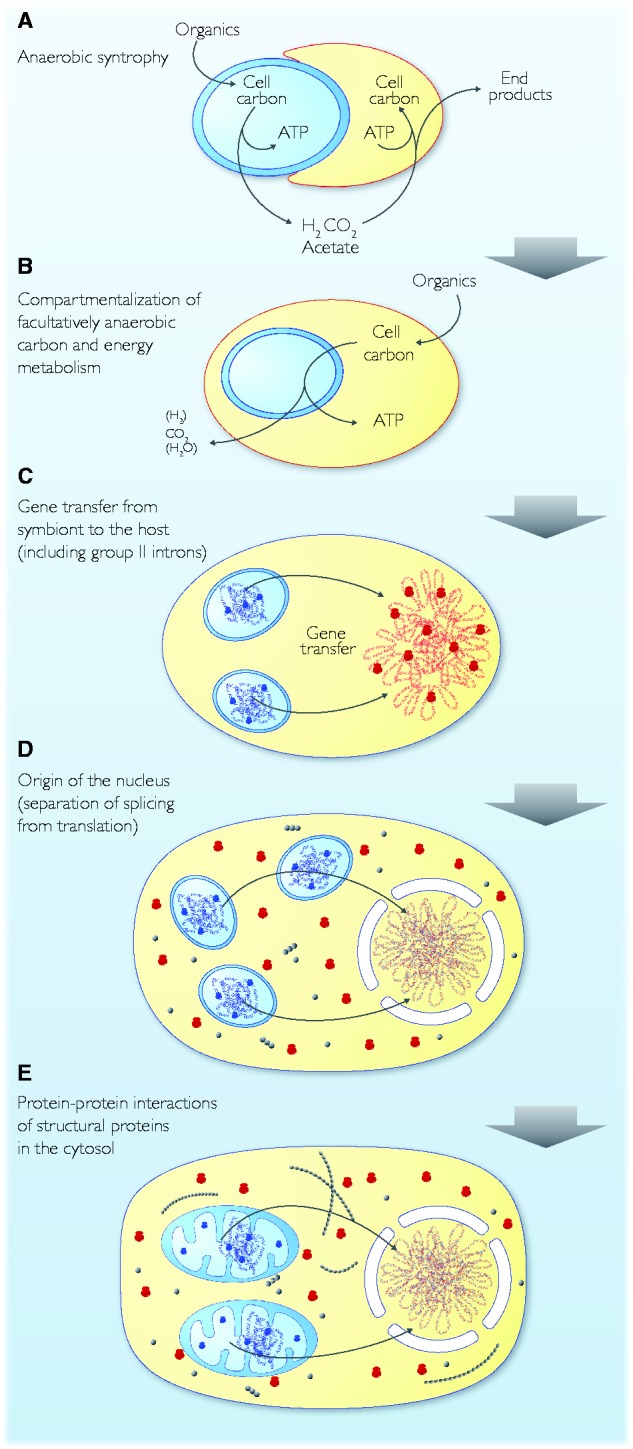
Steps en route from endosymbiont acquisition to an ancestral cell cycle. Blue, red and gray represent bacterial, archaeal and eukaryotic components, respectively (see text).
Stable symbiosis requires carbon and energy for both partners, which anaerobic syntrophy can provide. Eukaryotes conserve energy in the cytosol and in internalized bioenergetic organelles—mitochondria. The transfer of genes from the mitochondrial endosymbiont, a facultative anaerobe (Müller et al. 2012; Degli Esposti 2014) to the chromosomes of the host prior to the origin the mitochondrial protein import apparatus can account both for 1) the bacterial origin of the eukaryotic glycolytic pathway and 2) its cytosolic localization (Martin and Müller 1998). With importers for organic compounds in the host’s plasma membrane and a glycolytic pathway in the cytosol, the host compartment (the cytosol) has a source of net ATP synthesis (glycolysis) that is independent of chemiosmotic coupling at the plasma membrane, which is lost. To provide net ATP yield in the cytosol, glycolysis must proceed to the pyruvate-generating step. Transfer of the symbiont’s glycolytic pathway to the host’s cytosol and its carbon substrate importers to the host’s plasma membrane does not require inventions, it merely requires the transfer of genes from symbiont to host and the expression of bacterial genes in archaeal chromosomes (Martin and Müller 1998), evidence for which in modern archaeal genomes abounds (Nelson-Sathi et al. 2015).
The endosymbiont’s transition into an ATP-exporting organelle requires two things: 1) Ability to import pyruvate—which traverses membranes readily (Bakker and Van Dam 1974)—and oxidize it, yielding approximately five ATP per glucose (Müller et al. 2012) anaerobically or approximately 30 ATP per glucose aerobically (Rich and Maréchal 2010); and 2) ability to export ATP to the cytosol through the mitochondrial ADP/ATP carrier (AAC) (Whatley and Whatley 1979; Radzvilavicius and Blackstone 2015). Radzvilavicius and Blackstone (2015) have suggested that the AAC might even have been invented in, and originally expressed by, the symbiont’s genome, requiring no new protein import machinery, only the preexisting protein insertion machinery of the symbiont (fig. 2B and C), an intriguing idea.
From the energetic standpoint, the AAC consummates the symbiont-to-organelle transition (Whatley and Whatley 1979; Radzvilavicius and Blackstone 2015), although the invention of the mitochondrial protein import machinery—Translocase of the outer membrane (TOM) and Translocase of the Inner Membrane (TIM) complexes (Doležal et al. 2006)—and targeting signals on cytosolic precursor proteins (Garg et al. 2015) allows the symbiont to relinquish genes to the host’s chromosomes. Not all genes are relinquished however, those central to the electron transport chain in the inner mitochondrial membrane remain in the organelle, for reasons of redox balance (Allen 2015).
The mitochondrion alters the basic bioenergetic architecture of the cell and the amount of protein that the host compartment (cytosol) can afford to express (Lane and Martin 2010; Lane 2014). The conversion of an alphaproteobacterial endosymbiont with a heterogeneous genome (Martin 1999; Ku et al. 2015) into an ATP generating compartment furthermore coincided with the complete loss of chemiosmotic energy conservation (ATP synthesis) at the host’s plasma membrane (Gould et al. 2016). Eukaryotes have archaeal ribosomes in the cytosol, but the enzymes of eukaryotic carbon and energy metabolism stem from bacteria and trace to the eukaryote common ancestor (Blackstone 2013; Ku et al. 2015). As we see it, mitochondria change not only the bioenergetic state of the host compartment (Lane and Martin 2010) but also the physical content of the host’s cytosol through the addition of membrane vesicles consisting of bacterial lipids: Outer membrane vesicles produced by the mitochondrial symbiont (Gould et al. 2016). Both contributions of mitochondria, we contend, carried dramatic consequences for eukaryote evolution and account for the observation that only the cells that became genuinely complex have mitochondria or had them in their past.
For balance, we note here that some readers might not agree with the foregoing proposition. For example, population geneticists contend that mitochondria had nothing whatsoever to do with eukaryote origin (Lynch and Marinov 2015). The philosophically inclined might argue that eukaryote complexity, if it is not an illusion altogether, is not due to mitochondria at all, but to luck (Booth and Doolittle 2015). It is not our intent here to try to convince critics. There is certainly a role for population genetics at eukaryote origin, namely the small population size, increased drift, and reduced power of purifying selection (Nei 1987) obviously inherent to the single origin of both mitochondria and of eukaryotes as a group. But we think that the main hurdle at eukaryote origin is the origin of mitochondria, which (like plastids) is the result of endosymbiosis, not allele frequency changes. All organisms have population genetics, only eukaryotes have mitochondria. Is there a connection between mitochondria and complexity? We think so. Since its inception (Mereschkowsky 1905), endosymbiotic theory has always been a target of disparaging critique. Thus, we acknowledge criticisms divesting mitochondria (and endosymbiosis more generally) of evolutionary significance, and move on.
Gene Transfer, Introns, the Nucleus and Ploidy
All cells that have sex have a nucleus. How does the nucleus fit into eukaryote origin? The mitochondrial endosymbiont is more than a source of energy, it is a source of genes, lots of genes, and large scale chromosomal mutations. This process of gene transfer, from the bacterial symbiont to chromosomes of the archaeal host, is called endosymbiotic gene transfer, or EGT (Martin et al. 1993; Timmis et al. 2004), it is unidirectional, it still operates today involving insertion of whole organelle genomes hundreds of kilobases in length into nuclear DNA (Huang et al. 2005). The mechanism of DNA integration is nonhomologous end joining (Hazkani-Covo and Covo 2008), the mechanism of DNA release to the host is organelle lysis (Huang et al. 2004), the process of EGT has operated throughout eukaryotic history and is observable as an ongoing process, even during human evolution, with the most recent mitochondrion-to-nucleus transfers dating to the Tschernobyl incident (Hazkani-Covo et al. 2010). Eukaryotes are usually described as descendants of archaea (Cox et al. 2008; Williams et al. 2013), but if we look at the whole genome, bacterial genes vastly outnumber archaeal genes in eukaryotes (Esser et al. 2004; Thiergart et al. 2012), and genes that trace to the mitochondrion vastly outnumber those that trace to the host (Ku et al. 2015). At the outset, the host has no nucleus, and as long as cell division is not impaired, the symbiosis of prokaryotes is stable, as long as the environment supports growth.
The stability of the symbiosis changes however, probably as a consequence of EGT, a mutational mechanism that is specific to the eukaryotic lineage: Gene transfer from symbiont to host carries some fateful hitchhikers—self splicing group II introns. Group II introns are important, and their transition into spliceosomal introns could have precipitated the origin of the nucleus (Martin and Koonin 2006). How so? Group II introns occur in prokaryotic genomes (Lambowitz and Zimmerly 2011), they are mobile, they can spread to many copies per genome (Lambowitz and Zimmerly 2004) and they remove themselves through a self-splicing mechanism that involves the intron-encoded maturase (Matsuura et al. 1997). Their splicing mechanism is similar to that in spliceosomal intron removal (Lynch and Richardson 2002), for which reason they have long been viewed as the precursors of both 1) spliceosomal introns and 2) their cognate snRNAs in the spliceosome (Sharp 1985).
The crux of the intron hypothesis for nuclear origin (Martin and Koonin 2006) is that group II introns, which are mobile elements in prokaryotes (Lambowitz and Zimmerly 2011), entered the eukaryotic lineage through gene transfer from the mitochondrial endosymbiont to the archaeal host. In the host’s chromosomes they spread to many sites and underwent the transition to spliceosomal introns, as evidenced by the observation that many introns are located at conserved positions across eukaryotic supergroups (Rogozin et al. 2003) and by the presence of spliceosomes in the last eukaryote common ancestor (Collins and Penny 2005). The transition from group II introns to spliceosomal introns evokes a curious situation: Spliceosomal splicing is slow, on the order of minutes per intron (Audibert et al. 2002), whereas translation in ribosomes is fast, on the order of 10 peptide bonds per second (Sørensen et al. 1989). As the transition to spliceosomal introns set in, the host’s cytosol was still a prokaryotic compartment with cotranscriptional translation. With the origin of bona fide spliceosomes and spliceosomal splicing, nascent transcripts were being translated (ribosomes are fast) before they can be spliced (spliceosomes are slow). Translation of introns leads to defective gene expression at many loci simultaneously (though one essential locus would suffice), a lethal condition for the host unless immediately remedied.
The solution to this condition was, we posit, physical separation of the slow process of splicing from the fast process of translation so that the former could go to completion before the latter set in. Separation in cells usually involves membranes, and that is the central tenet of the intron hypothesis: The initial pressure that led to selection for nucleus–cytosol compartmentation (the origin of the nuclear membrane) was the requirement for physical exclusion of active ribosomes from nascent transcripts, to restore gene expression and intron-containing genes (fig. 2D). The primordial nuclear membrane allowed the slow process of splicing to go to completion around the chromosomes, thereby initially allowing distal diffusion, later specific export of processed mRNAs to the cytosol for translation, furthermore precipitating the origin of nonsense-mediated decay, a eukaryote-specific machinery that recognizes and inactivates intron-containing mRNAs in the cytosol (Martin and Koonin 2006) (fig. 2D).
The reader might protest that we have specified neither a mechanism nor a source for the vesicles that give rise to the nuclear membrane in the host’s archaeal cytosol. That is the topic of a separate paper (Gould et al. 2016), in which we outline how outer membrane vesicles produced by the mitochondrial endosymbiont in an archaeal host are likely both the physical source and the evolutionary origin of the eukaryotic endomembrane system.
A primitive nuclear membrane rescues gene expression, and DNA replication can continue to proceed as long as the cytosol supplies dNTP precursors. But there is no mechanism for chromosome segregation in place. Chromosomes replicate without division, polyploidy, extreme polyploidy in all likelihood ensues, and the symbiosis seems to be headed straight toward a dead end. But mitochondria can make a difference.
Protein–Protein Interactions in an Energy-Laden Cytosol
All cells that undergo sex divide their chromosomes with microtubules. Prokaryotes possess genes for tubulin precursors (Erickson 2007), but they do not make microtubules. Why not? Introns give rise to a cell that requires a nuclear membrane to express genes. That configuration is fine from the standpoint of stable gene expression to maintain carbon and energy metabolism. But sequestration of the host’s chromosomes within a nuclear compartment has two consequences of exceptional significance. First, though the nuclear membrane rescues gene expression, the chromosomes are no longer attached to the plasma membrane of the cell and segregation of the chromosomes (now contained within the nucleus) is no longer coupled to cell division. This is a problem of severe sorts. Our symbiotic consortium can satisfy its carbon and energy needs by virtue of compartmentalized carbon and energy metabolism between the cytosol and the mitochondrion. It can express intron-containing genes by virtue of a nuclear membrane, but it cannot segregate its chromosomes in the standard prokaryotic manner to produce progeny. Either a solution to the problem of chromosome partitioning is found or extinction is the alternative. The solution to chromosome partitioning stems, we propose, from the second consequence of nucleus–cytosol compartmentation.
The second consequence is that the nuclear membrane generates a fundamentally new kind of cell compartment in the biological realm of that day: A cytosol that is free of active chromatin. The eukaryotic cytosol is not only a compartment of protein–protein interactions (Martin and Koonin 2006), it can afford, energetically, to express the proteins that might interact (Lane and Martin 2010). The eukaryotic cytosol is unique in that it is a dedicated translation compartment where protein–protein interactions can take place at a magnitude never before possible in any prokaryotic cell (fig. 2E). Energy is crucial for that, because protein synthesis consumes about 75% of a cell’s energy budget (Harold 1986). It is also true that for the world of protein–protein interactions that emerged in the eukaryotic cytosol to materialize, orders of magnitude more ribosomes than typical of a prokaryotic cytosol need to be synthesized. This requires amplification of rDNA genes, which eukaryotes realize by various means, including the increase of rDNA genes to thousands of chromosomal copies (McGrath and Katz 2004), and the specific amplification of rDNA genes through rolling circle plasmids and other extrachromosomal elements in various eukaryotic lineages (Hourcade and Dressler 1973; McGrath and Katz 2004; Kobayashi 2011). Providing the cytosol with abundant protein requires not only abundant ATP but also very large numbers of ribosomes.
In addition to having a chromatin-free cytosol, hence a dedicated translation compartment, the stem eukaryote has, by virtue of mitochondria (Lane and Martin 2010), effectively unlimited ATP for protein synthesis. So despite being unable to divide in a well-coordinated manner, it can synthesize proteins (and ribosomes) in amounts unattainable by any prokaryotic cell, because of mitochondrial ATP synthesis. This enables the symbiotic consortium (the nascent eukaryote) to explore protein expression in a manner that no prokaryote could. Relative to prokaryotes, the existence of mitochondria in the nascent stem eukaryote enables energetically unpenalized protein overexpression. Mitochondria do not force an evolutionary transition, but they enable it. The stem eukaryote can overexpress virtually every protein, so countless protein expression experiments are possible.
Given the watchful eye of natural selection, which expression experiments might be successful? That is, which proteins might become expressed at high amounts? Massive overexpression of metabolic enzymes is not a viable option, as it will inevitably impair carbon and energy flux. In contrast, overexpression of enzymatically inert structural proteins such as tubulin, actin, and other structural proteins typical of the eukaryotic cytosol, but in their ancestral prokaryotic forms (FtsZ, MreB, Ta0583, CetZ, archaeal Cdv’s, the precursors of ESCRT complex proteins, etc.), will not alter metabolism. Expression of structural proteins will simply sink carbon and nitrogen into proteins that 1) can accumulate without interfering with carbon flux, and 2) that spontaneously assemble into higher order structures (Jékely 2014) while actually requiring ATP hydrolysis for their disaggregation. Cytoskeletal proteins aggregate spontaneously and consume ATP to depolymerize or disaggregate (Fleury-Aubusson 2003; Gould et al. 2011).
This is particularly interesting because it suggests that the origin of filamentous or otherwise aggregated cytoskeletal components was not a slow, stepwise evolutionary process, but rather that it was a spontaneous consequence of dramatically increased ATP availability (for protein synthesis), requiring a small additional supply for disassembly into monomers. A general underlying theme of eukaryotic cytoskeletal proteins is that—if synthesized in sufficient amounts—they spontaneously assemble into larger, ordered structures and require ATP hydrolysis for their disassembly or depolymerization. Cytoskeletal proteins could thus be seen as relicts of ancient overexpression experiments in which selection was acting to bring forth polymers that could undergo reversible self-assembly. Such experiments might also still be going on today, as this would explain why the intermediate filament proteins of various protist lineages seem not to share common ancestry with the intermediate filament proteins of animals and fungi (Gould et al. 2011), having arisen independently instead.
In short, at this stage in the prokaryote to eukaryote transition, the cytosol expresses proteins that make structures and move things through ATP and GTP, as opposed to converting substrates. ATP has to be in very abundant supply for that, otherwise the proteins could not be synthesized. At such a stage, the spectrum of eukaryotic-specific cytological novelties could have taken root in terms of becoming heritable and fixed. But there are still some unsolved problems with the process of heredity: Chromosome segregation.
Chromosome Division
Overexpressing cytoskeletal and other (novel) structural proteins is now an energetically affordable option for the mitochondrion bearing cell. Prokaryotic tubulin precursors are very similar in form and function to their eukaryotic counterparts (Erickson 2007), hence little in the way of protein sequence modification is required for tubulin monomers to assume new function, but rather the limitation is synthesizing them in large amounts. Synthesizing large amounts of tubulin monomers results in microtubules, and unconstrained (unregulated) polymerization of microtubules leads to a cytosol teeming with spontaneously polymerizing and GTP-dependent (ultimately ATP-dependent) depolymerizing microtubules: A world of molecular movement bearing the possibility of MDCS (fig. 3A). Obviously, chromosome movement requires attachment sites on the chromosomes: Primitive centromeric regions. Attachment sites are not a completely novel eukaryotic invention, because prokaryotic chromosomes attach to the plasma membrane, allowing them to be separated at cell division (Toro and Shapiro 2010). Indeed, the archaeal protein that serves as an attachment site for large plasmid segregation in Sulfolobus, ParB, is similar at the structural level to CenpA, which is a pivotal protein of eukaryotic microtubule chromosome attachment and chromosome segregation (Schumacher et al. 2015). So the basic machinery for attaching to DNA that was not bound to the plasma membrane (plasmids) was apparently in place in the host, and prokaryotic protein attachment sites for ParB-dependent segregation are present in prokaryotes (Mierzejewska and Jagura-Burdzy 2012), such that the initial process of physically segregating DNA with microtubules possibly hinged more upon merely being able to synthesize enough tubulin to get the job done than it did on evolving an orchestrated chromosome choreography. Synthesizing large amounts of protein required mitochondria.
Fig. 3.

(A) A coenocytic eukaryote common ancestor(Coeca) with multiple independently dividing nuclei. (B) Accidental budding off of spores using the archaeal vesicle secretion mechanisms. Only spores containing (at least) a complete genome enclosed within a nucleus and at least one (compatible) mitochondrion are viable. All other possibilities result in inviable spores, providing strong selection among spores for viable gene, chromosome and mitonuclear combinations.
Primitive MDCS likely occurred in the persistent presence of a nucleus (a closed mitosis state), because of the continued need to separate splicing from translation. The host’s chromosomes could interact with the inner leaf of the nuclear membrane through pre-existing chromosome-membrane attachment mechanisms (Toro and Shapiro 2010), whereas the microtubules could interact with primitive nuclear pore complexes (Zuccolo et al. 2007) that permitted diffusion of spliced mRNA from the nucleus to the cytosol, but excluded the diffusion of active ribosomes from the nucleus. Alternatively, microtubules might simply have formed within the nucleoplasm, attaching to chromosomes directly and pushing them and the nucleus apart in an ATP-dependent manner, as it occurs in Vaucheria (Takahashi et al. 2003), Bryopsis (McNaughton and Goff 1990), or diatoms (Pickett-Heaps et al. 1982; Cande and McDonald 1985). At the outset, some combination of both is not unlikely.
MDCS provides a means for, and leads to, unregulated division of continuously replicating chromosomes. But given the tools and the energy, chromosome segregation does not know when to stop (!) because there is neither a cell cycle nor a coordinated mitotic division process. Thus, the ability to move chromosomes apart, while initially en route to becoming a virtue, suddenly becomes a horrible vice: Microtubules continuously separate chromosomes, down to a state where no more segregation is possible (perhaps one chromosome or plasmid per nucleus).
Before going further, a short note about forces in chromosome segregation is in order. When we say MDCS, the reader might think that we mean the pulling apart of chromosomes via attachment of microtubuli to microtubule organizing centers at poles of the cell, or similar. Pulling is not what we have in mind. The simpler process of pushing chromosomes apart is what we have in mind. At this point in our inference, the cell has a nucleus but neither mitosis nor a cell cycle, so chromosome segregation and nuclear division are the issues, not coordinating nuclear division with cell division. In several lineages of eukaryotes that maintain their nuclear membrane intact at chromosome division, the chromosomes and nuclei are pushed apart by microtubules. This occurs in coenocytic eukaryotes such as Vaucheria (Takahashi et al. 2003) or Bryopsis (McNaughton and Goff 1990), where the central rod-like microtubule structure that pushes the chromosomes and nuclei apart is called the interzonal spindle. It occurs in trichomonads, where the central rod-like microtubule structure that pushes the flagellar apparatuses (and ultimately the chromosomes) apart is designated either as the central spindle (Raikov 1994) or as the paradesmosis (Bricheux et al. 2007). Pushing also occurs in Schizosachharomyces pombe, where the spindle pole bodies located within the nucleus are pushed apart by microtubules (Castagnetti et al. 2015). In diatoms, the shape and behavior of the spindle were shown to be highly suggestive of a pushing mechanism to separate chromosomes (Pickett-Heaps et al. 1982); purified, isolated diatom spindles were later directly shown to exert a pushing force in vitro upon addition of ATP (Cande and McDonald 1985). Thus, when we say chromosome segregation, we have an ATP-dependent pushing mechanism for chromosome segregation in mind. An overview of variation in mitotic types among eukaryotes is provided by Raikov (1994).
High ploidy (see Gene Transfer, Introns, the Nucleus and Ploidy section), which was inevitable before MDCS came into play, points to a possible reason why linear chromosomes, which the eukaryote ancestor certainly came to possess at some point, would be preferable or better suited to survival than circular chromosomes. If ploidy became high, linear chromosomes, which do occur in prokaryotes (Bentley et al. 2002), would be much easier to separate than circular chromosomes, which generate concatamers upon replication. Linear chromosomes would not require disentanglement of multiply replicated circles, and hence would appear advantageous for a primitive MDCS process.
This kind of chromosome division—a primitive microtubule-dependent division that is independent of plasma membrane movement and cell expansion and that segregates (linear) chromosomes out of heavily polyploid nuclei—is, we suggest, the evolutionary origin of reduction division, the cardinal event in meiosis. But because the segregation process does not know when to stop, the chromosome sets that emerge as products of this kind of uncontrolled reduction will strongly tend to lack sufficient chromosomes (or genes, or both) for stable heredity. At this stage, chromosomes have to be in nuclei for gene expression (because of spliceosomal splicing) and they can replicate without the need for invention, using preexisting prokaryotic replication machinery. Thanks to mitochondrial metabolic power, they can be pushed apart by microtubules in the presence of nuclei (corresponding to closed mitosis), but they are not pushed apart in a coordinated manner to start.
Cell Division? Things Actually Work Better without It
If (note the “if”) there is cell division going on concomitant with this kind of primitive and crude chromosome segregation, then many, most or all of the progeny from this “emergency solution” or “evolutionary loophole” to chromosome separation will not capable of continued reproduction for lack of chromosome sets that would permit self-sufficient and self-sustaining replicating progeny. Daughter cells might inherit enough active cytosolic protein from lost genes to keep them viable for days or months, but not enough genes to keep a lineage going.
In addition, continuous gene transfer from symbiont to host (Timmis et al. 2004; Hazkani-Covo et al. 2010) generates archaeal host chromosomes that are, both within and across individuals, heterogeneous with respect to bacterial chromosome insertions (Lane 2009,2014). This generates disrupted genes, DSBs, and chromosomes that are rapidly evolving in terms of gene content. The future does not look bright for this population of energetically overachieving but genetically underdeveloped cells. Short of a miracle, is this inference going to go anywhere? Probably not, were it not for two observations that come into play, each of which can potentially contribute to solving some very hard problems surrounding the origin of the eukaryotic lifestyle:
1. If these cells divide, a very curious property of the archaeal host cell could rescue progeny: Archaeal cells can fuse. That archaeal cells fuse has been reported for the crenarchaeote Sulfolobus (Schleper et al. 1995), for several Thermococcus species (Kuwabara et al. 2005), and the euryarchaeote Haloferax (Naor and Gophna 2014). It is thus a property found within both the crenarchaeal and the euryarchaeal groups, hence attributable to our host without need for invention. The ability to fuse is a preexisting property of the host, and if not lost during earlier phases of the transition, now fulfills a lifesaving function: It creates new combinations of chromosomes, chromosomes that can be very different in number and nature (the products of uncontrolled reduction). Fusion could, in principle, lead to restoration of viable gene and chromosome numbers at this stage, but a lot of fusion would have to be going on: Fusion rates would have to be roughly the same as division rates in order to keep the system going. It is possible, but it is a long shot. The ability to fuse is probably more important when it comes to closing the life cycle (syngamy), dealt with in a later section. That modern eukaryotes can undergo fusion (plasmogamy), probably to generate recombination, is documented for some amoebae (Tekle et al. 2014).
2. If these cells do not divide, but just grow in length, the basic machinery of host cell division being somehow impaired but symbiont division—requiring dynamins (Purkanti and Thattai 2015) and ftsZ (Beech et al. 2000)—remaining intact, the result is a filamentous cell having nucleus-surrounded chromosomes that are segregated within the cytosol alongside autonomously dividing mitochondria. This cellular habit, the syncytium or coenocytic state, with many dividing nuclei and organelles occurs in several eukaryotic groups, being perhaps best-known among fungi (Roper et al. 2011), green algae (Verbruggen et al. 2009), and algae with red secondary plastids like Vaucheria (Gavrilova and Rundanova 1999).
The decisive advantage of a syncytial habit over host cell fusion at this stage is 2-fold: 1) In a syncytium, many different nuclei with deficient chromosome sets can “complement each other” simultaneously through mRNA in the cytosol, keeping the coenocyte alive; and 2) in a syncytium, nuclei can fuse (karyogamy: every eukaryote with sex does it) and divide. Chromosomes can undergo replication, recombination, segregation, and reduction, while remaining heavily buffered from selection because defects are rescued through complementing mRNAs in the cytosol. A complete set of essential genes (or many sets thereof) can be expressed in the syncytium, but from chromosomes dispersed across many different individual nuclei, some (many?) containing perhaps only one chromosome. The concept of a syncytial eukaryote common ancestor (fig. 4A) has many virtues. Coexisting defective nuclei of the kind that we have in mind have been observed within contemporary syncytia among the charophytes (Hasitschka-Jenschke 1960).
Fig. 4.
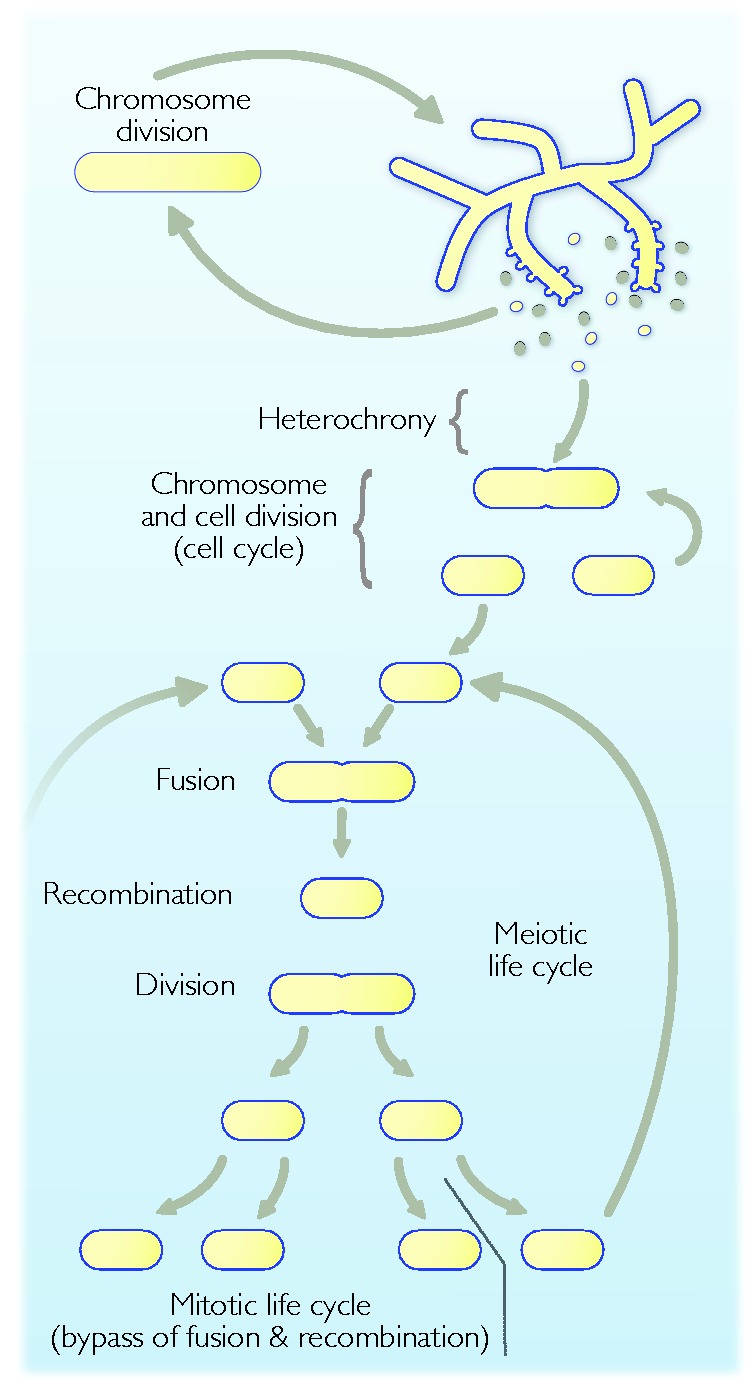
Possible life cycles of a coenocytic eukaryotic common ancestor. Viable meiospores (yellow) that bud off the syncytium have two possibilities. They can either 1) germinate to a new syncytium and continue with a coenocytic life cycle with multiple nuclear divisions or 2) undergo cell division (spore secretion) immediately after nuclear division, a case of heterochrony (spore secretion before filament formation). If this results in viable progeny, they can undergo fusion, recombination and division, which are characteristic of meiotic lifecycles. Mitotic life cycles can be easily derived from bypassing the fusion (syngamy, karyogamy) and recombination phases of a meiotic life cycle (see also fig. 1).
A Syncytial Eukaryote Common Ancestor
Some readers might gasp at the seemingly radical notion of a syncytial eukaryote common ancestor. But the closer one inspects the idea, the more robust it appears. The reasons are as follows.
First, in a syncytium, there is no pressure to solve the evolutionary problems of inventing the very complex and very novel (relative to prokaryotes) eukaryotic solutions to coordinated chromosome division and coordinated cell division all at once. The syncytium not only allows defective or incomplete chromosome sets in individual nuclei to persist through mRNA-mediated complementation in the cytosol, but also allows primitive nuclear divisions and nuclear fusions (karyogamy) to generate continuously new and potentially useful chromosome combinations. In principle, a syncytium could become very long, possibly branched (many prokaryotes including cyanobacteria and actinomycetes can branch), and thus generate ample opportunity for selection through the physical separation of chromosome combinations. Because the cell wall is the host’s, cell fusion as in archaea is a possibility that could, in principle, generate further combinations of fit nuclei at growing tips.
Second, the syncytial state is better suited to sorting out cytonuclear interactions than uninucleate cells. The genetic interactions between mitochondria and the nucleus have come under intense interest of late, not only because they are important for modern biology but also because they were likely important very early in eukaryote evolution, also at the origin of sex (Lane 2005,2009,2014; Havird et al. 2015; Radzvilavicius and Blackstone 2015; Speijer 2015; Speijer et al. 2015) and at the origin of anisogamy (Allen 1996; de Paula et al. 2013). The syncytial state allows mitochondria and nuclei to mix and interact in myriad combinations, without requiring that viable offspring (packaged as single cells) be produced. The combination of mitochondria and nuclei into diaspores provides a very strong selective mechanism with which to select for compatible mitonuclear interactions, but from a multinucleated reserve that was genetically buffered against deleterious effects of single nuclei or mitochondria harboring incompatibilities. Syncytial buffering plays an important role at this major evolutionary transition.
Third, a syncytium provides time, nutrients, energy, and a spatially differentiated landscape (territories) for nuclei to undergo selection for refining the simple process of coordinated chromosome division. Better nuclear division means more nuclei, as progeny, within the syncytium, so a clear selective advantage for nuclei capable of increasingly refined nuclear division is apparent. From our inference, it is evident that repeated rounds of DNA replication prior to unrefined division seem more likely at the onset than a fully regimented replicate-fall-in-line-up-and-divide mode of chromosome segregation. With RecA-type homologous recombination going on between DNA strands, the overall habit of this kind of reductive chromosome division would have more in common with meiosis than mitosis (a segment of the cell cycle, which for lacking cytokinesis does not have much utility in a syncytium anyway). We do not endeavor here to offer an explanation for why eukaryotic chromosomes came to undergo condensation, pairing, and alignment prior to nuclear division (closed state, without dissolution of the nuclear membrane). However, we do suggest that the process of ordered chromosome and nuclear division arose in a syncytial state, independent of cell division processes. This has the advantage of lowering the barriers of evolutionary invention that the first eukaryote had to surmount simultaneously. That is, a syncytial intermediate breaks down the almost intractably complex process of mitotic division into simpler yet still selectable component parts: Coordinated division of nucleus + chromosome being a simpler problem to solve than coordinated division of nucleus + chromosome + mitochondrion + cell. Our suggestion that coordinated nuclear and chromosome division evolved in a syncytial common ancestor would also directly account for the observation that all eukaryotes share the same basic conserved pattern of microtubule dependence in chromosome segregation (Koshland 1994) whereas their processes of cell division within and across supergroups are varied, for example, longitudinal fission in Euglena, phragmoblast formation in higher plants, gamete formation in Acetabulariua, or budding in yeast.
Fourth, the coenocyte offers possible transitional state solutions to the problem of cell division: Budding (spores). This is sketched in figure 3B. Archaea and bacteria produce membrane vesicles (Deatherage and Cookson 2012; Schwechheimer and Kuehn 2015; Gould et al. 2016). In archaea, the vesicles are pinched off from the cell surface with proteins of the Cdv (for cell division) family, which are archaeal precursors of the eukaryotic ESCRT III (for Vps2 and Vps4) proteins. These proteins are involved in making vesicles that protrude outwards from the cytosol, not inwards (as in endocytic processes). Reasonably assuming that our archaeal host had Cdv proteins, these could generate vesicles at the plasma membrane (Ellen et al. 2010). If no nuclei or mitochondria become contained in such a vesicle, fine, no problem, but no progeny. If nuclei with incomplete chromosome sets or lacking mitochondria become contained, also fine, also no progeny. But if vesicles come to contain both a mitochondrion and a nucleus (possibly more than one to start) with chromosome sets that are sufficiently complete to permit the formation of a new coenocyte, and sufficient proteins to initiate growth, the vesicle is a diaspore. This provides a very effective and powerful system of selection for combinations of nuclei and mitochondria that can found a new syncytium.
But, the diaspores are clonal. That brings us back to the dreaded dead end street of Muller’s ratchet. Unless, that is, the diaspores can fuse, like archaeal cells can, so as to permit new combinations of chromosomes and genes. If that happens, then what started out as a hopeless symbiotic consortium has basically completed a meiotic cell cycle (fig. 1) with a syncytial “diploid” stage. The diaspores are homologous to meiospores, their fusion is homologous to syngamy, and karyogamy can take place either immediately or in the syncytial state.
Fifth, and finally in this section, a coenocytic eukaryote common ancestor would go a long way to explaining why all of the traits that are common to eukaryotes were assembled in the eukaryote common ancestor, without intermediate forms in the prokaryote to eukaryote transition: There were attempts at the spawning of intermediates through budding, but only 1) those diaspores that came to possess complete chromosome sets and 2) those that were able to fuse with other diaspores of different chromosome parentage were viable on the long term. Together, these considerations, though not tested by modeling but clearly modelable using the stochastic corrector framework (Grey et al. 1995), provide cause to pursue the idea of a coenocytic eukaryote common ancestor, or Coeca. Nonetheless, if we look across eukaryotic supergroups, there is little alternative to the view that the last eukaryote ancestor was unicellular and that it possessed sex and a cell cycle, which bring us to the next section.
Coupling Chromosome Division to Cell Division
It is notable that chromosome division and cell division are not tightly coupled in many modern eukaryotes (Parfrey et al. 2008). During early eukaryogenesis, before a bona fide cell cycle had evolved, chromosome and cell division might not have been coupled at all. The syncytial intermediate inferred so far could probably divide nuclei and chromosomes (karyokinesis) with some proficiency and produce spores corresponding to meiospores, some of which would be viable (fig. 3B). It must have possessed a basic machinery for the scission act of cell division, otherwise it would have been unable to cleave off spores. In the simplest scenario, spores would do what their parental coenocyte did: Grow into another coenocyte, getting better through selection at ordered chromosome and nuclear division (fig. 4). If however, heterochrony—phenotypic expression at the wrong stage of development, here a change in the time spent between sporulation and being a syncytium—sets in, such that the spore cleavage process took place very early in filament growth, our eukaryote might have had a chance to attain a primitive form of that which its prokaryotic ancestors took for granted: Binary cell division, but this time with nuclei and mitochondria. In this respect, the vesicle (spore) formation function of Cdvs in archaea and their ESCRT homologs in eukaryotes, which are involved in cell division (Ellen et al. 2010; Morita et al. 2010), remained conserved.
Bypassing the syncytial stage (spore formation from spores, possibly akin to budding is yeasts) would generate cells whose fitness would dramatically improve by mutations that led to a temporally coordinated regulation of chromosome and cell division. In principle, a fairly straightforward process of selection could have brought forth the basics of a cell cycle (next section) as the solution to that problem. The products of these divisions would however, be clonal, returning our attention to Muller’s ratchet and the need for recombination to avoid extinction. Fusion of cells would more or less correspond to gamete fusion (syngamy) in the sexual cycle of modern eukaryotes. Yet recombination does not take place until karyogamy has occurred, and the reader will note that in figure 4 we have not indicated nuclei. The reason is that there are many possibilities regarding the timing of karyogamy (immediate, dikaryon phase, multinucleated phase), recombination and reduction, in addition to the issue of whether the mitotic cells are haploid or diploid (compare figs. 1 and 4), we just leave it open. We note however that the requirement for recombination (sex) in our inference—recalling that the starting point for this essay was the origin of sex—clearly traces alternation of generations into the eukaryote common ancestor.
Also of note, the proteins that serve to condense chromosomes during the cell cycle and align homologous chromosomes during homologous recombination, members of the SMC family, for structural maintenance of chromatin (Jeppsson et al. 2014), are extremely important in the prokaryote to eukaryote transition. SMC homologs are present in prokaryotes (Soppa 2001; Soppa et al. 2002), so as a gene family they are not a eukaryote-specific invention, but their gene family diversification and their cell cycle-specific expression (Jeppsson et al. 2014) clearly are eukaryote-specific attributes. We suggest that, as in the case of tubulin and some other cytoskeletal proteins, energetically unpenalized overexpression of preexisting prokaryotic genes for structural proteins in the eukaryote ancestor led to very useful and highly conserved processes: Chromatin condensation during the cell cycle and homologous chromatid pairing during meiotic recombination. The advent of cohesins (members of the SMC family) and the origin of homologous pairing was clearly important in eukaryote evolution, even the key event in the origin of meiosis under the synapsis homolog model (Wilkins and Holliday 2008). However, like other models that start with a mitosing cell that lacks meiosis, the synapsis homolog model takes the origin of mitosis as a given, which in our inference is an explanandum, hence the two models address very different things. Our proposal lacks mitosing cells incapable of recombination.
The cells at the bottom of figure 4 could represent the last eukaryote common ancestor, as such they would have had all the many traits that the last eukaryote common ancestor had, including the energy metabolic repertoire of a facultative anaerobe (Müller et al. 2012), a nucleus plus complex endomembrane system (Gould et al. 2016), and nowhere mentioned so far nor drawn in any figure, a eukaryotic flagellum. Clearly, a flagellum would improve the fitness of all single-celled stages. We have nowhere referred to phagocytosis, because despite occasional staunch claims in the literature, it is by no means clear that the eukaryotic ancestor was phagocytotic (Gould et al. 2016; Sousa et al. 2016). Although the ancestral eukaryotic habit sketched in figure 4 looks very much like a chytridiomycete, the spores of which have a flagellum (James et al. 2006), and would have a similar physiology (facultative anaerobes), any resemblance is not by design. Some fungi are syncytial and fungi are not phagocytotic.
Steps En Route to a Cell Cycle
With a heritable means to segregate chromosomes, natural selection can improve the basic invention: Cells with better, more refined and more accurate chromosome segregation proliferate according to their fitness. Though the nucleus permits continuous gene expression, it decouples the process of metabolite accumulation from cell and chromosome division—which in prokaryotes are tightly linked. Without means to coordinate metabolism with division, no single-celled eukaryotes will arise. Cyclins, also an attribute of the eukaryote common ancestor, apparently solved this problem by establishing a hierarchy of cytosol-based sensing and decision making, so as to sense 1) when metabolites had been accumulated (G1 checkpoint), 2) when chromosomes had been replicated (S phase checkpoint), and 3) when cell division could be initiated (Norbury and Nurse 1992; Morgan 1997).
The cyclin/CDK system reflects the origin of nucleus–cytosol compartmentalization. In the prokaryotic cytosol, DNA-binding proteins like DnaA bind directly to the chromosome where they are sensed by the replication machinery (Mott and Berger 2007); when DNA is cytosolic, the concentration of DnaA is linked to metabolism. In the ancestral eukaryotic cell, however, this communication is disrupted by the presence of the nuclear membrane, precipitating the need for new sensing mechanisms and possibly marking the advent of nuclear DNA-binding proteins with cytosolic-binding partners. Cyclins are indeed homologous to archaeal TFIIB (Gibson et al. 1994) and CDKs are ser/thr kinases, which are common among prokaryotes (Pereira et al. 2011; Kennelly 2014). For the cyclin/CDK system, no fundamental inventions were required, but interactions across novel compartments.
A primitive cyclin/CDK system could have established communication between the chromosomes and the cell division machinery, which had become disrupted by the origin of the nuclear membrane. In yeast it is possible to drive both mitosis and meiotic cell cycles by a single-engineered cyclin–CDK complex (Gutiérrez-Escribano and Nurse 2015) suggesting that a simpler ancestral network consisting of a few essential components could, in principle, underlie the origin of a primitive eukaryotic cell cycle regulation network.
A consequence of the cell cycle is that eukaryotes condense chromosomes and shut down once per cell division. Bacteria can shut down gene expression globally by shutting down ribosomes, for example, in toxin-mediated plasmid responses (Van Melderen and Bast 2009; Bertram and Schuster 2014). Global gene expression shutdown in eukaryotes entails chromatin-modification (Ptashne 2005). Chromatin-based shut down of gene expression is a eukaryotic invention, its evolutionary onset likely accompanied cell cycle origin (Maurer-Alcala and Katz 2015). Of course, a well-regulated cell cycle need not arise, but if it does not, no progeny will ensue. Genetic variation favoring the fixation of basic regulatory mechanisms governed by cyclins could generate the basic fabric of a cell cycle.
A basic cell cycle affords the first eukaryotes a plethora of fundamentally new possibilities. With mitochondrial power and ATP-dependent cytosolic structural proteins that can move and do things in the cytosol, they can undergo extended phases of gene expression without the burden of continuously dividing chromosomes (as in prokaryotes). This decoupling of chromosome replication from gene expression, and the regular shutdown of gene expression once per cell division is a hallmark of eukaryote biology. It enables long phases of gene expression from chromosomes that are not dividing, but are specifically dedicated to the gene expression process. The cell cycle shuts gene expression down, initiates chromosome and cell division, and then allows gene expression anew. Processes of cell development became possible that unfolded from the shut-down-and-reboot nature of chromatin condensation at every cell division. Mitochondrial power allowed eukaryotes to explore new protein function and protein overexpression in the cytosol, where eukaryote complexity takes root.
With the basic sequence of cell division, gene expression, cell fusion, and recombination in place, a meiotic cell cycle (recombination at every division) becomes dispensable. Occasional recombination suffices to escape Muller’s ratchet (Muller 1964; Felsenstein 1974). From this more complex meiotic starting point, mitotic shortcuts in either the haploid or the diploid state are readily attained by shortening meiosis (fig. 1), as are variants extending the number of mitotic divisions between meiosis (fig. 4). Most cell divisions are once again clonal, as in prokaryotes, but with new chromosome segregation mechanisms. With the conserved core of meiosis and mitosis in place, ploidy phase variation was possible. Eukaryotes, especially protists, exhibit baroque diversity among ploidy cycles (Parfrey et al. 2008; Parfrey and Katz 2010), but they do not relinquish their genes for sex (Ramesh et al. 2005).
Some Consequences of a Primordial Coenocytic Model
Karyogamy and karyokinesis within the syncytial intermediate have a curious attribute: Together, they homogenize populations of individually unviable chromosome sets otherwise headed to extinction. This is of interest in several ways.
First, it fits very well with the checkpoints in the cell cycle that carefully monitor, hence insure, proper chromosome replication: Do not enter into chromosome and cell division until the chromosomes are fully replicated. This would have been an important milestone en route to achieving a regulated cell cycle of the type underpinning eukaryote cell division today. However, regulated chromosome division at the coenocytic state could have evolved without the need for simultaneously coupling regulated chromosome division to regulated cell division, because the content of spores resulting from scission (our suggested precursor to cell division) was initially random, viable contents being selected.
Second, at the syncytial stage, eukaryotes had solved their carbon and energy problems with the help of glycolysis in the cytosol and terminal oxidation plus ATP export in mitochondria. With their core metabolic problems solved in a virtually unimprovable manner (no known eukaryote has ever replaced or supercharged its mitochondria) members of the emergent eukaryotic lineage could no longer genuinely benefit from LGT with prokaryotes. They did not need new terminal oxidases in the inner mitochondrial membrane or NADH oxidizing enzymes in the cytosol. They needed maintenance and improvements in the regulation of their operational yet still clumsy cell and chromosome division. That is to say, what they needed for lineage survival they could not get from prokaryotes, only from other eukaryotic chromosomes, namely variants on the themes surrounding the formation and regulation of novel structures and processes in the cytosol that emerge from suddenly affordable ATP-costly protein overexpression in that compartment and ATP-dependent protein aggregation states and protein interaction states therein. The genes and proteins underpinning eukaryotic specific traits (nucleus, endomembrane system, and the like) arose in the eukaryotic ancestor, eukaryotic cells were thus the only existing source of newly emergent genes that characterized the eukaryotic lineage. The transition to sex marked the departure from the LGT mechanisms that impart recombination among prokaryotes, and simultaneously marked the advent of reciprocal recombination among kin as the mechanism to escape Müller’s ratchet in the eukaryotic lineage. The origin of eukaryotes was the origin of vertical lineage inheritance (Ku et al. 2015), and sex was required to keep vertically evolving lineages viable.
Third, recurrent genome fusions generate multiple gene copies and large gene families, much in the same way that whole-genome duplications do among eukaryotes today. On the one hand, this creates massive paralogy among ancestral eukaryotic genes. In the presence of mitochondria, which permit the new gene copies to be expressed as protein, it also creates large gene families for the genes specific to the eukaryotic lineage, thereby allowing experimentation, functional specialization and fixation of members within eukaryote-specific gene families involved in membrane traffic, cell structure, and signaling. Clearly, our proposal predicts the existence of massive paralogy in the eukaryote common ancestor.
Fourth and finally, the syncytial and meiospore fusion aspects help to explain why eukaryote origins left no intermediate forms. Only when the whole suite of genes required for survival as a nucleus-bearing cell was assembled, were the products of cell division (meiospore) genuinely self-sufficient to an extent that would allow diversification into descendant lineages.
Conclusion
Our proposal has it that eukaryotic sex arose as a necessary replacement for prokaryotic gene transfer mechanisms in a cell that had evolved a nucleus (as a consequence of mitochondrial group II introns) and that was consequently able to undergo the transition to MDCS as a fortuitous byproduct of mitochondrial energetics. The mitochondrion transformed its host, generating a cytosol 1) where protein could be synthesized in amounts no prokaryote had ever experienced, 2) where proteins could interact without interfering either with chromatin or with cell division, and 3) where spontaneous aggregation of structural proteins to higher order structures and ATP-dependent disaggregation became possible. Selection was acting upon the outcome of spontaneous protein interactions—spontaneous chemical processes were thus decisive at this major evolutionary transition.
Once it exists, the benefits of sex are manifold. The how and why of how sex came into existence are more challenging. The DNA damage hypothesis (Hörandl and Hadacek 2013) posits that meiosis evolved as a repair response to oxidative damage. Our model would not exclude a role for recombination in repair, since the enzymes involved in meiotic recombination are homologous to prokaryotic repair machineries (Camerini-Otero and Hsieh 1995; Ramesh et al. 2005), and because mitochondria (the source of reactive oxygen species in eukaryotes) are present in the cell that evolved sex. Homolog synapsis involving cohesins, members of the SMC protein family (Jeppsson et al. 2014), has been suggested as the key innovation that allowed for the evolution of meiosis from mitosis (Wilkins and Holliday 2008). In our proposal, the chromosomes are initially heterogeneous. Hence until a homogeneous, ancestral eukaryote genome with defined chromosome numbers having haploid and diploid states during an evolved cell cycle and life cycle had emerged, homologous pairing would not have been obviously beneficial. Congruent with that view, many prokaryotes possess clear homologs of the SMC proteins that promote eukaryote chromosome condensation and that lead to pairing of sister chromatids (Soppa 2001; Soppa et al. 2002), yet prokaryotes neither condense their chromatin at cell division nor do they possess sister chromatids.
A number of major evolutionary transitions in eukaryote evolution involve endosymbiosis: The origin of mitochondria, the origin of plastids, and the origin of major algal groups through secondary endosymbiosis. Each of those endosymbiotic transitions also left a major impact on the genome in the form of gene transfers from organelles to the nucleus (Martin and Müller 1998; Martin et al. 2002; Ku et al. 2015). But endosymbiosis is not readily accommodated either by mathematics or by the gradualist paradigm of population genetics, which is why mitochondria play no role whatsoever in population genetic approaches to understanding the prokaryote–eukaryote transition (Lynch and Conery 2003; Lynch 2006; Lynch and Marinov 2015). Indeed, population genetic investigations tend to recognize no difference at all between prokaryotes and eukaryotes, both of which appear to map out along an uninterrupted continuum by the measure of population genetic parameters (Lynch and Conery 2003). That is not a criticism of population genetics, it is a statement about the evolutionary divide separating eukaryotes from prokaryotes. Viewed solely through the looking glass of population genetics and allele frequencies, one would not even be able to tell the difference between a lion and a palm tree, because on the long term, both have sex and diploid genetics. Clearly, population genetics does not tell us everything that we need to know about eukaryote evolution, if we are interested in the physiological and cell biological differences that distinguish eukaryotes from prokaryotes.
At the prokaryote to eukaryote transition, many major changes took place: Mitochondria, endomembrane system, nucleus, meiosis, mitosis, and the origin of sex. We have endeavored here to account for those differences in one essay. In doing so we necessarily crossed a border from modern endosymbiotic theory, starting from an archaeaon as the host, to population genetics (sex). We were able to sketch, albeit in broad strokes, a general outline that bridged the evolutionary gap between prokaryotes and eukaryotes and that resulted in a mitochondriate, nucleated, sexually recombining and mitosing cell. Whether population genetic approaches would be able to predict the single origin of plastids or mitochondria during evolution remains an open question. Whether the eukaryotic cell cycle or endomembrane system could be modeled as a population genetic parameter is also an open question. When it comes to modeling the processes underlying the cell biological differences that distinguish eukaryotes from prokaryotes and taking them apart into simpler, conceptually tractable components, endosymbiotic theory works fairly well. Yet endosymbiotic theory is founded in comparative biology and comparative physiology, not in mathematics and statistics, hence as a field it does not interface well with population genetics. Maybe future progress will improve that circumstance.
Sex, the cell cycle, chromosome division, and alternation of generations are processes. Inferring the evolutionary origin of processes is arguably more difficult than inferring the origin of structures. Understanding the origin of organelles and structures that distinguish eukaryotes from prokaryotes, for example, the endomembrane system, is important, and a strong case can be made that the origin of mitochondria had the decisive role in endomembrane origin (Gould et al. 2016). A better understanding of eukaryote origin requires understanding the evolution of eukaryotic processes. The syncytial stage has many virtues as an evolutionary intermediate. In terms of structuring the problem of eukaryote process origins, it also helps to break down the complex eukaryotic cell cycle and life cycle (fig. 1) into simpler component processes (fig. 4), and in doing so draws attention to the largely (but not completely: Cavalier-Smith 2010) neglected issue of the origin of alternation of generation in eukaryotes. Students of biology have had to learn alternation of generations for over a century, yet without an evolutionary context that could account for the origin of the sexual processes (karyogamy and reduction division) that connect the alternating generations. We have made an effort here to put sex and the alternation of generations into the evolutionary context of modern endosymbiotic theory. Our evolutionary intermediates obtain new gene variants through karyogamy in a syncytium and through archaeal-type spore fusion. The end result of the inference is a population of free-living, unicellular, sexually recombining and mitosing cells that have a cell cycle, archaeal ribosomes in the cytosol, and mitochondria.
In recent years, views on the origin of eukaryotes have changed in that 1) the mitochondrion is now recognized to be ancestrally present in the eukaryote common ancestor and in that 2) the host is now considered to have been an archaeon (Martin and Müller 1998; Martin and Koonin 2006; Cox et al. 2008; Lane and Martin 2010; Williams et al. 2013; McInerney et al. 2014; Raymann et al. 2015; Spang et al. 2015; Sousa et al. 2016; Gould et al. 2016). Views on the origin of sex have not changed at the same pace, though there is increasing interest in the possible role(s) of mitochondria in promoting the establishment of meiotic recombination (Lane 2009,2015; Speijer et al. 2015; Havird et al. 2015; Radzvilavicius and Blackstone 2015). Here, we have considered the origin of sex on the basis of those newer premises.
In contrast to earlier theories, we do not assume that the cell in which sex arose had already evolved a mitotic cell cycle. Rather, we start from a symbiotic association of two prokaryotes, which led to the origin of mitochondria, and consider the evolutionary sequence of events. As the most notable departures from previous theories on the origin of eukaryotes, mitosis or sex, 1) we posit that mitotic division is evolutionarily derived from meiotic division; 2) we place both processes in their natural context of eukaryotic cell cycle origin; 3) we propose a coenocytic eukaryote common ancestor, Coeca, which allowed nuclei harboring defective in chromosome sets to complement each other through mRNA in the cytosol; 4) we suggest that the first form of eukaryotic cell division was budding of meiospores from the coenocyte, a process that selected viable combinations of chromosomes and mitochondria; and 5) we suggest that the ability of meiospores to fuse, a property of archaeal cells, allowed them to undergo homologous meiotic recombination, or sex. It is an observation that sex has been retained in all eukaryotic lineages for over 1.7 Gyr, we suggest that the reason for its retention is the same as the reason for its fixation at eukaryote origins: It saves eukaryotes from extinction through Muller’s ratchet. Our inference orders the origin of major evolutionary innovations in the eukaryotic lineage as follows: Mitochondria, followed by the nucleus and endomembrane system, MDCS, reduction division in a syncytial eukaryote common ancestor, meiospore production, division and fusion leading to a meiotic life cycle and cell cycle (sex), and finally mitosis through bypassing of recombination and reduction division during the life cycle. The syncytial nature of the eukaryote common ancestor is a particularly interesting, and possibly useful, element of our proposal. It is interesting because it decouples the processes of chromosome segregation, ploidy cycling, karyogamy, progeny generation, cell division, and the cell cycle from each other. It is useful because it allows one to consider the evolution of each of those actions as a tendentially independent biological process, which fits well with the diversity, or lack thereof, observed for each of these processes among contemporary eukaryotic groups.
Acknowledgments
The authors gratefully acknowledge funding from the European Research Council (ERC 666053 to W.F.M.). They thank James O. McInerney, Laura A. Katz, Nick Lane, Eors Szathmary, Sven B. Gould, Neil Blackstone, John F. Allen, John Logsdon, and especially Klaus V. Kowallik for constructive discussions. The authors gratefully acknowledge funding from the European Research Council (ERC 666053 to W.F.M.).
Literature Cited
- Allen JF. 1996. Separate sexes and the mitochondrial theory of ageing. J Theor Biol. 180:135–140. [DOI] [PubMed] [Google Scholar]
- Allen JF. 2015. Why chloroplasts and mitochondria retain their own genomes and genetic systems: colocation for redox regulation of gene expression. Proc Natl Acad Sci U S A. 112:10231–10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audibert A, Weil D, Dautry F. 2002. In vivo kinetics of mRNA splicing and transport in mammalian cells. Mol Cell Biol. 22:6706–6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker EP, Van Dam K. 1974. The movement of monocarboxylic acids across phospholipid membranes: evidence for an exchange diffusion between pyruvate and other monocarboxylate ions. Biochim Biophys Acta–Biomembranes. 339:285–289. [DOI] [PubMed] [Google Scholar]
- Beech PL., et al. 2000. Mitochondrial FtsZ in a chromophyte alga. Science 287:1276–1279. [DOI] [PubMed] [Google Scholar]
- Bell G. 1982. The masterpiece of nature: the evolution and genetics of sexuality. Berkeley: Univ. of California Press. [Google Scholar]
- Bentley SD., et al. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147. [DOI] [PubMed] [Google Scholar]
- Bernstein H, Byerly HC, Hopf FA, Michod RE. 1984. Origin of sex. J Theor Biol. 110:323–351. [DOI] [PubMed] [Google Scholar]
- Bertram R, Schuster CF. 2014. Post-transcriptional regulation of gene expression in bacterial pathogens by toxin-antitoxin systems. Front Cell Infect Microbiol. 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone NW. 2013. Why did eukaryotes evolve only once? Genetic and energetic aspects of conflict and conflict mediation. Phil Trans R Soc B. 368:20120266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV. 2011. Cell cycle arrest is not senescence. Aging 4:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield G. 2016. Atypical ploidy cycles, Spo11, and the evolution of meiosis. Sem Cell Dev Biol. 54:158–164. [DOI] [PubMed] [Google Scholar]
- Booth A, Doolittle WF. 2015. Eukaryogenesis, how special really? Proc Natl Acad Sci U S A. 112:10278–10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricheux G, Coffe G, Brugerolle G. 2007. Identification of a new protein in the centrosome-like “atractophore” of Trichomonas vaginalis. J Microbiol Immunol Infect. 153:133–140. [DOI] [PubMed] [Google Scholar]
- Esser C., et al. 2004. A genome phylogeny for mitochondria among alpha-proteobacteria and a predominantly eubacterial ancestry of yeast nuclear genes. Mol Biol Evol. 21:1643–1660. [DOI] [PubMed] [Google Scholar]
- Camerini-Otero RD, Hsieh P. 1995. Homologous recombination proteins in prokaryotes and eukaryotes. Annu Rev Genet. 29:509–552. [DOI] [PubMed] [Google Scholar]
- Cande WZ, McDonald KL. 1985. In vitro reactivation of anaphase spindle elongation using isolated diatom spindles. Nature 316:168–170. [DOI] [PubMed] [Google Scholar]
- Castagnetti S, Božič B, Svetina S. 2015. Mechanical and molecular basis for the symmetrical division of the fission yeast nuclear envelope. Phys Chem Chem Phys. 17:15629–15636. [DOI] [PubMed] [Google Scholar]
- Cavalier-smith T. 1975. The origin of nuclei and of eukaryotic cells. Nature 256:463–468. [DOI] [PubMed] [Google Scholar]
- Cavalier-Smith T. 2002. Origins of the machinery of recombination and sex. Heredity 88:125–141. [DOI] [PubMed] [Google Scholar]
- Cavalier-Smith T. 2010. Origin of the cell nucleus, mitosis and sex: roles of intracellular coevolution. Biol Direct. 5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-smith T. 2014. The neomuran revolution and phagotrophic origin of eukaryotes and cilia in the light of intracellular coevolution and a revised tree of life. Cold Spring Harb Perspect Biol. 6:a016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland LR. 1947. The origin and evolution of meiosis. Science 105:287–289. [DOI] [PubMed] [Google Scholar]
- Collins L, Penny D. 2005. Complex spliceosomal organization ancestral to extant eukaryotes. Mol Biol Evol. 22:1053–1066. [DOI] [PubMed] [Google Scholar]
- Cox CJ, Foster PG, Hirt RP, Harris SR, Embley TM. 2008. The archaebacterial origin of eukaryotes. Proc Natl Acad Sci U S A. 105:20356–20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross FR, Buchler NE, Skotheim JM. 2011. Evolution of networks and sequences in eukaryotic cell cycle control. Phil Trans R Soc B. 366:3532–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF. 2005. Timeline–Hermann Joseph Muller, evolutionist. Nat Rev Genet. 6:941–945. [DOI] [PubMed] [Google Scholar]
- de Duve C. 2007. The origin of eukaryotes: a reappraisal. Nat Rev Genet. 8:395–403. [DOI] [PubMed] [Google Scholar]
- de Paula WBM, Lucas CH, Agip ANA, Vizcay-Barrena G, Allen JF. 2013. Energy, ageing, fidelity and sex: oocyte mitochondrial DNA as a protected genetic template. Phil Trans R Soc B. 368:20120263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage BL, Cookson BT. 2012. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun. 80:1948–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli Esposti M. 2014. Bioenergetic evolution in proteobacteria and mitochondria. Genome Biol Evol. 6:3238–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doležal P, Likic V, Tachezy J, Lithgow T. 2006. Evolution of the molecular machines for protein import into mitochondria. Science 313:314–318. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. 1999. Phylogenetic classification and the universal tree. Science 284:2124–2128. [DOI] [PubMed] [Google Scholar]
- Ellen AF, Zolghadr B, Driessen AMJ, Albers S-V. 2010. Shaping the archaeal cell envelope. Archaea 2010:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embley TM, Martin W. 2006. Eukaryotic evolution, changes and challenges. Nature 440:623–630. [DOI] [PubMed] [Google Scholar]
- Erickson HP. 1995. FtsZ, a prokaryotic homolog of tubulin? Cell 80:367–370. [DOI] [PubMed] [Google Scholar]
- Erickson HP. 2007. Evolution of the cytoskeleton. BioEssays 29:668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. 1974. The evolutionary advantage of recombination. Genetics 78:737–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury-Aubusson A. 2003. “Novel cytoskeletal proteins in protists”: introductory remarks. J Eukaryot Microbiol. 50:3–8. [DOI] [PubMed] [Google Scholar]
- Garg S., et al. 2015. Conservation of transit peptide-independent protein import into the mitochondrial and hydrogenosomal matrix. Genome Biol Evol. 7:2716–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilova OV, Rundanova EE. 1999. Cell architecture in the morphogenesis of coenocytic alga Vaucheria sessilis. I. The morphology of germination and the behaviour of nuclei. Protistology 1:5–9. [Google Scholar]
- Gibson TJ, Thompson JD, Blocker A, Kouzarides T. 1994. Evidence for a protein domain superfamily shared by the cyclins, TFIIB and RB/p107. Nucleic Acids Res. 22:946–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough U, Heitman J. 2014. Origins of eukaryotic sexual reproduction. Cold Spring Harb Perspect Biol. 6:a016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould SB, Garg SG, Martin WF. 2016. Bacterial vesicle secretion and the evolutionary origin of the eukaryotic endomembrane system. Trends Microbiol. 24:525–534. [DOI] [PubMed] [Google Scholar]
- Gould SB, et al. 2011. Ciliate pellicular proteome identifies novel protein families with characteristic repeat motifs that are common to Alveolates. Mol Biol Evol. 28:1319–1331. [DOI] [PubMed] [Google Scholar]
- Grey D, Hutson V, Szathmáry E. 1995. A re-examination of the stochastic corrector model. Proc R Soc B. 262:29–35. [Google Scholar]
- Gutiérrez-Escribano P, Nurse P. 2015. A single cyclin-CDK complex is sufficient for both mitotic and meiotic progression in fission yeast. Nat Commun. 6:6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjivasiliou Z, Lane N, Seymour RM, Pomiankowski A. 2013. Dynamics of mitochondrial inheritance in the evolution of binary mating types and two sexes. Proc R Soc B. 280:20131920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halary S., et al. 2011. Conserved meiotic machinery in Glomus spp., a putatively ancient asexual fungal lineage. Genome Biol Evol. 3:950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand ML, Koltunow AMG. 2014. The genetic control of apomixis: asexual seed formation. Genetics 197:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold FM. 1986. The vital force. New York: WH Freeman. [Google Scholar]
- Hartwell LH, Weinert TA. 1989. Checkpoints: controls that ensure the order of cell cycle events. Science 246:629–634. [DOI] [PubMed] [Google Scholar]
- Hasitschka-Jenschke G. 1960. Beitrag zur Karyologie von Characeen. Österreichische Botanische Zeitschrift. 107:228–240. [Google Scholar]
- Havird JC, Hall MD, Dowling DK. 2015. The evolution of sex: a new hypothesis based on mitochondrial mutational erosion. Bioessays 37:951–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazkani-Covo E, Covo S. 2008. Numt-mediated double-strand break repair mitigates deletions during primate genome evolution. PLoS Genet. 4:e1000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazkani-Covo E, Zeller RM, Martin W. 2010. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 6:e1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hörandl E, Hadacek F. 2013. The oxidative damage initiation hypothesis for meiosis. Plant Reprod. 26:351–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourcade D, Dressler D. 1973. The amplification of ribosomal RNA genes involves a rolling circle intermediate. Proc Natl Acad Sci U S A. 70:2926–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Ayliffe MA, Timmis JN. 2004. Simple and complex nuclear loci created by newly transferred chloroplast DNA in tobacco. Proc Natl Acad Sci U S A. 101:9710–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Grünheit N, Ahmadinejad N, Timmis JN, Martin W. 2005. Mutational decay and age of chloroplast and mitochondrial genomes transferred recently to angiosperm nuclear chromosomes. Plant Physiol. 138:1723–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Nurse P. 1991. A note on the evolution of meiosis. J Theor Biol. 150:561–563. [DOI] [PubMed] [Google Scholar]
- Husnik F., et al. 2013. Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell 153:1567–1578. [DOI] [PubMed] [Google Scholar]
- James TY., et al. 2006. A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 98:860–871. [DOI] [PubMed] [Google Scholar]
- Jékely G. 2014. Origin and evolution of the self-organizing cytoskeleton in the network of eukaryotic organelles. Cold Spring Harb Perspect Biol. 6:a016030–a016030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppsson K, Kanno T, Shirahige K, Sjögren C. 2014. The maintenance of chromosome structure: positioning and functioning of SMC complexes. Nat Rev Mol Cell Biol. 15:601–614. [DOI] [PubMed] [Google Scholar]
- Jones D, Sneath PH. 1970. Genetic transfer and bacterial taxonomy. Bacteriol Rev. 34:40–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LA. 2012. Origin and diversification of eukaryotes. Annu Rev Microbiol. 66:411–427. [DOI] [PubMed] [Google Scholar]
- Kennelly PJ. 2014. Protein Ser/Thr/Tyr phosphorylation in the Archaea. J Biol Chem. 289:9480–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T. 2011. Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell Mol Life Sci. 68:1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobialka M, Michalik A, Walczak M, Junkiert L, Szklarzewicz T. 2016. Sulcia symbiont of the leafhopper Macrosteles laevis (Ribaut, 1927) (Insecta, Hemiptera, Cicadellidae: Deltocephalinae) harbors Arsenophonus bacteria. Protoplasma 253:903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov AS. 1994. The asexual ploidy cycle and the origin of sex. Nature 370:213–216. [DOI] [PubMed] [Google Scholar]
- Koonin EV. 2015. Origin of eukaryotes from within archaea, archaeal eukaryome and bursts of gene gain: eukaryogenesis just made easier? Phil Trans R Soc B. 370:20140333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland D. 1994. Mitosis: back to the basics. Cell 77:951–954. [DOI] [PubMed] [Google Scholar]
- Krylov DM, Nasmyth K, Koonin EV. 2003. Evolution of eukaryotic cell cycle regulation: stepwise addition of regulatory kinases and late advent of the CDKs. Curr Biol. 13:173–177. [DOI] [PubMed] [Google Scholar]
- Ku C., et al. 2015. Endosymbiotic origin and differential loss of eukaryotic genes. Nature 524:427–432. [DOI] [PubMed] [Google Scholar]
- Kuwabara T., et al. 2005. Thermococcus coalescens sp. nov., a cell-fusing hyperthermophilic archaeon from Suiyo Seamount. Int J Syst Evol Microbiol. 55:2507–2514. [DOI] [PubMed] [Google Scholar]
- Lambowitz AM, Zirnmerly S. 2004. Mobile group II introns. Annu Rev Genet. 38:1–35. [DOI] [PubMed] [Google Scholar]
- Lambowitz AM, Zimmerly S. 2011. Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol. 3:a003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N. 2005. Power, sex, suicide: mitochondria and the meaning of life. Oxford: Oxford University Press. [Google Scholar]
- Lane N. 2009. Life ascending. London: Profile Books. [Google Scholar]
- Lane N. 2014. Bioenergetic constraints on the evolution of complex life. Cold Spring Harb Perspect Biol. 6:a015982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N. 2015. The vital question. Why is life the way it is? London: Profile Books. [Google Scholar]
- Lane N, Martin W. 2010. The energetics of genome complexity. Nature 467:929–934. [DOI] [PubMed] [Google Scholar]
- Lane N, Martin WF. 2015. Eukaryotes really are special, and mitochondria are why. Proc Natl Acad Sci U S A. 112:E4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N, Martin WF. 2016. Mitochondria, complexity, and evolutionary deficit spending. Proc Natl Acad Sci U S A. 113:E666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AS, Zhaxybayeva O, Beatty JT. 2012. Gene transfer agents: phage-like elements of genetic exchange. Nat Rev Microbiol. 10:472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindås AC, Bernander R. 2013. The cell cycle of archaea. Nat Rev Microbiol. 11:627–638. [DOI] [PubMed] [Google Scholar]
- Lukjancenko O, Wassenaar TM, Ussery DW. 2010. Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol. 60:708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2006. The origins of eukaryotic gene structure. Mol Biol Evol. 23:450–468. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. 2003. The origins of genome complexity. Science 302:1401–1404. [DOI] [PubMed] [Google Scholar]
- Lynch M, Marinov GK. 2015. The bioenergetic costs of a gene. Proc Natl Acad Sci U S A. 112:15690–15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Richardson AO. 2002. The evolution of spliceosomal introns. Curr Opin Genet Dev. 12:701–710. [DOI] [PubMed] [Google Scholar]
- Martin W. 1999. Mosaic bacterial chromosomes: a challenge en route to a tree of genomes. BioEssays 21:99–104. [DOI] [PubMed] [Google Scholar]
- Martin W, Brinkmann H, Savonna C, Cerff R. 1993. Evidence for a chimeric nature of nuclear genomes: eubacterial origin of eukaryotic glyceraldehyde-3-phosphate dehydrogenase genes. Proc Natl Acad Sci U S A. 90:8692–8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W, Koonin EV. 2006. Introns and the origin of nucleus–cytosol compartmentalization. Nature 440:41–45. [DOI] [PubMed] [Google Scholar]
- Martin W, Müller M. 1998. The hydrogen hypothesis for the first eukaryote. Nature 392:37–41. [DOI] [PubMed] [Google Scholar]
- Martin W., et al. 2002. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc Natl Acad Sci U S A. 99:12246–12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura M, et al. 1997. A bacterial group II intron encoding reverse transcriptase, maturase, DNA endonuclease activities: biochemical demonstration of maturase activity and insertion of new genetic information within the intron. Genes Dev. 11:2910–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Alcala XX, Katz LA. 2015. An epigenetic toolkit allows for diverse genome architectures in eukaryotes. Curr Opin Genet Dev. 35:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J. 1978. The evolution of sex. Cambridge: Cambridge Univ. Press. [Google Scholar]
- Maynard Smith J. 1986. Evolution: contemplating life without sex. Nature 324:300–301. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J., Szathmary E. 1997. The major transitions in evolution. Oxford: Oxford Univ. Press. [Google Scholar]
- McCutcheon JP, McDonald BR, Moran NA. 2009. Convergent evolution of metabolic roles in bacterial co-symbionts of insects. Proc Natl Acad Sci U S A. 106:15394–15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath CL, Katz LA. 2004. Genome diversity in microbial eukaryotes. Trends Ecol Evol. 19:32–38. [DOI] [PubMed] [Google Scholar]
- McInerney JO, O'Connell MJ, Pisani D. 2014. The hybrid nature of the Eukaryota and a consilient view of life on Earth. Nat Rev Microbiol. 12:449–455. [DOI] [PubMed] [Google Scholar]
- McNaughton EE, Goff LJ. 1990. The role of microtubules in establishing nuclear spatial patterns in multinucleate green algae. Protoplasma 157:19–37. [Google Scholar]
- Mereschkowsky C. 1905. Über Natur und Ursprung der Chromatophoren im Pflanzenreiche. Biol Centralbl. 25:593–604. [English translation in Martin W, Kowallik KV (1999) Eur. J. Phycol. 34:287-295]. [Google Scholar]
- Mierzejewska J, Jagura-Burdzy G. 2012. Prokaryotic ParA-ParB-parS system links bacterial chromosome segregation with the cell cycle. Plasmid 67:1–14. [DOI] [PubMed] [Google Scholar]
- Mitchison JM. 1971. The biology of the cell cycle. London: Cambridge Univ. Press. [Google Scholar]
- Moran NA. 1996. Accelerated evolution and Muller's ratchet in endosymbiotic bacteria. Proc Natl Acad Sci U S A. 93:2873–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO. 1997. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 13:261–291. [DOI] [PubMed] [Google Scholar]
- Morita E., et al. 2010. Human ESCRT-III and VPS4 proteins are required for centrosome and spindle maintenance. Proc Natl Acad Sci U S A. 107:12889–12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott ML, Berger JM. 2007. DNA replication initiation: mechanisms and regulation in bacteria. Nat Rev Microbiol. 5:343–354. [DOI] [PubMed] [Google Scholar]
- Muller HJ. 1964. The relation of recombination to mutational advance. Mutat Res. 106:2–9. [DOI] [PubMed] [Google Scholar]
- Müller M., et al. 2012. Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol Mol Biol Rev. 76:444–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naor A, Gophna U. 2014. Cell fusion and hybrids in Archaea. Bioengineered 4:126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K. 1995. Evolution of the cell-cycle. Phil Trans R Soc B. 349: 271–281. [DOI] [PubMed] [Google Scholar]
- Nei M. 1987. Molecular evolutionary genetics. New York: Columbia University Press [Google Scholar]
- Neidhardt FC, Ingraham JL, Schaechter M. 1990. Physiology of the bacterial cell. Sunderland (MA): Sinauer Associates Incorporated [Google Scholar]
- Nelson-Sathi S., et al. 2015. Origins of major archaeal clades correspond to gene acquisitions from bacteria. Nature 517:77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E. 2000. Structural insights into microtubule function. Annu Rev Biochem. 69:277–302. [DOI] [PubMed] [Google Scholar]
- Norbury C, Nurse P. 1992. Animal cell cycles and their control. Annu Rev Biochem. 61:441–468. [DOI] [PubMed] [Google Scholar]
- Novak B, Csikasz-Nagy A, Gyorffy B, Nasmyth K, Tyson JJ. 1998. Model scenarios for evolution of the eukaryotic cell cycle. Phil Trans R Soc B. 353:2063–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley BR. 2000. An abundance of tubulins. Trends Cell Biol. 10:537–542. [DOI] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304. [DOI] [PubMed] [Google Scholar]
- Otto SP, Goldstein DB. 1992. Deleterious mutations and the origin of the meiotic ploidy cycle. Genetics 131:745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RT, Koenig JE, Rodriguez-Valera F, Doolittle WF. 2004. Frequent recombination in a saltern population of Halorubrum. Science 306:1928–1929. [DOI] [PubMed] [Google Scholar]
- Parfrey LW, Katz LA. 2010. Dynamic genomes of eukaryotes and the maintenance of genomic integrity. Microbe 5:156–163. [Google Scholar]
- Parfrey LW, Lahr DJG, Katz LA. 2008. The dynamic nature of eukaryotic genomes. Mol Biol Evol. 25:787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey LW, Lahr DJG, Knoll AH, Katz LA. 2011. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc Natl Acad Sci U S A. 108:13624–13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira SFF, Goss L, Dworkin J. 2011. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol Mol Biol Rev. 75:192–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett-Heaps JD, Tippit DH, Porter KR. 1982. Rethinking mitosis. Cell 29:729–744. [DOI] [PubMed] [Google Scholar]
- Ptashne M. 2005. Regulation of transcription: from lambda to eukaryotes. Trends Biochem Sci. 30:275–279. [DOI] [PubMed] [Google Scholar]
- Purkanti R, Thattai M. 2015. Ancient dynamin segments capture early stages of host–mitochondrial integration. Proc Natl Acad Sci U S A. 112:2800–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzvilavicius AL, Blackstone NW. 2015. Conflict and cooperation in eukaryogenesis: implications for the timing of endosymbiosis and the evolution of sex. J R Soc Interface 12:20150584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raikov IB. 1994. The diversity of forms of mitosis in protozoa–a comparative review. Eur J Protistol. 30:253–269. [Google Scholar]
- Ramesh MA, Malik S-B, Logsdon JM., Jr. 2005. A phylogenomic inventory of meiotic genes. Curr Biol. 15:185–191. [DOI] [PubMed] [Google Scholar]
- Rasko DA., et al. 2008. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol. 190:6881–6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymann K, Brochier-Armanet C, Gribaldo S. 2015. The two-domain tree of life is linked to a new root for the Archaea. Proc Natl Acad Sci U S A. 112:6670–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich PR, Maréchal A. 2010. The mitochondrial respiratory chain. Essays Biochem. 47:1–23. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Wolf YI, Sorokin AV, Mirkin BG, Koonin EV. 2003. Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr Biol. 13:1512–1517. [DOI] [PubMed] [Google Scholar]
- Roper M, Ellison C, Taylor JW, Glass NL. 2011. Nuclear and genome dynamics in multinucleate ascomycete fungi. Curr Biol. 21: R786–R793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougier N, Werb Z. 2001. Minireview: parthenogenesis in mammals. Mol Reprod Dev. 59:468–474. [DOI] [PubMed] [Google Scholar]
- Schleper C, Holz I, Janekovic D, Murphy J, Zillig W. 1995. A multicopy plasmid of the extremely thermophilic archaeon Sulfolobus effects its transfer to recipients by mating. J Bacteriol. 177:4417–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher MA., et al. 2015. Structures of archaeal DNA segregation machinery reveal bacterial and eukaryotic linkages. Science 349:1120–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwechheimer C, Kuehn MJ. 2015. Outer-membrane vesicles from gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol. 13:605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searcy DG. 2003. Metabolic integration during the evolutionary origin of mitochondria. Cell Res. 13:229–238. [DOI] [PubMed] [Google Scholar]
- Sharp PA. 1985. On the origin of RNA splicing and introns. Cell 42: 397–400. [DOI] [PubMed] [Google Scholar]
- Solari AJA. 2002. Primitive forms of meiosis: the possible evolution of meiosis. Biocell 26:1–13. [PubMed] [Google Scholar]
- Soppa J. 2001. Prokaryotic structural maintenance of chromosomes (SMC) proteins: distribution, phylogeny, and comparison with MukBs and additional prokaryotic and eukaryotic coiled-coil proteins. Gene 278:253–264. [DOI] [PubMed] [Google Scholar]
- Soppa J, et al. 2002. Discovery of two novel families of proteins that are proposed to interact with prokaryotic SMC proteins, and characterization of the Bacillus subtilis family members ScpA and ScpB. Mol Microbiol. 45:59–71. [DOI] [PubMed] [Google Scholar]
- Sørensen MA, Kurland CG, Pedersen S. 1989. Codon usage determines translation rate in Escherichia coli. J Mol Biol. 207:365–377. [DOI] [PubMed] [Google Scholar]
- Sousa FL, Neukirchen S, Allen JF, Lane N, Martin WF. 2016. Lokiarchaeon is hydrogen dependent. Nature Microbiol. 1:16034. [DOI] [PubMed] [Google Scholar]
- Spang A., et al. 2015. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speijer D. 2015. Birth of the eukaryotes by a set of reactive innovations: new insights force us to relinquish gradual models. Bioessays 37:1268–1276. [DOI] [PubMed] [Google Scholar]
- Speijer D, Lukes J, Elias M. 2015. Sex is a ubiquitous, ancient, and inherent attribute of eukaryotic life. Proc Natl Acad Sci U S A. 112:8827–8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouthamer AH. 1978. Energy-yielding pathways In: Gunsalus IC. editor. The bacteria vol VI: bacterial diversity. New York: Academic Press; p. 389–462. [Google Scholar]
- Takahashi F, Yamaguchi K, Hishinuma T, Kataoka H. 2003. Mitosis and mitotic wave propagation in the coenocytic alga, Vaucheria terrestris sensu Goetz. J Plant Res. 116:381–387. [DOI] [PubMed] [Google Scholar]
- Tekle YI, Anderson OR, Leckya AF. 2014. Evidence of parasexual activity in “asexual amoebae” Cochliopodium spp. (Amoebozoa): extensive cellular and nuclear fusion. Protist 165:676–687. [DOI] [PubMed] [Google Scholar]
- Thiergart T, Landan G, Schenk M, Dagan T, Martin WF. 2012. An evolutionary network of genes present in the eukaryote common ancestor polls genomes on eukaryotic and mitochondrial origin. Genome Biol Evol. 4:466–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmis JN, Ayliffe MA, Huang CY, Martin W. 2004. Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat Rev Genet. 5:123–135. [DOI] [PubMed] [Google Scholar]
- Toro E, Shapiro L. 2010. Bacterial chromosome organization and segregation. Cold Spring Harb Perspect Biol. 2:a000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyenoyama MK, Bengtsson BO. 1989. On the origin of meiotic reproduction: a genetic modifier model. Genetics 123:873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Melderen L., De Bast MS 2009. Bacterial toxin–antitoxin systems: more than selfish entities? PLoS Genet. 5:e1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbruggen H., et al. 2009. A multi-locus time-calibrated phylogeny of the siphonous green algae. Mol Phylogenet Evol. 50:642–653. [DOI] [PubMed] [Google Scholar]
- von Dohlen CD, Kohler S, Alsop ST, McManus WR. 2001. Mealybug beta-proteobacterial endosymbionts contain gamma-proteobacterial symbionts. Nature 412:433–436. [DOI] [PubMed] [Google Scholar]
- Welch DBM, Meselson M. 2000. Evidence for the evolution of bdelloid rotifers without sexual reproduction or genetic exchange. Science 288:1211–1215. [DOI] [PubMed] [Google Scholar]
- Whatley JM, John P, Whatley FR. 1979. From extracellular to intracellular: the establishment of mitochondria and chloroplasts. Proc R Soc B. 204:165–187. [DOI] [PubMed] [Google Scholar]
- Wilkins AS, Holliday R. 2008. The evolution of meiosis from mitosis. Genetics 181:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC. 1975. Sex and evolution. Princeton, NJ: Princeton Univ. Press. [Google Scholar]
- Williams TA, Foster PG, Cox CJ, Embley TM. 2013. An archaeal origin of eukaryotes supports only two primary domains of life. Nature 504:231–236. [DOI] [PubMed] [Google Scholar]
- Wujek DE. 1979. Intracellular bacteria in the blue-green alga Pleurocapsa minor. Trans Am Microsc Soc. 98:143–145. [Google Scholar]
- Zuccolo M., et al. 2007. The human Nup107–160 nuclear pore subcomplex contributes to proper kinetochore functions. EMBO J. 26:1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]