

ABSTRACT
The termite gut accomplishes key physiologic functions that underlie termite symbiosis and sociality. However, potential candidate functions of the host-symbiont holobiome have not yet been explored across seemingly divergent processes such as digestion, immunity, caste differentiation, and xenobiotic tolerance. This study took a meta-analysis approach for concurrently studying host and symbiont gut metatranscriptome responses of the lower termite Reticulitermes flavipes, which has ancestral characteristics and hosts a diverse mix of eukaryotic and bacterial symbionts. Thirteen treatments were compared from 5 categories (dietary, social, hormonal, immunological, and xenobiotic), revealing 3 main insights. First, each of the 5 tested colonies had distinct magnitudes of transcriptome response, likely as a result of unique symbiont profiles, which highlights the uniqueness of individual termite colonies. Second, after normalization to standardize colony response magnitudes, unique treatment-linked metatranscriptome topologies became apparent. Third, despite colony and topology differences, 4 co-opted master genes emerged that were universally responsive across diverse treatments. These master genes encode host functions related to protein translation and symbiont functions related to protein degradation and pore formation in microbial cell walls. Three of the 4 master genes were from co-evolved protist symbionts, highlighting potentially co-evolved roles for gut symbiota in coordinating functional responses of the collective host-symbiont holobiome. Lastly, for host genes identified, these results provide annotations of recent termite genome sequences. By revealing conserved domain genes, as well as apparent roles for gut symbiota in holobiome regulation, this study provides new insights into co-opted eusocial genes and symbiont roles in termite sociobiology.
KEYWORDS: microbiome, microbial ecology, protist, septicolysin, social evolution, SPRY, TRMT, ubiquitin ligase
Introduction
Termites (Order Blattodea) are eusocial insects with complex, multifaceted life histories and lifestyles. The key processes underlying termite sociobiology and evolution include intricate developmental pathways, mutualisms with gut symbiota, and reliance on nutrient-poor lignocellulose diets. There are 3 general caste phenotypes in termites (i.e., worker, soldier, and reproductive) that arise from common genotypes; however, caste proportions vary according to environmental and social cues. In lower termites, the worker caste is a sterile, developmentally plastic immature stage that comprises the majority of termite colonies.1 Lower termite workers also perform the majority of lignocellulose feeding and digestion, and they house many thousands of species of eukaryotic and prokaryotic symbiota within their guts. The worker termite digestive tract thus underlies many aspects of termite social living.
Two factors that pose key challenges to termite societies are maintenance of symbiont populations and group living in pathogen-rich soil environments. On the first point, workers of lower termites molt on a regular basis, losing gut symbiota. However, living in close proximity to siblings and the eusocial practice of trophallaxis serve to replenish gut symbiota and ensure uniform symbiont populations among nestmates.2 Second, group living in soil environments poses significant pathogenic challenges to termites. Furthermore, the exhibited stomatodeal and proctodeal feeding are behaviors ideally suited for disease transmission. Pathogen challenges are effectively met by allogrooming, social immunity, and to a lesser degree, innate immune responses at the individual level.3,4 However, the potentially intersecting roles of the host-symbiont holobiome in immune, digestive, caste differentiation, and xenobiotic response functions are poorly understood.
A fundamental idea in insect social evolution is that during the transition from solitary to social lifestyles, key traits of solitary ancestors were co-opted for new eusocial functions.5–7 Two of the first-identified examples of such traits were honey bee vitellogenins, which are egg yolk proteins that also play roles in reproductive caste regulation, and termite hexamerins, which are storage proteins that also physiologically regulate caste differentiation.8–10 Other features predicted in bees relate to increasingly complex gene regulatory networks in correlation with increasing social complexity.11 Additional examples of co-opted social mechanisms in termites await discovery. Moreover, potentially co-opted master-regulatory functions of termite gut microbiota remain essentially unexplored. Genomic resources recently have been developed for many termite species, but as yet, they have not been applied to address complex sociobiology questions such as gene or symbiota co-option.12
The goal of this study was to using existing data sets from 3 prior published studies17–19 to identify potentially co-opted master genes of the lower termite Reticulitermes flavipes and its gut symbiota. To do this, we used a microarray meta-analysis to experimentally compare gut metatranscriptome composition across 13 treatments from 5 broad categories (dietary, social, hormonal, immunological, and xenobiotic) and their interactions. Our hypothesis was that key co-opted and/or master genes would respond similarly across multiple treatment categories. Our working definition of a candidate “co-opted” gene in this case is a gene having translated homologs of known function in solitary organisms. In total, 65 individual array hybridizations were performed using 5 replicate termite colonies, followed by clustering, topology, and co-option analyses. Findings reveal unique topology profiles among treatment categories, 4 candidate “master” genes shared among treatments, and possible regulatory roles for eukaryotic gut symbiota.
Results
Overview of approaches and normalization strategies
Data were generated from a meta-analysis of 64 individual microarrays (as detailed in prior papers17–19) hybridized concurrently with poly-A enriched host-gut and symbiont cDNAs from worker termites (Fig. 1). Thirteen treatments were compared that included: feeding on wood or cellulose paper (W or C); exposures to juvenile hormone, soldier head extract, live soldiers, live neotenic reproductive, or acetone controls (JH, SH, LS, LR or A); and exposures to bacteria (Serratia marcescens), fungi (Metharizium anisopliae), imidacloprid, fungi + imidacloprid, bacteria + imidacloprid or DMSO solvent controls (B, F, I, FI, BI, or S). Five replicate colonies were tested for each treatment (each based on a sample of 20 worker termite guts per replicate colony), and array results were independently validated by RT-qPCR. Only one replicate microarray was excluded due to inconsistent signal intensity (treatment W, colony #3).
Figure 1.
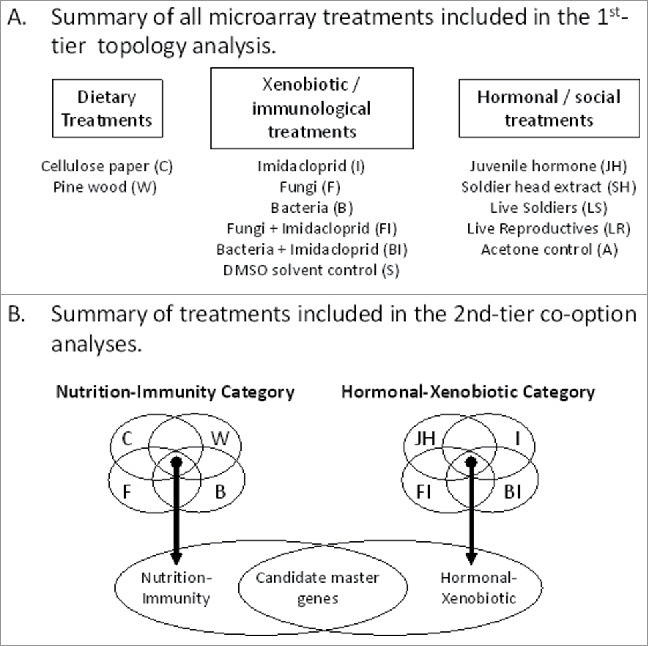
Overview of treatments and experimental analyses. (A) Summary of all microarray treatments used in the present study for the 1st-tier topology analysis. (B) Summary of treatments selected for the 2nd-tier co-option analysis and the stepwise approach used. The goal of the topology analysis shown in A was to determine relationships of treatments to one another based on gene expression, while the goal of the analysis outlined in B to identify candidate master genes responsive across treatment categories. See panel A for treatment abbreviations shown in B.
Different normalization strategies were compared for their relative ability to correct for colony differences in absolute array intensities. Normalizations were performed by scaling individual log2 fold-changes so that each data set had comparable median and lower and upper quartile values. Box plots of the data sets before and after normalization are shown in Fig. S1. This normalization step was based on the assumption that the distributions of gene expression follow the same statistical law independent of treatments or colony origin, which may not be entirely valid in the present analysis. We thus present both non-normalized and normalized data summaries, as well as 2 different normalization strategies that include normalization within (i) each treatment or (ii) each treatment category (Table 1).
Table 1.
Numbers of differentially expressed array positions at various significance levels when comparing each treatment with a mixed reference using different normalization strategies. *The column denoted by an asterisk indicates the normalization strategy and p-value cutoff used for co-option analyses.
| Non-normalized |
Normalized by treatment or treatment category (identical results) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.01 level |
0.05 level |
0.01 level |
0.05 level |
||||||
| Treatment category | Treatment | P | Q | P | Q | P | Q | P* | Q |
| Nutritional | C | 1123 | 0 | 3386 | 29 | 1382 | 66 | 3287 | 446 |
| W | 235 | 0 | 1020 | 0 | 259 | 0 | 1116 | 0 | |
| Social | JH | 831 | 0 | 2374 | 17 | 901 | 1 | 2435 | 5 |
| LS | 195 | 4 | 942 | 14 | 213 | 0 | 975 | 18 | |
| A (control) | 249 | 1 | 906 | 20 | 216 | 0 | 853 | 0 | |
| SH | 139 | 0 | 530 | 0 | 146 | 0 | 547 | 0 | |
| LR | 114 | 0 | 495 | 0 | 111 | 0 | 481 | 0 | |
| Xenobiotic / Immunological | B+I | 2534 | 28 | 4768 | 1887 | 2764 | 5 | 4791 | 2508 |
| F+I | 1714 | 0 | 4133 | 425 | 2326 | 0 | 4405 | 1643 | |
| S (control) | 778 | 3 | 2096 | 22 | 697 | 0 | 2084 | 0 | |
| F | 259 | 1 | 1111 | 4 | 298 | 0 | 1141 | 0 | |
| I | 283 | 1 | 1115 | 29 | 297 | 0 | 1140 | 0 | |
| B | 217 | 3 | 993 | 15 | 244 | 0 | 1069 | 0 | |
Table 1 lists the numbers of differentially expressed (DE) array positions (i.e., gene probes) among treatments, relative to a mixed/common reference, using different normalization settings. These analyses used one-sample Student's t-tests with P and Q values13 at different significance levels; i.e., p-values of 0.01 or 0.05 indicate that 1% or 5% of all t-tests will result in false positives, whereas q-values of the same values indicate that minimum false discovery rates of 1% or 5% are incurred when calling tests significant.9 Normalizing samples by either treatment or treatment category produced identical results. Also, although the absolute number of significantly different array positions varied between the normalized and non-normalized analyses, the general trends were similar.
Clustering analysis of colony-level effects
Non-normalized log2 fold-change data were used to assess the influence of termite colony backgrounds. Each of the 64 microarray profiles was treated as a vector and hierarchical clustering was performed, with single-link clustering being adopted based on Euclidian distances. Clustering results are shown in Fig. 2. Rather than treatment replicates clustering together, samples from each of 5 replicate colonies formed distinct clusters. Repeating this analysis as a principal component analysis (PCA) revealed the same trend (Fig. S2). These results indicate that colony effects contribute more to crude/absolute magnitudes of gene expression than does any individual treatment; as a result, normalized data were used for the analyses described hereafter (but non-normalized analyses were also done and are presented in Supplementary Figs. S4-S6).
Figure 2.
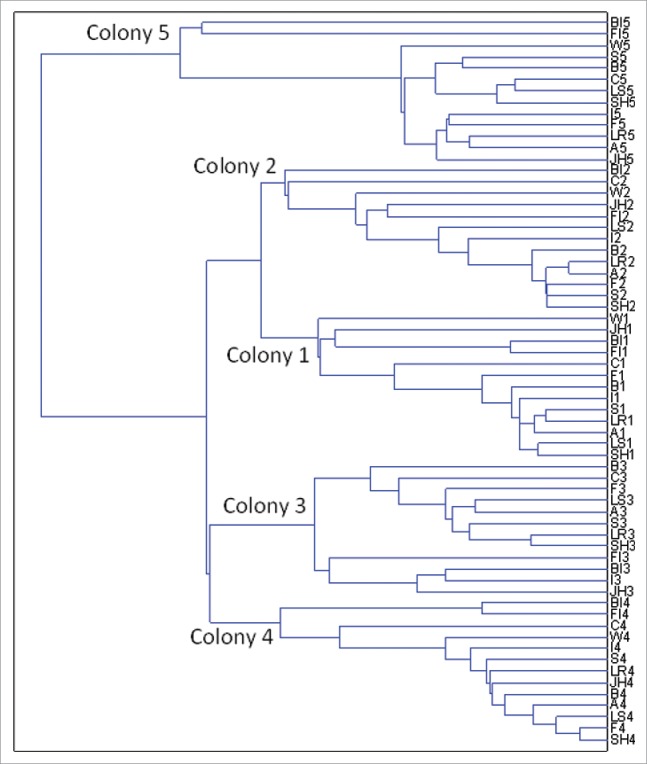
Cluster analysis results showing significant colony effects that supersede treatment effects when non-normalized data are used. See Figure 1 for treatment abbreviations and details.
Topology survey
A Euclidian distance-based topology survey was performed for the purpose of determining the similarity of global gene expression responses among treatments. To correct for colony effects, a matrix of pairwise comparisons was performed across treatments within each colony to compute Euclidean distances. Thus, there were 5 pairwise distances averaged for each pair of treatments compared (one from each colony with the exception of the W treatment; see above). The mean ± std deviation pairwise distance sets across all treatments are shown as a 13 × 13 distance matrix (Fig. S3). Next, by applying the ISOMAP algorithm14 on the obtained distance matrix, the relationships of the 13 treatments were projected into 3-dimensional space to reveal similarity relationships among treatments (Fig. 3 top). The topology can be summarized as follows: (i) the LS, LR, S, A, and SH treatments are closest to each other and form a core group; (ii) the F and I treatments are distinct but are closest to the core group; (iii) the B, BI, FI, W, JH, and C treatments are distant from the center; and (iv) the BI and FI treatments are furthest from all other treatments but closest to each other. Dividing the various controls and treatment groups and examining them in more detail provides additional topology resolution (Fig. 3a-d). Complementary analyses were also done for validation purposes, i.e., correlation distance, multidimensional scaling and PCA. Although different distance matrices were obtained from these analyses, topology relationships were still maintained (e.g., Fig. S3).
Figure 3.
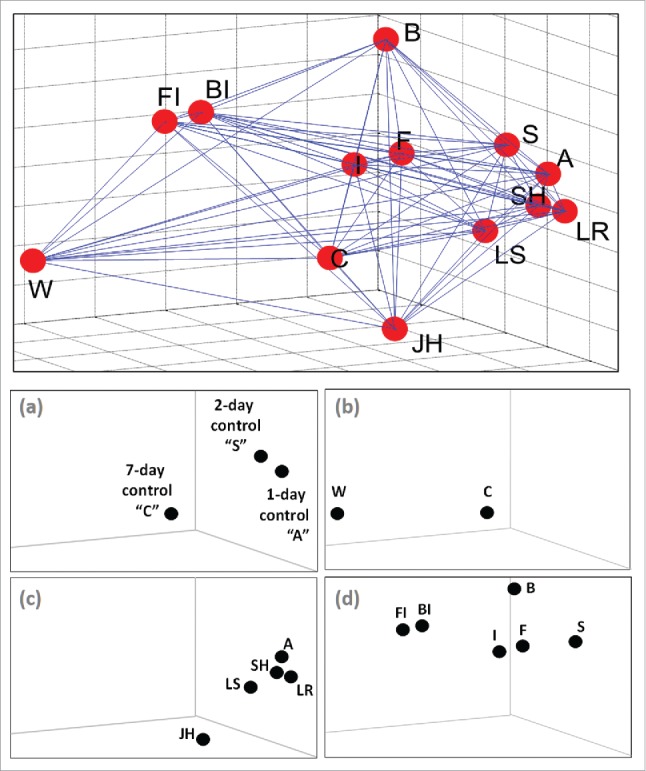
Topology profiles based on a Euclidian distance analyses of normalized microarray data. Top: A 3-dimensional plot of all data. Bottom: (a) control treatments, (b) diet/nutritional treatments, (c) hormonal/social treatments, (d) xenobiotic/immunological treatments. Abbreviations: S, DMSO solvent control; A, acetone control; C, cellulose paper; W, pine wood; JH, juvenile hormone; LS, live soldiers; SH, soldier head extract; LR, live reproductives; F, fungi; B, bacteria; I, imidacloprid; FI, fungi+imidacloprid; BI, bacteria+imidacloprid.
Co-option analyses based on common differentially expressed (DE) genes
To identify candidate major-effect genes potentially co-opted for key social functions, we looked for common DE genes shared among treatment categories. DE genes normalized by treatment relative to a common mixed reference (P < 0.05) were used for this analysis (Table 1). Specifically, we searched within and then between the nutrition-immunity and hormonal-xenobiotic treatment categories (Fig. 1). Treatments compared in the nutrition-immunity category were W, C, B, and F; in the hormonal-xenobiotic category, they were JH, I, FI, and BI (Fig. 4A).
Figure 4.
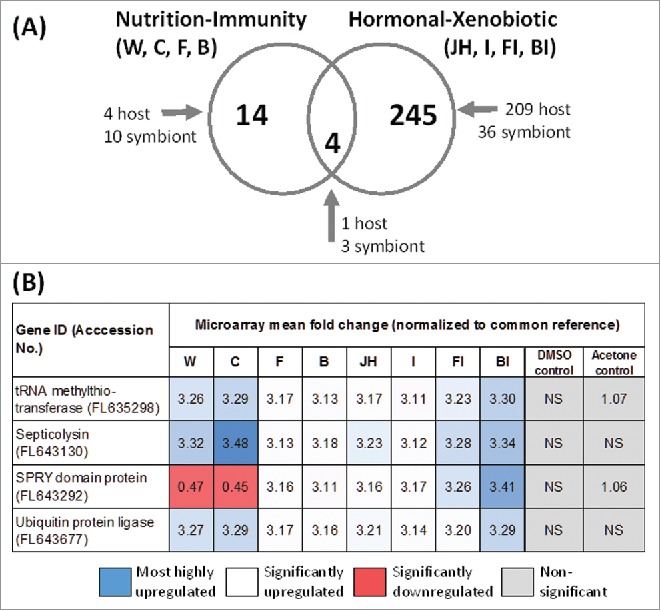
Identification of candidate co-opted genes. (A) Venn diagram showing numbers of shared passing genes in the nutrition-immunity (left) and hormonal-xenobiotic categories (right), and putative master genes shared among both categories (center), along with host or symbiont origins. See Fig. 1A for treatment abbreviations. (B) Identities and accession numbers of putative master genes along with fold change values across microarray treatments and controls relative to a common reference.
In the nutrition-immunity category, there were 14 common DE genes shared among treatments, of which 10 were from symbiont origins and 4 from host termite gut tissue (Table S1). By contrast, in the hormonal-xenobiotic category there were 245 common DE genes shared across treatments, of which the majority were of host origin (209 host and 36 symbiont) (Table S2). Many of the best- translated hits from these analyses are to Zootermopsis angusticollis genome sequences (Tables S2 and S3).
As a last step, the nutrition-immunity and hormonal-xenobiotic DE lists were merged to identify shared DE genes across all 8 treatment categories (Table S3, Fig. 4A). This analysis yielded 4 candidate “master” genes encoding proteins potentially co-opted for unique social functions: (i) a host tRNA methylthiotransferase, (ii) a symbiont septicolysin-like superfamily member, (iii) a symbiont SPRY superfamily member, and (iv) a symbiont ubiquitin protein ligase. Comparison of expression levels for these 4 genes across all nutrition-immunity and hormonal-xenobiotic categories reveals that all of the upregulated transcripts were inducible >3-fold (Fig. 4B). The only exception is the SPRY domain protein, which was significantly downregulated by more than 2-fold in the C and W treatments. In the 2 control treatments DMSO (S) and Acetone (A), all 4 genes either had non-significant expression changes, or when significant, only 1.06–1.07-fold upregulation.
Discussion
Overview
Termites are global pests of structures but also perform important ecosystem services in biomass recycling. Lower termites are distinguished by the immature status of their worker caste and the presence of protist and bacterial symbionts in their digestive tracts. Contributions of the symbiota to digestion and intermediary metabolism have been well studied, but intersecting contributions of host and gut symbiota to other, more ultimate processes have not been explored. We used an experimental metatranscriptome approach to analyze gene expression topologies across diverse challenge treatments and to look for “master” genes that responded across diverse treatment categories. Our assumption at the outset, based on prior work,17–19 was that such genes would be of broad functional significance rather than simply being unimportant or associated with adequate organismal function when expressed at any level.
Here we report on an unprecedented meta-analysis of 65 individual microarrays hybridized with host-gut and symbiont cDNAs obtained from the guts of non-reproductive, mostly-altruistic worker termites (Fig. 1). The focus species was the lower termite R. flavipes, which hosts gut symbiota that include a combination of 11 eukaryotic protists21 and >4,000 species level bacterial OTUs.15,16 Gut cDNAs were synthesized concurrently from total eukaryotic gut mRNA obtained after dietary, social, hormonal, immunological, or xenobiotic bioassay treatments as detailed previously.17–19 These prior studies elucidated details of responsive genes unique to each treatment category. The goal of the current study was to investigate for commonalities across treatment categories, and for potentially co-opted master genes. Thirteen treatments were compared that included feeding on wood or cellulose paper; exposures to juvenile hormone, soldier head extract, live soldiers, or live neotenic reproductives; and exposures to bacteria, fungi, imidacloprid, fungi+imidacloprid, or bacteria+imidacloprid. Acetone was included as the control for hormonal/social treatments and DMSO for immunological/xenobiotic treatments. Cellulose paper feeding was used as a control in a diet-focused study,17 but because of its distinct effects relative to the other controls (Fig. 3a), it was considered here as a treatment. Five independently replicated colonies were tested for each treatment, and only one microarray was excluded due to inconsistent signal intensity (treatment W, colony #3).
Due to significant intercolony variation in raw array signal intensities (Fig. 2), normalized data were used for both the topology and co-option analyses. There are several reasons why microarray signal values can require normalization, including unequal quantities of starting RNA, differences in labeling or detection efficiencies of fluorophores used, and/or systematic biases in measured expression levels.20 However, because in our case (i) only a single array was excluded due to inconsistent signal intensity and (ii) dye swaps were performed to correct for dye-associated variations in signal intensity, the chief factor contributing to signal variability across arrays was intercolony variation. Likely factors contributing to intercolony variation include different microbial profiles among the termite colonies used, impacts of local environment on bacterial microbiota composition, caste composition as influenced by colony semiochemicals, social structure as influenced by breeding structure, and/or colony genetics as influenced by founding pair genotypes.1,10,16,18
The topology and co-option analyses (discussed in the following sections) were done independently but provide complementary insights into co-option themes and “master” genes apparently co-evolved for broad functionality. The two analysis approaches were distinct; whereas the topology analysis considered coordinated responses of the entire meta-transcriptome, the co-option analysis considered individual responsive genes.
Topology
Topology results provide unprecedented insights into the molecular-level relatedness of various aspects of termite social living and associated physiologic processes. Treatments in this analysis segregated into several distinct clusters, namely (i) LS, LR, S, A, and SH; (ii) JH, C, I, F, and B; (iii) W; and (iv) FI and BI (Fig. 3 top). Dividing the various controls and treatment groups and examining them in more detail provided additional topology resolution (Fig. 3a-d). Among the various controls, which received identical cellulose diets but for differing lengths of time, more pronounced changes occurred as time passed (Fig. 3a). This temporal trend suggests effects on symbiont composition and/or gene expression by transitioning from large to small group living. In nutritional treatments (Fig. 3b), wood feeding had more substantial impacts than cellulose (paper), most likely due to the lignin/phenolic content of wood.19,22 Among the social and hormonal treatments (Fig. 3c), JH had the most pronounced impacts, whereas soldier head extracts and live soldiers were surprisingly distinct from each other, suggesting that additional non-extractable chemical cues were produced by soldiers.23 Finally, among the pathogen-xenobiotic interaction treatments (Fig. 3d), the bacterial treatment was most unique, while treatments of fungi and the xenobiotic imidacloprid were more similar. The dual B + I and F + I treatments were the most similar of all treatments, but at the same time, they had the most unique effects relative to all other treatments.
More generally, although some treatment groups were more distant from each other (e.g., W and B) the number of DE probes was generally similar across treatments. However, the composition of DE genes across treatments was more diverse, and therefore, there are few genes that are more represented in all samples likely due in part to sample heterogeneity. To the contrary, a larger number of gene probes were DE in the C treatment relative to other treatments such as S, SH, LS, and JH, indicating that the impact of cellulose feeding is consistent for all samples from different colonies. Because very few DE genes were shared among all treatments, this provided a unique opportunity to search for universally responsive co-option gene candidates that would be identifiable based on their shared significance across treatment categories.
Co-option
The co-option analysis was done by searching for common DE genes shared among the nutrition-immunity and hormonal-xenobiotic treatment categories (Fig. 4A). The rationale for investigating co-opted genes in the first category is the convergent carbohydrate structures between plant and microbial cell walls, i.e., β1–4 glucans in plants and β1–3 glucans in microbes.24 The rationale for including the FI and BI treatments in the hormonal-xenobiotic co-option category came from previous findings showing signatures of endocrine-linked gut remodeling in response to imidacloprid + pathogen challenges.19 Alternatively, the LS, SH, and LR treatments were excluded from the co-option analyses because of the relatively small number of significant DE genes present in these treatment categories (Fig. 3 and 18).
Contrary to conventional perceptions on the relative importance of digestion and immunity, the hormonal-xenobiotic data set shared 17.5x more responsive genes in common (n = 245) than did the nutrition-immunity data set (n = 14). In terms of host vs. symbiont origins for common DE genes, the relative proportions between the two data sets contrasted somewhat. The nutrition-immunity data set, although much smaller, had 2.5x more symbiont than host genes represented. Conversely, the hormonal-xenobiotic category had a 5.8x more host than symbiont genes represented. These trends suggest that eukaryotic gut symbiota might play a larger role in the interplay between nutrition and immune responses, whereas the host termite might play a larger role in mounting responses to hormonal cues and xenobiotic challenges.
One unique responsive gene shared across treatments in the nutrition-immunity category is a glycosyl hydrolase family 7 cellulase from the protist symbiont community. This enzyme family confers cellobiohydrolase and endoglucanase activity against cellulose polymers,25,26 and it was downregulated up to ∼1000x before the onset of mortality after lethal imidacloprid + fungal pathogen challenges.19 This enzyme family is absent from higher termite metagenomes and is poorly represented in the tri-partite symbiotic system associated with fungus-farming higher termites.27–28 A shared domain found in 3 of the 14 co-opted nutrition-immunity genes was an actin homolog NBD sugar kinase/HSP70/actin superfamily domain of unknown significance (Table S1). In the hormonal-xenobiotic data set, common domains appearing repeatedly included α-crystallin-type small heat shock protein, cytochrome C and P450, EF hand calcium signal modulator, glutamine amidotransferase, NABD Rossman metabolic dehydrogenase, peptidase, P-loop NTPase, ribosomal proteins, Thioredoxin-like superfamily, Toxin_37 superfamily antifungal peptide, and ubiquitin-associated proteins (among others; Table S2).
Finally, after merging the nutrition-immunity and hormonal-xenobiotic DE data sets, 4 universally responsive “master” genes emerged. The 4 genes were significantly homologous to tRNA methylthiotransferase, Septicolysin, SPRY domain protein, and Ubiquitin protein ligase (Table S3, Fig. 4B).
Although it has highest homology to a bacterial tRNA methylthiotransferase (TRMT), the TRMT gene is the only host-derived gene out of the 4 universally responsive master genes identified. TRMTs are involved in stress-response modifications of tRNAs (tRNAs) that impact the production of stress response proteins and ultimately, transcripts whose translation is influenced by “wobble base” tRNA modification.29 When human TRMT genes are mutated to produce a truncated TRMT protein, a syndrome of young-onset diabetes, short stature, microcephaly, and intellectual disability occurs. More significantly, complete TRMT silencing causes apoptosis of insulin-producing pancreatic β cells.30 These findings suggest the identified termite TRMT plays a central role in responses to stress, possibly with links to insulin signaling, which has documented links to termite caste differentiation and physiology.18,31–33
The second master gene encodes a symbiont-derived Septicolysin protein toxin. Although the range of functions for septicolysins are unknown, they are designated as cholesterol-dependent cytolysins. Septicolysins have been identified in multi-drug resistant pathogenic bacteria, where they aid in tissue and/or cell invasion by acting as pore-forming toxins.34,35 In the current study, septicolysin gene expression was upregulated across all 8 treatments compared. This consistent upregulation suggests interesting antimicrobial potential for the septicolysins in the gut microbial environment, particularly with respect to the dramatic microbial declines that occur in the R. flavipes gut after JH, dietary, xenobiotic, and pathogen challenges.17–19
The last 2 genes, SPRY domain protein and Ubiquitin protein ligase, both are symbiont-derived and are linked to proteasome-dependent protein degradation. First, the SPRY domain gene is unique among the 4 master genes in that it was the only one showing downregulation in any treatments, which occurred exclusively in wood and cellulose dietary/feeding treatments (it was upregulated along with the other 3 master genes in all other treatments; Fig. 4B). The name “SPRY” is derived based on homologous sequence repeats initially discovered in splA kinases and ryanodine receptors. Of these 2 families, ryanodine receptors are an important target site for diamide insecticides that are highly active against R. flavipes.36,37 In animals, SPRY domain proteins are activated by signaling factors in invading pathogenic microbes, before they bind to pattern-recognition receptors that include transmembrane Toll-like receptors38 (which were not DE in entomopathogen array treatments19). Downstream effects of SPRY activation include protein-ubiquitin conjugation, protein degradation, and ultimately, suppression of signaling. The last gene, Ubiquitin ligase, is a member of a protein family that conjugates ubiquitin residues onto proteins, targeting them for proteasomal degradation.39 Thus, it seems highly significant that the SPRY domain gene is upregulated in all hormone and pathogen challenge treatments, downregulated in the cellulose/lignocellulose dietary treatments, and apparently works in synchrony with ubiquitin ligase to achieve protein degradation. These expression profiles seem particularly relevant when considering the microbe-rich environment of the termite gut, where substantial protist disappearance occurs following some challenge treatments and during the caste differentiation process, but not in association with feeding.17–19,40
Conclusions
With its meta-analysis of 64 individual microarrays and comparison of 13 individual challenge treatments, the depth and breadth of this study are unprecedented in termite research. Four outcomes emerged that give new glimpses into termite ecology, sociality and symbiosis. The first was the unexpected level of intercolony variation that was observed, most likely due to significant environment-associated microbiota differences that existed among the study colonies.16 Second, an unprecedented topology analysis revealed new insights into the molecular-level relatedness of various aspects of termite social living and underlying physiologic processes. In particular, the topology analysis revealed broad differences in gut metatranscriptome composition after hormonal, social, dietary, pathogen, and xenobiotic challenges. Third, despite the varying metatranscriptome compositions identified through the topology analysis, the co-option analysis revealed 4 apparent master genes that were universally responsive across diverse treatment categories. Of the 4 master genes identified, it is possible that the symbiont SPRY domain gene (with homology to microbial-responsive antimicrobial genes of solitary organisms) is a legitimate co-option candidate and could play a central role in regulating gut symbiont numbers and/or composition, as well as host processes.
Another trend emerging from the co-option analysis is that gut symbiota seem to play a larger role in the interplay between nutrition and immunity functions, while the host termite plays a larger role in mounting responses to hormonal and xenobiotic challenges. This trend is logical when considering that plant feeding and microbial ecology/competition are evolutionarily distinct processes as compared with processes in higher organisms such as xenobiotic defense and hormone signaling. Lastly, many of the best-translated hits from the various co-option analyses are to Zootermopsis angusticollis genome sequences (ref. 41; Tables S2 and S3), which provides new annotations for many previously unannotated or weakly annotated termite genes. These findings thus reveal new insights into host-symbiont interactions that underlie various aspects of termite biology as well as important co-opted genes that direct socially relevant biochemical and physiologic processes in the termite gut. The identified responsive genes and networks identified will be excellent targets for termite-inspired and termite-targeted biotechnology,42 as well as functional sociogenomic research.12 These findings are also consistent with earlier research findings showing trends for increased complexity of gene networks in association with increasingly eusocial lifestyles,11 as well as altruistic adaptations for group living in unicellular organisms.7
Methods
Details of experiments are repeated here from preceding reports.17–19
Termites
Termites used for microarrays originated from 5 established laboratory colonies at the University of Florida, Entomology and Nematology Department, in Gainesville, FL: (1) B1#1 (established 05/20/2009); (2) B2 (06/03/2010); (3) K2 (07/11/2007); (4) K5 (08/02/2008); and (5) K9 (06/29/2010). All colonies were verified as R. flavipes by mitochondrial 16s rRNA sequencing and soldier morphology. Colonies were maintained in darkness in sealed plastic boxes containing moist pine wood shims and brown paper towels, within an environmental chamber kept at 22°C and 60–100% relative humidity (RH). Bioassays were conducted in darkness at 27°C and 60–100% RH.
Microarray design
Custom cDNA oligonucleotide microarrays were designed using the eARRAY platform and printed on glass slides in 8×15,000 formats (Agilent Technologies, Santa Clara, CA). Each array position contained an identical pool of 60-base oligonucleotides that represented longer ESTs from gut and symbiont, as well as whole-body EST and gene sequences from R. flavipes. In particular, Tartar et al.,43 previously sequenced from the transcriptomes of host gut and protist symbiont fractions, separately, to yield 2 EST libraries enriched for termite/host specific transcripts (5,872 sequences) and symbiont/protist transcripts (4,739 sequences). Along with these 2 libraries were added whole-body library ESTs, including 93 randomly sequenced transcripts,44 88 worker- or soldier-biased transcripts,45 96 reproductive-biased transcripts,46 and 91 presoldier-biased transcripts47 Additionally, 11 immune and neuropeptide-coding genes/transcripts were included (refs. 48–49 and Xu & Chen, unpublished; Genbank Accession Nos. FJ184563, FJ184567, FJ184572, FJ184577, FJ184582, FJ184587, FJ184591, FJ184596 and FJ184600). All of the above array positions were printed with a 3′ bias. Additionally, 3,960 eukaryotic gut-symbiont ESTs were also printed with 5′ bias to enhance gene discovery from the protist symbiont EST pools. Finally, in addition to the entire pool of 14,950 60-mer oligonucleotides noted above, ca. 50 positive and negative control spots (Agilent) were also printed on the arrays. In total, 10,990 unique termite and symbiont gene and EST sequences were represented on arrays.
Bioassay treatments
Bioassays for microarray studies were performed in August–September, 2010. With the exception of live soldier and neotenic treatments, bioassays included 20 worker termites exclusively. All assays were conducted in 3.5-cm diameter Petri dishes in darkness at 27°C and 60–100% RH. Dietary treatments consisted of pine wood (i.e., complex lignocellulose) or 98% pure cellulose paper and lasted for 7 d.17 Hormonal bioassays lasted 24-hr and included cellulose paper substrates treated with either JH III, SHE (soldier head extracts), or 150 μl acetone as a control.18 JH III (93% purity; Sigma; St. Louis, MO, USA) was applied on one filter paper disk per assay and at 150 μg per disk in 150 μl acetone. SHE was prepared by homogenizing 10 soldier heads in 1.5-mL acetone with a Tenbroeck glass homogenizer and applied at 2 soldier head equivalents per disk in 150 μl acetone. Social treatments lasted 24-hr and consisted of 20 workers held with either 2 soldiers or 2 neotenic reproductives originating from the same colony, and were provided filter paper discs as food. Xenobiotic and immunological treatments lasted 48-hr and included the insecticide imidacloprid, the pathogenic fungus Metharizium anisopliae, the pathogenic bacteria Serratia marcescens, paired treatments of imidacloprid + fungus or imidacloprid + bacteria, and the solvent carrier DMSO as a control.19 For fungal treatments, M. anisopliae spores (isolate Ma1630) were collected from in vitro cultures 10–12 d after inoculation and diluted with water to a final concentration of 105 spores/ml. Termites were exposed to fungi by placing them in a steel mesh specimen basket (16×8 mm) and submersing in 5 ml of spore suspension for 20-sec before draining excess liquid and placing in assay dishes. Termites were exposed to S. marcescens cells (isolate “New Zealand May 18”) that were harvested from liquid cultures by centrifugation. Cells were suspended in sterilized saline, and cell concentration adjusted to 6.5×109 cells/ml before 150µl was applied filter paper assay discs (final dose = 2.35 × 108 cells/cm2). For insecticide treatments, filter paper discs were treated with a 0.0001% aqueous solution of imidacloprid (97.5% purity, Bayer, Pittsburgh, PA; initially dissolved at 1% w/v in dimethyl sulfoxide, DMSO) and allowed to air-dry before use. Termites in dual treatments were exposed to imidacloprid plus either M. anisopliae or S. marcescens as detailed above. Discs for control treatments were pretreated with 0.0001% aqueous DMSO and moistened with 150 µl saline.
RNA isolation and microarray hybridizations
After bioassays termites were removed from treatment dishes, cold-immobilized, surface-sterilized by a serial rinse in 0.3% sodium hypochlorite (1x) and sterilized water (2x), and dissected on Parafilm® to collect digestive tracts, including salivary glands. Digestive tracts were transferred into RLA Lysis Buffer (Promega, Fitchburg, WI, USA) and stored at −70°C until RNA isolation. RNA extraction from whole guts using the SV Total RNA Isolation kit (Promega, Fitchburg, WI, USA) following the manufacturer protocol. RNA quantity and quality were assessed using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). cDNA for hybridizing to microarrays was synthesized from total gut RNA using the RNA input linear amplification kit (Agilent Technologies, Santa Clara, CA).
A type II microarray design was used with a common reference strategy.50–51 The common reference consisted of a normalized blend of all RNA samples included in the experiment. This common reference was co-hybridized against each replicate sample on single microarrays. Dye swaps.46,47 were performed between replicate samples and references to check for potential dye impacts on spot intensity.50–51 Sixty-five total microarray hybridizations were performed, which consisted of 5 colonies each exposed to the treatments detailed above. Microarray data are provided as supporting information in Tables S4–S16. These data are summarized by array position for each treatment, normalized to mixed reference hybridizations, and include negative and positive controls. GenBank accession numbers for sequences at each microarray position are provided in Tables S17–S18 and references.43–49
Validation of microarray results by quantitative real-time PCR
Fold-change data from microarrays were validated by performing sets of Quantitative Real-Time PCRs (qRT-PCR) with a CFX-96 Real-time System (Bio-Rad, Hercules, CA) using the SYBR green detection method (SensiMix SYBR & Fluorescein one-step PCR reagent; Bioline; Taunton, MA). cDNA templates for qPCR validations were synthesized using the iScript cDNA kit (Bio-Rad; Hercules, CA) from original microarray RNA samples according to manufacturer instructions. As detailed in preceding reports,17–19 microarray results were independently verified in all instances by regressing microarray fold change data (x-axis) against qRT-PCR fold change data (y-axis).
First-pass bioinformatic analyses
Because individual termite transcripts could be represented by ESTs at multiple array positions, contigs were generated within each treatment category (i.e., dietary, hormonal/social and xenobiotic/immunological) from sequence pools of significantly differentially expressed array positions. All sequences corresponding to array positions having +/− 1.2-fold change and p-values <0.05 were processed using Sequencher (Gene Codes Corporation, Ann Arbor, MI) with a 95% minimum match. The poly-A tail and the poly-G cap (an artifact cDNA Library Construction43) were removed before assembly, and all generated contigs were manually checked for artifacts. The generated contigs and the remaining orphan sequences were used for further analyses. The selected contigs and orphan sequences were analyzed using the program BLAST2GO for identification and annotation.52 By using the inbuilt BLASTx algorithm, these sequences were used as queries in BLASTx searches against the GenBank non-redundant (nr) database with an e-value cut-off of ≤ 1e-03.
Statistical analysis
The Matlab bioinformatics and statistics toolboxes (MathWorks, Natick, MA) were used for statistical analysis of microarray intensity data of 64 samples with 13 different treatments. Before comparative analysis, the individual signal intensity values obtained from the microarray probes were log-transformed (using 2 as the base) and normalized between all individual samples included in the study. We performed normalization by scaling the individual log-transformed signal intensities so that each data set had comparable lower, median and upper quartile values.20 After data normalization, Student's t-tests were performed considering a probe-by-probe comparison between pairs of comparison groups. In each comparison, a p-value and fold change were computed for each gene locus. In addition to p-values, q-values also were computed. While the p-value measures the minimum statistical false positive rate incurred when setting a threshold for test significance, the q-value measures the minimum false discovery rate incurred when calling that test significant.13
Hierarchical cluster analysis and ISOMAP
A hierarchical cluster analysis was performed to assess the impacts of treatments and colonies on overall gene expression values. Euclidean distances of each pair of samples were computed to measure their similarities. The pairwise distances were then fed into the single-linkage-based hierarchical clustering algorithm to generate a dendrogram plot. We also performed an analysis using the ISOMAP algorithm14 to evaluate how different treatments affected gene expression values of the samples. First, the 64 samples were partitioned into 13 subgroups based on the treatments they received. Then, the Euclidean distances of a pair of samples from 2 groups were calculated and averaged to represent the pairwise distance between 2 groups. ISOMAP was applied to the obtained 13×13 pairwise distance matrix to project the data into a 3-dimensional space to visualize the impact of different treatments on the gene expression values of the samples.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Funding was provided by USDA-NIFA-AFRI grants 2009–05245 and 2010–65106–30727 to MES and DGB, by NSF grant 1233484CBET to MES, and by the O.W. Rollins/Orkin Endowment in the Department of Entomology at Purdue University.
References
- [1].Korb J. Juvenile hormone: a central regulator of termite caste polyphenism, In: Advances in Insect Physiology (2015) Vol. 48, pp. 131-161. Ed. by Zayed A, Kent CF. Academic Press, Oxford. [Google Scholar]
- [2].Brune A. Symbiotic digestion of lignocellulose in termite guts. Nat Rev Microbiol 2014; 12:168-80; PMID:24487819; http://dx.doi.org/ 10.1038/nrmicro3182 [DOI] [PubMed] [Google Scholar]
- [3].Rosengaus RB, Traniello JFA, Bulmer MS. Ecology, behavior and evolution of disease resistance in termites In, Biology of termites: a modern synthesis, (2011) pp 165-191. Ed. by Bignell DE, Roisin Y, Lo N. Springer, Dordrecht. [Google Scholar]
- [4].Peterson BF, Scharf ME. Lower termite associations with microbes: synergy, protection, and interplay. Front Microbiol. 2016; 7:422; PMID:27092110; http://dx.doi.org/ 10.3389/fmicb.2016.00422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].West-Eberhard MJ. Developmental Plasticity and Evolution (2003). Oxford Univ. Press, Oxford. [Google Scholar]
- [6].Toth A, Robinson GE. Evo-devo and the evolution of social behavior. Trends Genet 2007; 23:334-41; PMID:17509723; http://dx.doi.org/ 10.1016/j.tig.2007.05.001 [DOI] [PubMed] [Google Scholar]
- [7].Nedelcu AM. Environmentally induced responses co-opted for reproductive altruism. Biol lett 2009; 5:805-808; PMID:19578098; http://dx.doi.org/ 10.1098/rsbl.2009.0334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nelson CM, Ihle KE, Fondrk MK, Page RE, Amdam GV. The gene vitellogenin has multiple coordinating effects on social organization. PLoS Biol 2007; 5:e62; PMID:17341131; http://dx.doi.org/ 10.1371/journal.pbio.0050062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wheeler MM. Brain gene expression changes elicited by peripheral vitellogenin knockdown in the honey bee et al. Insect Mol Biol 2013; 22:562-73; PMID:23889463; http://dx.doi.org/ 10.1111/imb.12043 [DOI] [PubMed] [Google Scholar]
- [10].Miura T, Scharf ME. Molecular basis underlying caste differentiation in termites In: Biology of Termites: A Modern Synthesis, (2011) pp. 211-253. Ed. by Bignell DE, Roisin Y, Lo N. Springer, Dordrecht. [Google Scholar]
- [11].Kapheim KM, Pan H, Li C, Salzberg SL, Puiu D, Magoc T, Robertson HM, Hudson ME, Venkat A, Fischman BJ, et al.. Genomic signatures of evolutionary transitions from solitary to group living. Science 2015; 384:1139-43; http://dx.doi.org/ 10.1126/science.aaa4788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Scharf ME. Omics research in termites: an overview and a roadmap. Front Genet 2015. 6:76; PMID:25821456; http://dx.doi.org/ 10.3389/fgene.2015.00076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Storey JB. A direct approach to false discovery rates. J R Stat Soc Series B Stat Methodol 2002; 64:479-98; http://dx.doi.org/ 10.1111/1467-9868.00346 [DOI] [Google Scholar]
- [14].Tenenbaum JB, de Silva V, Langford JC. A global geometric framework for nonlinear dimensionality reduction. Science 2000; 290:2319-23; PMID:11125149; http://dx.doi.org/ 10.1126/science.290.5500.2319 [DOI] [PubMed] [Google Scholar]
- [15].Lewis JH, Forschler BT. Protist communities from four castes and three species of Reticulitermes. Ann Entomol Soc Am 2004; 97:1242-51; http://dx.doi.org/ 10.1603/0013-8746(2004)097[1242:PCFFCA]2.0.CO;2 [DOI] [Google Scholar]
- [16].Boucias DG, Cai Y, Sun Y, Lietze VU, Sen R, Raychoudhury R, Scharf ME. The hindgut lumen prokaryotic microbiota of the termite Reticulitermes flavipes and its responses to dietary lignocellulose composition. Mol Ecol 2013; 22:1836-1853; PMID:23379767; http://dx.doi.org/ 10.1111/mec.12230 [DOI] [PubMed] [Google Scholar]
- [17].Raychoudhury R, Sen R, Cai Y, Sun Y, Lietze VU, Boucias DG, Scharf ME. Comparative metatranscriptomic signatures of wood and paper feeding in the gut of the termite Reticulitermes flavipes. Insect Mol Biol 2013; 22:155-71; PMID:23294456; http://dx.doi.org/ 10.1111/imb.12011 [DOI] [PubMed] [Google Scholar]
- [18].Sen R, Raychoudhury R, Cai Y, Sun Y, Lietze VU, Boucias DG, Scharf ME. Differential impacts of juvenile hormone, soldier head extract and alternate caste phenotypes on host and symbiont transcriptome composition in the gut of the termite Reticulitermes flavipes. BMC Genomics 2013; 14:491; PMID:23870282; http://dx.doi.org/ 10.1186/1471-2164-14-491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sen R, Raychoudhury R, Cai Y, Sun Y, Lietze VU, Peterson BF, Scharf ME, Boucias DG. Molecular signatures of nicotinoid-pathogen synergy in the termite gut. PLoS One 2015; 10(4):e0123391; PMID:25837376; http://dx.doi.org/ 10.1371/journal.pone.0123391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Quackenbush J. Microarray data normalization and transformation. Nat Genet Suppl 2002; 32:496-501; http://dx.doi.org/ 10.1038/ng1032 [DOI] [PubMed] [Google Scholar]
- [21].Tai V, James ER, Nalepa CA, Scheffrahn RH, Perlman SJ, Keeling PJ. The role of host phylogeny varies in shaping microbial diversity in the hindguts of lower termites. Appl Environ Microbiol 2015; 81:1059-70; PMID:25452280; http://dx.doi.org/ 10.1128/AEM.02945-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sethi A, Slack JM, Kovaleva ES, Buchman GW, Scharf ME. Lignin-associated metagene expression in a lignocellulose-digesting termite. Insect Biochem Mol Biol 2013; 43:91-101; PMID:23108206; http://dx.doi.org/ 10.1016/j.ibmb.2012.10.001 [DOI] [PubMed] [Google Scholar]
- [23].Tarver MR, Schmelz EA, Scharf ME. Soldier caste influences on candidate primer pheromone levels and juvenile hormone-dependent caste differentiation in workers of the termite Reticulitermes flavipes. J Insect Physiol 2011; 57:771-7; PMID:21356212; http://dx.doi.org/ 10.1016/j.jinsphys.2011.02.015 [DOI] [PubMed] [Google Scholar]
- [24].Pauchet Y, Freitak D, Heidel-Fischer HM, Heckel DG, Vogel H. Immunity or digestion: glucanase activity in a glucan-binding protein family from Lepidoptera. J Biol Chem 2009; 284:2214-24; PMID:19033442; http://dx.doi.org/ 10.1074/jbc.M806204200 [DOI] [PubMed] [Google Scholar]
- [25].Sethi A, Kovaleva ES, Slack JM, Brown S, Buchman GW, Scharf ME. A GHF7 cellulase from the protist symbiont community of Reticulitermes flavipes enables more efficient lignocellulose processing by host enzymes. Arch Insect Biochem Physiol 2013; 84:175-93; PMID:24186432; http://dx.doi.org/ 10.1002/arch.21135 [DOI] [PubMed] [Google Scholar]
- [26].Todaka N, Moriya S, Saita K, Hondo T, Kiuchi I, Takasu H, Ohkuma M, Piero C, Hayashizaki Y, Kudo T. Environmental cDNA analysis of the genes involved in lignocellulose digestion in the symbiotic protist community of Reticulitermes speratus. FEMS Microbiol Ecol 2007; 59:592-9; PMID:17239084; http://dx.doi.org/ 10.1111/j.1574-6941.2006.00237.x [DOI] [PubMed] [Google Scholar]
- [27].Warnecke F, Luginbühl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, et al.. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 2007; 450:560-5; PMID:18033299; http://dx.doi.org/ 10.1038/nature06269 [DOI] [PubMed] [Google Scholar]
- [28].Poulsen M, Hu H, Li C, Chen Z, Xu L, Otani S, Nygaard S, Nobre T, Klaubauf S, Schindler PM, et al.. Complementary symbiont contributions to plant decomposition in a fungus-farming termite. Proc Natl Acad Sci U S A 2014; 111:14500-5; PMID:25246537; http://dx.doi.org/ 10.1073/pnas.1319718111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Endres L, Dedon PC, Begley TJ. Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol 2015; 12:603-14; PMID:25892531; http://dx.doi.org/ 10.1080/15476286.2015.1031947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Igoillo-Esteve M, Genin A, Lambert N, Désir J, Pirson I, Abdulkarim B, Simonis N, Drielsma A, Marselli L, Marchetti P, et al.. tRNA methyltransferase homolog gene TRMT10A mutation in young onset diabetes and primary microcephaly in humans. PLoS Genet 2013; 9(10):e1003888; PMID:24204302; http://dx.doi.org/ 10.1371/journal.pgen.1003888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hattori A, Sugime Y, Sasa C, Miyakawa H, Ishikawa Y, Miyazaki S, Okada Y, Cornette R, Lavine LC, Emlen DJ, et al.. Soldier morphogenesis in the damp-wood termite is regulated by the insulin signaling pathway. J Exp Zool B Mol Dev Evol 2013; 320:295-306; PMID:23703784; http://dx.doi.org/ 10.1002/jez.b.22501 [DOI] [PubMed] [Google Scholar]
- [32].Lavine L, Gotoh H, Brent CS, Dworkin I, Emlen DJ. Exaggerated trait growth in insects. Annu Rev Entomol 2015; 60:453-72; PMID:25341090; http://dx.doi.org/ 10.1146/annurev-ento-010814-021045 [DOI] [PubMed] [Google Scholar]
- [33].Christie AE. In silico prediction of a neuropeptidome for the eusocial insect Mastotermes darwiniensis. Gen Comp Endocrinol 2015; 224:69-83; PMID:26095226; http://dx.doi.org/ 10.1016/j.ygcen.2015.06.006 [DOI] [PubMed] [Google Scholar]
- [34].Rosado CJ, Kondos S, Bull TE, Kuiper MJ, Law RH, Buckle AM, Voskoboinik I, Bird PI, Trapani JA, Whis stock JC, et al.. The MACPF/CDC family of pore-forming toxins. Cell Microbiol 2008; 10:1765-74; PMID:18564372;http://dx.doi.org/ 10.1111/j.1462-5822.2008.01191.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Acosta J, Merino M, Viedma E, Poza M, Sanz F, Otero JR, Chaves F, Bou G. Multidrug-resistant Acinetobacter baumannii Harboring OXA-24 carbapenemase, Spain. Emerg Infect Dis 2011; 17:1064-7; PMID:21749771; http://dx.doi.org/ 10.3201/eid/1706.091866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wan PJ, Guo WY, Yang Y, Lü FG, Lu WP, Li GQ. RNAi suppression of the ryanodine receptor gene results in decreased susceptibility to chlorantraniliprole in Colorado potato beetle Leptinotarsa decemlineata. J Insect Physiol 2014; 63:48-55; PMID:24607641; http://dx.doi.org/ 10.1016/j.jinsphys.2014.02.009 [DOI] [PubMed] [Google Scholar]
- [37].Buczkowski G, Scherer CW, Bennett GW. Toxicity and horizontal transfer of chlorantraniliprole in the eastern subterranean termite. J Econ Entomol 2012; 105:1736-45; PMID:23156171; http://dx.doi.org/ 10.1603/EC12038 [DOI] [PubMed] [Google Scholar]
- [38].D'Cruz AA, Babon JJ, Norton RS, Nicola NA, Nicholson SE. Structure and function of the SPRY/B30.2 domain proteins involved in innate immunity. Protein Sci 2013; 22:1-10; PMID:23139046; http://dx.doi.org/ 10.1002/pro.2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wilson VG, Heaton PR. Ubiquitin proteolytic system: focus on SUMO. Expert Rev Proteomics 2008; 5:121-35; PMID:18282128;http://dx.doi.org/ 10.1586/14789450.5.1.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brugerolle G, Radek R. Symbiotic protozoa of termites (2006) pp 243-269. In, Soil Biology vol 6, Ed. by Varma A, Konig H. Springer-Verlag, Berlin. [Google Scholar]
- [41].Terrapon N, Li C, Robertson HM, Ji L, Meng X, Booth W, Chen Z, Childers CP, Glastad KM, Gokhale K, et al.. Molecular traces of alternative social organization in a termite genome. Nat Commun 2014; 5:3636; PMID:24845553; http://dx.doi.org/ 10.1038/ncomms4636 [DOI] [PubMed] [Google Scholar]
- [42].Scharf ME. Termites as targets and models for biotechnology. Annu Rev Entomol 2015; 60:77-102; PMID:25341102; http://dx.doi.org/ 10.1146/annurev-ento-010814-020902 [DOI] [PubMed] [Google Scholar]
- [43].Tartar A, Wheeler MM, Zhou X, Coy MR, Boucias DG, Scharf ME. Parallel metatranscriptome analyses of host and symbiont gene expression in the gut of the termite Reticulitermes flavipes. Biotechnol Biofuels 2009; 2:25; ; http://dx.doi.org/ 10.1186/1754-6834-2-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wu-Scharf D, Scharf ME, Pittendrigh BR, Bennett GW. Expressed sequence tags from a polyphenic Reticulitermes flavipes cDNA library. Sociobiology 2003; 41:479-90. [Google Scholar]
- [45].Scharf ME, Wu-Scharf D, Pittendrigh BR, Bennett GW. Caste- and development-associated gene expression in a lower termite. Genome Biol 2003; 4:10; http://dx.doi.org/ 10.1186/gb-2003-4-10-r62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Scharf ME, Wu-Scharf D, Zhou X, Pittendrigh BR, Bennett GW. Gene expression profiles among immature and adult reproductive castes of the termite Reticulitermes flavipes. Insect Mol Biol 2005; 14:31-44; PMID:15663773; http://dx.doi.org/ 10.1111/j.1365-2583.2004.00527.x [DOI] [PubMed] [Google Scholar]
- [47].Zhou X, Song C, Grzymala TL, Oi FM, Scharf ME. Juvenile hormone and colony conditions differentially influence cytochrome P450 gene expression in the termite Reticulitermes flavipes. Insect Molec Biol 2006; 15:749-61; http://dx.doi.org/ 10.1111/j.1365-2583.2006.00675.x [DOI] [PubMed] [Google Scholar]
- [48].Elliott KL, Hehman GL, Stay B. Isolation of the gene for the precursor of Phe-Gly-Leu-amide allatostatins in the termite Reticulitermes flavipes. Peptides 2009; 30:855-60; http://dx.doi.org/ 10.1016/j.peptides.2009.01.009 [DOI] [PubMed] [Google Scholar]
- [49].Nuss AB, Forschler BT, Crim JW, TeBrugge V, Pohl J, Brown MR. Molecular characterization of neuropeptide F from the eastern subterranean termite Reticulitermes flavipes. Peptides 2010; 31:419-28; http://dx.doi.org/ 10.1016/j.peptides.2009.09.001 [DOI] [PubMed] [Google Scholar]
- [50].Gibson G, Muse SV. A primer of genome science. Sunderland: Sinauer; (2002). [Google Scholar]
- [51].Eisen MB, Brown PO. DNA arrays for analysis of gene expression. Meth Enzymol 1999; 303:179-205; PMID:10349646 [DOI] [PubMed] [Google Scholar]
- [52].Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al.. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25: 25-29; PMID:10802651; http://dx.doi.org/ 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.