

Abstract
Right ventricular (RV) function determines prognosis in pulmonary arterial hypertension (PAH). We hypothesize that ischemia causes RV dysfunction in PAH by triggering dynamin-related protein 1 (Drp1)-mediated mitochondrial fission. RV function was compared in control rats (n=50) versus rats with monocrotaline-induced PAH (MCT- PAH; n=60) both in vivo (echocardiography) and ex vivo (RV Langendorff). Mitochondrial membrane potential and morphology and RV function were assessed before or after two cycles of ischemia-reperfusion injury challenge (RV-IR). The effects of Mdivi-1 (25 μM), a Drp1 GTPase inhibitor and P110 (1 μM), a peptide inhibitor of Drp1–Fis1 interaction were studied. We found that MCT caused RV hypertrophy, RV vascular rarefaction and RV dysfunction. Prior to IR, the mitochondria in MCT-PAH RV were depolarized and swollen with increased Drp1 content and reduced aconitase activity. RV-IR increased RV end diastolic pressure (RVEDP) and mitochondrial Drp1 expression in both control and MCT-PAH RVs. IR depolarized mitochondria in control RV but did not exacerbate the basally depolarized MCT-PAH RV mitochondria. During RV IR mdivi-1 and P110 reduced Drp1 translocation to mitochondria, improved mitochondrial structure and function, and reduced RVEDP. In conclusion, RV ischemia occurs in PAH and causes Drp1-Fis1-mediated fission leading to diastolic dysfunction. Inhibition of mitochondrial fission preserves RV function in RV-IR.
Keywords: ischemia-reperfusion injury, pulmonary arterial hypertension, diastolic dysfunction, mitochondrial membrane potential, Mdivi-1, mitochondrial division inhibitor 1
INTRODUCTION
Right ventricular (RV) ischemia is a feature of pulmonary arterial hypertension (PAH), which contributes to RV failure. Evidence of RV ischemia in PAH patients includes abnormal coronary artery flow reserve [1] and elevated plasma troponin levels [2]. In PAH, RV ischemia can result from microvascular rarefaction [3,4], reduced epicardial coronary artery perfusion pressure [5,6], or both. In addition, PAH patients are exposed to periodic episodes of hypotension, arrhythmia and/or surgical procedures that may expose the RV to ischemia-reperfusion injury (IR) [7]. The effects of basal ischemia and IR on the RV in health and in PAH are unknown.
In the left ventricle (LV), IR impairs systolic function indicated by reducing developed pressure and diastolic function indicated by elevating end diastolic pressure (EDP) [8,9]. In LV-IR, production of reactive oxygen species (ROS) and the resulting LV dysfunction largely result from fission of mitochondria in cardiac myocytes [10–13].
Dynamin-related protein 1 (Drp1) normally resides in the cytosol, but upon activation, translocates to the mitochondria where it associates with binding partners located on the outer mitochondrial membrane (OMM), including fission protein 1 (Fis1), mitochondrial fission factor (MFF), and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51) [14–16]. Drp1 and its binding partners create a multimeric fission apparatus that constricts and divides the mitochondria (reviewed in [17]). Drp1 can be activated by dephosphorylation of serine 637, as occurs in IR [10,11,18], or phosphorylation at serine 616 [19,20].
The small molecule, mitochondrial division inhibitor 1 (Mdivi-1), which is a Drp1 GTPase inhibitor, preserves mitochondrial morphology in ischemic LV cardiac myocytes [10,12]. In global LV-IR, Mdivi-1 reduces ROS production, preserves mitochondrial morphology and improves pulse pressure development [10]. In vivo, Mdivi-1 ameliorates aortic banding-induced heart failure [21], reduces myocardial infarct size [12], and improves survival in a mouse cardiac arrest model [11]. Preventing the interaction between Drp1 and Fis1, using the disruptive peptide P110, also preserves mitochondrial morphology and myocyte function in neonatal rat cardiac myocytes [13].
Here, we demonstrate that ischemia, whether occurring in vivo due to the vascular rarefaction that occurs in MCT-PAH or induced ex vivo in the RV Langendorff model, causes mitochondrial fission. Increased fission in RV myocytes from PAH RV causes RV diastolic dysfunction. Ischemia-induced fission involves Drp1 translocation to the mitochondria. Interaction of Drp1 and Fis1 mediate the resulting RV injury. RV function is preserved ex vivo by inhibiting ischemia-induced mitochondrial fission. This research has relevance for PAH, a condition in which RV function, determined in part by RV ischemia, is a major determinant of prognosis.
MATERIALS AND METHODS
Experiments were conducted in accordance with published guidelines of the Canadian Council on Animal Care and approved by the Queen’s University Animal Care Committee.
Reagents
Monocrotaline (MCT), Mdivi-1, dimethyl sulfoxide (DMSO), Krebs-Henseleit (KH) buffer, sodium bicarbonate, calcium chloride dehydrate, heparin sodium salt and albumin-fluorescein isothiocyanate conjugate (albumin-FITC) were purchased from Sigma (St. Louis, MO, USA). The P110 peptide includes TAT to enhance membrane permeability. Both P110 and the peptide control sequence, TAT, were purchased from United Peptide (Herndon, VA, USA). Phosphate buffered saline (PBS) was purchased from Fisher Scientific (Fair Lawn, NJ, USA).
Monocrotaline-induced PAH
Experimental procedures were conducted on male Sprague-Dawley rats (Charles River, QC, Canada). Animals were initially pair-housed in a colony room (12/12 h light cycle; light off at 19:00) with ad libitum access to food and water. All experimental procedures took place between the time of 9:00 and 18:00. At the weight of ~ 250 g, rats received a single subcutaneous injection of MCT (60 mg/kg, SC) or PBS.
Echocardiography
Doppler, 2-dimensional, and M-mode echocardiography was performed 6–24 hours prior to the RV Langendorff surgery, using a high-frequency ultrasound system (Vevo 2100; Visual Sonics, Toronto, ON, Canada), as described [22]. Pulmonary artery acceleration time (PAAT), pulmonary artery diameter at the level of the pulmonary outflow tract during mid-systole, diastolic and systolic thickness of the RV free wall (RVFW), and tricuspid annular plane systolic excursion (TAPSE) were measured. RVFW systolic thickening and cardiac output (CO) were calculated as described [22,23].
Treadmill distance
Exercise capacity was evaluated 6–24 hours prior to the sacrifice of animals by measuring the maximal distance run on a motor-driven treadmill (Simplex II Instrument, Columbus Instruments; Columbus, OH, USA), as described [24]. The treadmill belt speed was set at 10 m/min for the initial 5 minutes, and increased by 5 m/min every 5 minutes. The test was terminated within 30 minutes or when the rat exhibited signs of exhaustion.
Langendorff RV-IR model
The Langendorff preparation used an isolated perfused heart system (Radnoti; Monrovia, CA, USA). The model was performed as described previously for the LV [10,25], except the ventricular balloon was placed in the RV. Rats were anesthetized with Ketamine 75 mg/kg plus Xylazine 10 mg/kg (IP). Blood was heparinized and their hearts were rapidly removed. The aorta was mounted on a Langendorff apparatus and perfused with oxygenated KH buffer (95% O2 + 5% CO2) at 37°C at a constant pressure of 85 cmH2O. RV pressure was measured by a fluid-filled balloon in the RV, which was connected to a pressure transducer (Harvard Apparatus; Holliston, MA, USA). High fidelity signals were recorded using PowerLab 8/35 data acquisition hardware and analyzed using LabChart Pro 8 software (AD Instruments; Colorado Springs, CO, USA).
IR protocol
Hearts were stabilized for 10–20 min before beginning the experimental protocol. Mdivi-1 (25 μM) or P110 (1 μM) was added to the perfusate 10 min prior to ischemia. DMSO and TAT were used as vehicle control for Mdivi-1 and P110, respectively. Two consecutive cycles of 15 min of global ischemia followed by 15 min of reperfusion were performed in the RV Langendorff preparation. In hearts used solely as a source of tissue for immunoblotting, aconitase activity assay or transmission electron microscopy (TEM), either a single cycle of IR or 45 min of perfusion without IR was performed.
Immunoblotting
At the end of the Langendorff RV-IR experiment, the RV was dissected free from the LV plus septum (LV+S) and weighed. The tissues were then preserved at −80 °C for subsequent immunoblotting. RV proteins were extracted from the various subcellular compartments using a cytoplasmic, mitochondrial and nuclear protein extraction kit (Cat #: MCN-002, ZmTech Scientifique; Montreal, QC, Canada). The following antibodies were used: anti-Drp 1 antibody (1:500; Cat #: ab56788, Abcam; Cambridge, MA, USA), anti-β-tubulin antibody (1:1000; Cat #: 32–2600, Invitrogen; Grand Island, NY, USA), anti-voltage-dependent anion channel (VDAC) antibody (1:1000; Cat #: 4866, Cell Signaling; Whitby, ON, Canada), anti-phospho-Drp 1 (serine 616) antibody (1:1000; Cat #: 3455S, Cell signaling; Whitby, ON, Canada), and anti-Fis 1 antibody (1:500; Cat #: 10956-1-AP, ProteinTech; Rosemont, IL, USA). β-tubulin and VDAC were used as cytoplasmic and mitochondrial loading controls, respectively.
Transmission electron microscopy (TEM)
After the Langendorff experiment, the RV was immediately fixed in 4% formaldehyde and 1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) overnight or longer. The tissue was then immersed in 2% sodium cocodylate buffer solution and post-fixed in 1% osmium tetroxide in 0.1 M phosphate buffer for 1 hour. After standard dehydration, embedding and sectioning, the grids with tissue sections were stained with 5% uranyl acetate for 15 min followed by staining with 3% lead citrate for 5 min. Images were collected using a transmission electron microscope (Tecnai Osiris; FEI, Hillsboro, OR, USA) at 80 kV with a Gatan charge-coupled device camera. Mitochondria were traced manually for area measurement using Leica imaging software (Version 4.7, Leica Microsystems; Wetzlar, Germany). Mitochondrial ultrastructure was graded and assigned a numerical score as described previously [26]. A score of 0 indicates a normal mitochondrion, whilst numbers 1 through 3 indicate progressive swelling of mitochondria and disintegration of cristae, with higher numbers reflecting more severe swelling and disintegration. A minimum of 120 mitochondria/group was analyzed.
Confocal microscopy on mitochondrial membrane potential of RV tissue
Mitochondrial membrane potential was measured in RV myocytes using tetramethylrhodamine methyl ester (TMRM; Cat #: T668, Lifetechnologies; Carlsbad, CA, USA), a potentiometric dye which is more fluorescent when membrane potential is more negative. Membrane potential was measured in RVs post Langendorff experiments and in RVs freshly harvested from rats that did not undergo a Langendorff experiment. In both cases the RV was immediately incubated in KH buffer containing NucBlue® Live ReadyProbes® Reagent (2 drops/mL) following the manufacturer’s protocol (Cat #: R37605, Life Technologies; Carlsbad, CA, USA) and TMRM (250 nM) at 37 °C for 30~45 min. The tissue was then imaged using a Leica SP8 confocal, laser-scanning microscope (Leica Microsystems; Wetzlar, Germany) with a 1.40NA, 63X oil immersion objective. A frame was taken in every ~16 seconds over a 5~10 min period. TMRM intensity was measured using ImageJ software (National Institutes of Health; Bethesda, MD, USA).
Confocal microscopy of RV vasculature
After rats were anesthetized with Ketamine 75mg/kg plus Xylazine 10mg/kg (IP), the right internal jugular vein was cannulated for infusion of 1 mL PBS containing 10 mg of albumin-FITC (Cat #: A9771, Sigma, St. Louis, MO, USA). 30 seconds after the infusion, the chest was opened and the main pulmonary artery, ascending aorta, superior and inferior vena cava, and mitral valve were occluded with suture to prevent the leaking of the blood out of the heart vasculature. The whole heart was then isolated and dissected for imaging of the RV free wall using a Leica SP8 confocal laser scanning microscope (Leica Microsystems; Wetzlar, Germany) with a 1.40NA, 40X oil immersion objective. Z-stacking to a depth of 12 μm over an area of ~ 1 mm × ~ 1 mm from 1~3 spots per each RV was performed. The Z-stacked images were then projected to view the vessels that contain albumin–FITC. The intensity of fluorescent signal for albumin–FITC was quantified using ImageJ software (National Institutes of Health; Bethesda, MD, USA) to quantify patent, perfused vessel. For each spot, 5~11 regions of interest with a size of 200 μm × 200 μm were analyzed and averaged to obtain a mean intensity.
Aconitase activity assay
The aconitase activity assay was performed according to the manufacturer’s protocol. Briefly, isolated mitochondrial pellets obtained from the RVs after the Langendorff experiment were disrupted via sonication and extracted using Aconitase Activity Assay kit components (Cat #: ab110169, Abcam; Cambridge, MA, USA). Enzyme activity was measured by quantifying the conversion of citrate to isocitrate. Isocitrate was further processed and resulted in a product that converts a colorless probe to a colored product in proportion to aconitase activity.
Statistical analysis
All of the data are reported as mean ± standard error of the mean (SEM). A two-tailed, Student’s t-test was used for comparison between 2 groups. One-way analysis of variance (ANOVA) was used for comparison between > 2 groups, followed by Tukey multiple- comparison post hoc test. A p < 0.05 was considered statistically significant.
RESULTS
PAH and RVH in MCT rats were confirmed by echocardiography
At week 4 post-injection of MCT or PBS, body weight was significantly less in MCT vs. control rats (330±11 vs. 516±9 g, p < 0.001). Compared to control, MCT rats had shorter PAAT (19±0.5 vs. 33±1 ms, p < 0.001; Fig. 1A) and reduced CO (73±4 vs. 136±6 ml/min, p < 0.001; Fig. 1B), findings which are consistent with PAH. Likewise, MCT caused RV hypertrophy (RVH), as indicated by increased RVFW diastolic thickness (1.42±0.05 vs. 0.63±0.02 mm, p < 0.001; Fig. 1C) and Fulton index (RV/LV+S) (0.58±0.04 vs. 0.26±0.04, p < 0.001; Fig. 1D) in MCT vs. control rats. RVFW systolic thickening and TAPSE were reduced in MCT rats compared to control (31±3 vs. 100±5, p < 0.001; Fig. 1E, and 1.6±0.1 vs. 2.9±0.1 mm, p < 0.001; Fig. 1F, respectively), consistent with RV dysfunction. Treadmill distance was also reduced in MCT vs. control rats (32±8 vs. 193±18 m, p < 0.001; Fig. 1G).
Figure 1. Cardiac ultrasound confirms the presence of severe PAH and RVH in the MCT rat model.
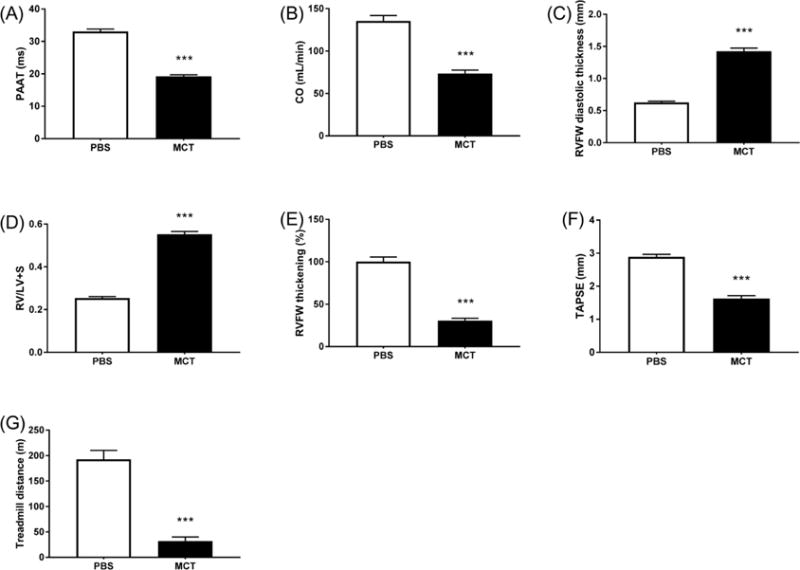
PAAT, CO, TAPSE and treadmill distance were reduced in MCT vs. PBS rats (A, B, F, G). RVFW diastolic thickness and RV/LV+S were increased and RVFW thickening was reduced in MCT vs. PBS rats (C, D, E). n > 15/group. MCT, monocrotaline. *** p < 0.001 vs. PBS.
MCT-PAH RV displays microvascular rarefaction
The number of small blood vessels (5~15 μm in diameter) was significantly less in MCT versus control RV, as indicated by the fluorescent intensity over a line of 500 μm (Fig. 2A). Confirmation of this finding was evidenced by the significantly reduced area intensity of the blood vessels’ fluorescent signal in MCT RVs compared to the control (Fig. 2B).
Figure 2. MCT rat RV displays microvascular rarefaction.
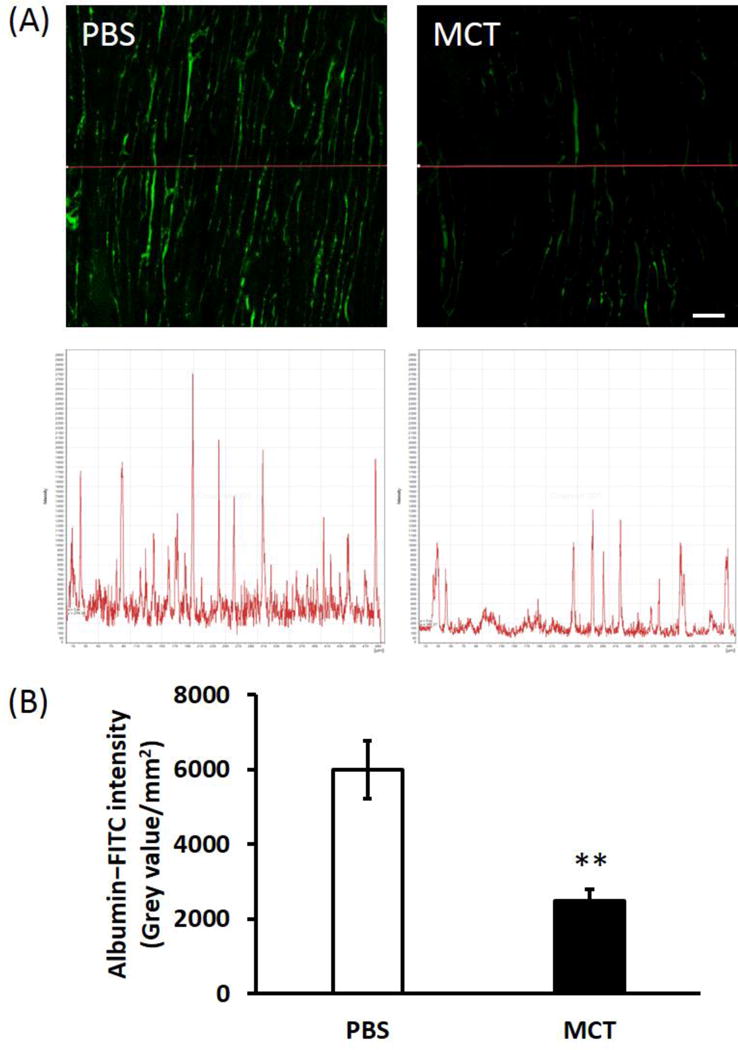
(A) Representative confocal microscopic images of RV blood vasculature (green color) and the fluorescent intensity over a line of 500 μm. Scale bar = 50 μm; (B) Summary of the area intensity of fluorescent signal for albumin-FITC. n = 7~8/group. MCT, monocrotaline. ** p < 0.01 vs. PBS.
Mdivi-1 and P110 attenuate IR-induced RV diastolic dysfunction
RVs from MCT rats developed greater RV systolic pressure (RVSP) than the control RVs, but had similar RVEDP at baseline (Fig. 3). Following IR, both RVSP and RVEDP increased in both control and MCT RVs (Fig. 3A&C). Neither of the vehicle controls (DMSO or TAT) had any hemodynamic effect (Supplemental Fig. S1), so these two groups were combined and reported as a vehicle control group in all the experimental studies. Both Mdivi-1 and P110 significantly reduced RVEDP post-IR compared to vehicle control in control and MCT RVs (Fig. 3B&D). Interestingly, while P110 had no effect on RVSP, Mdivi-1 tended to reduce post reperfusion RVSP in control RVs, though not significantly (Fig. 3B).
Figure 3. Mdivi-1 and P110 reduce the IR-induced elevation in RVEDP in both control and PAH.
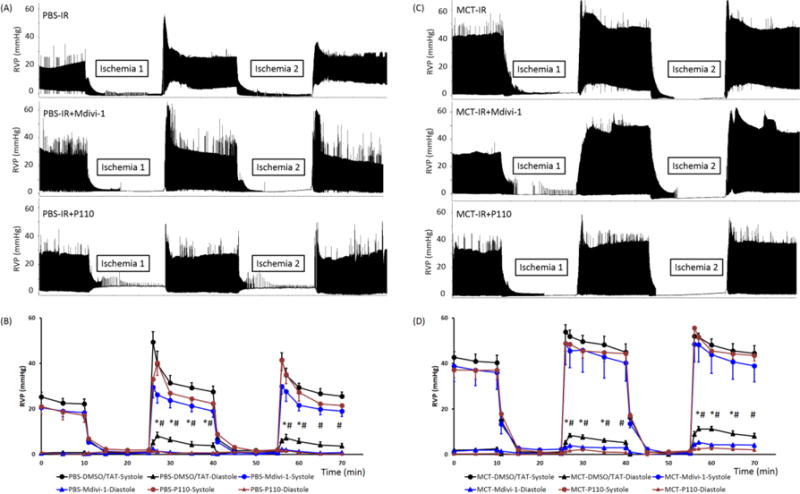
(A, C) Representative traces showing experiments in which two cycles of IR were performed in the RV Langendorff model with treatment of vehicle, Mdivi-1 and P110. Vehicle control (DMSO or TAT) or treatment (Mdivi-1 or P110) was given in the baseline 10 min prior to the first IR challenge. (B, D) Mean±SEM of RVSP and RVEDP in PBS and MCT hearts in the Langendorff experiment. RVP at the time of 2, 5, 10 and 15 min after reperfusion were compared between groups. IR caused an elevation in RVEDP and Midivi-1 or P110 reduced it significantly. n = 8~11/group. RVP, RV pressure. MCT, monocrotaline. * p < 0.05 for Mdivi-1 vs. vehicle control; # p < 0.05 for P110 vs. vehicle control. p = 0.05~0.09 for Mdivi- 1 vs. vehicle control at time 65 and 70 min for PBS RVs and at time 40 and 70 min for MCT RVs.
Mdivi-1 and P110 protect mitochondrial network structure and membrane potential from IR
Control RVs that did not undergo IR had mitochondria that were organized in regular grids and were strongly polarized, evident from high uptake of TMRM (Fig. 4A; Supplemental video S1). In contrast in MCT RVs that did not undergo IR, mitochondria were fragmented and TMRM intensity was significantly reduced (Fig. 4; Supplemental video S5). IR caused a disruption in the mitochondrial network of control RVs, but did not exacerbate the already fragmented mitochondrial network of MCT RVs. Both Mdivi- 1 and P110 improved the mitochondria network in control and MCT RVs with IR (Fig. 4). Following IR, TMRM intensity decreased in control RVs (p < 0.05), but did not further decreased in the already depolarized mitochondria of MCT RVs. Both Mdivi-1 and P110 increased TMRM intensity in both control and MCT RVs compared to the vehicle control (p < 0.05; Fig. 4B; Supplemental videos).
Figure 4. Both Mdivi-1 and P110 preserve mitochondrial membrane potential in both control and PAH RVs with IR.
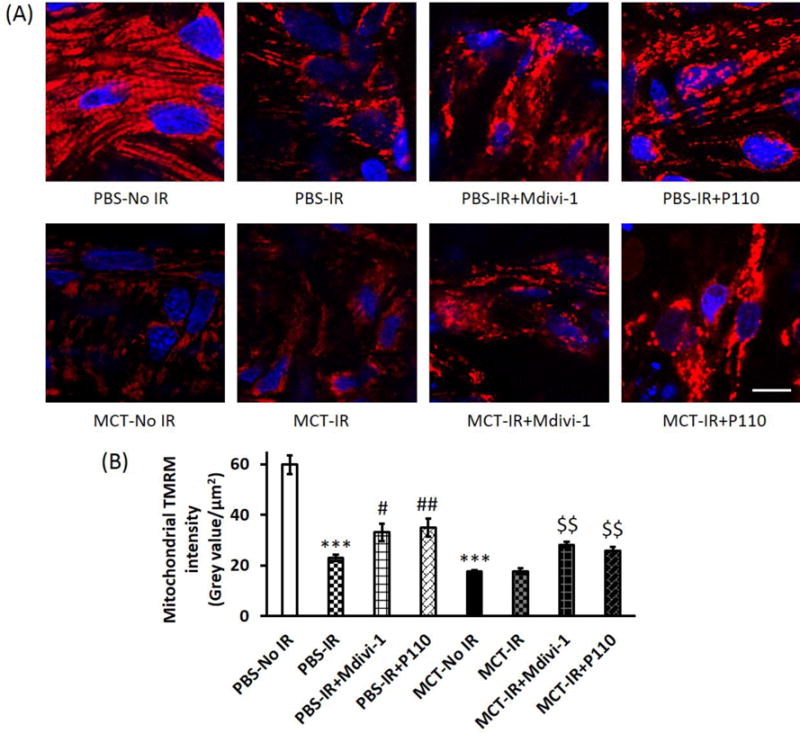
(A) Representative confocal microscopic images of RV tissue stained with TMRM. Scale bar = 10 μm; (B) Summary of TMRM intensity. TMRM intensity of MCT RVs at baseline (i.e., no IR) was significantly lower than PBS RVs. IR caused a decrease in TMRM intensity in PBS RVs but not in MCT RVs. Mdivi-1 and P110 increased TMRM intensity in both PBS and MCT RVs with IR. n > 11/group. MCT, monocrotaline. *** p < 0.001 vs. PBS-No IR; # p < 0.05 and ## p < 0.01 vs. PBS-IR; $$ p < 0.01 vs. MCT-IR.
Mdivi-1 and P110 preserve mitochondrial morphology and ultrastructure
Without IR, mitochondria in MCT RVs already displayed swelling with less dense cristae on TEM compared to control RVs, as indicated by greater mitochondrial area and a lower percentage of morphologically normal mitochondria (Fig. 5; Supplemental Fig. S2). Following IR, mitochondria in both control and MCT RVs were swollen with less dense cristae. Both Mdivi-1 and P110 preserved mitochondrial morphology and ultrastructure, reducing mitochondrial area and increasing the % of mitochondria that were scored as normal, in both control and MCT RVs (Fig. 5; Supplemental Fig. S2).
Figure 5. Both Mdivi-1 and P110 preserve mitochondrial morphology and ultrastructure in both control and PAH RVs with IR.

(A) Representative TEM images at a magnification of × 13500; (B) Summary of mitochondrial area. At baseline without IR, mitochondrial area in MCT RVs was significantly greater than that in PBS RVs; IR caused swelling of mitochondria and Mdivi-1 or P110 reduced the mitochondrial area in both PBS and MCT RVs with IR; (C) Summary of normal mitochondrial percentage. Normal mitochondria at baseline without IR were less in MCT vs. PBS RVs; IR decreased the normal mitochondrial percentage and Mdivi-1 or P110 restored it. n > 120 mitochondria from 1~2 RV tissues/group. MCT, monocrotaline. *** p < 0.001 vs. PBS-No IR; # p < 0.05 and ### p < 0.001vs. PBS-IR; $ p < 0.05 vs. MCT-IR.
Aconitase activity was reduced by IR in control RV to levels seen in PAH RV
Without IR, aconitase activity was depressed in MCT RVs compared to control RVs (p < 0.05; Fig. 6). Following IR, aconitase activity was significantly decreased in control RVs to levels seen at baseline in MCT RVs (Fig. 6). The treatment of Mdivi-1 or P110 did not restore the aconitase activity for either control or MCT RVs with IR (Supplemental Fig. S3).
Figure 6. Aconitase activity is reduced in PBS RVs with IR to levels seen in MCT RVs at baseline without IR.
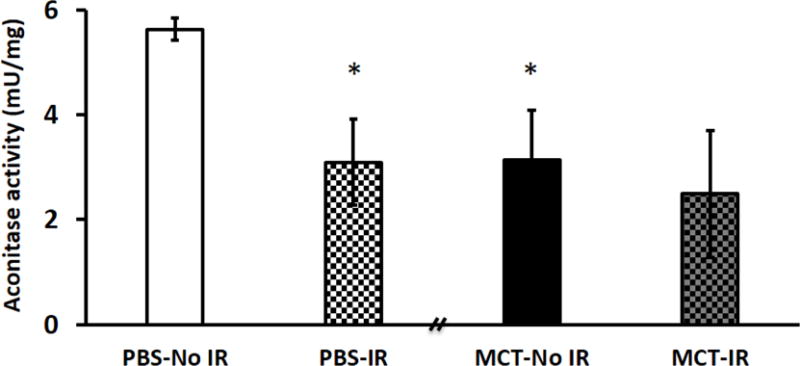
A reduction but not significant in aconitase activity was observed in MCT RVs with IR compared to MCT RVs at baseline without IR. n = 4~6/group. MCT, monocrotaline. * p < 0.05 vs. PBS-No IR.
Mdivi-1 and P110 decreased mitochondrial Drp1 expression
There was increased Drp1 in the mitochondrial fraction of MCT RVs at baseline (Fig. 7), whereas cytoplasmic Drp1 levels tended to be lower in MCT vs. control RVs (Supplemental Figs. 4A). In both control and MCT RVs, IR increased mitochondrial translocation of Drp1 (p < 0.05; Fig. 7). Drp1 phosphorylation at serine 616 in mitochondria increased significantly in control RVs with IR (p < 0.05; Supplemental Fig. S4B) but not in MCT RVs with IR (data not shown). Treatment with either Mdivi-1 or P110 reduced mitochondrial translocation of Drp1, though this was statistically significant only in MCT RVs (Fig. 7). Finally, mitochondrial Fis1 expression was not significantly different between control and MCT RVs, even with IR (Supplemental Fig. S4C).
Figure 7. Both Mdivi-1 and P110 decrease mitochondrial Drp1 expression in both control and PAH RVs with IR.
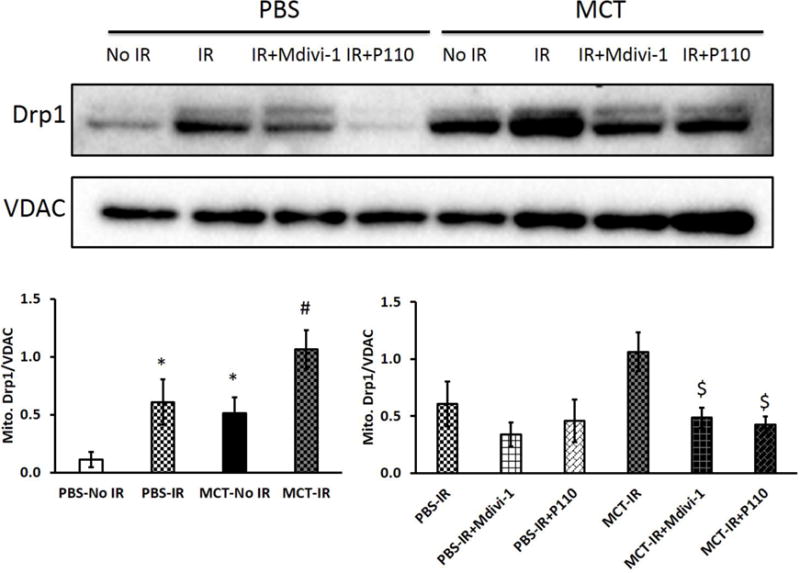
Translocation of Drp1 to the mitochondria was greater in MCT RVs than in PBS RVs at baseline. Mitochondrial Drp1 expression increased in both PBS and MCT RVs following IR. Both Mdivi-1 and P110 reduced the translocation of Drp1 to the mitochondria though only significantly in MCT RVs with IR but not in PBS RV with IR. n = 3~5/group. MCT, monocrotaline. * p < 0.05 vs. PBS-No IR; # p < 0.05 vs. MCT-No IR; $ p < 0.05 vs. MCT-IR.
DISCUSSION
This study describes a role for ischemia-mediated mitochondrial fission in causing diastolic dysfunction of the RV and impaired functional capacity in PAH. RV ischemia in PAH may occur for several reasons. In vivo, it may result from capillary rarefaction or impaired coronary perfusion pressure in the right coronary artery. In this study, we assessed the consequences of a chronic ischemic RV burden in PAH (by examining the RV microvasculature and RV function in MCT-PAH in vivo) and the resilience of the RV to more extreme ischemia (assessed using global ischemic challenges in the ex vivo RV Langendorff model). In both instances, ischemia leads to mitochondrial fission and depolarization in RV myocytes and this change in mitochondrial dynamics ultimately causes RV diastolic dysfunction (ex vivo) (Fig. 3) and functional impairment (in vivo) (Fig. 1).
This study revealed four significant findings. First, when exposed to IR, the RV, whether from normal rats or MCT-PAH rats, develops diastolic dysfunction, manifesting as an elevation of RVEDP. Second, this RV-IR response is due to the activation and translocation of the fission mediator Drp1 to the mitochondria where it causes mitochondrial fission, depolarization, swelling, and oxidative damage. Third, in the RV of MCT-PAH rats, the baseline mitochondrial phenotype (increased mitochondrial Drp1 expression, disruption of cristae, and depolarization) resembles that induced by global IR in the normal RV. This is consistent with the interpretation that ischemic mitochondrial fission in vivo creates a basal mitochondriopathy that contributes to RV dysfunction in the MCT-PAH model. Fourth, we demonstrate that two chemically and mechanistically distinct Drp1 inhibitors (Mdivi-1 and P110) preserve diastolic function post RV-IR via inhibiting Drp1-induced fission. In aggregate, these discoveries highlight the importance of the Drp1-Fis1 axis in mediating RV dysfunction in PAH.
Compared to control, MCT rats had significantly elevated pulmonary arterial pressures, as indicated by reduced PAAT (Fig. 1A). As expected, there was substantial RVH (both RVFW diastolic thickness and RV mass were increased, Fig. 1C&D). MCT RVs displayed microvasculature rarefaction (Fig. 2), consistent with previous studies [3,4]. Our group has previously shown that compared to the control, MCT RVs exhibited reduced coronary flow measured in the RV Langendorff experiment, reduced expression of vascular endothelial growth factor, and increased lactate production [4]. In aggregate these data indicate that the MCT RV is indeed ischemic. Although these are not novel findings, we document that they resulted in RV dysfunction, evident from the significantly reduced CO (Fig. 1B), decreased RVFW thickening, and decreased TAPSE (Fig. 1E&F). The impairment of RV function was associated with reduced functional capacity, apparent as shorter treadmill distances in MCT vs. control rats (Fig. 1G). In the ex vivo Langendorff RV-IR model, MCT rats developed greater RV systolic pressures than the control. This increased capacity for pressure development in a load-independent model relates primarily to the increase in RV mass in MCT-PAH. In aggregate, these data show a phenotype of RV failure in the MCT model similar to that seen in PAH patients.
Our findings in RV-IR are consistent with but extend prior work on the role of Drp 1- mediated fission in LV-IR. Sharp et al. [10] previously reported the IR-induced rise in LVEDP in normal rats, assessed using an ex vivo LV Langendorff model. Our observations of the RV-IR-induced increase in EDP are consistent with those of the LV- IR [10]. However, both normal and hypertrophied RVs retained systolic contractility despite two cycles of IR (Fig. 3), which contrasts to the LV following IR, which demonstrates decreased contractile capacity [10]. Inhibiting mitochondrial fission using Mdivi-1 or P110 ex vivo or in vivo protects the LV from many insults, including global IR in the Langendorff model [10,13], coronary artery occlusion [12,13], aortic banding [21], and KC1-induced cardiac arrest [11]. We have extended these studies, demonstrating the benefits of Mdivi-1 and P110 in RV-IR, (Fig. 3). We have also demonstrated the mechanism by which ischemia occurs and show that it is the interaction of Drp1 and Fis 1, which is critical in causing RV dysfunction.
IR activates Drp1 in many tissues, including rodent LV cardiac myocytes, renal proximal tubular cells, and retinal ganglion cells [10,11,13,27,28]. Drp1 is also activated in pulmonary artery smooth muscle cells in human and experimental PAH [20]. In the heart Drp1 activation appears relevant as a mechanism of myocyte injury, likely because fission increases ROS production [10]. In contrast, Drp1 activation in the pulmonary vasculature in PAH is reflective of pathologic rates of mitotic fission, which supports accelerated mitosis (reviewed in [17]). One might expect that inhibiting Drp1 in PAH would benefit the RV (providing protection from ischemia) and the pulmonary vasculature (serving as an antiproliferative agent).
Drp1 activation can result either from an increase in phosphorylation at serine 616 or a decrease in phosphorylation at serine 637. After LV-IR or cardiac arrest, a decrease in Drp1 phosphorylation at serine 637 appears pathologically important [10,11,27]. Serine 637 dephosphorylation results from calcium-induced activation of the phosphatase, calcineurin [10]. However, increased Drp1 phosphorylation at serine 616 in pulmonary artery smooth muscle cells appears to be the predominant mechanism of Drp1 activation in the vasculature [20]. This phosphoform seems particularly important in enhancing mitotic fission and participates in the hyperproliferative phenotype of these cells. As expected with ischemia-induced Drp1 translocation to the mitochondria, the cytoplasmic (inactive) form of the enzyme was reduced in MCT-PAH RV (Fig. 7; Supplemental Fig. S4). Likewise, in the normal RV, IR increased the accumulation of activated Drp1, including the activated serine 616 isoform, at the OMM while depleting the cytoplasmic Drp1 pool (Fig. 7; Supplemental Fig. S4). However, we did not compare the roles of these two phosphoforms of Drp1 (i.e., serine 616 and 637) in RV-IR.
IR not only causes mitochondrial fission; several other changes occur, notably mitochondrial depolarization and swelling. Ikeda et al. found that adenovirus-mediated Drp1 overexpression is sufficient to depolarize mitochondrial membrane potential in mouse cardiomyocytes [29]. Consistent with this, we demonstrate that MCT RV myocytes not only have an increased mitochondrial Drp 1 pool, but also have markedly depolarized mitochondria, as measured with live cell imaging (Fig. 4). Indeed, the degree of mitochondrial depolarization at baseline in MCT-PAH RV is qualitatively similar to that observed in normal RV following IR (Fig. 4). These findings in the RV are consistent with studies of LV-derived adult murine and neonatal rat cardiomyocytes, in which IR depolarizes mitochondrial membrane potential [12,30]. Mitochondrial depolarization is a prelude to myocyte death, and inhibiting Drp1 can prevent depolarization stimulated by apoptosis-inducers such as cisplatin, staurosporine and etoposide [31].
IR also alters mitochondrial morphology and ultrastructure, causing mitochondrial swelling, loss of matrix density and cristae disintegration [26,32]. We observed these mitochondrial changes in RV myocyte mitochondria, both at baseline, in the MCT-PAH model, and with IR in the normal RVs (Fig. 5). In contrast, mitochondrial size (cross-sectional area) decreased with IR in rat LV ex vivo [13] and with cardiac arrest in mouse LV in vivo [11]. Such morphologic discrepancies may reflect chamber-specific differences in mitochondrial sensitivity to IR or could possibly due to the difference in the experimental protocol since both these LV protocols had much longer periods of reperfusion prior to imaging mitochondrial morphology.
Finally, in the LV, ischemia causes mitochondrial oxidant damage with increased production of ROS, as indicated by reduced LV aconitase activity [11,30]. Consistent with these studies, we demonstrate the loss of aconitase activity in normal RV with IR. As with many of the measures of mitochondrial health, MCT-RV derived mitochondria had basal depression of aconitase activity similar in severity to that induced in normal RVs following global IR (Fig. 6).
We want to emphasize from these data that MCT RV at baseline (without IR) shares many of the abnormalities of that occur in normal RV with IR, including Drp1 translocation to mitochondria, mitochondrial fission, mitochondrial depolarization, mitochondrial swelling, and a decrease in aconitase activity. This is consistent with the studies on mitochondrial fission in RV fibroblast from MCT rats [33] and pulmonary artery smooth muscle cells and fibroblasts from human PAH patients [20,34,35] and on mitochondrial Drp1 translocation in pulmonary artery smooth muscle cells with PAH [20]. The MCT RV, without an ex vivo IR challenge, is already ischemic due to RV microvasculature rarefaction (Fig. 2). Moreover, mitochondria from MCT RV are already at a maximally impaired level such that experimental RV-IR does not cause further mitochondrial damage (Fig. 4, 5&6); however, mitochondrial Drp1 levels are further increased (Fig. 7).
We demonstrate the therapeutic benefit of inhibiting Drp1, using Mdivi-1, or inhibiting the Drp1-Fis1 interaction, using P110. These therapies not only reduce RVEDP but also restore the mitochondrial structure and function, counteracting IR damage. Several studies have found that Mdivi-1 and P110 can restore mitochondrial Drp1 level and preserve mitochondrial structure and function [10,11,13]. We extended these studies, demonstrating that both Mdivi-1 and P110 effectively restore mitochondrial membrane potential and mitochondrial cristae integration and prevent mitochondrial swelling (Fig. 4&5) by preventing the translocation of Drp1 to mitochondria (Fig. 7), resulting in the improvement of RV diastolic function ex vivo in both normal and PAH rats (Fig. 3).
In addition, while Mdivi-1 inhibits Drp1 GTPase activity, and does not help us distinguish which binding partners are relevant to ischemic injury, P110 only blocks the interaction between Drp1 and Fis1. The observation that P110 is as effective as Mdivi-1 in restoring mitochondrial Drp1 level (Fig. 7), preserving mitochondrial structure and function (Fig. 4&5) and improving RV diastolic function with IR (Fig. 3), identifies Fisl as the relevant Drpl binding partner in RV-IR and excludes a crucial role for MFF, MiD49, and MiD51. This is consistent with an immunoprecipitation study of LV myocyte exposed to IR, which found that Drpl binds to Fisl, but not MFF or MiD51 [13].
Taking all of these findings together, a proposed mechanistic pathway for Drpl activation along with the intervention of Mdivi-1 and P110 is presented in Fig. 8. The RV is ischemic in PAH [1,2] and Drpl plays a role in the ischemia-induced RV diastolic dysfunction (Fig. 8). Therefore, Drpl is a promising therapeutic target. Inhibition of Drpl GTPase activity, via drugs such as Mdivi-1, or Drp1-Fis1 interaction, via disruptive peptides such as P110, can improve mitochondrial structure and function and thus improve RV function in IR injury. Targeting Drpl or Drpl-Fisl interaction to prevent fission may be a novel cardioprotective strategy in PAH.
Figure 8. Proposed mechanistic pathway of Drp1 activation in the RV-IR.
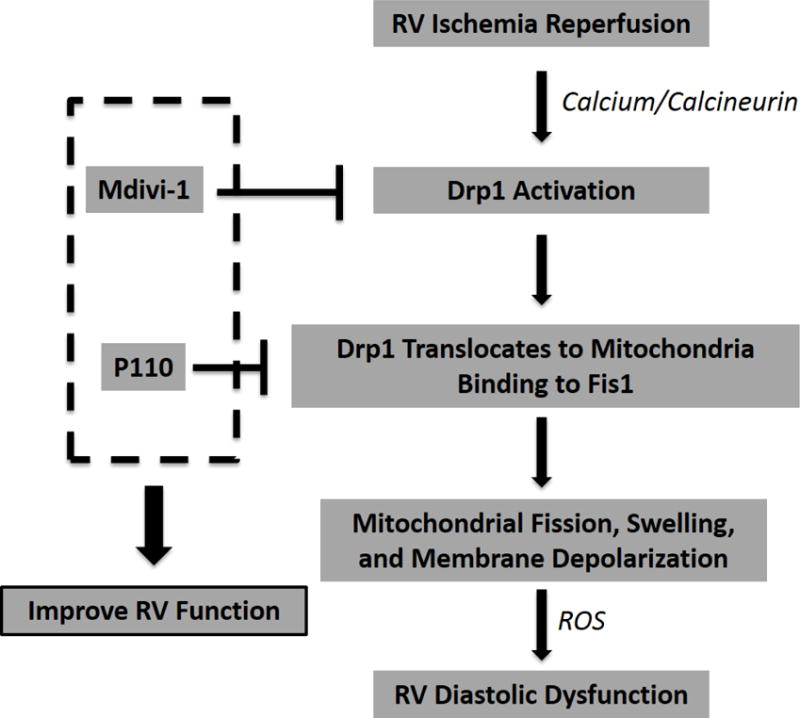
Drp1, dynamin-related protein 1; Fis1, fission protein 1; ROS, reactive oxygen species.
Limitations
Several limitations are acknowledged. First, the effects of Mdivi-1 or P110 were not studied in vivo in MCT PAH. However, we have previously demonstrated benefits from Mdivi-1 on the pulmonary vasculature in both MCT [20] and chronic hypoxia-SU5416 rat models [34] of PAH. In addition, several studies have shown the benefits of Mdivi-1 and P110 in the LV subjected to IR injury or cardiac arrest [11–13]. Thus further study is required to evaluate whether Mdivi-1 and P110 improve RV function in vivo in PAH. Second, we observed an ischemia-induced increase in Drp1 phosphorylation at serine 616 only in the normal RV but not in the MCT-PAH RV with IR. The basis for this is uncertain and further studies to measure phosphorylation of serine 637 and 616 may be useful. Third, although aconitase activity is an indicator of ROS production, direct measurement of ROS production was not performed. Fourth, we did not perform immunoprecipitation studies on RV myocytes to corroborate the findings of our P110 experiments. These additional experiments might help confirm the importance of Fis1 as the unique Drp1 binding partner in RV ischemia, although the specificity of P110 is compelling. Fifth, no single model replicates all features of the human PAH [36], and even in human PAH there is phenotypic and histological variability amongst individual patients [37]. We chose MCT rat model of PAH because this model captures the aspect of the phenotype of human PAH which was the main focus of our research, namely the development of RV failure due to maladaptive RVH and ischemia [37]. We speculate that chronic hypoxia-SU5416 rats (especially the Fischer rats) [36] would also develop ischemic RV failure. Sixth, we have previously shown that compared to the control, MCT RVs displayed decreased oxygen consumption, increased glycolysis, and increased 2-fluoro-2-deoxy-D-glucose uptake. This reflects upregulation of uncoupled glycolysis, due in part to inhibitory phosphorylation of pyruvate dehydrogenase (PDH) [23]. We have also shown that dichloroacetate (DCA) increased glucose oxidation and improved cardiac function in MCT rats, consistent to the study by Ussher et al. who demonstrated protection against IR resulted from DCA-mediated activation of PDH [38]. While the metabolic function in RV is important to cardiac function, the focus of the current study is the upstream mediator, namely Drp1, which regulates mitochondrial fission and triggers RV dysfunction in the MCT RV. Fission-mediated dysfunction of the RV is likely reinforced by metabolic dysregulation. Finally, we acknowledge that our finding of mitochondrial depolarization in MCT RV differs from studies by Nagendran et al. [39] and Sutendra et al. [40], who, using similar optical techniques and TMRM, found that RV myocyte mitochondrial membrane potential was hyperpolarized in MCT RV. The reason for this discrepancy is unknown.
Conclusions
We conclude that in MCT-induced PAH and ex vivo IR, Drp1 activation impairs RV lusitropy. Ischemia-induced RV mitochondrial fission primarily results from fission caused by interaction between activated Drp1 and Fis1. Inhibition of Drp1 GTPase activity or Drp1-Fis1 interaction can improve RV diastolic function. Drp1 inhibition is a promising therapeutic strategy to improve RV diastolic function in PAH or other conditions in which the RV is exposed to IR, such as pulmonary embolism or cardiopulmonary bypass.
Supplementary Material
Key messages.
Right ventricular ischemia reperfusion (RV-IR) causes RV diastolic dysfunction.
IR-induced mitochondrial fission causes RV diastolic dysfunction.
In RV-IR Drp1 translocates to mitochondria, binds Fis1 and causes fission and injury.
A baseline RV mitochondriopathy in MCT PAH resembles IR-induced mitochondrial injury.
Drp1 inhibitors (Mdivi-1 and P110) preserve RV diastolic function post RV-IR.
Acknowledgments
This study was supported in part by U.S. National Institutes of Health (NIH) grants NIH R01HL113003 (S.L.A.), NIH R01HL071115 (S.L.A.), and NIH 1R01HL133675–01 (W.W.S), Canada Foundation for Innovation 229252 and 33012 (S.L.A.), Tier 1 Canada Research Chair in Mitochondrial Dynamics and Translational Medicine 950–229252 (S.L.A.), the William J. Henderson Foundation (S.L.A.), and Canadian Vascular Network Scholar Award (L.T.; A.D.; K.D.S.; D.W.).
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest related to the study.
References
- 1.Vogel-Claussen J, Skrok J, Shehata ML, Singh S, Sibley CT, Boyce DM, Lechtzin N, Girgis RE, Mathai SC, Goldstein TA, et al. Right and left ventricular myocardial perfusion reserves correlate with right ventricular function and pulmonary hemodynamics in patients with pulmonary arterial hypertension. Radiology. 2011;258(1):119–127. doi: 10.1148/radiol.10100725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torbicki A, Kurzyna M, Kuca P, Fijalkowska A, Sikora J, Florczyk M, Pruszczyk P, Burakowski J, Wawrzynska L. Detectable serum cardiac troponin T as a marker of poor prognosis among patients with chronic precapillary pulmonary hypertension. Circulation. 2003;108(7):844–848. doi: 10.1161/01.CIR.0000084544.54513.E2. [DOI] [PubMed] [Google Scholar]
- 3.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120(20):1951–1960. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 4.Piao L, Fang YH, Parikh K, Ryan JJ, Toth PT, Archer SL. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med (Berl) 2013;91(10):1185–1197. doi: 10.1007/s00109-013-1064-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bian X, Williams AG, Jr, Gwirtz PA, Downey HF. Right coronary autoregulation in conscious, chronically instrumented dogs. Am J Physiol. 1998;275(1 Pt 2):H169–175. doi: 10.1152/ajpheart.1998.275.1.H169. [DOI] [PubMed] [Google Scholar]
- 6.Vlahakes GJ, Baer RW, Uhlig PN, Verrier ED, Bristow JD, Hoffmann JI. Adrenergic influence in the coronary circulation of conscious dogs during maximal vasodilation with adenosine. Circ res. 1982;51(3):371–384. doi: 10.1161/01.res.51.3.371. [DOI] [PubMed] [Google Scholar]
- 7.Minai OA, Yared JP, Kaw R, Subramaniam K, Hill NS. Perioperative risk and management in patients with pulmonary hypertension. Chest. 2013;144(1):329–340. doi: 10.1378/chest.12-1752. [DOI] [PubMed] [Google Scholar]
- 8.Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Bottiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC, et al. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation. 2008;118(23):2452–2483. doi: 10.1161/CIRCULATIONAHA.108.190652. [DOI] [PubMed] [Google Scholar]
- 9.Zia A, Kern KB. Management of postcardiac arrest myocardial dysfunction. Curr Opin Crit Care. 2011;17(3):241–246. doi: 10.1097/MCC.0b013e3283447759. [DOI] [PubMed] [Google Scholar]
- 10.Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J: official publication of the Federation of American Societies for Experimental Biology. 2014;28(1):316–326. doi: 10.1096/fj.12-226225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharp WW, Beiser DG, Fang YH, Han M, Piao L, Varughese J, Archer SL. Inhibition of the mitochondrial fission protein dynamin-related protein 1 improves survival in a murine cardiac arrest model. Crit Care Med. 2015;43(2):e38–47. doi: 10.1097/CCM.0000000000000817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121(18):2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 13.Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X, Mochly-Rosen D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc. 2013;2(5):e000461. doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24(5):659–667. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191(6):1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M. Human MTEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30(14):2762–2778. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. New Engl J Med. 2013;369(23):2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 18.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105(41):15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282(15):11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 20.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110(11):1484–1497. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Givvimani S, Munjal C, Tyagi N, Sen U, Metreveli N, Tyagi SC. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PloS One. 2012;7(3):e32388. doi: 10.1371/journal.pone.0032388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urboniene D, Haber I, Fang YH, Thenappan T, Archer SL. Validation of highresolution echocardiography and magnetic resonance imaging vs. high-fidelity catheterization in experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2010;299(3):L401–412. doi: 10.1152/ajplung.00114.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med (Berl) 2010;88(1):47–60. doi: 10.1007/s00109-009-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, Rehman J, Archer SL. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. J Mol Med (Berl) 2012;90(1):31–43. doi: 10.1007/s00109-011-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henry TD, Archer SL, Nelson D, Weir EK, From AH. Enhanced chemiluminescence as a measure of oxygen-derived free radical generation during ischemia and reperfusion. Circ Res. 1990;67(6):1453–1461. doi: 10.1161/01.res.67.6.1453. [DOI] [PubMed] [Google Scholar]
- 26.Edoute Y, van der Merwe E, Sanan D, Kotze JC, Steinmann C, Lochner A. Normothermic ischemic cardiac arrest of the isolated working rat heart. Effects of time and reperfusion on myocardial ultrastructure, mitochondrial oxidative function, and mechanical recovery. Circ Res. 1983;53(5):663–678. doi: 10.1161/01.res.53.5.663. [DOI] [PubMed] [Google Scholar]
- 27.Cho SG, Du Q, Huang S, Dong Z. Drp1 dephosphorylation in ATP depletion-induced mitochondrial injury and tubular cell apoptosis. Am J Physiol Renal Physiol. 2010;299(1):F199–206. doi: 10.1152/ajprenal.00716.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park SW, Kim KY, Lindsey JD, Dai Y, Heo H, Nguyen DH, Ellisman MH, Weinreb RN, Ju WK. A selective inhibitor of drp1, mdivi-1, increases retinal ganglion cell survival in acute ischemic mouse retina. Invest Ophthalmol Vis Sci. 2011;52(5):2837–2843. doi: 10.1167/iovs.09-5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116(2):264–278. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 30.Dezfulian C, Shiva S, Alekseyenko A, Pendyal A, Beiser DG, Munasinghe JP, Anderson SA, Chesley CF, Vanden Hoek TL, Gladwin MT. Nitrite therapy after cardiac arrest reduces reactive oxygen species generation, improves cardiac and neurological function, and enhances survival via reversible inhibition of mitochondrial complex I. Circulation. 2009;120(10):897–905. doi: 10.1161/CIRCULATIONAHA.109.853267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1(4):515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 32.Yeh ST, Lee HL, Aune SE, Chen CL, Chen YR, Angelos MG. Preservation of mitochondrial function with cardiopulmonary resuscitation in prolonged cardiac arrest in rats. J Mol Cell Cardiol. 2009;47(6):789–797. doi: 10.1016/j.yjmcc.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian L, Hong Z, Feng MG, Chen KH, Dasgupta AASL. Paper presented at the The PVRI Annual World Congress. Guangzhou, China: 2015. Role of mitochondrial-metabolic changes in the right ventricular fibroblasts in experimental pulmonary arterial hypertension. [Google Scholar]
- 34.Ryan JJ, Marsboom G, Fang YH, Toth PT, Morrow E, Luo N, Piao L, Hong Z, Ericson K, Zhang HJ, et al. PGC1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;187(8):865–878. doi: 10.1164/rccm.201209-1687OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plecita-Hlavata L, Tauber J, Li M, Zhang H, Flockton AR, Pullamsetti SS, Chelladurai P, D’Alessandro A, El Kasmi KC, et al. Constitutive Reprogramming of Fibroblast Mitochondrial Metabolism in Pulmonary Hypertension. Am J Respir Cell Mol Biol. 2016;55(1):47–57. doi: 10.1165/rcmb.2015-0142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang B, Deng Y, Suen C, Taha M, Chaudhary KR, Courtman DW, Stewart DJ. Marked Strain-Specific Differences in the SU5416 Rat Model of Severe Pulmonary Arterial Hypertension. Am J Respir Cell Mol Biol. 2016;54(4):461–468. doi: 10.1165/rcmb.2014-0488OC. [DOI] [PubMed] [Google Scholar]
- 37.Ryan JJ, Marsboom G, Archer SL. Rodent models of group 1 pulmonary hypertension. Handb Exp Pharmacol. 2013;218:105–149. doi: 10.1007/978-3-642-38664-0_5. [DOI] [PubMed] [Google Scholar]
- 38.Ussher JR, Wang W, Gandhi M, Keung W, Samokhvalov V, Oka T, Wagg CS, Jaswal JS, Harris RA, Clanachan AS, et al. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res. 2012;94(2):359–369. doi: 10.1093/cvr/cvs129. [DOI] [PubMed] [Google Scholar]
- 39.Nagendran J, Gurtu V, Fu DZ, Dyck JR, Haromy A, Ross DB, Rebeyka IM, Michelakis ED. A dynamic and chamber-specific mitochondrial remodeling in right ventricular hypertrophy can be therapeutically targeted. J Thorac Cardiovasc Surg. 2008;136(1):168–178. 178 e161–163. doi: 10.1016/j.jtcvs.2008.01.040. [DOI] [PubMed] [Google Scholar]
- 40.Sutendra G, Dromparis P, Paulin R, Zeevopoulos S, Haromy A, Nagendran J, Michelakis ED. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med (Berl) 2013;91(11):1315–1327. doi: 10.1007/s00109-013-1059-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.