

Abstract
A series of 7-substituted 1,1-diphenyl-hexahydro-oxazolo[3,4-a]pyrazin-3-ones were synthesized and tested for Neuropeptide S (NPS) antagonist activity. A concise synthetic route was developed, which features a DMAP catalyzed carbamate formation. 4-Fluorobenzyl urea (1c) and benzyl urea (1d) were identified as the most potent antagonists among the compounds examined. Structure-activity relationships (SARs) demonstrate that a 7-position urea functionality is required for potent antagonist activity and alkylation of the urea nitrogen (1e) or replacement with carbon or oxygen (5a) results in reduced potency. In addition, compounds with alpha-methyl substitution (1b) or elongated alkyl chains (1h and 1j) had reduced potency, indicating a limited tolerance for 7-position substituents.
Graphical abstract
Description: Structural features important for Neuropeptide S antagonist activity have been identified though the synthesis and testing of a series of 7-substituted 1,1-diphenyl-hexahydro-oxazolo[3,4-a]pyrazin-3-ones.
The coupling of putative transmitters with orphan receptors has led to the identification of several interesting ligand-receptor pairings that are now beginning to show novel pharmacology.1 In particular, the neuropeptide S receptor (NPSR) system deorphanized by Sato and co-workers2 has recently been shown to modulate a variety of physiological states such as sleep,3 feeding,4,5 anxiety,3,6,7 drug abuse,8 and inflammation.9
The NPS receptor is activated by an endogenous 20 amino acid peptide (Figure 1) resulting in elevated intracellular calcium via its cognate proteins Gq and Gs.3 Amino acids important for NPS agonist activity have been identified through Ala scanning mutagenesis. In particular, Phe 2, Arg 3, Asn 4, and Val 6 are critical for agonist activity whereas residues 5–13 are hypothesized to form an α-helical recognition sequence.10 NMR and circular dichroism studies on NPS have indicated a significant degree of flexibility among the amino acids critical for receptor activation, thus making identification of the bioactive NPS conformer difficult using currently available data.10,11
Figure 1. Neuropeptide S and previously described NPS antagonists.

Several single nucleotide-polymorphisms (SNP) of the NPS receptor have been shown to exist. In particular, NPSR Asn107Ile and NPSR C-alt have been fully characterized for agonist sensitivity.12 Radioligand binding of [125I]Tyr10-NPS is unaltered among receptor variants, however, a 5–10-fold enhancement in functional sensitivity using calcium flux is observed for the Ile107 variant.12 Importantly, the SNP (Asn107 to Ile107) in the NPS receptor has been implicated as a risk factor for asthma and has been shown to modulate the functional properties of NPS.9
The unique pharmacological properties of NPS were first described by Xu and colleagues who determined that NPS was involved in both arousal and anxiety.3 Administration of NPS (i.c.v.) increased locomotor activity in both habituated and naïve mice. NPS-treated mice also demonstrated anxiolytic-like behaviors in the elevated plus maze, light dark box, and marble burying paradigm. In more recent studies, Rizzi and co-workers confirmed the arousal and anxiolytic promoting properties of NPS using stress-induced hypothermia, which is a behavioral model insensitive to alterations in locomotor activity.6
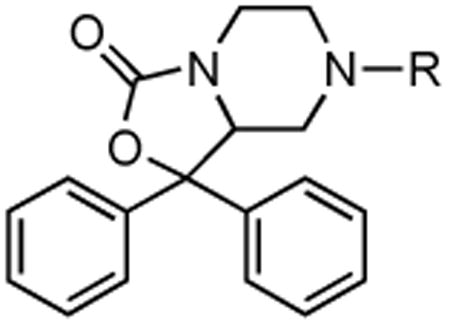
Recognizing that small molecule probes would greatly aid the pharmacological characterization of NPS receptors, our laboratory undertook a program to identify small molecule antagonists. The development of small molecule antagonists based on a computationally generated structure of the endogenous peptide was not considered reliable due to the significant conformational flexibility inherent in the peptide.11 Therefore, our laboratory chose to employ a previously patented scaffold as a lead structure.13 Although no functional or receptor binding data was disclosed in the patent, structural features important for functional activity appeared to center around 7-position derivatives of 1,1-diphenyl-hexahydro-oxazolo[3,4-a]pyrazin-3-ones. Therefore, our laboratory synthesized and evaluated a diverse series of 7-substituted analogs (Tables 1 and 2). During the course of preparing this manuscript, two small molecules described in the Takeda patent were also synthesized and tested by Okamura et al.14 These compounds (SHA-66, SHA-68) were shown to be potent and selective antagonists of both the Asn107 and Ile107 variants of the NPS receptor (Figure 1).
Table 1. NPS Antagonist Activity for Urea Derivatives.
| ||||
|---|---|---|---|---|
|
| ||||
| Compounda | R | NPS Ile107 Ke | NPS Asn107 Ke | Asn/Ile |
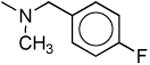
| 1b |
|
214 ± 42 | 588 ± 150 | 2.8 |
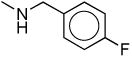
| 1cb |
|
13.7 ± 2.8 | 52 ± 2 | 3.8 |
| 1dc |
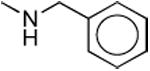
|
9.9 ± 1.6 | 47 ± 15 | 4.8 |
| 1e |
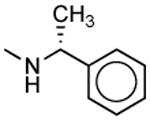
|
317 ± 5 | 229 ± 75 | 0.7 |
| 1f |
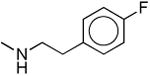
|
321 ± 120 | 326 ± 140 | 1.0 |
| 1g |
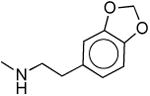
|
567 ± 190 | 690 ± 390 | 1.2 |
| 1h |
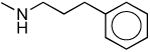
|
309 ± 52 | 417 ± 71 | 1.3 |
| 1i |
|
537 ± 190 | 437 ± 150 | 0.8 |
| 1j |
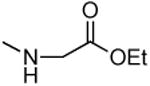
|
726 ± 107 | 1260 ± 250 | 1.7 |
| 1k |
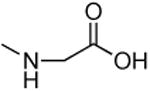
|
> 4 mM | > 4 mM | -- |
| 1l |
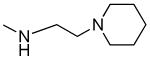
|
109 ± 23 | 490 ± 173 | 4.5 |
All target compounds were fully characterized using NMR and Mass Spectrometry.
SHA-68.
SHA-66.
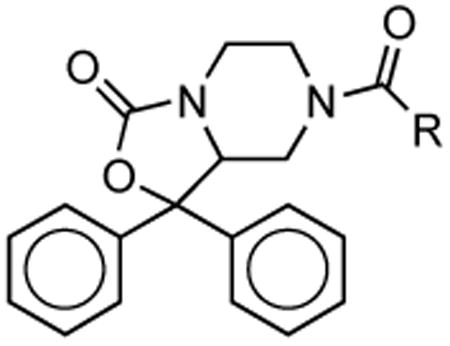
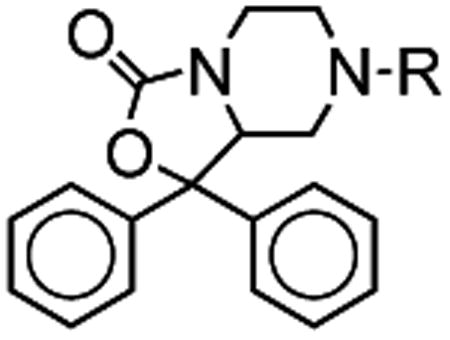
Table 2. NPS Antagonist Activity for Alternate 7-Substitution.
| ||||
|---|---|---|---|---|
|
| ||||
| Compound | R | NPS Ile107 Ke | NPS Asn107 Ke | Asn/Ile |
| 5a |
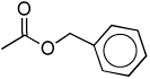
|
354 ± 51 | 353 ± 22 | 1.0 |
| 1m |
|
339 ± 76 | 460 ± 130 | 1.4 |
| 1n |
|
244 ± 42 | 267 ± 41 | 1.1 |
| 1o |
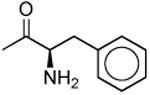
|
911 ± 130 | 4640 ± 1400 | 5.1 |
| 1p |
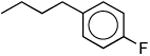
|
1110 ± 30 | 1100 ± 300 | 1.0 |
| 1q | −CH3 | > 4 mM | > 4 mM | -- |
| 1r |
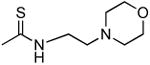
|
3760 ± 470 | 2560 ± 70 | 0.7 |
| 1s |
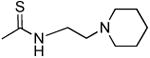
|
280 ± 41 | 354 ± 96 | 1.3 |
All target compounds were fully characterized using NMR and Mass Spectrometry.
Target compounds (1b–1s), designed to identify structural features important for NPS antagonist activity, were prepared using the methods described in Schemes 1 and 2. The key intermediate 1,1-diphenyl-hexahydro-oxazolo[3,4-a]pyrazin-3-one (1a) was synthesized following a concise route as depicted in Scheme 1. Treatment of commercially available dibenzyl ester 2 with excess phenyllithium at -78 °C afforded (1,4-dib enzyl-piperazin-2-yl)-diphenyl-methanol in moderate yield. Subsequent debenzylation using palladium on barium sulfate under a hydrogen atmosphere with two equivalents of hydrochloric acid at 60 °C afforded 3 in nearly quantitative yield. Reprotection of the resulting amino groups as carbamates seemed ideal, since the desired cyclic carbamate 5 could be readily prepared upon subsequent heating of 4 with base. Based on this hypothesis, carboxybenzyl (Cbz), and 2-(trimethylsilyl)ethoxycarbonyl (Teoc) groups were investigated. Compounds 4a and 4b were obtained as planned and cyclization with sodium hydride in dimethyl formamide proceeded smoothly to provide mono-protected derivatives 5a and 5b. Unfortunately, removal of either protecting group proved problematic. Under normal deprotection conditions both cyclic carbamates 5a and 5b either collapsed to give 3 or decomposed. In an effort to circumvent these issues an alternate protecting group was explored. Knölker and colleagues reported that 1,2-aminoalcohols, when treated with di-tert-butyl dicarbonate (Boc2O) and dimethylaminopyridine (DMAP), readily cyclize to provide oxazolidin-2-ones.15 Using this rationale, diamine 3 was treated with Boc2O, triethylamine, and DMAP in tetrahydrofuran to afford compound 5c in excellent yield. Key intermediate 1a was then obtained in quantitative yield by treatment of 5c with trifluoroacetic acid (TFA) in dichloromethane. This approach now allows for the preparation of key intermediate 1a from diamine 3 in two high yielding steps.
Scheme 1. Synthesis of key intermediate 1a.

Reagents and Conditions. Synthesis of amine 1a: (a) PhLi, THF, -78 °C; (b) H2, 40 psi, Pd/BaSO4, EtOH, 2eq, HCl, 60 °C ; (c) 4a, CbzCl, K2CO3, H2O/THF, 4b, Teoc-Succinimide, Et3N, CH3CN; (d) NaH, DMF; (e) Boc2O, DMAP, Et3N, THF; (f) TFA,CH2Cl2.
Scheme 2. Synthesis of target compounds 1b–1s.
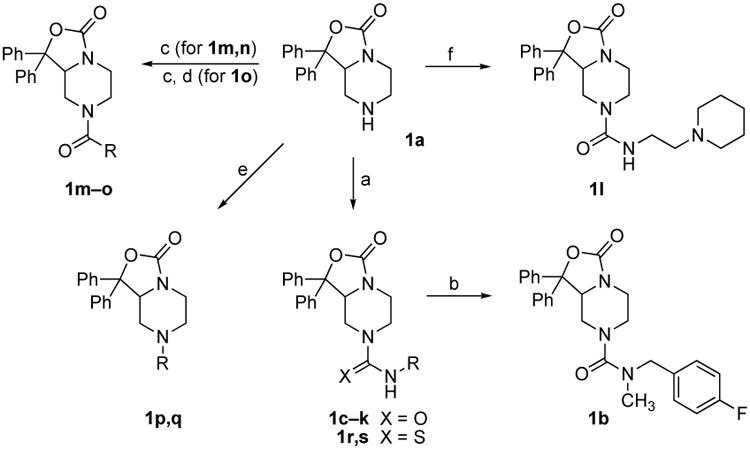
Reagents and Conditions. Synthesis of target compounds 1b–s: (a) Appropriate isocyanate or isothiocyanate, CH2Cl2; (b) Mel, NaHMDS, THF; (c) Appropriate acid, BOP, Et3H, THF; (d) TFA, CH2Cl2; (e) Appropriate aldehyde, NaB(OAc)3H, dichloroethane; (f) N-(2-aminoethyl)piperidine, triphosgene, Et3N, CH2Cl2.
The four-step procedure described in Scheme 1 provides key intermediate 1a in good yield. Although the yield of step a in Scheme 1 occurs in moderate yield (≈ 50%), steps b,e and f are accomplished in almost quantitative yield. Compared to the procedure adopted by both Takeda Pharmaceuticals13 and Okamura et al.,14 orthogonal protection of the two amino groups was not needed in our approach. In addition, the DMAP mediated cyclization of 3 to 5c makes this route an attractive alternative.
Target ureas 1b–l and thioureas 1r and 1s were prepared by reacting 1a with various commercially available isocyanates or isothiocyanates (Scheme 2). For 1l, where the isocyanate was not available, reaction of 1a with triphosgene and N-(2-aminoethyl)piperidine proceeded well to give 1l in moderate yield. Alternatively, 1a was coupled to a number of acids with benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (BOP) in tetrahydrofuran to provide amides 1m–o. The N-alkyl amines 1p and 1q were prepared by reductive amination of 1a with 4-(4-fluorophenyl)-butanal, which was obtained through oxidation of 4-(4-fluorophenyl)butanol16 with pyridinium chlorochromate in dichloromethane, or formaldehyde using sodium triacetoxyborohydride in dichloroethane.17 Target compounds were fully characterized by NMR and mass spectroscopy.
Considering the potential pharmacological significance of small molecule NPS receptor antagonists, we have defined key structural features of 7-substituted 1,1-diphenyl-hexahydro-oxazolo[3,4-a]pyrazin-3-ones required for antagonist activity. In keeping with earlier reports,12,14 NPS was more potent at the Ile107 variant (EC50 of 2.5 nM) than at the Asn107 variant (EC50 16.7 nM) using a calcium flux functional assay.18 The number of spare receptors in an expression system can affect potency, however, taken together, these data support there being a functional difference between the two variants. Compound 1c and a closely related 1,2,3,6-tetrahydro-pyridine analog of 1l were resolved into their individual enantiomers in the Takeda Patent although no indication of biological activity was provided (Table 1).13 Okamura and colleagues recently reported Kb's of 27.9 nM (Ile107) and 16.9 nM (Asn107) for racemic 1c and 32.6 nM (Ile107) and 21.7 nM (Asn107) for the desfluoro analog 1d, respectively.14 In our hands, racemic 1c (Ke = 13.7 and 51 nM) and 1d (Ke = 9.9 and 47 nM) were the most potent NPS antagonists among the compounds described herein at both the Ile107 and Asn107 variants, respectively. In contrast to Okamura, we found these compounds to be slightly more selective for the Ile107 variant. Methylation of urea 1c (1b, Ke = 214 and 588 nM) reduced potency by 16-fold at Ile107 and 12-fold at Asn107. Similarly, replacement of the urea nitrogen with carbon (1m, Ke = 339 and 460 nM) or oxygen (5a, Ke = 354 and 353 nM) significantly reduced potency at both receptor variants. This suggests a hydrogen on the 7-position urea is critical for antagonist activity. In order to determine if a hydrogen bond donor placed in close proximity to the urea hydrogen could serve as a suitable surrogate, phenylalanine derivative 1o was prepared. A 100-fold reduction in potency was observed for 1o (Ke = 911 nM and 4640 nM). Because the urea functionality appeared critical for NPS antagonist activity, we began to evaluate alternate substitution of the 7-position urea. Addition of an alpha methyl group (1e, Ke = 317 and 229 nM) reduced potency by 32- and 48-fold compared to 1d. Lengthening the alkyl spacer between the terminal phenyl ring to two carbons (1f, Ke = 321 nM) reduced potency by 25-fold compared to 1c. Further elongation of the alkyl spacer to three (1h, Ke = 309 nM) and four carbons (1i, Ke = 537 nM) significantly reduced potency compared to the parent 1d. These results suggest that the hypothetical receptor pocket has specific size requirements. In order to assess if hydrophilic or hydrophobic character of the urea substituent was essential for antagonist potency, carboxylic acid 1k and piperidine 1l were prepared. Intermediate ester 1j (Ke = 726 nM) and target acid 1k (Ke > 4 μM) were both inactive as antagonists. Interestingly, 2-piperidinoethyl analog 1l had intermediate potency at the Ile107 receptor variant (Ke = 109 nM) indicating bulky amine substituents are tolerated, albeit at slightly reduced potency. Converting urea 1l to thiourea 1s moderately reduced potency (Ke = 280 nM) whereas replacement of the piperidine with a more water soluble morpholino group (1r, Ke = 3760 nM) dramatically reduced potency. Finally, evaluation of 4-(4-fluorophenyl)butyl analog 1p (Ke = 1100) and methyl analog 1q (Ke > 4 μM) were inactive as antagonists. In general, it appears that tolerance to 7-substitution is limited.
This study demonstrates that antagonists were generally more potent at the Ile107 variant (1–5 fold) with limited exceptions (Tables 1 and 2). This suggests that future development of antagonists selective for one receptor variant may be possible. In addition, none of the compounds presented in this study possessed measurable intrinsic activity at 10 μM in the NPS Ile107 cell line.
In conclusion, we have provided a novel synthetic route to 7-substituted 1,1-diphenyl-hexahydro-oxazolo[3,4-a]pyrazin-3-ones and begun to identify the key structural features required for NPS antagonist activity. In particular, we have demonstrated the importance of the urea functionality possessing a free hydrogen and that ethylpiperidine (1l) can serve as a substitute for the benzyl group of 1d. The combination of these results provides a basis for the design and testing of novel scaffolds with enhanced potency and drug-like properties for the NPS receptor.
Acknowledgments
This work was supported by a United States National Institute of Mental Health research grant (MH081247-01). We also wish to thank Mr. Keith Warner, Ms. Tiffany Langston and Ms. Kathleen Kitsopoulos for their valuable technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Civelli O. GPCR deorphanizations: the novel, the known and the unexpected transmitters. Trends Pharmacol Sci. 2005;26:15. doi: 10.1016/j.tips.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Sato S, S Y, Miyajima N, Yoshimura K. Novel G-protein coupled receptor protein and DNA thereof. World Patent Application WO 02/31145 A1. 2002
- 3.Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, Brucher FA, Zeng J, Ly NK, Henriksen SJ, de Lecea L, Civelli O. Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron. 2004;43:487. doi: 10.1016/j.neuron.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Smith KL, Patterson M, Dhillo WS, Patel SR, Semjonous NM, Gardiner JV, Ghatei MA, Bloom SR. Neuropeptide S stimulates the hypothalamo-pituitary-adrenal axis and inhibits food intake. Endocrinology. 2006;147:3510. doi: 10.1210/en.2005-1280. [DOI] [PubMed] [Google Scholar]
- 5.Beck B, Fernette B, Stricker-Krongrad A. Peptide S is a novel potent inhibitor of voluntary and fast-induced food intake in rats. Biochem Biophys Res Commun. 2005;332:859. doi: 10.1016/j.bbrc.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 6.Rizzi A, Vergura R, Marzola G, Ruzza C, Guerrini R, Salvadori S, Regoli D, Calo G. Neuropeptide S is a stimulatory anxiolytic agent: a behavioural study in mice. Br J Pharmacol. 2008 doi: 10.1038/bjp.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leonard SK, Dwyer JM, Sukoff Rizzo SJ, Platt B, Logue SF, Neal SJ, Malberg JE, Beyer CE, Schechter LE, Rosenzweig-Lipson S, Ring RH. Pharmacology of neuropeptide S in mice: therapeutic relevance to anxiety disorders. Psychopharmacology (Berl) 2008;197:601. doi: 10.1007/s00213-008-1080-4. [DOI] [PubMed] [Google Scholar]
- 8.Cioccioppo R, Economidou D, Cannella N, Braconi S, Stopponi S. In Society for Neuroscience. Vol. 2007. San Diego; 2007. Neuropeptide s system activation facilitates conditioned reinstatement of cocaine-seeking in the rat; p. 271.18/Z1. [Google Scholar]
- 9.Laitinen T, Polvi A, Rydman P, Vendelin J, Pulkkinen V, Salmikangas P, Makela S, Rehn M, Pirskanen A, Rautanen A, Zucchelli M, Gullsten H, Leino M, Alenius H, Petays T, Haahtela T, Laitinen A, Laprise C, Hudson TJ, Laitinen LA, Kere J. Characterization of a common susceptibility locus for asthma-related traits. Science. 2004;304:300. doi: 10.1126/science.1090010. [DOI] [PubMed] [Google Scholar]
- 10.Bernier V, Stocco R, Bogusky MJ, Joyce JG, Parachoniak C, Grenier K, Arget M, Mathieu MC, O'Neill GP, Slipetz D, Crackower MA, Tan CM, Therien AG. Structure-function relationships in the neuropeptide S receptor: molecular consequences of the asthma-associated mutation N107I. J Biol Chem. 2006;281:24704. doi: 10.1074/jbc.M603691200. [DOI] [PubMed] [Google Scholar]
- 11.Tancredi T, Guerrini R, Marzola E, Trapella C, Calo G, Regoli D, Reinscheid RK, Camarda V, Salvadori S, Temussi PA. Conformation-activity relationship of neuropeptide S and some structural mutants: helicity affects their interaction with the receptor. J Med Chem. 2007;50:4501. doi: 10.1021/jm0706822. [DOI] [PubMed] [Google Scholar]
- 12.Reinscheid RK, Xu YL, Okamura N, Zeng J, Chung S, Pai R, Wang Z, Civelli O. Pharmacological characterization of human and murine neuropeptide s receptor variants. J Pharmacol Exp Ther. 2005;315:1338. doi: 10.1124/jpet.105.093427. [DOI] [PubMed] [Google Scholar]
- 13.Fukatsu K, Nakayama Y, Tarui N, Mori M, Matsumoto H, Kurasawa O, Banno H. Bicyclic Piperazine Compound And Use Thereof. 2006 Jun 7; Patent Application No.: PCT/JP2004/012683 (EPO 4772639.3) [Google Scholar]
- 14.Okamura N, Habay SA, Zeng J, Chamberlin AR, Reinscheid RK. Synthesis and pharmacological in vitro and in vivo profile of SHA 68 (3-Oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide), a selective antagonist of the Neuropeptide S receptor. J Pharmacol Exp Ther. 2008 doi: 10.1124/jpet.107.135103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knölker HJ, Braxmeier T. Isocyanates. Part 5. Synthesis of chiral oxazolidin-2-ones and imidazolidin-2-ones via DMAP-catalyzed isocyanation of amines with di-tert-butyl dicarbonate. Tetrahedron Lett. 1998;39:9407. [Google Scholar]
- 16.Wan AS, Ngiam TL, Leung SL, Go ML, Heng PW, Natarajan PN, Shafiee A, Vossoghi M, Savabi F, Francisco CG, et al. Long-acting contraceptive agents: norethisterone esters of arylcarboxylic acids. Steroids. 1983;41:309. doi: 10.1016/0039-128x(83)90101-0. [DOI] [PubMed] [Google Scholar]
- 17.Matthews JM, Dyatkin AB, Evangelisto M, Gauthier DA, Hecker LR, Hoekstra WJ, Liu F, Poulter BL, Sorgi KL, Maryanoff BE. Synthesis, resolution, and absolute configuration of novel tricyclic benzodiazepines. Tetrahedron. 2004;15:1259. [Google Scholar]
- 18.Functional Determinations: Identification of functional agonists as well as antagonists at the NPS receptor utilized RD-HGA16 cells (Molecular Devices), a CHO cell line stably over-expressing the promiscuous Gq-protein Gα16. Two individual cell lines were created that stably express each NPS receptor variant (NPS Ile107 and Asn107). Cells are loaded with the calcium sensitive dye calcium3 (Molecular Devices) for 1 h and compounds are assayed in separate experiments for intrinsic activity and for the ability to inhibit NPS activity as measured by calcium mobilization in the FlexStation assay. Test compound Ke values were determined by running an 8-point half-log NPS concentration response curve in the presence and absence of a single concentration of test compound. EC50 values were calculated for NPS (A) and NPS + test compound (A′), and these used to calculate the test compound Ke. A three-parameter logistic equation was fit to the concentration response data with Prism (v5 for Windows, GraphPad Software; San Diego, CA) to calculate the EC50 values. At least two different concentrations of test compound were used for these experiments, and these were chosen such that they at least caused a four-fold rightward shift in the NPS EC50. The Ke was calculated from the formula: Ke = [L]/(DR-1), where [L] equals the concentration of test compound in the assay and DR equals the dose ratio (A′/A). The data represent the mean ± SE from at least three independent experiments.