

Abstract
Chemokine-induced leukocyte migration into the vessel wall is an early pathological event in the progression of atherosclerosis, the underlying cause of myocardial infarction. The immune-inflammatory response, mediated by both the innate and adaptive immune cells, is involved in the initiation, recruitment, and resolution phases of cardiovascular disease progression. Activation of leukocytes via inflammatory mediators such as chemokines, cytokines, and adhesion molecules is instrumental in these processes. In this review, we highlight leukocyte activation with the main focus being on the mechanisms of chemokine-mediated recruitment in atherosclerosis and the response postmyocardial infarction with key examples from experimental models of cardiovascular inflammation.
1. Atherosclerosis
Atherosclerosis is a chronic inflammatory condition affecting the medium- and large-sized arteries, characterised by the progressive development of lesions consisting of lipid, fibrosis, and inflammatory cell infiltrate within the tunica media [1]. The progression of these lesions into complex atherosclerotic plaques is in turn associated with vessel stenosis and plaque rupture with the generation of atheromatous thromboembolism [2]. It is the major pathological process underlying several cardiovascular diseases, such as myocardial infarction, and is thus a leading contributor to morbidity and mortality in the Western world [3].
The established link between hypercholesterolaemia and atherosclerosis led to the characterisation of atherosclerosis as primarily a disorder of lipids, a hypothesis that would dominate thinking for much of the 20th century. However, the last few decades have seen the establishment of inflammation as a key part of atherosclerosis and the important contribution of leukocytes to the initiation and progression of atherosclerotic plaques [1].
This section will focus on the role of leukocytes in atherosclerosis and specifically their migration and trafficking with a focus on the most recent developments in the field.
1.1. The Role of Leukocytes in Atherosclerosis
The macrophage is one of the defining cells present in an atherosclerotic lesion in addition to the macrophage-derived foam cell, an enlarged macrophage characterised by accumulation of oxLDL and cholesterol crystals [4]. Derived from circulating monocytes [5], the macrophage is a phagocytic cell that detects, processes, and clears pathogens and cell debris in addition to generating appropriate inflammatory responses.
Macrophage activation in atherosclerotic lesions is associated with the local release of inflammatory cytokines and reactive oxygen species, both of which contribute to the continued recruitment and activation of leukocytes. Activation is also associated with the release of plaque-destabilizing molecules such as matrix metalloproteinases [1]. Finally, upon transformation into a foam cell, the macrophage may undergo necrosis further releasing inflammatory stimuli and creating the necrotic core of advanced lesions [6].
Activation of macrophages is dependent upon ligand interactions with pattern recognition receptors [7]. In atherosclerosis, the study of a subset of these receptors, the toll-like receptors (TLRs), has allowed the key role of the macrophage to be demonstrated. In the atheromatous lesion, there is upregulation of TLR1, TLR2, TLR4, and TLR5 with oxLDL (a constituent component of atheromatous lesions) specifically being shown to upregulate TLR4 mRNA levels in vitro [8]. Deletion of TLR4 in ApoE−/− (apolipoprotein E knockout) mice leads to significantly marked reductions in lesion formation [9], and this is also seen in TLR2−/−ApoE−/− mice [10], whilst ApoE−/− mice with inactivating mutations of M-CSF (macrophage colony stimulating factor) also show marked reductions in both lesion size and macrophage content [11].
Lymphocytes, particularly T-lymphocytes, are also present in atherosclerotic lesions albeit in smaller numbers than macrophages [12]. However, their role is important as highlighted in lymphocyte-deficient animal models of atherosclerosis displaying reduced lesion burden [13]. Lymphocytes are recruited into the subendothelial space in a similar manner to monocytes and subsequently become activated by locally present cytokines, such as IL-12 and IL-18 [13]. Clonal expansion of T-cells and a dependence on CD40 ligand has suggested that T-cell activation can also occur due to antigen recognition, most likely an auto-antigen present in the atheroma [14].
In turn, T-cell activation plays a role in atherosclerosis with interferon-gamma, the signature cytokine of T helper 1 (TH1) cells, being present in human atherosclerotic lesions and in turn being associated with enhanced cytokine activity, increased macrophage activation, and reduced collagen formation [14]. The role of other CD4+ T-cell types as well as CD8+ T-cells is less clear, whilst B-cells appear to play an athero-protective role [15]. B-cells are present in plaques with B-cell transfer in immune-deficient mice or administration of anti-oxLDL antibody reducing lesion burden, although no correlation between antibody titres and atherosclerosis has been found in humans [15].
1.2. Initiation: The Recruitment of Leukocytes in Atherosclerosis
Atherosclerosis occurs at specific sites in the arterial tree where turbulent flow is present, such as the aortic root and arch, in humans as well as animal models of atherosclerosis with “fatty streak” lesions present from the first decade of life in humans [2]. Whilst numerous pathological observations demonstrate the capacity of endothelial denudation to cause lesion formation, it is rather endothelial dysfunction thought to be critical in the initiation of atherosclerosis [16].
Further to this, our understanding of vascular biology has come to demonstrate the important interactions of endothelial cells in trafficking leukocytes to areas of local inflammation and injury. Indeed, the expression of adhesion molecules on their surface allows subsequent interaction between those molecules and leukocyte selectins to enable rolling and extravasation [16] (Figure 1). As a result, normal endothelium resists leukocyte adherence whilst dysfunctional or activated endothelium demonstrates firm leukocyte attachment.
Figure 1.

The formation of atherosclerotic plaques is characterised by the recruitment of monocytes to the artery wall directed by chemokines, followed by adherence and transmigration across the endothelium. The accumulation of lipid deposits containing oxLDL in the intima is then taken up by plaque macrophages to form foam cells resulting in fatty streak lesion formation.
The expression of several proteins in endothelial cells is linked to the levels of shear stress. Early en-face immunohistochemical staining in animal studies of atherosclerosis demonstrates the upregulation of VCAM-1 (vascular cell adhesion molecule-1) in turbulent flow sites in hyperlipidaemic animals compared to wild-type animals [17]. In addition, ApoE−/− mice with hypomorphic variants of VCAM-1 display reduced lesion formation [18]. Concurrently, ICAM-1 (intracellular adhesion molecule 1) is upregulated in these sites; however, this upregulation also occurs in control animals [17]. Both VCAM-1 and ICAM-1 serve to recruit leukocytes to the endothelial surface.
Conversely, in sites of laminar flow, there is upregulation of several “athero-protective” genes such as eNOS (endothelial nitric oxide synthase), with the subsequent nitric oxide release serving to combat oxidative stress as well as to directly limit VCAM-1 expression through the inhibition of NF-κB [19]. Indeed, the athero-protective role of eNOS is demonstrated by the ApoE−/−eNOS−/− mouse which shows increased lesion burden, VCAM-1 expression, and plaque macrophage content [20]. Conversely, the overexpression of eNOS in a ApoE−/− mouse, in conjunction with the overexpression of the rate limiting enzyme (GTPCH1) which is involved in its cofactor synthesis, to prevent eNOS uncoupling, shows markedly reduced lesion burden [21].
Thus, it appears that in endothelial sites of turbulent flow, there is upregulation of adhesion molecules with enhanced upregulation in the presence of hyperlipidaemia and relative downregulation of endothelial genes important in preventing atherogenesis. This initial process and the subsequent recruitment of leukocytes as well as lipid into the subendothelial spaces of turbulent flow sites lead to the creation of a local inflammatory environment, where lipid is oxidised and leukocytes are activated, which is critical in the development of lesions [1].
1.3. Leukocyte Migration and Retention
At the sites of inflammation, leukocyte attraction and retention not only requires the local expression of endothelial adhesion molecules but also the activation of leukocytes and subsequent conversion into a high affinity state. This is typically achieved through the local release of chemical mediators, principally the chemokines (chemotactic cytokines), from local tissue-resident cells [22].
In the initial lesion, the entrapment of LDL (low density lipoprotein) and its subsequent oxidation leads to localised inflammation and the release of chemokines with other inflammatory mediators; this synergises with shear stress-induced expression of adhesion molecules to recruit leukocytes which become activated by and then contribute to the inflammatory environment [1].
Chemokines are 8–12 kDa proteins that signal through their respective G-protein-coupled receptors (GPCRs). They are released from endothelial cells, platelets, macrophages, and lymphocytes following activation by inflammation and cardinal inflammatory mediators, such as TNF-α. Upon release, chemokines bind to glycosaminoglycans on cell surfaces to establish local gradients that are able to activate and then direct leukocytes to specific sites [15].
Chemokines and chemokine receptors are significantly implicated in atherosclerosis with chemokine receptors known to be expressed within animal and human plaques and their expression being correlated to progression [23].
Although the chemokine system displays a great deal of redundancy, the development of chemokine receptor knockouts in atherosclerosis murine models has given insight into their role. This was first demonstrated in the ApoE−/−CCR2−/− murine model which shows significant reductions in macrophage content and plaque content compared to ApoE−/− mice [24]. Additionally, the knockout of CCR5 in ApoE−/− displays striking reductions of at least 50% of lesion area as well as greater reductions in macrophage content and an increase in smooth muscle content [25].
In vivo antichemokine strategies have also been successful in reducing plaque burden. The antagonism of RANTES (regulated upon activation, normal T-cell expressed and secreted) with met-RANTES in LDLR−/− (LDL receptor) mice for 14 weeks resulted in significant lesion area reduction as well as a reduction in CCR2 expression suggesting an anti-inflammatory and antiatherogenic effect [26]. The use of both adenoviral and lentiviral vectors to express vCCI (viral chemokine inhibitor), a broad-spectrum chemokine inhibitor, also produces a marked antiatherogenic effect in ApoE−/− mice [27].
However, chemokines do not appear to be universally proatherogenic, with the ApoE−/−CCR1−/− murine model demonstrating an increase in lesion area with increased levels of T-cell infiltration, providing further evidence of the contribution of T-lymphocytes [25].
Chemokines are also known to play an apparent role in the retention of leukocytes within the plaque, although this has not been as comprehensively studied as the process of leukocyte recruitment in atherosclerosis. For example, the chemokine CX3CL1 promotes macrophage adhesion to smooth muscle cells (SMCs) within coronary artery plaques and its expression is elevated in hyperlipidaemia. Conversely, a reduction in CX3CL1 expression reduces macrophage adherence to SMCs [28].
Perhaps most striking is the apparent role of chemokines in leukocyte migration in regression models of atherosclerosis. In the surgical model, whereby regression in an affected artery is induced by transfer to a normo-lipidaemic animal, there is a reduction in VCAM-1 levels in the atherosclerotic artery but, conversely, an increase in CCR7 levels. Further to this, when recipient animals were treated with antibodies against the CCR7 ligands, the regressing lesions were markedly impaired, with retention of leukocyte content seen as well, suggesting a failure of migration [29].
Finally, in addition to the principal role of chemokines in leukocyte migration in atherosclerosis, other molecules such as chemotaxins may also play a role. Leukocyte cell-derived chemotaxin 2 has recently been shown to correlate with atherosclerotic lesion burden and macrophage content in human tissue samples [30], and further to this, LECT-2 treatment of human umbilical vein endothelial cell culture has been shown to produce c-Jun N-terminal kinase-mediated increases in ICAM-1 and MCP-1 expressions [31] which would both serve to enhance leukocyte recruitment. Further in vivo studies with LECT-2 and other chemotaxins may well demonstrate important contributions to this aspect of atherosclerosis.
1.4. Leukocyte Tracking in Experimental Models of Cardiovascular Disease
One of the more recent developments in atherosclerosis research has been the production of murine models that facilitate tracking of myeloid cells thus providing new insights into myeloid cell biology beyond the work previously discussed regarding the role of chemokines and shear stress-linked regulation of genes.
It has been established, for instance, that the macrophages and foam cells in murine experimental plaques are mainly recruited from circulating monocytes as opposed to local resident cells [5]. An example is seen in a murine model expressing an alternative isoform of CD45, which after full engraftment of a bone marrow transplant demonstrates a dominance of donor myeloid cells in plaques induced by arterial injury [5]. A further example is seen with the adoptive transfer of radioactively labelled monocytes into ApoE−/− animals which demonstrates a dominance of donor myeloid cells in the plaque [32].
It has also been possible to elucidate the role of different macrophage subtypes. The creation of a murine model expressing GFP under the CX3CR1 promoter revealed the existence of low-expression and high-expression populations with expression of CX3CR1 being negatively correlated to the expression of Ly6C and Gr-1. It was subsequently found that CX3CR1lo/Ly6Chi monocytes preferentially migrate to the sites of acute inflammation whereas the CX3CR1hi/Ly6Clo population does not [33].
Further to this, it is now understood that the peripheral blood monocytosis that develops in ApoE−/− mice, when induced through diet to develop hypercholesterolaemia, is dominated by the CX3CR1lo/Ly6Chi population. Indeed, in this model, the CX3CR1lo/Ly6Chi population doubled every month on a high-fat diet and is correlated with lipid levels; conversely, the monocytosis of this population is attenuated by statin treatment [34]. Additionally, recent data utilising spleen transplantation experiments has highlighted the role of the spleen as an additional source of CX3CR1lo/Ly6Chi monocytes proving not only to be a reservoir but also an active source of monocyte generation [35].
This work has also been applied to further demonstrate the role of chemokines in leukocyte migration in atherosclerosis. The monocytosis of the CX3CR1lo/Ly6Chi population is not seen in animals on high-fat diet when CCR2 is knocked out for instance [36]. In addition, the use of radioactive bead labelling in different monocyte subsets in a CCR2 null murine model that sees an atherosclerotic aortic arch transplanted into it demonstrated the dependence of CX3CR1lo/Ly6Chi monocytes on CCR2 for migration [37].
It has also offered further insights into the role of adhesion molecules. For example, the CX3CR1lo/Ly6Chi monocyte population is now known to express higher levels of PSGL-1 (P-selectin glycoprotein ligand-1), an adhesion molecule, compared to CX3CR1hi/Ly6Clo monocytes. In turn, the PSGL-1 null mouse displays a reduced migration of CX3CR1lo/Ly6Chi monocytes to atherosclerotic plaques with a concordant reduction in lesion area [38].
Finally, a murine model expressing GFP under the control of the human CD68 promoter has recently been created which drives GFP expression in all monocytes of the blood, spleen, and bone marrow. Most promisingly, these monocytes retain high levels of GFP expression for at least 72 hours after differentiation into macrophages and so, unlike the CX3CR1GFP model previously discussed, enables the study of monocyte fate as well as initial trafficking and recruitment [39].
Indeed, this model has already been utilised in atherosclerosis research with the generation of CD68GFP ApoE−/− mice, where the expression of GFP had no significant effect on the atherosclerosis process and allowed clear visualisation of plaque macrophages, with quantification by GFP correlating with traditional macrophage quantification methods [40]. Adoptive transfer of bone marrow monocytes into ApoE−/− with established plaques demonstrated clear visualisation of GFP+ cells 72 hours posttransfer further confirming the ongoing recruitment of peripheral monocytes as the source of the plaque macrophage population [40]. In contrast to traditional methods of assessing monocyte kinetics and fate, this model appears to not profoundly alter plaque biology and thus promises to offer new and exciting insights in the field. New experimental models of cardiovascular disease not only provide a better understanding of the processes behind leukocyte recruitment and retention but also enable the discovery of novel therapeutic targets in cardiovascular inflammation.
2. Myocardial Infarction and Heart Failure
According to the American Heart Association, myocardial infarction (MI) and congestive heart failure (CHF) mortalities are the major contributors of the cardiovascular diseases that are the leading causes of death in the Western world [41]. MI describes the death of cardiac tissue due to prolonged ischaemia (20 minutes plus) or through the occlusion of a coronary vessel commonly by lumen-reducing atherosclerotic plaques and subsequent thrombosis formation [42].
Factors that increase the lifetime risk of an acute (i.e., MI) or chronic (CHF) coronary syndrome, which manifest over decades, are a high-lipid diet (atherosclerosis), low levels of exercise, chronic diseases (diabetes), smoking, and genetic predisposition (familial hypercholesterolaemia) [43]. The average age for the first MI is 65 years for women and 56 years for men, of which an increasing number of patients survive post-MI due to the improvements in reperfusion techniques and acute coronary care [44]. However, surviving patients are faced with restricting morbidities, an increased mortality risk of 8–12% [45] and a further ischaemic event risk of 1 in every 2, both within the first year [46]. Clinical management post-MI is improving, but the mechanisms leading to heart failure and the possibility for prevention offer a therapeutic target for cardiovascular disease research.
2.1. Myocardial Infarction
Cardiovascular disease was first described in 1772 by Herberden [47]; symptoms of breast pain were later diagnosed as angina pectoris (the temporary reduction in blood flow to the heart causing chest pain after exercise) with leukocytosis documented in fibrosed left ventricles of prolonged angina mortalities. However, MI was not identified until 1912 [48], once the relationship between coronary artery disease, acute/prolonged angina, and coronary thrombosis had been established, via the advent of echocardiography (ECG) in defining the pathological conditions. Since then, scientists have attempted to elucidate the mechanisms that underlie MI, that is, the potential role of leukocytes and inflammation. However, in the clinic, emergency reperfusion by fibrolytic agents targeting the thrombosis was and remains the staple treatment post-MI for the survival of patients [49], which can result in adverse left ventricle remodelling and heart failure.
The most frequent coronary vessel occluded in MI is the left anterior descending artery (LAD) which nourishes the left ventricle (LV), and the ischaemia that follows impairs the endothelial vascular integrity leading to leukocyte infiltration [50]. The cardiomyocytes, tissue-resident macrophages, and fibroblasts, deprived of nutrients, undergo necrosis within the first 30 minutes [51], passively releasing damage-associated molecular patterns (DAMPs), such as ATP and hyaluronic acid by breakdown of the extracellular matrix (ECM). TLRs 2 and 4 on local macrophages and neutrophils are activated by these DAMPS and stimulate a proinflammatory immune response via chemokines, cytokines, and vascular adhesion molecules, through intracellular MAPK (mitogen-activated protein kinases) and NF-κB signalling [51]. The tissue macrophage release of CXC chemokines, TNF-α (activating endothelial cells and increasing vessel permeability), and IL-1β/IL-18 stimulates neutrophil recruitment within 12–24 hours post-MI. The positive self-feedback loop of CCL8 secretion recruits further neutrophils to the infarct for the phagocytosis of necrotic, autophagic, or apoptosed cardiac tissue [51]. Neutrophils not only release MMPs to breakdown the ECM and the cardiomyocytes within the infarct but also produce soluble IL-6 which activates endothelial cells to express chemokine ligand CCL2 and VCAM1 for monocyte recruitment [52]. Neutrophil clearance (at around day 7) manifests via high concentrations of the aforementioned proinflammatory chemokines and the release of death signals, for inhibition of neutrophil migration/prophagocytosis, such as lactoferrin and annexin A1 [53].
2.2. The Biphasic Immune Response to MI
The immune response to acute MI can be divided between the inflammatory phase at days 1–4 and the healing phase from days 5–14 from which point the scar matures [54] (Figure 2). Certain monocytes dominate these different phases and are masters of the adverse LV remodelling and heart failure post-MI as highlighted by Nahrendorf et al. [55]. This key study identified two subsets of monocytes that were alternatively “recruited” throughout MI inflammation and healing, with different but complementary functions: proinflammatory Ly6Chi monocytes (recruited via CCR2) and the prohealing Ly6Clo monocytes (recruited via CX3CR1), differentiating to various subsets of macrophages within the classically activated M1 and the alternatively activated M2 phenotypes [56]. In humans, the proinflammatory monocytes express CD14++CD16− and the prohealing monocytes express CD14+CD16++, resembling their murine counterparts and allowing the study of MI in mice to simulate the human response [57].
Figure 2.
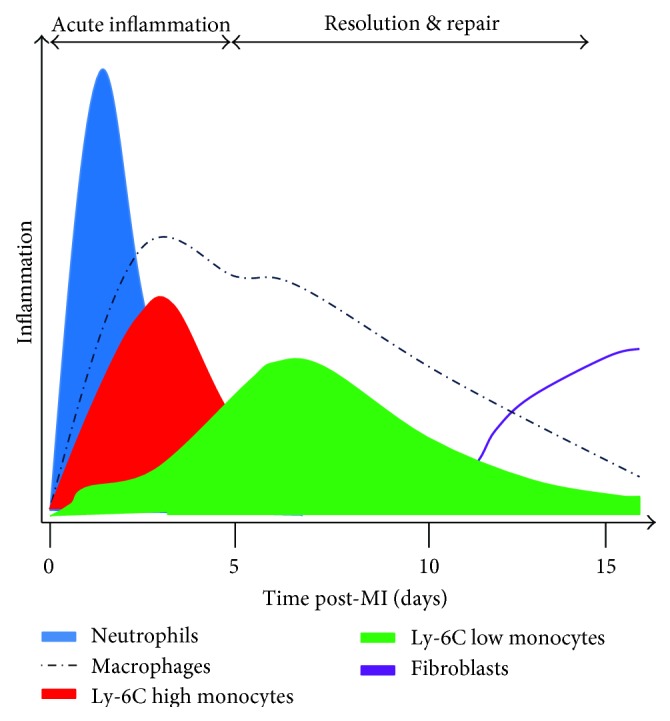
Track of innate immune system inflammatory cell variance over the time course of 15 days within the ischaemic left ventricle, postmyocardial infarction in mice.
With the innate immune response underway, tissue-resident macrophages are proliferating under the control of M-CSF, from activated endothelial cells, fibroblasts, and macrophages, and secrete increasing levels of CCL2. CCL2 stimulates the recruitment of the Ly6Chi monocytes from the blood pool via their receptor CCR2 and initiates HSC (haematopoietic stem cell) progenitor proliferation in the bone marrow [58].
Furthermore, MI activates the neuro-humoral axis via sympathetic innervations of adrenaline secretion for β3-adrenergic receptors in the bone marrow, reducing levels of CXCL12 (the “monocyte progenitor egress” inhibitory cytokine) and allowing HSC relocation to the spleen for proliferation and maturation [54]. Limiting the immediate fibroblast-mediated ECM remodelling and infarct healing is the result of M1 macrophages NLRP3 inflammasome activation (releasing and converting pro-IL-1B to IL-1B by caspase-1) via M1 engulfing of cholesterol crystals, promoting fibroblast proteolytic enzyme release to assist in the ECM degradation [59]. Splenic monocyte recruitment is activated via angiotensin II signalling, with increased levels due to the release of ACE (angiotensin-converting enzyme) from dead cardiac monocytes, suggesting the benefit of post-MI therapy via ACE inhibitors to reduce the size and leukocyte populations in the infarct [60].
Extravasation of Ly6Chi monocytes to the infarct continues the acute phase response and the efferocytosis of dead cardiac tissue including the apoptotic neutrophils, preparing the infarct site for new ECM and collagen deposition by fibroblasts and Ly6Clo monocytes. Prolonged inflammation via the Ly6Chi monocytes is associated with dilative remodelling (LV thinning) and systolic dysfunction, ultimately leading to heart failure when the inflammatory phase is not resolved and Ly6Chi cells repopulate the LV weeks to months postinfarct healing [61].
The mechanisms of monocyte recruitment in MI were first challenged by Kaikita et al. using CCR2−/− mice [62]. The authors noted a great impairment of macrophage accumulation in the infarcted region within 7 days post-MI and a critical reduction in LV remodelling. At the time, they were unaware of the biphasic immune response but they concluded on the importance of CCR2 for monocyte-macrophage recruitment, suggesting the remainder of monocytes unaffected by CCR2−/− could be due to CCR1 and CCR5 low affinity for CCL2 released by the infarct.
Further studies have shown that a balance between proinflammation versus healing is key for cardiac remodelling as CCR2−/− animals have impaired necrotic cell clearance, collagen deposition, and angiogenesis with the predisposition for cardiac rupture and immediate death [63]. Overactivation of the healing phase leads to concentric remodelling and diastolic dysfunction with LV stiffness (predisposing heart failure) [64].
Building on the work of Hanna et al. [65], which described the orphan nuclear hormone receptor, Nr4a1, as a vital regulator of monocyte development in the bone marrow affecting the levels of Ly6Clo monocyte populations, with decreased peripheral levels compared to the Ly6Chi populations (in Nr4a1−/−), Hilgendorf et al. [66] identified that the recruitment of the prohealing, blood-patrolling, Ly6Clo monocytes is amplified by a Ly6Chi monocyte to Ly6Clo macrophage conversion, with Nr4a1 levels fluctuating between the two cell phenotypes with high levels dampening the proinflammatory macrophage secretions.
The previous finding that ACE inhibitors reduced the leukocyte populations can now be explained by not just inhibiting Ly6Chi monocyte recruitment but its subsequent differentiation to the infarct macrophage populations. Additionally, Ly6Clo monocytes are also recruited to the infarct from the blood via CX3CL1 release through endothelial cells [56]. With the accumulation of dead neutrophils, inflammatory resolution is stimulated by neutrophil “eat me” signals (phosphatidylserine) as well as their expression of lipoxins and resolvins, engaging the M2 phenotype conversion [67]. Phagocytosis of these neutrophils by macrophages encourages the expression and secretion of IL-10, the anti-inflammatory cytokine, expressed in greater proportions from the Ly6Clo cells as well as TGF-β, a stimulator for ECM gene expression by cardiac myofibroblasts. Angiogenic factors are also secreted by the Ly6Clo cells such as VEGF (vascular endothelial growth factor) to restore oxygen supply by neovascularisation and to establish the granulation tissue for scar formation/maturation [57]. Noncontractile scar formation is the result of myofibroblast-depositing collagen following activation via TGF-β (transforming growth factor beta) signalling and TCRP6 (transient receptor potential 6) expression to switch from cardiac fibroblasts [68].
3. Concluding Remarks
From a clinical perspective of leukocyte migration in atherosclerosis, the chemokine system represents a particularly excellent candidate for exploring therapeutic options. Whilst the chemokine system displays a great deal of redundancy, there are clear and powerful effects seen in specific murine knockouts of key chemokine receptors which might be replicated. Indeed, the chemokine system could be targeted through several different avenues, such as classical GPCR antagonists, for example. Due to the important role of chemokines in wound healing and infection control, long-term therapy would likely be associated with adverse effects. However, utilising chemokine inhibition to stabilise atherosclerotic plaques in particular circumstances could well provide clinical benefit. Such examples might include delivery of therapy to stabilise plaques following myocardial infarction to prevent further rupture or vessel stenosis or delivery of therapeutics in transient ischaemic attacks to reduce stroke risk due to carotid artery atherosclerosis. With several drug targets against chemokines already identified in drug discovery programmes, this field bears considerable future potential [69]. In addition, the major targets for MI and heart failure research are to regulate the inflammatory monocyte infiltration phase and to reduce the potential for adverse remodelling and future heart failure. Given that the clinical trials of anti-inflammatory medications are failing (anti-TNF/IL-1β) [70], the mechanisms that underlie adverse and normal MI healing are yet to be uncovered.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Acknowledgments
The authors would like to acknowledge the support from the BHF Centre of Research Excellence, Oxford (RE/13/1/30181).
Authors' Contributions
Daniel P. Jones and Harry D. True contributed equally to this work.
References
- 1.Ross R. Atherosclerosis—an inflammatory disease. The New England Journal of Medicine. 1999;340(2):115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Croce K., Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Current Opinion in Hematology. 2007;14(1):55–61. doi: 10.1097/00062752-200701000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Dahlof B. Cardiovascular disease risk factors: epidemiology and risk assessment. The American Journal of Cardiology. 2010;105(1 Supplement):3A–9A. doi: 10.1016/j.amjcard.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Napoli C., Glass C. K., Witztum J. L., Deutsch R., D’Armiento F. P., Palinski W. Influence of maternal hypercholesterolaemia during pregnancy on progression of early atherosclerotic lesions in childhood: Fate of Early Lesions in Children (FELIC) study. Lancet. 1999;354(9186):1234–1241. doi: 10.1016/S0140-6736(99)02131-5. [DOI] [PubMed] [Google Scholar]
- 5.Lessner S. M., Prado H. L., Waller E. K., Galis Z. S. Atherosclerotic lesions grow through recruitment and proliferation of circulating monocytes in a murine model. The American Journal of Pathology. 2002;160(6):2145–2155. doi: 10.1016/S0002-9440(10)61163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansson G. K., Robertson A. K., Soderberg-Naucler C. Inflammation and atherosclerosis. Annual Review of Pathology. 2006;1(1):297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- 7.Janeway C. A., Jr., Medzhitov R. Innate immune recognition. Annual Review of Immunology. 2002;20(1):197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 8.Xu X. H., Shah P. K., Faure E., et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104(25):3103–3108. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 9.Michelsen K. S., Wong M. H., Shah P. K., et al. Lack of toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(29):10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mullick A. E., Tobias P. S., Curtiss L. K. Modulation of atherosclerosis in mice by toll-like receptor 2. The Journal of Clinical Investigation. 2005;115(11):3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiao J. H., Tripathi J., Mishra N. K., et al. Role of macrophage colony-stimulating factor in atherosclerosis: studies of osteopetrotic mice. The American Journal of Pathology. 1997;150(5):1687–1699. [PMC free article] [PubMed] [Google Scholar]
- 12.Hansson G. K. Immune mechanisms in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(12):1876–1890. doi: 10.1161/hq1201.100220. [DOI] [PubMed] [Google Scholar]
- 13.Reardon C. A., Blachowicz L., White T., et al. Effect of immune deficiency on lipoproteins and atherosclerosis in male apolipoprotein E-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(6):1011–6. doi: 10.1161/01.ATV.21.6.1011. [DOI] [PubMed] [Google Scholar]
- 14.Liuzzo G., Goronzy J. J., Yang H., et al. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation. 2000;101(25):2883–2888. doi: 10.1161/01.CIR.101.25.2883. [DOI] [PubMed] [Google Scholar]
- 15.Hansson G. K., Hermansson A. The immune system in atherosclerosis. Nature Immunology. 2011;12(3):204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 16.Vestweber D. Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunological Reviews. 2007;218(1):178–196. doi: 10.1111/j.1600-065X.2007.00533.x. [DOI] [PubMed] [Google Scholar]
- 17.Iiyama K., Hajra L., Iiyama M., et al. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circulation Research. 1999;85(2):199–207. doi: 10.1161/01.RES.85.2.199. [DOI] [PubMed] [Google Scholar]
- 18.Cybulsky M. I., Iiyama K., Li H., et al. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. The Journal of Clinical Investigation. 2001;107(10):1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolfrum S., Teupser D., Tan M., Chen K. Y., Breslow J. L. The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-kappaB target genes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(47):18601–18606. doi: 10.1073/pnas.0709011104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ponnuswamy P., Schröttle A., Ostermeier E., et al. eNOS protects from atherosclerosis despite relevant superoxide production by the enzyme in apoE mice. PloS One. 2012;7(1, article e30193) doi: 10.1371/journal.pone.0030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takaya T., Hirata K., Yamashita T., et al. A specific role for eNOS-derived reactive oxygen species in atherosclerosis progression. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(7):1632–7. doi: 10.1161/ATVBAHA.107.142182. [DOI] [PubMed] [Google Scholar]
- 22.Charo I. F., Ransohoff R. M. The many roles of chemokines and chemokine receptors in inflammation. The New England Journal of Medicine. 2006;354(6):610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 23.Braunersreuther V., Mach F., Steffens S. The specific role of chemokines in atherosclerosis. Thrombosis and Haemostasis. 2007;97(5):714–721. [PubMed] [Google Scholar]
- 24.Boring L., Gosling J., Cleary M., Charo I. F. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394(6696):894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 25.Braunersreuther V., Zernecke A., Arnaud C., et al. Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(2):373–379. doi: 10.1161/01.ATV.0000253886.44609.ae. [DOI] [PubMed] [Google Scholar]
- 26.Veillard N. R., Kwak B., Pelli G., et al. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circulation Research. 2003;94(2):253–261. doi: 10.1161/01.RES.0000109793.17591.4E. [DOI] [PubMed] [Google Scholar]
- 27.Bursill C. A., Choudhury R. P., Ali Z., Greaves D. R., Channon K. M. Broad-spectrum CC-chemokine blockade by gene transfer inhibits macrophage recruitment and atherosclerotic plaque formation in apolipoprotein E-knockout mice. Circulation. 2004;110(16):2460–2466. doi: 10.1161/01.CIR.0000145122.58420.CO. [DOI] [PubMed] [Google Scholar]
- 28.Barlic J., Zhang Y., Foley J. F., Murphy P. M. Oxidized lipid-driven chemokine receptor switch, CCR2 to CX3CR1, mediates adhesion of human macrophages to coronary artery smooth muscle cells through a peroxisome proliferator-activated receptor gamma-dependent pathway. Circulation. 2006;114(8):807–819. doi: 10.1161/CIRCULATIONAHA.105.602359. [DOI] [PubMed] [Google Scholar]
- 29.Trogan E., Feig J. E., Dogan S., et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(10):3781–3786. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonmez F. C., Yildiz P., Akhtar M. S., et al. New markers in atherosclerosis: thrombospondin-2 (THBS-2) and leukocyte cell-derived chemotaxin-2 (LECT-2). An immunohistochemical study. Medical Science Monitor. 2016;22(1):5234–5239. doi: 10.12659/MSM.898889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang H. J., Jung T. W., Hong H. C., et al. LECT2 induces atherosclerotic inflammatory reaction via CD209 receptor-mediated JNK phosphorylation in human endothelial cells. Metabolism. 2015;64(9):1175–1182. doi: 10.1016/j.metabol.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 32.Kircher M. F., Grimm J., Swirski F. K., et al. Noninvasive in vivo imaging of monocyte trafficking to atherosclerotic lesions. Circulation. 2008;117(3):388–395. doi: 10.1161/CIRCULATIONAHA.107.719765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geissmann F., Jung S., Littman D. R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. doi: 10.1016/S1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 34.Swirski F. K., Libby P., Aikawa E., et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. The Journal of Clinical Investigation. 2007;117(1):195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chinetti-Gbaguidi G., Colin S., Staels B. Macrophage subsets in atherosclerosis. Nature Reviews Cardiology. 2015;12(1):10–17. doi: 10.1038/nrcardio.2014.173. [DOI] [PubMed] [Google Scholar]
- 36.Tsou C. L., Peters W., Si Y., et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. The Journal of Clinical Investigation. 2007;117(4):902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tacke F., Alvarez D., Kaplan T. J., et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. The Journal of Clinical Investigation. 2007;117(1):185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.An G., Wang H., Tang R., et al. P-selectin glycoprotein ligand-1 is highly expressed on Ly-6Chi monocytes and a major determinant for Ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation. 2008;117(25):3227–3237. doi: 10.1161/CIRCULATIONAHA.108.771048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iqbal A. J., McNeill E., Kapellos T. S., et al. Human CD68 promoter GFP transgenic mice allow analysis of monocyte to macrophage differentiation in vivo. Blood. 2014;124(15):e33–e44. doi: 10.1182/blood-2014-04-568691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNeill E., Iqbal A. J., Jones D., et al. Tracking monocyte recruitment and macrophage accumulation in atherosclerotic plaque progression using a novel hCD68GFP/ApoE−/− reporter mouse-brief report. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37(2):258–263. doi: 10.1161/ATVBAHA.116.308367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roger V. L., Go A. S., Lloyd-Jones D. M., et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thygesen K., Alpert J. S., Jaffe A. S., et al. Third universal definition of myocardial infarction. Circulation. 2012;126(16):2020–2035. doi: 10.1161/CIR.0b013e31826e1058. [DOI] [PubMed] [Google Scholar]
- 43.Anand S. S., Islam S., Rosengren A., et al. Risk factors for myocardial infarction in women and men: insights from the INTERHEART study. European Heart Journal. 2008;29(7):932–940. doi: 10.1093/eurheartj/ehn018. [DOI] [PubMed] [Google Scholar]
- 44.Movahed M. R., John J., Hashemzadeh M., Jamal M. M., Hashemzadeh M. Trends in the age adjusted mortality from acute ST segment elevation myocardial infarction in the United States (1988-2004) based on race, gender, infarct location and comorbidities. The American Journal of Cardiology. 2009;104(8):1030–1034. doi: 10.1016/j.amjcard.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 45.Roe M. T., Ohman E. M. A new era in secondary prevention after acute coronary syndrome. The New England Journal of Medicine. 2012;366(1):85–87. doi: 10.1056/NEJMe1112770. [DOI] [PubMed] [Google Scholar]
- 46.Jernberg T., Hasvold P., Henriksson M., Hjelm H., Thuresson M., Janzon M. Cardiovascular risk in post-myocardial infarction patients: nationwide real world data demonstrate the importance of a long-term perspective. European Heart Journal. 2015;36(19):1163–1170. doi: 10.1093/eurheartj/ehu505. [DOI] [PubMed] [Google Scholar]
- 47.Conti C. R. Re-thinking angina. Clinical Cardiology. 2007;30(2) Supplement 1:I1–I3. doi: 10.1002/clc.20041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martins e silva J. Ischemic myocardial disease as an example of a thrombotic event. A historical note. Revista Portuguesa de Cardiologia. 2006;25(5):537–543. [PubMed] [Google Scholar]
- 49.Van de Werf F. The history of coronary reperfusion. European Heart Journal. 2014;35(37):2510–2515. doi: 10.1093/eurheartj/ehu268. [DOI] [PubMed] [Google Scholar]
- 50.Eltzschig H. K., Eckle T. Ischemia and reperfusion—from mechanism to translation. Nature Medicine. 2011;17(11):1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frangogiannis N. G. Regulation of the inflammatory response in cardiac repair. Circulation Research. 2012;110(1):159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang M., Yang D., Xiang M., Wang J. Role of interleukin-6 in regulation of immune responses to remodeling after myocardial infarction. Heart Failure Reviews. 2015;20(1):25–38. doi: 10.1007/s10741-014-9431-1. [DOI] [PubMed] [Google Scholar]
- 53.Ortega-Gomez A., Perretti M., Soehnlein O. Resolution of inflammation: an integrated view. EMBO Molecular Medicine. 2013;5(5):661–674. doi: 10.1002/emmm.201202382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nahrendorf M., Swirski F. K. Innate immune cells in ischaemic heart disease: does myocardial infarction beget myocardial infarction? European Heart Journal. 2016;37(11):868–872. doi: 10.1093/eurheartj/ehv453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nahrendorf M., Swirski F. K., Aikawa E., et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. The Journal of Experimental Medicine. 2007;204(12):3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinez F. O., Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Reports. 2014;6(1):p. 13. doi: 10.1007/978-94-024-1057-0_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hulsmans M., Sam F., Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. Journal of Molecular and Cellular Cardiology. 2016;93(1):149–155. doi: 10.1016/j.yjmcc.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dutta P., Nahrendorf M. Monocytes in myocardial infarction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2015;35(5):1066–1070. doi: 10.1161/ATVBAHA.114.304652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saxena A., Chen W., Su Y., et al. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. Journal of Immunology. 2013;191(9):4838–4848. doi: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leuschner F., Panizzi P., Chico-Calero I., et al. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circulation Research. 2010;107(11):1364–1373. doi: 10.1161/CIRCRESAHA.110.227454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ismahil M. A., Hamid T., Bansal S. S., Patel B., Kingery J. R., Prabhu S. D. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circulation Research. 2014;114(2):266–282. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaikita K., Hayasaki T., Okuma T., Kuziel W. A., Ogawa H., Takeya M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. The American Journal of Pathology. 2004;165(2):439–447. doi: 10.1016/S0002-9440(10)63309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Amerongen M. J., Harmsen M. C., van Rooijen N., Petersen A. H., van Luyn M. J. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. The American Journal of Pathology. 2007;170(3):818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Glezeva N., Horgan S., Baugh J. A. Monocyte and macrophage subsets along the continuum to heart failure: misguided heroes or targetable villains? Journal of Molecular and Cellular Cardiology. 2015;89(Pt B):136–145. doi: 10.1016/j.yjmcc.2015.10.029. [DOI] [PubMed] [Google Scholar]
- 65.Hanna R. N., Carlin L. M., Hubbeling H. G., et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nature Immunology. 2011;12(8):778–785. doi: 10.1038/ni.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hilgendorf I., Gerhardt L. M., Tan T. C., et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circulation Research. 2014;114(10):1611–1622. doi: 10.1161/CIRCRESAHA.114.303204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prabhu S. D., Frangogiannis N. G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circulation Research. 2016;119(1):91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen W., Frangogiannis N. G. Fibroblasts in post-infarction inflammation and cardiac repair. Biochimica et Biophysica Acta. 2013;1833(4):945–953. doi: 10.1016/j.bbamcr.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mackay C. R. Moving targets: cell migration inhibitors as new anti-inflammatory therapies. Nature Immunology. 2008;9(9):988–998. doi: 10.1038/ni.f.210. [DOI] [PubMed] [Google Scholar]
- 70.Mann D. L. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circulation Research. 2015;116(7):1254–1268. doi: 10.1161/CIRCRESAHA.116.302317. [DOI] [PMC free article] [PubMed] [Google Scholar]