

Abstract
Thyroid cancer is a major component cancer of Cowden syndrome (CS), a disorder typically associated with germline mutations in PTEN. Germline variants in succinate dehydrogenase genes (SDHx) co-occurring with PTEN germline mutations confer a 2-fold increased prevalence (OR 2.7) of thyroid cancer compared to PTEN-associated CS but 50% decreased prevalence (OR 0.54) of thyroid cancer compared to SDHx-associated CS. We have previously shown that CS-associated SDHD variants G12S and H50R induce PTEN oxidation and nuclear accumulation in thyroid cancer. Our current study shows that SDHD-G12S and -H50R variants cause down-regulation of autophagy, demonstrating a role for SDHD in autophagy-associated pathogenesis of differentiated thyroid cancer. These findings could explain the increased prevalence of thyroid cancer in CS patients with SDHx germline mutations compared to those with PTEN mutations alone. Importantly, we demonstrate the dependence of this process on functional wild-type PTEN with reversal of decreased autophagy after PTEN knockdown. The latter could explain the clinically observed decrease in thyroid cancer prevalence in patients with co-existent PTEN mutations and SDHx variants. We also show that SDHD-G12S/H50R promotes mono-ubiquitination of PTEN, causing its translocation into the nucleus, upregulation of AKT and consequent phosphorylation of FOXO3a. Furthermore, SDHD-G12S/H50R-mediated increase in acetylation of FOXO3a further enhances AKT-associated phosphorylation of FOXO3a. This combination of phosphorylation and acetylation of FOXO3a results in its nuclear export for degradation and consequent down-regulation of FOXO3a-target autophagy-related gene (ATG) expression. Overall, our study reveals a novel mechanism of crosstalk amongst SDHD, PTEN and autophagy pathways and their potential roles in thyroid carcinogenesis.
Introduction
Thyroid cancer in the general population has been increasing in incidence over the last 10 years, without clear etiology but likely in part related to increased detection. It is also being increasingly recognized that thyroid cancer can often occur in families, including in inherited neoplasia syndromes such as Cowden syndrome (CS). The thyroid is one of the major organs affected in CS with pathologies including follicular thyroid adenomas, thyroid hamartomas, Hashimoto thyroiditis and differentiated thyroid carcinoma. CS is a difficult-to-detect, under-diagnosed autosomal dominant inherited disorder characterized by multiple hamartomas affecting derivatives of all three germ layers and a high risk of solid tumors, such as those of the thyroid, breast and kidney. Germline mutations in the gene encoding phosphatase and tensin homolog deleted on chromosome 10 (PTEN) tumor suppressor are found in approximately 25% of classic CS patients and CS-like patients, combined (1). Germline variants of genes encoding the different subunits of succinate dehydrogenase (SDHB-D) have been found (and subsequently validated) in ∼10% of PTEN mutation negative CS/CS-like individuals (2,3). Notably, SDHB-D variants can co-exist with in germline PTEN mutations in approximately 6–8% of CS/CSL individuals and appear to modify the risk of both breast and thyroid cancer (2).
Succinate dehydrogenase (SDH) or complex II of the mitochondrial respiratory chain is a unique membrane-bound enzyme that plays dual functions in both the Krebs cycle for succinate oxidation and the respiratory chain for electron transfer to the terminal acceptor ubiquinone. SDH is a highly conserved hetero-tetrameric protein, with SDHA and SDHB as catalytic subunits that protrude into the mitochondrial matrix and SDHC and SDHD as structural anchors on the inner membrane. Each of the genes encoding the four subunits of SDH (SDHA-D) is considered to be a classic tumor suppressor gene (4). Germline SDHB-D variants have been shown to upregulate the AKT and MAPK pathways, similar to that seen in PTEN dysfunction (3). Compared to PTEN mutation carriers, SDHx variant carriers have significantly higher frequencies of breast, thyroid, and renal cell carcinomas (2,3). The two most common SDHx variants in CS/CS-like individuals are SDHD-G12S and SDHD-H50R (2,3). We have recently shown that SDHD-G12S and SDHD-H50R result in upregulation of reactive oxygen species (ROS) resulting in oxidized PTEN (thus, loss-of function) which accumulates in the nucleus in follicular thyroid cancer cell lines (5). However, the functional interactions between the SDH and PTEN pathways are not completely understood.
Accumulating evidence supports the critical role of autophagy in cancer development from initiation to progression and metastasis, including in thyroid carcinoma. Autophagy is a catabolic process present in all eukaryotic cells, involving degradation of the cell’s own components through the lysosomal machinery. Basal levels of autophagy are fundamental to maintaining cellular homeostasis and play an integral part in cell growth and development. Inability of the cell to up-regulate autophagy, for instance due to defective autophagy-related genes (ATGs), can result in accumulation of defective molecules or excessive ROS, which in turn impair cellular functions and DNA stability resulting in cancers (6). Whether autophagy negatively or positively regulates the development of thyroid cancer is not clear yet (7). The paradoxical role of autophagy in prevention of cell transformation or promotion of cancer cell survival depends on its regulation by various signaling pathways, like the MAPK and PI3K/AKT pathways. Based on these known pathways for autophagy and previous observations by our group stated above, we sought to address our hypothesis that CS-associated SDHD variants (G12S, H50R) cross-talk with the PTEN pathway to influence autophagy and hence predispose thyroid cells to carcinogenesis.
Results
Cell lines expressing SDHD-G12S, or -H50R variants show increased cell growth compared to SDHD-wildtype-expressing cells
Nthy-Ori-3.1 ‘normal’ thyroid follicle and TPC1 papillary thyroid cancer cell lines stably expressing Flag-tagged SDHD-WT, -G12S, or -H50R constructs were analyzed. Growth curves obtained via daily cell counts indicate that SDHD-G12S or -H50R expressing Nthy-Ori-3.1 and TPC1 cells grow faster than SDHD-WT cells (Supplementary Material, Fig. S1A and B). We observed that equal numbers of plated Nthy-Ori-3.1 cells expressing SDHD-G12S or SDHD-H50R were completely confluent on day 5 and 6 of culture, respectively, whereas SDHD-WT expressing cells showed only 50% confluence by day 6. Similar results were observed in TPC1 cells where cell numbers of the SDHD-G12S or -H50R expressing cell lines were double that of SDHD-WT expressing cells on day 5 of culture.
Figure 1.

SDHD-G12S or -H50R inhibit autophagy in HEK293, Nthy-Ori-3.1 and TPC1 cells. Western blot for LC3BII and p62 autophagy markers in (A) HEK293, (B) Nthy-Ori-3.1 and (C) TPC1 cells stably transfected with SDHD-WT, SDHD-G12S or SDHD-H50R. GAPDH was blotted as loading control (left panels). Note that quantification of intensity of LC3BII and p62 was normalized to GAPDH from three independent experiments (* indicates P < 0.05 and *** indicates P < 0.01) (right panels).
SDHD-G12S or -H50R variants suppress autophagy in HEK293, Nthy-Ori-3.1 and TPC1 cells
To investigate if SDHD variants affect autophagy, two autophagy markers were assessed by Western blotting. LC3B is an ubiquitin-like protein that is necessary for the formation of autophagosomes. It is recruited from the cytosol and associated with autophagophores during the process of autophagy. With increased levels of autophagy, LC3B I (16KD) is cleaved into LC3B II (14KD) (8). Therefore, increased expression levels of LC3B II can serve as a marker for increased autophagy. In contrast, p62 is an ubiquitin-binding scaffold protein degraded by autophagy. Since p62 accumulates when autophagy is inhibited, decreases in p62 levels can serve as a marker for increased induction of autophagy (9). Western blot analysis of lysates from HEK293, Nthy-Ori-3.1 and TPC1 cells with stable expression of SDHD-G12S or SDHD-H50R constructs showed decreased LC3B II levels and increased p62 accumulation compared to the SDHD-WT expressing cells (Fig. 1A–C, respectively). No increase in p62 levels was observed in SDHD-G12S or SDHD-H50R expressing cells compared to SDHD-WT expressing cells (Supplementary Material, Fig. S2). Cyto-ID staining for vesicles produced during autophagy showed more intense vesicle staining (green) in SDHD-WT cells than that in the cell lines expressing SDHD-G12S or SDHD-H50R (Fig. 2). Using three experimental read-outs, our data demonstrate that SDHD-G12S and SDHD-H50R variants decrease autophagy levels in HEK293 cells and in ‘normal’ follicular as well as malignant thyroid cell lines.
Figure 2.

SDHD-G12S or -H50R inhibit autophagy vesicle formation in HEK293, Nthy-Ori-3.1 and TPC1 cells. Cyto-ID staining (Green) for autophagosomes reveals increased staining in HEK293, Nthy-Ori-3.1 and TPC1 cells expressing SDHD-WT (left panels) compared to those expressing SDHD-G12S (middle panels) or -H50R (right panels). Blue staining represents nuclei. Comparative quantification of Cyto-ID staining (Green) is shown as a histogram of signal intensity (X-axis) vs signal area (Y-axis) for SDHD-WT (Blue), SDHD-G12S (Red) and SDHD-H50R (Green) expressing cells. Magnification for all panels: 10 × 40.
SDHD-G12S/H50R variants induce degradation of FOXO3a
As a master regulator of autophagy, FOXO3a expression was investigated and compared between SDHD-WT-expressing cells and SDHD-G12S or SDHD-H50R-expressing cells in HEK293, Nthy-Ori-3.1 and TPC1 lines. In all three cell lines, Western analysis showed that FOXO3a levels decreased in cells expressing SDHD-G12S or SDHD-H50R compared to SDHD-WT expressing cells (Fig. 3, left panels). Since protein levels of FOXO3a are mainly regulated by proteasome-mediated degradation, we next investigated the ubiquitination status of FOXO3a in all stable cell lines. We found that ubiquitination of FOXO3a increased in SDHD-G12S and SDHD-H50R expressing cells compared with that in SDHD-WT-expressing cells across all the three cell lines (Fig. 3, right panels), suggesting that SDHD-variants may down-regulate FOXO3a through an increase in its ubiquitination.
Figure 3.

SDHD-G12S or SDHD-H50R decrease FOXO3a protein levels through proteasome-mediated degradation in HEK293, Nthy-Ori-3 and TPC1 cells. Western blot for total FOXO3a expression (left panels) in HEK293 (A), Nthy-Ori-3 (B) and TPC1 (C) and Immunoprecipitation (IP) Western blots for FOXO3a-ubiquitination in the corresponding cell lines treated with proteasome inhibitor MG132 (right panels). Blots shown are best representatives of three separate experiments.
SDHD-G12S/H50R variants promote phosphorylation of nuclear FOXO3a
Localization of FOXO3a is critical for its activity and is determined by its phosphorylation levels (10). Upon AKT-mediated phosphorylation at Serine-253, phosphorylated FOXO3a is translocated to the cytoplasm and subsequently binds to 14-3-3 leading to its degradation by proteasomes (10). In contrast, dephosphorylation of FOXO3a by PP2A, facilitates FOXO3a relocation into the nucleus, thereby activating its target genes (10). Therefore, we analyzed FOXO3a protein localization through subcellular fractionation followed by Western blot in the cell lines expressing SDHD-WT, SDHD-G12S, or SDHD-H50R. Although cytosolic FOXO3 phosphorylation was similar between the three transfectants in each cell line, in all cases, elevated nuclear FOXO3a phosphorylation was observed in the SDHD-G12S and SDHD-H50R-expressing cells compared to SDHD-WT (Fig. 4, left panels).
Figure 4.

SDHD-G12S or -H50R induce PTEN mono-ubiquitination accompanied by increased nuclear phosphorylation of FOXO3a and AKT. Western analyses for phosphorylation of FOXO3a or AKT in nuclear and cytoplasmic fractions from HEK293 (A), Nthy-Ori-3 (B) and TPC1 (C). GAPDH and PARP-1 were used as cytoplasmic and nuclear loading controls, respectively (left panel). Assay for mono-ubiquitination of PTEN was performed by PTEN immunoprecipitation followed by Western blot for ubiquitin in the presence (+) or absence (-) of MG132 in the context of stable expression of either SDHD-WT or SDHD-G12S/H50R (right panels). Numbers below each set of bands represents the intensity ratio of mono-ubiquitinated PTEN to total PTEN, the latter as loading control. All blots shown are best representatives of three separate experiments.
In addition, we observed increased levels of phosphorylated AKT in the nucleus of all three cell lines expressing SDHD-G12S or -H50R compared to those expressing SDHD-WT (Fig. 4, left panels). Since PTEN is a major negative regulator of AKT, we assessed total PTEN protein levels by Western blot but found no difference between cells expressing SDHD-WT and those expressing SDHD-G12S/H50R (data not shown). Previous studies have reported that PTEN nuclear import is mediated by its mono-ubiquitination (11). To explain increased phosphorylation of nuclear AKT, we measured mono-ubiquitination of PTEN. Indeed, we observed increased mono-ubiquitinated PTEN in HEK293, Nthy-Ori-3.1 cells and TPC1 cells expressing SDHD-G12S or -H50R but not in SDHD-WT expressing cells (Fig. 4, right panels).
Taken together, our data suggest that SDHD-G12S or SDHD-H50R cause(s) mono-ubiquitination of PTEN, with consequent translocation into the nucleus accompanied by upregulation of AKT and further promotes phosphorylation of nuclear FOXO3a (also, see below).
SDHD-G12S/H50R variants promote acetylation of nuclear FOXO3a
It is accepted that the acetylation status of FOXO transcription factors influence transcriptional activity (10). To investigate if SDHD variants affect FOXO3a acetylation, immunoprecipitation of FOXO3a with Acetyl-Lys was performed in HEK293, Nthy-Ori-3 and TPC1 cells expressing SDHD-WT, SDHD-G12S or SDHD-H50R. Augmented acetylation of FOXO3a was observed in all three cell lines expressing SDHD-G12S or SDHD-H50R compared to their corresponding SDHD-WT-expressing cells (Fig. 5, left panels).
Figure 5.

SDHD-G12S or SDHD-H50R increases FOXO3a acetylation through up-regulation of p300. Acetylation of FOXO3a was assayed by immunoprecipitation of FOXO3a followed by anti-Lysine-acetylation Western blot in HEK293 (A), Nthy-Ori-3 (B) and TPC1 (C) cells expressing SDHD-WT, SDHD-G12S or SDHD-H50R (left panels); Western blot for acetyl-transferase (p300) and two relevant deacetyltransferases (SIRT1, SIRT3) were performed in the three corresponding cell lines (right panels). Blots shown are best representatives of three separate experiments.
Since the SIRT1 deacetylase and p300 acetytransferase are responsible for the acetylation status of FOXO transcription factors (12–14), we investigated whether the SDHD variants affect the expression of these two enzymes. Ectopic expression of SDHD-G12S or SDHD-H50R in HEK293, Nthy-Ori-3 and TPC1 cell lines is accompanied by an increased p300 expression but no decrease of SIRT1 compared to cells expressing SDHD-WT (Fig. 5, right panels). These observations suggest that SDHD variants induce FOXO3a acetylation in association with up-regulation of expression of p300-acetyltransferase.
SDHD-G12S/H50R variants down-regulate expression of FOXO3a target genes
To further investigate the effect of SDHD variants on FOXO3a activity, mRNA and protein expressed from two ‘read out’ FOXO3a target genes, BECN1 and ATG12, were analyzed. In HEK293 and TPC1 cells, decreased BECN1 and ATG12 mRNA levels were observed in SDHD variant-expressing cells compared to SDHD-WT-expressing cells (Fig. 6A and C, left panel). In Nthy-Ori-3 cells, we observed decreased BECN1, but not ATG12, mRNA levels in SDHD-variant expressing cells compared to SDHD-WT controls (Fig. 6B, left Panel). Notably, expression of SDHD-G12S or H50R, but not SDHD-WT, in each of the three cell lines, HEK293, Nthy-Ori-3 and TPC1, showed decreased BECLIN1 and ATG12 protein expression by Western analysis (Fig. 6A–C, right panels). Taken together, our observations suggest that SDHD-G12S and -H50R variants suppress FOXO3a activity resulting in decreased expression of autophagy-related genes.
Figure 6.

SDHD-G12S or SDHD-H50R downregulates FOXO3a autophagy-related target gene expression. qRT- PCR for FOXO3a target genes BECLIN1 and ATG12 in HEK293 (A), Nthy-Ori-3 (B) and TPC1 (C) cells expression SDHD-WT, SDHD-G12S or SDHD-H50R (* indicates P < 0.05) (left panels); Western blots for BECLIN1 and ATG12 in the three corresponding cell lines (A-C, right panels).
PTEN is necessary for SDHD variant-associated inhibition of autophagy
To further examine if PTEN is required for SDHD variant-associated autophagy inhibition, PTEN was knocked down in Nthy-Ori-3 and TPC1 cells transfected with SDHD-WT, SDHD-G12S or SDHD-H50R (Fig. 7, left panels). Western blots for autophagy markers LC3B and p62 were performed to determine autophagy levels. The SDHD-G12S/H50R-mediated inhibition of autophagy was abrogated when PTEN was knocked down (Fig. 7, right panels). These data suggest that PTEN expression is necessary for SDHD-variant-associated autophagy suppression.
Figure 7.

SDHD-G12S or SDHD-H50R suppressing autophagy is PTEN-dependent. Western blot showing PTEN knockdown in Nthy-Ori-3 (A) and TPC1 (B) thyroid cells with stable expression of SDHD-WT or SDHD variants G12S or H50R (left panels). Western blot reveals autophagy marker (LC3BII and p62) levels in (A) Nthy-Ori-3 and (B) TPC1 cells stably expressing SDHD-WT, SDHD-G12S or SDHD-H50R after PTEN knockdown (right panels). Blots shown are best representatives of three separate experiments.
Discussion
It has been established that germline SDHD variants and somatic SDHD/B alterations are associated with heritable and sporadic thyroid cancer, respectively (15–17). We have also demonstrated previously that SDHD variants promote apoptosis in follicular thyroid cancer cells (5). Apoptosis and autophagy are intricately linked processes where modulation of autophagy controls increase or decrease in apoptosis as per the requirements of the cell to survive or perish (18,19). In this study, we show data confirming that SDHD variants regulate autophagy in both benign and malignant thyroid cells, suggesting that this function may be involved in SDH variant-associated thyroid cancer. Here, we show that expression of SDHD-G12S and SDHD-H50R lead to increased nuclear phosphorylation of AKT, and subsequent phosphorylation of the transcription factor FOXO3a, the master regulator of autophagy. It has been shown that AKT-mediated phosphorylation at Ser-253 on FOXO3a, leads to its cytoplasmic localization and degradation by proteasomes (20). In addition, we observed that SDHD variants increased acetylation of FOXO3a in association with p300 overexpression. Therefore, phosphorylation and acetylation of FOXO3a together lead to its nuclear export and binding to 14-3-3 for degradation. This ultimately results in suppression of autophagy via down-regulation of FOXO3a autophagy-related, target genes such as BECN1 and ATG12 (Fig. 8).
Figure 8.
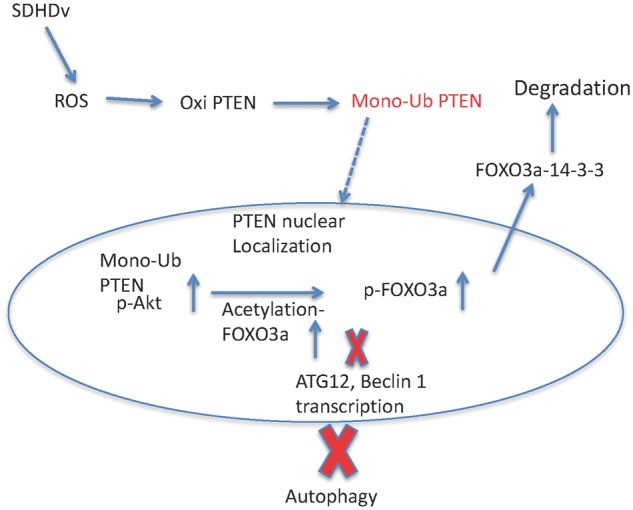
Schematic diagram depicting the signaling pathway for the germline SDHD variants affecting autophagy via PTEN mono-ubiquitination.
There is increasing evidence of crosstalk between PTEN and mitochondrial signaling pathways, but the precise mechanism of how dysfunctional mitochondrial pathways or mutations in mitochondrial tumor suppressor genes such as SDHx lead to PTEN dysfunction, and result in neoplastic transformation is not fully understood. Our current observations elucidate a novel mechanism that links CS-associated SDHD variants (SDHD-G12S, SDHD-H50R) to PTEN function. In addition to inducing oxidized PTEN, as suggested by our earlier studies, we now show that SDHD-G12S or -H50R stimulate mono-ubiquitination of PTEN, which leads to PTEN dysfunction with consequent upregulation of pAKT, especially of nuclear pAKT. Notably, we observed that this inhibition of autophagy by SDHD variants -G12S or -H50R was dependent on functional PTEN (Fig. 7). Although we recognize that our data are limited to cell lines, our observation that knocking down PTEN abrogates SDHD-G12S or -H50R suppressed autophagy might explain a puzzling clinical genetic observation in CS/CS-like individuals. The lifetime risk of epithelial thyroid cancer in PTEN mutation positive persons is 35% (21). Our initial observations of evaluable CS/CS-like research participants revealed an odds ratio (OR) of three (95% CI 1.47–6.19; P < 0.002) for thyroid cancer in individuals with SDHx variants compared to those with PTEN mutations alone (2). Individuals with both PTEN mutations and SDHx variants have an OR 0.34 (0.12–1.08; P = 0.038) compared to those with SDHx variants alone, and an OR 1.08 (0.39–3.05; P = 0.21) compared to those with germline PTEN mutations alone (2). We have subsequently accrued an independent validation series. Compared to PTEN mutation only, OR for thyroid cancer was 4.95 in CS/CSL with SDHx variants only (P < 0.001) and 2.70 for those carrying PTEN mutation and SDHx variant (P = 0.003; Table 1). Compared to SDHx variants only, OR for thyroid cancer for those carrying both PTEN mutation and SDHx variant was 0.54. SDHx-variant-associated suppression of autophagy could help explain the increased risk of thyroid cancers in our CS/CS-like patients with SDHx variants. Our current observation that PTEN knockdown reverses/rescues SDHx-variant-associated autophagy suppression might help explain the decreased prevalence of thyroid cancers when both PTEN mutation and SDHx variants co-exist compared to those with only SDHx variants.
Table 1.
Comparison of cancer frequencies of adult SDHx variant carriers to PTEN mutation carriers (Fisher’s two-tailed exact test)
| Female Breast Cancer (/total number) | Thyroid Cancer (/total number) | Renal Cancer (/total number) | ||
|---|---|---|---|---|
| PTENMT+ | 111/302 (36.7%) | 65/551 (11.8%) | 29/551 (5.2%) | |
| SDHMT+ | 70/115 (60.8%) | 53/133 (39.8%) | 21/133 (15.8%) | |
| PTENMT+/SDHMT+ | 29/46 (63.0%) | 17/64 (26.6%) | 1/64 (1.5%) | |
| SDHMT+ vs PTENMT+ | P-value | <0.001 | <0.001 | <0.001 |
| Odds Ratio (95% CI) | 2.67 (1.68, 4.26) | 4.95 (3.14, 7.82) | 3.375 (1.78, 6.38) | |
| PTENMT+/SDHMT+ vs SDHMT+ | P-value | 0.86 | 0.08 | 0.003 |
| Odds Ratio (95% CI) | 1.09 (0.51, 2.36) | 0.54 (0.27, 1.10) | 0.10 (0.01, 0.61) | |
| PTENMT+ /SDHMT+ vs PTENMT+ | P-value | <0.001 | 0.003 | 0.35 |
| Odds Ratio (95% CI) | 3.93 (1.48, 5.87) | 2.70 (1.40, 5.18) | 0.29 (0.01, 2.01) | |
That SDHD-G12S/H50R-mediated suppression of autophagy requires wildtype (WT) PTEN suggests that SDHD variants increase phosphorylation of nuclear AKT only when intact PTEN is present, and that nuclear AKT is activated by mono-ubiquitinated PTEN. The important role of nuclear AKT in multiple cellular processes such as apoptosis, cell cycle, DNA repair, cell differentiation and tumorigenesis are well known (22–24). Previous work from our laboratory demonstrated that PTEN is necessary and sufficient for inhibiting AKT activation in the nucleus through its intact lipid phosphatase activity and proper subcellular localization (25). Ubiquitination of PTEN inhibits its activity (26). Mono- and poly-ubiquitination appear to have separate effects on PTEN function, with mono- and poly-ubiquitination targeting PTEN for nuclear localization and proteasome degradation, respectively. In the current study, we show that SDHD variants G12S and H50R induce mono-ubiquitination of PTEN, which translocates into the nucleus, where it functions as a dominant negative PTEN, resulting in AKT upregulation and subsequent tumorigenesis. Overall ubiquitination status of PTEN is determined by both ubiquitination and de-ubiquitination. Many E3 ligases, such as NEDD4-1, WWP2, XIAP and RFP, and de-ubiquitylase, such as HAUSP/USP7, OTUD3, and USP13, have been identified (27). XIAP (X-Linked Inhibitor of Apoptosis), an anti-apoptotic protein, is involved in mono- and poly-ubiquitination of PTEN and knockdown of XIAP prevented nuclear translocation of PTEN (28). Though XIAP is a good candidate E3 ligase for SDHD variant-mediated mono-ubiquitination of PTEN, identification of the precise E3 ligase needs to be further investigated.
Our current data show increased phosphorylation of nuclear FOXO3a at S253 in SDHD-G12S/H50R transfected cells in association with an increase in phosphorylation of nuclear AKT. AKT is an important kinase downstream of PTEN and that directly phosphorylates FOXO3a at T32, S253 and S315 (29). S253 is the crucial residue regulating the nuclear-cytoplasmic shuttling of FOXO3a. As a transcription factor, FOXO3a is regulated by posttranslational modifications such phosphorylation, acetylation and ubiquitination (30). Phosphorylation of FOXO3a prevents its localization in the nucleus and induces its interaction with 14-3-3 nuclear export protein, resulting in FOXO3a’s degradation and functional inactivation by the proteasome pathway. This is consistent with our observation of lower total FOXO3a protein level and elevated ubiquitination of FOXO3a in SDHD-G12S/H50R expressing cells. As a generic E3 ligase for FOXO protein, MDM2 is likely involved in ubiquitination of FOXO3a (31). In addition, we observed the two CS-associated SDHD variants (SDHD-G12S, SDHD-H50R) inducing FOXO3a acetylation associated with up-regulation of the acetylase P300. Thus, it seems likely that the deacetylase SIRT1/3 is not involved in the process of acetylation of FOXO3a in the presence of the two SDHD variants.
The role of autophagy in cancer development, including thyroid cancer, is complicated where it may act as a cancer suppressor or promoter, depending on the cancer type and stage of progression (32–36). Modulation of autophagy has been suggested as an effective treatment modality against tumors and is also highly dependent on the type of tumor and its mutation status (35).We observed impaired autophagy in ‘normal’ or non-transformed thyroid follicular cells (Nthy-Ori-3.1) and papillary thyroid cancer cells (TPC1) when overexpressing either SDHD variant, highlighting the suppressive role of autophagy in cancer development. Our results are in agreement with the model that autophagy exerts its tumor suppressive protective effects during cancer initiation promoted by conditions of stress or oncogene activation. In contrast, once cancer is established, activation of autophagy promotes tumor development by helping the cancer cells meet energy requirements in unfavorable environments. However, even at this stage, excessive activation of autophagy can lead to cell death (18,19,36,37). Our findings on reduced autophagy levels as a result of germline SDHD variants, provide a potential transformation mechanism linking host genetic background to aberrant autophagy levels and predisposition to cancer. The novel mechanistic link between SDHx variants, autophagy and PTEN status in thyroid carcinomas described here provides insights that may impact thyroid cancer diagnosis, prevention, and treatment in heritable and potentially sporadic settings.
Materials and Methods
Cell culture
HEK293 embryonic kidney cells were grown in DMEM plus 10% fetal bovine serum at 37ºC with 5% CO2. Nthy-Ori-3 thyroid follicle cells were grown in RPMI supplemented with 10% fetal bovine serum and 2mM Glutamine and incubated at 37ºC with 5% CO2. HEK293T cell line was originally purchased from the American Type Culture Collection (ATCC, Manassas, VA) in 2011 and obtained in 2014 from the Cleveland Clinic Lerner Research Institute Cell Culture Core. The Nthy-ori 3-1 human thyroid follicular epithelium cell line (catalog number EC90011609, lot number 09C008 purchased in 2014 from Sigma, St. Louis, MO) was authenticated through STR PCR (AmpFLSTR SGM Plus PCR Amplification Kit, Life Technologies) by European Collection of Cell Cultures (ECACC, original source of the cell line, test data 14/04/2009). TPC1 papillary thyroid carcinoma cell line, established by Dr. Nobuo Sato (Cancer Research Institute, Kanazawa University, Japan) and housed and authenticated by MDR, are grown in DMEM with 10% fetal bovine serum and NEAA (non-essential amino acids). All cells were used at passage 1 to 6 for all experiments.
Generation of stable cell lines
Flag-tagged SDHD WT, SDHD-G12S, and SDHD-H50R plasmids, or shPTEN were transfected into HEK293, Nthy-Ori-3 and TPC1 cells with LipofectamineTM 3000 transfection reagent (Thermo Fisher, Waltham, MA). Fresh medium was replaced 8 h post transfection. Stably transfected cells were selected using G418 (50 μg for HEK293 cells, 300μg for Nthy-Ori-3 cells) in culture for over 3 weeks. Cell colonies were identified for a positive transfection by Western blot analysis
Immunoprecipitation for ubiquitination and acetylation
Cell lysates were collected in RIPA buffer and incubated with PTEN antibody (Clone 6H2.1, Cascade Biosciences, Winchester, MA) or FOXO3a antibody (Clone D19A7, Cell Signaling Technology, Danvers, MA) overnight at 4ºC followed by protein A/G beads incubation for an additional 2 h. After centrifugation at 800 g for 5 min, the supernatants were removed and cell pellets washed in RIPA buffer, incubated at 4ºC for 10 min, then centrifuged again at 800 g for 5 min. Pellets were washed three times and re-suspended in protein loading buffer. Samples were boiled for 5 min and centrifuged at maximum speed for 1 min. Supernatants were collected for Western blot analysis.
Nuclear-cytoplasmic fractionation
Nuclear-cytoplasmic fractionation was performed as previously described (6). Briefly, cytoplasmic proteins were isolated in buffer A (10 mM HEPES, 1.5 mM Potassium Chloride, 1% Triton X-100, 1 mM Dithiothreitol, protease inhibitor cocktail and phosphatase inhibitor) and nuclear proteins were isolated in Nuclear Extraction Reagent (Pierce, Rockford, IL).
Western blotting
Whole cell lysates were harvested in M-PER buffer (Pierce, Rockford, IL) with protease and phosphatase inhibitors. Samples were electrophoresed on 4–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to nitrocellulose membranes and probed with protein specific primary antibodies followed by incubation with secondary antibody. Target proteins were detected using the Odyssey infrared imaging system (Li-COR, Lincoln, NE). The following primary antibodies were used for detection of specific proteins:FOXO3a, Ubiquitin and LC3B specific antibodies were used at 1:1000 dilution (Cell signaling); P62 specific antibodies were used at 1:500 dilution (Santa Cruz Biotech, Dallas, TX). Best representative blots of results from three separate experiments have been shown in the manuscript for all Western blotting results
Quantitative-reverse transcription (RT) PCR for detection of FOXO3a target gene expression
Total RNA was extracted from transfected cells using the GeneJET RNA Purification Kit (Thermo Scientific, Waltham, MA) according to the manufacturer’s protocol and subsequently treated with DNase I (Invitrogen, Carlsbad, CA). DNase-treated total RNA was reverse-transcribed into cDNA using qScript cDNA SuperMix (Quanta BioSciences, Inc., Gaithersburg, MD) as specified by the manufacturer. Quantitative PCR was performed on a LightCycler 480 (Roche Diagnostics, Basel, Switzerland) using TaqMan Probe ready mix for BECLIN1 and ATG12, and GAPDH as internal control. Primers for BECLIN1 mRNA: forward sequence: 5’-TGGGGAGGTTAGG ATTTGGGA-3’, reverse sequence: 5’-GAGCCGTAGGGTGGAAAGC-3’. Primers for ATG12 mRNA: forward sequence: 5’-TAGAGCGAACACGAACCATCC-3’, reverse sequence: 5’-CACTGCCAAAACACTCATAGAGA-3’.
Cyto-ID staining for autophagy
Cyto-ID staining kit (Enzo Life Sciences, Farmingdale, NY) was used to detect autophagy in HEK293, Nthy-Ori-3 and TPC1 cells according to the manufacturer’s protocol. Briefly, cells were plated on cover slips at 70% confluency and allowed to attach overnight. Cell medium was aspirated followed by carefully washing twice using 1x Assay Buffer with 5% FBS. Cells were incubated with Cyto-ID Green Detection Reagent for 30 min at 37ºC in the dark. Cells were carefully washed twice with 1X Assay Buffer to remove excess dye and observed immediately under a confocal microscope.
Statistical analysis
Statistical analysis was performed using Prism for Macintosh (GraphPad Software, La Jolla, CA) using the two-tailed t-test. Data are expressed as means ± SEM as indicated. Results were considered statistically significant at a P value of < 0.05.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
M.D.R. is the Kurtz Family Chair in Cancer Research at The Ohio State University. C.E. is the Sondra J. and Stephen R. Hardis Endowed Chair of Cancer Genomic Medicine at the Cleveland Clinic and an ACS Clinical Research Professor.
Conflict of Interest statement. None declared.
Funding
National Cancer Institute (P01CA124570 to M.D.R. and C.E.), Bethesda, MD; Breast Cancer Research Foundation (to C.E.). Funding to pay the Open Access publication charges for this article was provided by NCI grant P01CA124570.
References
- 1. Tan M.H., Mester J., Peterson C., Yang Y., Chen J.L., Rybicki L.A., Milas K., Pederson H., Remzi B., Orloff M.S., Eng C. (2011) A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet., 88, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ni Y., He X., Chen J., Moline J., Mester J., Orloff M.S., Ringel M.D., Eng C. (2012) Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependant destabilization of p53. Hum. Mol. Genet., 21, 300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ni Y., Zbuk K.M., Sadler T., Patocs A., Lobo G., Edelman E., Platzer P., Orloff M.S., Waite K.A., Eng C. (2008) Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am. J. Hum. Genet., 83, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gimm O., Armanios M., Dziema H., Neumann H.P., Eng C. (2000) Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Cancer Res., 60, 6822–6825. [PubMed] [Google Scholar]
- 5. Yu W., He X., Ni Y., Ngeow J., Eng C. (2015) Cowden syndrome-associated germline SDHD variants alter PTEN nuclear translocation through SRC-induced PTEN oxidation. Hum. Mol. Genet., 24, 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dewaele M., Maes H., Agostinis P. (2010) ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy, 6, 838–854. [DOI] [PubMed] [Google Scholar]
- 7. White E., DiPaola R.S. (2009) The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res., 15, 5308–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. embo J., 19, 5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem., 82, 24131–24145. [DOI] [PubMed] [Google Scholar]
- 10. Nho R.S., Hergert P. (2014) FoxO3a and disease progression. World J. Biol. Chem., 5, 346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trotman L.C., Wang X., Alimonti A., Chen Z., Teruya-Feldstein J., Yang H., Pavletich N.P., Carver B.S., Cordon-Cardo C., Erdjument-Bromage H., et al. (2007) Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell, 128, 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van der Heide L.P., Smidt M.P. (2005) Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem. Sci., 30, 81–86. [DOI] [PubMed] [Google Scholar]
- 13. Giannakou M.E., Partridge L. (2004) The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol., 14, 408–412. [DOI] [PubMed] [Google Scholar]
- 14. Frescas D., Valenti L., Accili D. (2005) Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem., 280, 20589–20595. [DOI] [PubMed] [Google Scholar]
- 15. von Dobschuetz E., Leijon H., Schalin-Jäntti C., Schiavi F., Brauckhoff M., Peczkowska M., Spiazzi G., Demattè S., Cecchini M.E., Sartorato P., et al. (2015) A registry-based study of thyroid paraganglioma: histological and genetic characteristics. Endocr. Relat. Cancer, 22, 191–204. [DOI] [PubMed] [Google Scholar]
- 16. Opocher G., Schiavi F., Iacobone M., Toniato A., Sattarova S., Erlic Z., Martella M., Mian C., Merante Boschin I., Zambonin L., et al. (2006) Familial nonsyndromic pheochromocytoma. Ann. N. Y. Acad. Sci., 1073, 149–155. [DOI] [PubMed] [Google Scholar]
- 17. Ni Y., Seballos S., Ganapathi S., Gurin D., Fletcher B., Ngeow J., Nagy R., Kloos R.T., Ringel M.D., LaFramboise T., Eng C. (2015) Germline and somatic SDHx alterations in apparently sporadic differentiated thyroid cancer. Endocr. Relat. Cancer, 22, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishida K., Yamaguchi O., Otsu K. (2008) Crosstalk between autophagy and apoptosis in heart disease. Circ. Res., 103, 343–351. [DOI] [PubMed] [Google Scholar]
- 19. Maiuri M.C., Zalckvar E., Kimchi A., Kroemer G. (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol., 8, 741–752. [DOI] [PubMed] [Google Scholar]
- 20. Plas D.R., Thompson C.B. (2003) Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J. Biol. Chem., 278, 12361–12366. [DOI] [PubMed] [Google Scholar]
- 21. Tan M.H., Mester J.L., Ngeow J., Rybicki L.A., Orloff M.S., Eng C. (2012) Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res., 18, 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martelli A.M., Tabellini G., Bressanin D., Ognibene A., Goto K., Cocco L., Evangelisti C. (2012) The emerging multiple roles of nuclear Akt. Biochem. Biophys. Acta, 1823, 2168–2178. [DOI] [PubMed] [Google Scholar]
- 23. Miyamoto S., Rubio M., Sussman M.A. (2009) Nuclear and mitochondrial signalling Akts in cardiomyocytes. Cardiovasc. Res., 82, 272–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanaka K., Horiguchi K., Yoshida T., Takeda M., Fujisawa H., Takeuchi K., Umeda M., Kato S., Ihara S., Nagata S., Fukui Y. (1999) Evidence that a phosphatidylinositol 3,4,5-trisphosphate-binding protein can function in nucleus. J. Biol. Chem., 274, 3919–3922. [DOI] [PubMed] [Google Scholar]
- 25. He X., Saji M., Radhakrishnan D., Romigh T., Ngeow J., Yu Q., Wang Y., Ringel M.D., Eng C. (2012) PTEN lipid phosphatase activity and proper subcellular localization are necessary and sufficient for down-regulating AKT phosphorylation in the nucleus in Cowden syndrome. J. Clin. Endocrinol. Metab., 97, 2179–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maccario H., Perera N.M., Gray A., Downes C.P., Leslie N.R. (2010) Ubiquitination of PTEN (phosphatase and tensin homolog) inhibits phosphatase activity and is enhanced by membrane targeting and hyperosmotic stress. J. Biol. Chem., 285, 12620–12628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gupta A., Leslie N.R. (2016) Controlling PTEN (Phosphatase and Tensin Homolog) Stability: A dominant role for Lysine 66. J. Biol. Chem., 291, 18465–18473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Themsche C., Leblanc V., Parent S., Asselin E. (2009) X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J. Biol. Chem., 284, 20462–20466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daitoku H., Sakamaki J., Fukamizu A. (2011) Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochem. Biophys. Acta, 1813, 1954–1960. [DOI] [PubMed] [Google Scholar]
- 30. Vogt P.K., Jiang H., Aoki M. (2005) Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle, 4, 908–913. [DOI] [PubMed] [Google Scholar]
- 31. Huang H., Tindall D.J. (2011) Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim Biophys Acta, 1813, 1961–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bretz J.D., Mezosi E., Giordano T.J., Gauger P.G., Thompson N.W., Baker J.R. Jr. (2002) Inflammatory cytokine regulation of TRAIL-mediated apoptosis in thyroid epithelial cells. Cell Death Differ., 9, 274–286. [DOI] [PubMed] [Google Scholar]
- 33. Mitsiades C.S., Poulaki V., Mitsiades N. (2003) The role of apoptosis-inducing receptors of the tumor necrosis factor family in thyroid cancer. J. Endocrinol., 178, 205–216. [DOI] [PubMed] [Google Scholar]
- 34. Li L.C., Liu G.D., Zhang X.J., Li Y.B. (2014) Autophagy, a novel target for chemotherapeutic intervention of thyroid cancer. Cancer Chemother. Pharmacol., 73, 439–449. [DOI] [PubMed] [Google Scholar]
- 35. Yi H., Long B., Ye X., Zhang L., Liu X., Zhang C. (2014) Autophagy: A potential target for thyroid cancer therapy (Review). Mol. Clin. Oncol., 2, 661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Netea-Maier R.T., Plantinga T.S., van de Veerdonk F.L., Smit J.W., Netea M.G. (2015) Modulation of inflammation by autophagy: consequences for human disease. Autophagy, 12, 245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Netea-Maier R.T., Klück V., Plantinga T.S., Smit J.W. (2015) Autophagy in thyroid cancer: present knowledge and future perspectives. Front. Endocrinol. (Lausanne), 22 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.