

Abstract
Craniofacial development is a complex process that involves sequential growth and fusion of the facial prominences. When these processes fail, congenital craniofacial anomalies can occur. For example, choanal atresia (CA) is a congenital craniofacial anomaly in which the connection between the nasal airway and nasopharynx is completely blocked. CA occurs in approximately 1/5000 live births and is a frequent component of congenital disorders such as CHARGE, Treacher Collins, Crouzon and Pfeiffer syndromes. However, the detailed cellular and molecular mechanisms underpinning the etiology and pathogenesis of CA remain elusive. In this study, we discovered that mice with mutations in retinol dehydrogenase 10 (Rdh10), which perturbs Vitamin A metabolism and retinoid signaling, exhibit fully penetrant CA. Interestingly, we demonstrate Rdh10 is specifically required in non-neural crest cells prior to E10.5 for proper choanae formation, and that in the absence of Rdh10, Fgf8 is ectopically expressed in the nasal fin. Furthermore, we found that defects in choanae development are associated with decreased cell proliferation and increased cell death in the epithelium of the developing nasal cavity, which retards invagination of the nasal cavity, and thus appears to contribute to the pathogenesis of CA. Taken together, our findings demonstrate that RDH10 is essential during the early stages of facial morphogenesis for the formation of a functional nasal airway, and furthermore establish Rdh10 mutant mice as an important model system to study CA.
Introduction
Craniofacial malformations comprise one of the most frequent classes of congenital anomalies in live human births (1). The high incidence is attributable to the complexity of craniofacial development, which depends on multiple precise biological events, including growth and fusion of the facial prominences and pharyngeal arches (2,3). Perturbation of these events can result in a wide variety of congenital craniofacial deformities, such as cleft lip and/or palate (4). Choanal atresia (CA), which is characterized by complete blockage of the connection between the nasal airway and nasopharynx (5), occurs in 1/5000–7000 human live births, and requires immediate airway management to prevent respiratory problems (6). CA is often a frequent feature of complex craniofacial disorders including CHARGE, Treacher Collins, Pfeiffer and Crouzon syndromes (7–9).
A number of mechanisms (10) have been hypothesized to underlie the pathogenesis of CA including (i) persistent buccopharyngeal membrane from the forgut; (ii), abnormal persistence or location of mesoderm forming adhesions in the nasochoanal region; (iii), abnormal persistence of the nasobuccal membrane of Hochstetter; and (iv), misdirection of neural crest cell migration (11–14). However, the detailed cellular and molecular mechanisms underpinning the etiology and pathogenesis of CA remain to be elucidated. In particular, we still do not know the critical period or tissues that are responsible for the pathogenesis of CA and a lack of relevant model for studying this condition contributes to our lack of knowledge.
In an ENU mutagenesis screen, which was aimed at identifying novel alleles that played critical roles in murine craniofacial development, we identified a mutant called trex which exhibited severe craniofacial anomalies including a midline facial cleft in association with mid-gestational lethality (15,16). We determined that the trex mice carried a mutation in retinol dehydrogenase 10 (Rdh10) and showed that RDH10 is responsible for oxidizing Vitamin A (retinol) to retinal (15), which is a rate-limiting step in the synthesis of retinoic acid (17,18). Commensurate with these discoveries, we demonstrated that Rdh10trex/trex mutant embryos exhibit deficiencies in retinoid signaling in association with their specific developmental anomalies (15,19–22), and furthermore that supplementation with the Vitamin A metabolic intermediate retinal, enabled Rdh10trex/trex mutant embryos to survive to birth (17). However, despite the remarkable overall prevention of their developmental anomalies, newborn Rdh10trex/trex pups often died shortly after birth, possibly as a result of a defect in oronasal development such as CA.
In order to investigate the precise role of Rdh10 and retinoid signaling in oronasal development and in the etiology and pathogenesis of CA, we utilized an allelic series of Rdh10 mutant mice including, Rdh10trex/trex and an Rdh10 conditional knockout line (Rdh10flox) (18) crossed with a tamoxifen-inducible ubiquitous Cre mouse (23) so that Rdh10 could be knocked out in a stage-specific manner (Cre-ERT2; Rdh10flox/flox) or tissue specific manner (Wnt1-Cre;Rdh10flox/flox) during embryogenesis. In the present study, we discovered that when tamoxifen was administered at E7.5 to induce excision of Rdh10 by E8.5, Cre-ERT2; Rdh10flox/flox mutant embryos exhibited CA mimicking the phenotype observed in humans, including an obstructed airway with ectopic bone formation in the maxillary region. Mechanistically, we demonstrate that the expression of Rdh10 in non-neural crest cells prior to E10.5 is critical for connecting the nasal airway and nasopharynx during normal choanae development. Furthermore, we show that Fgf8 is ectopically expressed in the nasal epithelium in association with elevated cell death and reduced cell proliferation in Rdh10 mutants, which perturbs morphogenesis of the nasal cavity, underpinning the pathogenesis of CA. Collectively, our findings reveal novel genetic pathways and cellular mechanisms underlying the etiology and pathogenesis of CA.
Results
Rdh10 expression and retinoid signaling are present in the developing nasal cavity
As a first step in investigating the potential role for Rdh10 and retinoid signaling in oronasal development and in the etiology of CA, it was important to define the spatiotemporal activity of Rdh10 and retinoid signaling during early craniofacial development. Therefore, we performed in situ hybridization using a riboprobe for Rdh10 in concert with β-galactosidase staining of retinoic acid response element (RARE) -LacZ reporter mice to document the distribution of retinoid signaling during embryogenesis (24). In E10.5 embryos, Rdh10 mRNA was detected in the distal and lateral epithelium of the developing nasal pit (Fig. 1A), while an overlapping but somewhat broader domain of activity was observed with RARE-lacZ (Fig. 1E). As the primitive choanae begin to form around E11.5, Rdh10 became more broadly expressed proximo-distally throughout the nasal epithelium in a pattern that was again overlapped by RARE-LacZ activity (Fig. 1B and F, red arrowhead). Histological sections confirmed that expression of Rdh10 was largely confined to the nasal epithelia (Fig. 1C, dotted line, asterisk), whereas the domain of RARE-LacZ occupied both the epithelium, which includes the oronasal membrane (Fig. 1G, red arrowhead), and the adjacent mesenchyme of the medial nasal and maxillary prominences. As the nasal cavity continued to develop through E12.5, strong expression of Rdh10 and staining for RARE-LacZ continued in both the proximal (red arrowheads) and distal tissues (Fig. 1D and H). Collectively, these data place Rdh10 and retinoid signaling in the precise location at the appropriate time to play a prominent role in oronasal development and choanae formation.
Figure 1.

Expression of Rdh10 and activity of RARE-LacZ during craniofacial development. (A,B,D) Ventral view of Rdh10 expression in serial embryonic stages in wild-type mice. (E,F,H) Activity of RARE-LacZ reporter during embryonic maxillary development. Red arrowheads indicate the position of primitive choanae in B and F, invaginating nasal cavity in D and H. (C and G) Frontal section of E11.5 control embryo showing expression of Rdh10 (C) and activity of RARE-LacZ (G). Red arrowhead indicates oronasal membrane in C and G, and black dotted line indicates the boundary of epithelial cells lining the nasal cavity. MNP, medial nasal process. LNP, lateral nasal process. MXP, maxillary process. MN, mandibular process. Scale bar = 200 μm.
Efficient deletion of Rdh10 activity in conditional knockout mice
In order to demonstrate a functional requirement for Rdh10 and retinoid signaling during oronasal development and in the etiology of CA, we temporally deleted Rdh10 throughout the entire embryo during early stages of development (18). We crossed Rdh10flox/flox mice to Cre-ERT2 mice (Cre-ERT2; Rdh10flox/flox) and administered tamoxifen to pregnant dams at E7.5 (Fig. 2A). First, we confirmed the efficiency of this conditional knock out allele by performing in situ hybridization using an Exon2 specific riboprobe for Rdh10. In wild type embryos at E10.5, Rdh10 is expressed in a tissue-specific manner in the nasal pit amongst other tissues (Fig. 2B). In contrast, Cre-ERT2; Rdh10flox/flox mutant embryos generated by exposure to tamoxifen at E7.5 exhibited a substantial reduction of Rdh10 expression throughout the entire body as expected and particularly in the nasal region (Fig. 2D). This phenomenon is similar to the loss of Rdh10 expression previously reported in Rdh10trex/trex mutant embryos (15). Furthermore, the expression of Rdh10 in the choanal region was considerably reduced in E11.5 Cre-ERT2; Rdh10flox/flox embryos when tamoxifen was administered at E7.5, compared to control littermates (Fig. 2C and E). Collectively, this demonstrates the efficient temporal generation of an Rdh10 loss-of-function model which together with Rdh10trex/trex mice provides an allelic series for studying the role of Rdh10 and retinoid signaling in oronasal development and the pathogenesis of CA.
Figure 2.

Confirmation of reduction of Rdh10 mRNA in mice with conditional null allele. (A) Schematic drawing of the method used to generate the Rdh10 conditional null allele (modified from reference (18). (B–E) In situ hybridization of Rdh10 using an Exon2 specific riboprobe. E10.5 and E11.5 control embryos (B and C) compared with the same stage of Cre-ERT2; Rdh10flox/flox embryos (D and E) which were administered tamoxifen at E7.5. Red arrowhead indicates the lambdoidal region in B,D; and primitive choanae in C,E. TMX E7.5, tamoxifen was administered at E7.5. E, eye. MXP, maxillary process. MN, mandibular process. MNP, medial nasal process. LNP, lateral nasal process. FL, forelimb. Scale bar = 500 μm in B,D and 200 μm in C,E.
Rdh10 loss-of-function results in CA with ectopic bone formation during craniofacial development
The effect of Rdh10 loss-of-function was initially determined morphologically via whole mount nuclear fluorescent (pseudo-SEM) imaging (25). In E11.5 control embryos, the primitive choanae starts to develop from the posterior region of the medial nasal process and maxillary process, as indicated by an epithelial invagination (Fig. 3A, red arrowhead). Rdh10trex embryos, which survive until around this stage without retinal supplementation, exhibit an absence of epithelial invagination, indicating that choanal development was impaired (Fig. 3E, red arrowhead). In E12.5, control embryos, oronasal development has progressed to the extent that the connection of the nasal cavity to the airway is complete (Fig. 3B, red arrowhead). In contrast, E12.5 Cre-ERT2; Rdh10flox/flox embryos, which were administered tamoxifen at E7.5, exhibit bilateral CA (Fig. 3F, red arrowhead) together with other defects such as cleft lip (Fig. 3F, yellow arrowhead). Alizarin red and alcian blue staining of the craniofacial skeleton of E15.5 Cre-ERT2; Rdh10flox/flox embryos revealed ectopic bone formation in the maxillary region (Fig. 3C and G). Frontal histological sections also revealed a substantially enlarged bone mass in the anterior maxillary region of E15.5 Cre-ERT2; Rdh10flox/flox embryos (Fig. 3H, asterisk) compared to controls (Fig. 3D, asterisk). In addition, the nasal airway failed to connect to the nasopharynx, and the shape of the nasal septum was severely distorted in Cre-ERT2;Rdh10flox/flox embryos compared to wild-type littermates (Fig. 3D and H). Taken together, these data demonstrate that Rdh10 plays a critical role in oronasal development and that Rdh10 loss-of-function can underpin the pathogenesis of CA.
Figure 3.

Morphological and histological analysis of Rdh10 mutant maxilla. Nuclear fluorescent staining of control (A,B) and Rdh10trex (E), and Cre-ERT2; Rdh10flox/flox mice to which tamoxifen was administered at E7.5 (F). Red arrowhead indicates the position of primitive choana(e) in (A and B), and their absence (E and F). Yellow arrowhead in (F) shows partial cleft lip. Skeletal preparation of E15.5 control (C) and Cre-ERT2; Rdh10flox/flox embryos (G). Hematoxylin and eosin staining of frontal section of E15.5 control (D) and Cre-ERT2; Rdh10flox/flox (H) embryos. Yellow asterisk demarcates the premaxillary bone. TMX E7.5, administered tamoxifen at E7.5. MNP, medial nasal process. LNP, lateral nasal process. MXP, maxillary process. MN, mandibular process. PMX, premaxillary bone. MX, maxillary bone. P, palatine bone. NS, nasal septum. Scale bar = 200 μm in A,E and 500 μm in B-D, F-H.
Rdh10 loss-of-function results in ectopic Fgf8 expression, reduced epithelial cell proliferation and elevated cell death
To better understand the molecular mechanisms by which diminished Rdh10 causes CA, we performed comprehensive comparative transcriptome analyses of the developing maxillary complex in E11.5 wild-type and mutant Cre-ERT2;Rdh10flox/flox embryos via RNAseq. We initially focused on the Fgf signaling pathway because it is critical for craniofacial development and also known to associate with the etiology of CA (7,26). Among the Fgf signaling related genes expressed in the developing facial area (27), Fgf8 showed the most significant difference between control and Cre-ERT2;Rdh10flox/flox embryos that were treated with tamoxifen at E7.5 (Supplementary Material, Table S1). To validate any spatiotemporal difference in the pattern of Fgf8 expression between control and Cre-ERT2; Rdh10flox/flox mice, we performed in situ hybridization for Fgf8 in the maxillary complex of E11.5 wild-type and mutant embryos. In control E11.5 embryos, Fgf8 was not detected in the nasal fin, which is the thin mass of tissue separating the medial nasal and maxillary prominences (Fig. 4A and B, red arrowhead). In contrast, E11.5 Cre-ERT2;Rdh10flox/flox embryos, which were administered tamoxifen at E7.5, exhibited ectopic Fgf8 expression in the nasal fin (Fig. 4C and D, red arrowhead). Since FGF8 signaling is known to influence cellular activities such as cell proliferation and cell death during embryonic craniofacial development (28,29), we performed immunostaining with phosphohistone H3 (PHH3), to assess for alterations in cell proliferation, and TUNEL staining to detect any changes in apoptosis. PHH3 immunostaining revealed a significant reduction in proliferating epithelial cells in the nasal fin of mutant embryos as compared to controls. Furthermore, the region of diminished proliferation coincided with the domain of ectopic Fgf8 expression (Fig. 4E–G and E white arrowhead). At the same time, TUNEL staining revealed a significant elevation of cell death in the corresponding region in mutant embryos when compared to controls (Fig. 4E–G and F white arrow). It is well established that cell proliferation and cell death play critical roles during embryonic morphogenesis. Thus ectopic FGF8 signaling in association with elevated apoptosis and diminished proliferation in the nasal epithelium may contribute to malformation of the nasal cavity which appears to underlie the pathogenesis of CA. In addition to elevated expression of Fgf8, we discovered other genes that exhibited significant expression level changes in Cre-ERT2; Rdh10flox/flox mutant embryos. Distal-less gene family members Dlx1 and Dlx2 (Supplementary Material, Table S1), which are known to play important roles in craniofacial development (30), were also elevated.
Figure 4.

Excision of Rdh10 resulted in elevation of Fgf8 expression and altered cell proliferation and cell death. (A–D) In situ hybridization for Fgf8 in E11.5 embryos. (B and D) Frontal section of E11.5 embryos hybridized with an Fgf8 riboprobe. Red arrowheads indicate the nasal fin. (E and F) Immunohistochemistry of frontal sections from E11.5 embryos. Proliferating cells are stained red and cells undergoing cell death are stained green (E and F). White arrowheads indicate proliferating cells and white arrows indicate TUNEL-positive cell death in the nasal fin. Dotted line indicates the extent of the nasal epithelium. (G) Statistical analysis showed significant reduction in number of proliferating cell and elevation of cell death in Cre-ERT2; Rdh10flox/flox nasal fin. TMX E7.5, administered tamoxifen at E7.5. MNP, medial nasal process. LNP, lateral nasal process. N, nasal cavity. PHH3, phosphohistone H3. C, control mice. M, mutant (Cre-ERT2; Rdh10flox/flox) mice. *P < 0.05.
Spatiotemporal requirement for Rdh10 for primitive choanae formation
Neural crest cells play critical roles in regulating craniofacial morphogenesis and it was important to determine whether Rdh10 was intrinsically or extrinsically required in neural crest cells for normal oronasal development. Therefore, we used Wnt1-Cre mice (Supplementary Material, Fig. S1) to conditionally delete Rdh10 from presumptive neural crest cells and their derivatives during embryogenesis. Nuclear fluorescent staining (pseudo-SEM imaging) clearly revealed the primitive choanae formation in both E11.5 control and Wnt1Cre;Rdh10flox/flox mutant embryos (Fig. 5A and B). This indicates that Rdh10 is not required in neural crest cells for proper primitive choanae development. Next we determined the precise developmental period in which Rdh10 was required for primitive choanae formation. E12.5 Cre-ERT2;Rdh10flox/flox embryos that were administered tamoxifen at E10.5 exhibited narrower choanae formation when compared to littermate controls which resembles choanal stenosis in humans (Fig. 5C and D, red arrowhead). Nonetheless, examination of sections of E13.5 wild-type and mutant embryos revealed that a connection does indeed form between the nasal airway and nasopharynx in Cre-ERT2;Rdh10flox/flox embryos (Supplementary Material, Fig. S2). Taken together, these results demonstrate that the formation of connection between the nasal airway and nasopharynx prior to E10.5. After E10.5 Rdh10 is not needed for airway connection, but remains important for continued choanal development (Fig. 5E).
Figure 5.
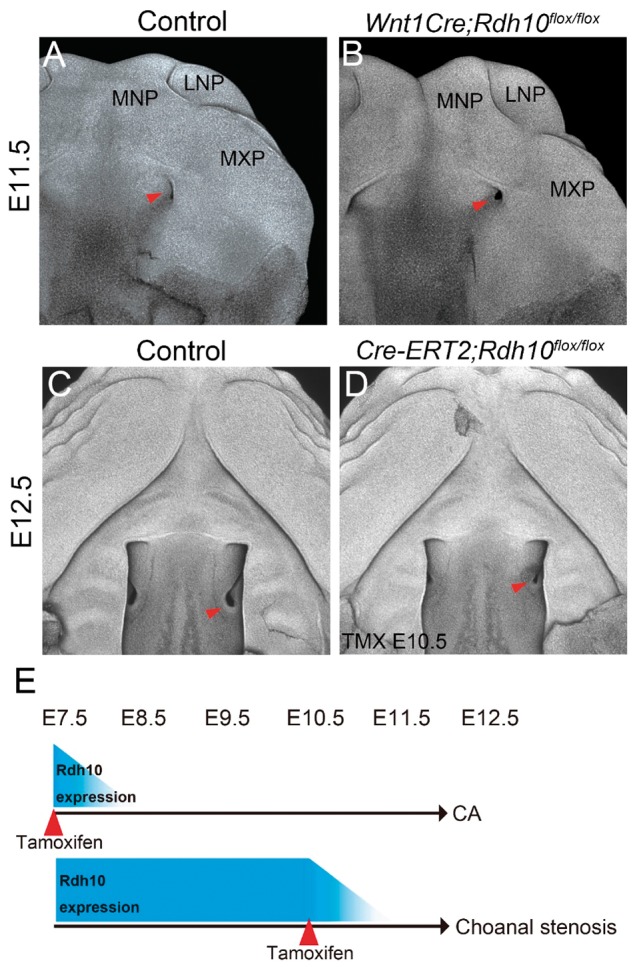
Morphological analysis following conditional deletion of Rdh10 during craniofacial development. Nuclear fluorescent image of the maxillary complex in E11.5 control mouse embryos (A). Red arrowhead indicates the position of the primitive choana, which still forms in Wnt1Cre;Rdh10flox/flox (neural crest-specific deletion of Rdh10) mice (B). At E12.5, the nasal cavity invaginates posteriorly in control mice (C, red arrowhead), but is impaired and hypoplastic in E10.5 tamoxifen (TMX)-administered embryos (D, red arrowhead). (E) Schematic drawing of the phenotype of Cre-ERT2; Rdh10flox/flox embryos at E12.5 according to when the tamoxifen is administrated. TMX E10.5, administered tamoxifen at E10.5. MNP, medial nasal process. LNP, lateral nasal process. MXP, maxillary process.
Retinal supplementation ameliorates the pathogenesis of CA in Rdh10 deficient mice
We have previously shown that the malformations observed in Rdh10trex/trex embryos can be largely rescued through maternal supplementation with retinal or retinoic acid during early embryogenesis (15,17,31,32). However, detailed examination of choana formation in embryos developing with retinal supplementation has not been performed. In order to characterize which aspects of choana development can be rescued by maternal retinal supplementation, we administered 12.5 mg/kg of retinal by oral gavage to Rdh10trex/+ pregnant mothers, twice daily between E7.5 and E11, and subsequently harvested Rdh10trex/trex embryos and littermate controls at E18.5. Examination of frontal sections through the nasal septum and nasal cavity of wild-type and mutant embryos revealed differences in the shape of the nasal cavity. The nasal cavity in Rdh10trex/trex embryos was slightly wider and shorter than in littermate controls (Fig. 6A and B). However, from serial sections we observed that there was indeed complete penetration of the nasal cavity to the airway in retinal supplemented Rdh10trex/trex mice (Fig. 6C and D). When compared to the phenotype of non-supplemented Rdh10trex/trex embryos, as well as Cre-ERT2;Rdh10flox/flox embryos treated with tamoxifen at E7.5, this indicates that choanal defects in Rdh10 mutant embryos result from insufficient production of retinal and addition of retinal by maternal administration is sufficient to promote connection of the airway.
Figure 6.

All-trans retinal rescued CA in Rdh10trex/trex mutants. (A) Hematoxylin and eosin staining of frontal section of anterior position of E18.5 control mouse and (B) Rdh10trex mutant treated with retinal. At the posterior position, the nasal cavity and pharynx are clearly connected in the control (C, red arrowhead) and in the Rdh10trex mutant treated with retinal (D, red arrowhead).
Discussion
Rdh10 and retinoid signaling play critical roles in the developing maxillary complex
It is well established that retinoid signaling plays an indispensable role in the development and patterning of various tissues and organs including craniofacial structures during embryogenesis (33–35). Retinoic acid is the metabolic byproduct of vitamin A generated by the two-step oxidation process. Excessive maternal intake of vitamin A is known to cause a significant elevation of retinoid signaling during embryogenesis, which results in craniofacial defects such as facial clefting in mammals and avians (36,37). Rdh10 represents a nodal point in the feedback regulation of retinoid signaling and is thus a rate limiting factor in the first oxidation reaction which converts vitamin A (retinol) to retinal (15,17,18). Retinoic acid can either activate or repress the transcription of downstream target genes by binding to nuclear retinoic acid receptors (RARs), which in turn directly bind to specific DNA sequences called RAREs (33,38). In this study, we characterized the spatiotemporal patterns of Rdh10 expression and retinoid signaling (RARE-lacZ) during mouse embryonic craniofacial development from stage E10.5 onwards with a particular emphasis on the oronasal region. In situ hybridization in E10.5 embryos revealed that Rdh10 is strongly expressed in the epithelial lining of the developing nasal cavity, and is encompassed by a broader domain of active retinoid signaling, as shown by RARE-lacZ activity. The overlapping domains of Rdh10 expression and RARE-lacZ activity continued through E11.5 and were maintained in the maxillary region, particularly where the primitive choanae develop. Rdh10 is predominantly expressed in epithelial cells (Fig. 1C), while RARE-lacZ expression was detected in both epithelial and mesenchymal cells (Fig. 1G), which indicates that while retinoic acid might be produced primarily in epithelial tissue, the surrounding tissues actively respond to retinoid signaling. The intense pattern of RARE-lacZ staining observed in histological sections of the oronasal membrane (Fig. 1G, red arrowhead), suggests that retinoid signaling may play an important role in connecting the nasal airway and nasopharynx.
Excising Rdh10 during maxillary complex morphogenesis in mice leads to a phenotype resembling CA in humans
Previous studies revealed that germline mutation of Rdh10 results in severe developmental defects, including craniofacial abnormalities and embryonic lethality around E11.5 (15,31). Here, we employed an allelic series of Rdh10 mutant mice including conditional knockouts to investigate the role of Rdh10 during craniofacial development and particularly in oronasal development and the pathogenesis of CA. Tamoxifen administration at E7.5 in combination with Cre-ERT2 (39) resulted in the specific temporal and global deletion of Rdh10 during early embryogenesis (Fig. 2C and E). These Cre-ERT2; Rdh10flox/flox embryos survived longer than Rdh10trex/trex embryos allowing for an investigation of the role of Rdh10 in craniofacial development after E11.5, during which nasal cavity invagination and the primitive choanae formation occurs. In E11.5 Rdh10trex/trex embryos, epithelial invagination and the primitive choanae formation fails to occur resulting in CA (Fig. 3A,B,E and F).
These results clearly demonstrate that Rdh10 is critically required between E7.5 and E10.5 for proper development of the primitive choanae, which is an important morphological step in connecting the nasal airway and nasopharynx. Furthermore, our histological analyses revealed ectopic bone formation in this obstructed area. Interestingly, the bone which was enlarged in Cre-ERT2; Rdh10flox/flox embryos seems to be part of developing premaxilla bone, while pterygoid plates and vomer bone are the bones affected in human CA. However, given the similarity in primitive choanae development between mice and humans (40), the allelic series of Rdh10 mutant mice described in this study still provide useful models for investigating the etiology and pathogenesis of CA in humans.
Disruption of retinoid and FGF8 signaling underpins the etiology of CA
Relatively few studies have investigated the cellular and molecular mechanisms underpinning CA using animal models. However, in support of our findings of a potential link between retinoid signaling and CA, mutations in Aldh1a3 also cause CA. Aldh1a3 plays a critical role in the second oxidation step of Vitamin A metabolism by converting retinal into retinoic acid. Furthermore, Aldh1a3 is expressed in the developing nasal placode (41) and Aldh1a3 loss-of-function mouse mutants exhibit CA in association with diminished retinoid signaling (26).
Interestingly, the phenotype of Cre-ERT2; Rdh10flox/flox embryos that were administered tamoxifen at E7.5 exhibited a wider spectrum of craniofacial deformities, including cleft lip and/or palate (CLP) (Fig. 3F), than Aldh1a3 mutant embryos. CA in humans is known to be often associated with other craniofacial abnormalities including facial clefts (7,42), and there are other animal models which exhibit CA in combination with a facial cleft (26,43). The fact that Rdh10 mutants exhibit either a midfacial cleft (15) or cleft lip (Fig. 3F) in combination with CA suggests that retinoid signaling mediated by Rdh10 may contribute to the etiology of facial clefts in association with CA.
To further investigate the molecular basis of CA we undertook RNA-seq transcriptional analyses and identified the Fibroblast growth factor (FGF) signaling pathway as being altered in the maxillary complex of Rdh10 mutant embryos (Supplementary Material, Table S1). Fgf8 exhibited the most significant difference in expression level between control and Cre-ERT2;Rdh10flox/flox embryos that were treated with tamoxifen at E7.5 (Supplementary Material, Table S1). More importantly, we determined that Fgf8 was ectopically expressed in the nasal fin of tamoxifen-administered Cre-ERT2; Rdh10flox/flox embryos (Fig. 4C and D).
Similar to our results reported here of ectopic Fgf8 expression in Rdh10 mutants, persistent Fgf8 expression has also been observed in the nasal fin of Aldh1A3 mutant embryos (26). Thus, elevated Fgf8 as a consequence of reduced retinoid production may constitute a common mechanism leading to the manifestation of CA. These results indicate that CA in Rdh10 and Aldh1A3 mutant embryos shares a similar molecular etiology that starts with reduced retinoid signaling, and is followed by ectopic or elevated FGF8 signaling specifically in the nasal fin. Our working model therefore is that retinoid signaling is necessary to repress the activity of Fgf8 during normal oronasal development. The role of retinoic acid in repressing Fgf8 in oronasal development is therefore similar to the molecular competition between retinoid and FGF8 signaling that occurs during somitogenesis and in limb development (19–21). Moreover, studies of chick craniofacial development have demonstrated the disruption of Fgf8 expression that occurs under conditions of reduced retinoid signaling (44,45). Furthermore, syndromes such as Pfeiffer syndrome, Crouzon syndrome, Apert syndrome, Beare–Stevenson syndrome and Antley-Bixler syndrome (46–48) exhibit CA in association with other craniofacial anomalies, and each has been linked to exaggerated FGF signaling. Finally, a recent study revealed that elevated Fgf8 expression in cranial neural crest cells results in perturbation of nasal cavity invagination (49). This provides additional evidence supporting our observations and conclusions of the importance of retinoid and FGF signaling integration in the etiology of CA in Rdh10 mutants.
FGF8 signaling is well known to play critical roles during craniofacial development through interactions with other signaling pathways and by regulating cellular activities such as cell proliferation and survival (49,50). In the present study, we discovered reduced cell proliferation and increased cell death in the nasal fin epithelium of Rdh10 mutant embryos in association with ectopic FGF8 signaling (Fig. 4).
Mitotic cell rounding is known to be associated with epithelial invagination (51,52) and thus it’s tempting to speculate that reduced proliferation in the nasal epithelium of Cre-ERT2; Rdh10flox/flox mutant embryos is associated with a reduction in mitotic cell rounding, resulting in impaired invagination and CA (Fig. 7). Furthermore, the increased cell death observed in Cre-ERT2; Rdh10flox/flox mutant embryos may actually then eliminate the cells that are important for invagination of the nasal cavity and primitive choanae development (Fig. 7).
Figure 7.

Schematic drawing of the process of nasal cavity invagination and primitive choanae formation. Nasal cavity invagination takes place to connect the nasal cavity to the airway by rupturing the oronasal membrane during normal development (A). In Rdh10 mutants, the number of proliferating epithelial cells is reduced, and the nasal cavity cannot connect to the airway, resulting in CA (B).
In addition to elevated expression of Fgf8, we noted elevated expression of Dlx1 and Dlx2, two established targets of FGF8 signaling (53,54). The observation that FGF8 target genes are elevated in addition to Fgf8 strengthens the conclusion that ectopic FGF8 signaling plays an active role in abnormal facial morphogenesis in Rdh10 mutant embryos. The possible roles or interaction of those genes with retinoid signaling during craniofacial development and in the pathogenesis of choanal atresia will be pursued in the future.
Rdh10 is spatiotemporally required for proper nasal cavity and primitive choanae development
Although we detected Rdh10 expression in both the epithelium and mesenchyme of the developing nasal cavity, and know that crosstalk between epithelial and mesenchymal tissues are critical for normal organogenesis, it remained to be determined when and in which tissue Rdh10 was critical required for proper primitive choanae formation. We found no obvious evidence for CA in embryos in which Rdh10 was conditionally deleted in neural crest cells or temporally deleted throughout the embryo at E10.5. Collectively, these data strongly suggest that Rdh10 is critically required between E7.5 and E10.5 in the non-neural crest cell derived nasal epithelium in order to connect the nasal airway and nasopharynx.
Mutant mice, which were administered tamoxifen at E10.5 exhibited narrower choanae which could mimic choanal stenosis in humans (Fig. 5D). This suggests that a common mechanism could underlie choanal atresia and choanal stenosis with choanal atresia being a more severe form of choanal stenosis in cases of disrupted retinoid signaling.
There is, however, clear evidence that defects in cranial neural crest cell development can also contribute to the etiology of CA as exemplified by the phenotype of Treacher-Collins syndrome which is classified as a neurocristopathy (2,9). These results imply that different signaling pathway may work in a tissue specific manner to promote the proper development of a functional airway.
Craniofacial morphogenesis is a complex process requiring growth and fusion of the medial and lateral nasal processes, together with nasal cavity invagination for proper oronasal development (55). Interestingly, the embryonic craniofacial development is a highly conserved process as has been shown through principal component analyses of comparative anatomy (56). This morphological conservation in combination with our results suggests that Rdh10 is an important gene whose expression needs to be tightly regulated in a spatiotemporal manner for proper orofacial development.
Maternal retinal supplementation could restore the CA phenotype in Rdh10 mutant
Consistent with the idea of a precise spatiotemporal requirement for Rdh10 during oronasal development, we showed that the pathogenesis of CA in Rdh10trex/trex mice could be prevented by administering retinal from E7.5 through E11.5. Taken together with the temporal deletion of Rdh10 in Cre-ERT2; Rdh10flox/flox embryos following tamoxifen administration, these results again indicate that Rdh10 and retinoid signaling is critically required between E7.5-E10.5 for choanae formation and for connecting the nasal airway and nasopharynx. Furthermore, this is consistent with our previous studies showing that maternal retinal or retinoic acid supplementation can rescue the gross morphology and embryonic lethality of Rdh10 mutant embryos (15,31,32).
Materials and Methods
Animals
The Rdh10trex and Rdh10flox mice lines used in this study were generated and maintained as described previously (15,18). Briefly, the Rdh10flox mice were derived from ES cells generated through KOMP and is equivalent to C57BL/6N-Rdh10tm1a(KOMP)Wtsi. Cre-ERT2 (B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J, Jax stock #008463), and Wnt1Cre mice (Tg(Wnt1-cre)11Rth Tg(Wnt1-GAL4)11Rth/J, Jax stock #003829) were obtained from the Jackson Laboratory and maintained as previously described (39,57,58). Cre driver mouse lines were crossed with Rdh10flox mice in order to eliminate RDH10 function in neural crest cells (Wnt1Cre) or in all embryonic cells following treatment with tamoxifen (Cre-ERT2). The homozygous Rdh10 flox alleles in combination with either Cre-ERT2 or Wnt1Cre are respectively termed Cre-ERT2; Rdh10flox/flox and Wnt1Cre; Rdh10flox/flox in this manuscript, and either wild-type or heterozygous littermates were used as controls. RARE-LacZ (Tg(RARE-Hspa1b/lacZ)12Jrt/J, Jax stock #008477) and Rosa 26 Reporter (R26R) (FVB.129S4(B6)-Gt(ROSA)26Sortm1Sor/J, Jax stock #009427) reporter mice were also obtained from The Jackson Laboratory and maintained as described previously (24,59). For embryonic staging, the morning of identification of the vaginal plug was defined as E0.5.
Administration of tamoxifen
To activate the Cre protein at defined stages of embryonic development, we administered 5 mg of tamoxifen and 1 mg of progesterone in 100 μl of corn oil by oral gavage to individual pregnant female Rdh10fllox/flox mice at defined time points after they had been successfully mated with an Ert2Cre:Rdh10 fllox/flox male.
In situ hybridization
Whole-mount in situ hybridization and subsequent sectioning of stained embryos were performed as described previously (3). Exon2 of Rdh10 was amplified using the following set of primers: forward 5' CTGGAAAAGGTGAGGAGGAAATC 3', reverse 5' CCAGAAGTGTGCGTGGCAGTTG 3', and cloned into the TOPO vector (Invitrogen) to produce an RNA riboprobe.
Whole-mount nuclear fluorescent imaging
To analyze embryonic morphology, a nuclear fluorescent imaging technique called ‘Pseudo SEM’ was performed as previously described (25).
Skeletal preparation
Embryos were collected at E16.5 and stained with Alizarin red and alcian blue to visualize bone and cartilage, respectively, as previously described (60).
Immunohistochemistry and TUNEL staining
Embryos were dissected at various defined developmental stages, fixed in 4% PFA at 4 °C overnight, equilibrated through a sucrose gradient to 30%, and mounted in Tissue Tek O.C.T. 10-μm sections were cut, mounted on glass slides and stored at −30°c until required. Immunohistochemistry was performed with an M.O.M. Immunodetection Kit (VECTOR) according to the manufacturer’s protocol. After the sections were incubated with the primary antibody (anti-phospoho-Histone-3 (Millipore)), they were washed and incubated with a fluorescent secondary antibody and counterstained with DAPI (DAKO). To detect the presence of apoptosis, we used an In Situ Cell Death Detection Kit Fluorescein (Roche) according to the manufacturer’s instructions.
β- g alactosidase staining
β-Galactosidase staining of RARE-LacZ and Wnt1Cre;R26R mouse embryos was performed as described previously (3). For detailed analysis, some of the stained embryos were processed into 18-μm-thick frozen sections.
Maternal supplementation with retinal
Rdh10trex/+ pregnant female mice that had been mated with Rdh10trex/+ males were administered twice per day (morning and evening) by oral gavage, 12.5 mg/kg weight retinal from E7.5 through E11.5.
RNAseq analysis
The maxillary complex (including medial nasal, lateral nasal and maxillary processes) (3) was isolated from E11.5 Cre-ERT2; Rdh10flox/flox mouse embryos (n = 3) and their littermate controls (n = 3) that had been treated with tamoxifen via oral gavage of their mothers at E7.5. Total RNA was extracted using an RNeasy Kit (Qiagen) according to the manufacturer’s protocol and RNA-seq was performed using 2 of each samples which showed a similar overall expression profile to analyze the data. Sequences of each RNA sample were aligned using TopHat (v2.0.10) (61). The reference genome and annotation files used for the alignment were from UCSC mm10 genome and ensemble build 75. We generated read counts that were mapped to the exons of genes using BEDTOOLs. Normalized gene expression (RPKMs) is calculated using the rpkm function after the TMM normalization with R package edgeR (62).
Statistics
P values in Figure 4E were adjusted using the Benjamini-Hochberg procedure to control the false discovery rate. An R package, edgeR, was used to find differentially expressed genes between control and mutant samples. t-test that assumes non-equal variances were performed for data represented in Figure 4H. Adjusted P-values of less than 0.05 were considered significant in all experiments.
Study approval
Mice were housed either in an animal facility in Department of Dentistry, Osaka University or the Laboratory Animal Services Facility at the Stowers Institute for Medical Research. Welfare guidelines and procedures were performed with the approval of Osaka University Graduate School of Dentistry animal committee and the Stowers Institute for Medical Research IACUC.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors thank the members of the Trainor lab and of the Department of Orthodontics and Dentofacial Orthopedics, Graduate School of Dentistry, Osaka University, for their insights and constructive comments throughout the course of this project and completion of this manuscript. We are indebted to Melissa Childers for excellent care and maintenance of our mouse colony; Nancy Thomas, Nannette Marsh and Karen Smith for histological work; and Karin Zueckert-Gaudenz, Anoja Perera and Hua Li for transcriptome analysis.
Conflict of Interest statement. None declared.
Funding
JSPS KAKENHI Grants 15H05687 and 16K15836 (HK), and the National Institute for Dental and Craniofacial Research (DE DE016082), Stowers Institute for Medical Research (PAT). Funding to pay the Open Access publication charges for this article was provided by Osaka University.
References
- 1. Trainor P.A., Andrews B.T. (2013) Facial dysostoses, Etiology, pathogenesis and management. Am. J. Med. Genet. C. Semin. Med. Genet., 163C, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Trainor P.A. (2010) Craniofacial birth defects, The role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am. J. Med. Genet. A, 152A, 2984–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kurosaka H., Iulianella A., Williams T., Trainor P.A. (2014) Disrupting hedgehog and WNT signaling interactions promotes cleft lip pathogenesis. J. Clin. Invest., 124, 1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dixon M.J., Marazita M.L., Beaty T.H., Murray J.C. (2011) Cleft lip and palate, understanding genetic and environmental influences. Nat. Rev. Genet., 12, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kwong K.M. (2015) Current Updates on Choanal Atresia. Front. Pediatr., 3, 52.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramsden J.D., Campisi P., Forte V. (2009) Choanal atresia and choanal stenosis. Otolaryngol. Clin. North. Am, 42, 339–352, x. [DOI] [PubMed] [Google Scholar]
- 7. Burrow T.A., Saal H.M., de Alarcon A., Martin L.J., Cotton R.T., Hopkin R.J. (2009) Characterization of congenital anomalies in individuals with choanal atresia. Arch. Otolaryngol. Head. Neck. Surg., 135, 543–547. [DOI] [PubMed] [Google Scholar]
- 8. Stieve M., Kempf H.G., Lenarz T. (2009) Management of choanal atresia in cases of craniofacial malformation. J. Maxillofac. Oral. Surg., 8, 52–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Andrade E.C., Júnior V.S., Didoni A.L., Freitas P.Z., Carneiro A.F., Yoshimoto F.R. (2005) Treacher Collins Syndrome with choanal atresia, a case report and review of disease features. Braz. J. Otorhinolaryngol., 71, 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hengerer A.S., Brickman T.M., Jeyakumar A. (2008) Choanal atresia, embryologic analysis and evolution of treatment, a 30-year experience. Laryngoscope, 118, 862–866. [DOI] [PubMed] [Google Scholar]
- 11. Som P.M., Naidich T.P. (2013) Illustrated review of the embryology and development of the facial region, part 1, Early face and lateral nasal cavities. AJNR. Am. J. Neuroradiol., 34, 2233–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim C.H., Park H.W., Kim K., Yoon J.H. (2004) Early development of the nose in human embryos, a stereomicroscopic and histologic analysis. Laryngoscope, 114, 1791–1800. [DOI] [PubMed] [Google Scholar]
- 13. Flake C.G., Ferguson C.F. (1964) Congenital choanal atresia in infants and children. Ann. Otol. Rhinol. Laryngol., 73, 458–473. [DOI] [PubMed] [Google Scholar]
- 14. Hengerer A.S., Strome M. (1982) Choanal atresia, a new embryologic theory and its influence on surgical management. Laryngoscope, 92, 913–921. [PubMed] [Google Scholar]
- 15. Sandell L.L., Sanderson B.W., Moiseyev G., Johnson T., Mushegian A., Young K., Rey J.P., Ma J.X., Staehling-Hampton K., Trainor P.A. (2007) RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes. Dev., 21, 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sandell L.L., Iulianella A., Melton K.R., Lynn M., Walker M., Inman K.E., Bhatt S., Leroux-Berger M., Crawford M., Jones N.C., et al. (2011) A phenotype-driven ENU mutagenesis screen identifies novel alleles with functional roles in early mouse craniofacial development. Genesis, 49, 342–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Farjo K.M., Moiseyev G., Nikolaeva O., Sandell L.L., Trainor P.A., Ma J.X. (2011) RDH10 is the primary enzyme responsible for the first step of embryonic Vitamin A metabolism and retinoic acid synthesis. Dev. Biol., 357, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sandell L.L., Lynn M.L., Inman K.E., McDowell W., Trainor P.A. (2012) RDH10 oxidation of Vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PLoS. One, 7, e30698.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cunningham T.J., Chatzi C., Sandell L.L., Trainor P.A., Duester G. (2011) Rdh10 mutants deficient in limb field retinoic acid signaling exhibit normal limb patterning but display interdigital webbing. Dev. Dyn., 240, 1142–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cunningham T.J., Zhao X., Sandell L.L., Evans S.M., Trainor P.A., Duester G. (2013) Antagonism between retinoic acid and fibroblast growth factor signaling during limb development. Cell. Rep., 3, 1503–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cunningham T.J., Brade T., Sandell L.L., Lewandoski M., Trainor P.A., Colas A., Mercola M., Duester G. (2015) Retinoic Acid Activity in Undifferentiated Neural Progenitors Is Sufficient to Fulfill Its Role in Restricting Fgf8 Expression for Somitogenesis. PLoS. One, 10, e0137894.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lenti E., Farinello D., Yokoyama K.K., Penkov D., Castagnaro L., Lavorgna G., Wuputra K., Sandell L.L., Tjaden N.E., Bernassola F., et al. (2016) Transcription factor TLX1 controls retinoic acid signaling to ensure spleen development. J. Clin. Invest., 126, 2452–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hayashi S., McMahon A.P. (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre, a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol., 244, 305–318. [DOI] [PubMed] [Google Scholar]
- 24. Rossant J., Zirngibl R., Cado D., Shago M., Giguère V. (1991) Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes. Dev., 5, 1333–1344. [DOI] [PubMed] [Google Scholar]
- 25. Sandell L.L., Kurosaka H., Trainor P.A. (2012) Whole mount nuclear fluorescent imaging, convenient documentation of embryo morphology. Genesis, 50, 844–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dupé V., Matt N., Garnier J.M., Chambon P., Mark M., Ghyselinck N.B. (2003) A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. U S A, 100, 14036–14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bachler M., Neubüser A. (2001) Expression of members of the Fgf family and their receptors during midfacial development. Mech. Dev., 100, 313–316. [DOI] [PubMed] [Google Scholar]
- 28. Green R.M., Feng W., Phang T., Fish J.L., Li H., Spritz R.A., Marcucio R.S., Hooper J., Jamniczky H., Hallgrímsson B., et al. (2015) Tfap2a-dependent changes in mouse facial morphology result in clefting that can be ameliorated by a reduction in Fgf8 gene dosage. Dis. Model. Mech., 8, 31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trumpp A., Depew M.J., Rubenstein J.L., Bishop J.M., Martin G.R. (1999) Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes. Dev., 13, 3136–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gordon C.T., Brinas I.M., Rodda F.A., Bendall A.J., Farlie P.G. (2010) Role of Dlx genes in craniofacial morphogenesis, Dlx2 influences skeletal patterning by inducing ectomesenchymal aggregation in ovo. Evol. Dev., 12, 459–473. [DOI] [PubMed] [Google Scholar]
- 31. Rhinn M., Schuhbaur B., Niederreither K., Dollé P. (2011) Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proc. Natl. Acad. Sci. U S A, 108, 16687–16692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wright D.M., Buenger D.E., Abashev T.M., Lindeman R.P., Ding J., Sandell L.L. (2015) Retinoic acid regulates embryonic development of mammalian submandibular salivary glands. Dev. Biol., 407, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Niederreither K., Dollé P. (2008) Retinoic acid in development, towards an integrated view. Nat. Rev. Genet., 9, 541–553. [DOI] [PubMed] [Google Scholar]
- 34. Tamarin A., Crawley A., Lee J., Tickle C. (1984) Analysis of upper beak defects in chicken embryos following with retinoic acid. J. Embryol. Exp. Morphol., 84, 105–123. [PubMed] [Google Scholar]
- 35. Wedden S.E., Tickle C. (1986) Quantitative analysis of the effect of retinoids on facial morphogenesis. J. Craniofac. Genet. Dev. Biol. Suppl., 2, 169–178. [PubMed] [Google Scholar]
- 36. Abbott B.D., Harris M.W., Birnbaum L.S. (1989) Etiology of retinoic acid-induced cleft palate varies with the embryonic stage. Teratology, 40, 533–553. [DOI] [PubMed] [Google Scholar]
- 37. Richman J.M., Delgado J.L. (1995) Locally released retinoic acid leads to facial clefts in the chick embryo but does not alter the expression of receptors for fibroblast growth factor. J. Craniofac. Genet. Dev. Biol., 15, 190–204. [PubMed] [Google Scholar]
- 38. Cunningham T.J., Duester G. (2015) Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell. Biol., 16, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ventura A., Kirsch D.G., McLaughlin M.E., Tuveson D.A., Grimm J., Lintault L., Newman J., Reczek E.E., Weissleder R., Jacks T. (2007) Restoration of p53 function leads to tumour regression in vivo. Nature, 445, 661–665. [DOI] [PubMed] [Google Scholar]
- 40. Abramyan J., Thivichon-Prince B., Richman J.M. (2015) Diversity in primary palate ontogeny of amniotes revealed with 3D imaging. J. Anat., 226, 420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mic F.A., Molotkov A., Fan X., Cuenca A.E., Duester G. (2000) RALDH3, a retinaldehyde dehydrogenase that generates retinoic acid, is expressed in the ventral retina, otic vesicle and olfactory pit during mouse development. Mech. Dev., 97, 227–230. [DOI] [PubMed] [Google Scholar]
- 42. Bonafos G., Capon-Degardin N., Fayoux P., Pellerin P. (2004) Choanal atresia and rare craniofacial clefts, report of three cases with a review of the literature. Cleft. Palate. Craniofac J., 41, 78–83. [DOI] [PubMed] [Google Scholar]
- 43. Pauws E., Hoshino A., Bentley L., Prajapati S., Keller C., Hammond P., Martinez-Barbera J.P., Moore G.E., Stanier P. (2009) Tbx22null mice have a submucous cleft palate due to reduced palatal bone formation and also display ankyloglossia and choanal atresia phenotypes. Hum. Mol. Genet., 18, 4171–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schneider R.A., Hu D., Rubenstein J.L., Maden M., Helms J.A. (2001) Local retinoid signaling coordinates forebrain and facial morphogenesis by maintaining FGF8 and SHH. Development, 128, 2755–2767. [DOI] [PubMed] [Google Scholar]
- 45. Song Y., Hui J.N., Fu K.K., Richman J.M. (2004) Control of retinoic acid synthesis and FGF expression in the nasal pit is required to pattern the craniofacial skeleton. Dev. Biol., 276, 313–329. [DOI] [PubMed] [Google Scholar]
- 46. Xie C., De S., Selby A. (2016) Management of the Airway in Apert Syndrome. J. Craniofac. Surg., 27, 137–141. [DOI] [PubMed] [Google Scholar]
- 47. Upmeyer S., Bothwell M., Tobias J.D. (2005) Perioperative care of a patient with Beare-Stevenson syndrome. Paediatr. Anaesth., 15, 1131–1136. [DOI] [PubMed] [Google Scholar]
- 48. Escobar L.F., Bixler D., Sadove M., Bull M.J. (1988) Antley-Bixler syndrome from a prognostic perspective, report of a case and review of the literature. Am. J. Med. Genet., 29, 829–836. [DOI] [PubMed] [Google Scholar]
- 49. Shao M., Liu C., Song Y., Ye W., He W., Yuan G., Gu S., Lin C., Ma L., Zhang Y., et al. (2015) FGF8 signaling sustains progenitor status and multipotency of cranial neural crest-derived mesenchymal cells in vivo and in vitro. J. Mol. Cell. Biol., 7, 441–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Griffin J.N., Compagnucci C., Hu D., Fish J., Klein O., Marcucio R., Depew M.J. (2013) Fgf8 dosage determines midfacial integration and polarity within the nasal and optic capsules. Dev. Biol., 374, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kondo T., Hayashi S. (2013) Mitotic cell rounding accelerates epithelial invagination. Nature, 494, 125–129. [DOI] [PubMed] [Google Scholar]
- 52. Kondo T., Hayashi S. (2015) Mechanisms of cell height changes that mediate epithelial invagination. Dev. Growth. Differ., 57, 313–323. [DOI] [PubMed] [Google Scholar]
- 53. Thomas B.L., Liu J.K., Rubenstein J.L., Sharpe P.T. (2000) Independent regulation of Dlx2 expression in the epithelium and mesenchyme of the first branchial arch. Development, 127, 217–224. [DOI] [PubMed] [Google Scholar]
- 54. Bei M., Maas R. (1998) FGFs and BMP4 induce both Msx1-independent and Msx1-dependent signaling pathways in early tooth development. Development, 125, 4325–4333. [DOI] [PubMed] [Google Scholar]
- 55. Kurosaka H. (2015) The Roles of Hedgehog Signaling in Upper Lip Formation. Biomed. Res. Int., 2015, 901041.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Young N.M., Hu D., Lainoff A.J., Smith F.J., Diaz R., Tucker A.S., Trainor P.A., Schneider R.A., Hallgrímsson B., Marcucio R.S. (2014) Embryonic bauplans and the developmental origins of facial diversity and constraint. Development, 141, 1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jiang X., Rowitch D.H., Soriano P., McMahon A.P., Sucov H.M. (2000) Fate of the mammalian cardiac neural crest. Development, 127, 1607–1616. [DOI] [PubMed] [Google Scholar]
- 58. Chai Y., Jiang X., Ito Y., Bringas P., Han J., Rowitch D.H., Soriano P., McMahon A.P., Sucov H.M. (2000) Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development, 127, 1671–1679. [DOI] [PubMed] [Google Scholar]
- 59. Soriano P. (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet., 21, 70–71. [DOI] [PubMed] [Google Scholar]
- 60. Dennis J.F., Kurosaka H., Iulianella A., Pace J., Thomas N., Beckham S., Williams T., Trainor P.A. (2012) Mutations in Hedgehog acyltransferase (Hhat) perturb Hedgehog signaling, resulting in severe acrania-holoprosencephaly-agnathia craniofacial defects. PLoS. Genet., 8, e1002927.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Trapnell C., Pachter L., Salzberg S.L. (2009) TopHat, discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Robinson M.D., McCarthy D.J., Smyth G.K. (2010) edgeR, a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.