

Abstract
Key points
Gap junction channels are essential for the formation and regulation of physiological units in tissues by allowing the lateral cell‐to‐cell diffusion of ions, metabolites and second messengers.
Stimulation of the adenosine receptor subtype A2B increases the gap junction coupling in the human blood–brain barrier endothelial cell line hCMEC/D3.
Although the increased gap junction coupling is cAMP‐dependent, neither the protein kinase A nor the exchange protein directly activated by cAMP were involved in this increase.
We found that cAMP activates cyclic nucleotide‐gated (CNG) channels and thereby induces a Ca2+ influx, which leads to the increase in gap junction coupling.
The report identifies CNG channels as a possible physiological link between adenosine receptors and the regulation of gap junction channels in endothelial cells of the blood–brain barrier.
Abstract
The human cerebral microvascular endothelial cell line hCMEC/D3 was used to characterize the physiological link between adenosine receptors and the gap junction coupling in endothelial cells of the blood–brain barrier. Expressed adenosine receptor subtypes and connexin (Cx) isoforms were identified by RT‐PCR. Scrape loading/dye transfer was used to evaluate the impact of the A2A and A2B adenosine receptor subtype agonist 2‐phenylaminoadenosine (2‐PAA) on the gap junction coupling. We found that 2‐PAA stimulated cAMP synthesis and enhanced gap junction coupling in a concentration‐dependent manner. This enhancement was accompanied by an increase in gap junction plaques formed by Cx43. Inhibition of protein kinase A did not affect the 2‐PAA‐related enhancement of gap junction coupling. In contrast, the cyclic nucleotide‐gated (CNG) channel inhibitor l‐cis‐diltiazem, as well as the chelation of intracellular Ca2+ with BAPTA, or the absence of external Ca2+, suppressed the 2‐PAA‐related enhancement of gap junction coupling. Moreover, we observed a 2‐PAA‐dependent activation of CNG channels by a combination of electrophysiology and pharmacology. In conclusion, the stimulation of adenosine receptors in hCMEC/D3 cells induces a Ca2+ influx by opening CNG channels in a cAMP‐dependent manner. Ca2+ in turn induces the formation of new gap junction plaques and a consecutive sustained enhancement of gap junction coupling. The report identifies CNG channels as a physiological link that integrates gap junction coupling into the adenosine receptor‐dependent signalling of endothelial cells of the blood–brain barrier.
Keywords: adenosine receptor, blood‐brain barrier, Ca2+ influx, CNG channel, cyclic adenosine monophosphate (cAMP), endothelial cell, gap junction
Key points
Gap junction channels are essential for the formation and regulation of physiological units in tissues by allowing the lateral cell‐to‐cell diffusion of ions, metabolites and second messengers.
Stimulation of the adenosine receptor subtype A2B increases the gap junction coupling in the human blood–brain barrier endothelial cell line hCMEC/D3.
Although the increased gap junction coupling is cAMP‐dependent, neither the protein kinase A nor the exchange protein directly activated by cAMP were involved in this increase.
We found that cAMP activates cyclic nucleotide‐gated (CNG) channels and thereby induces a Ca2+ influx, which leads to the increase in gap junction coupling.
The report identifies CNG channels as a possible physiological link between adenosine receptors and the regulation of gap junction channels in endothelial cells of the blood–brain barrier.
Abbreviations
- 2‐PAA
2‐phenylaminoadenosine
- BBB
blood–brain barrier
- CNG
cyclic nucleotide‐gated
- CNS
central nervous system
- Cx
connexin
- db‐cGMP
dibutyryl‐cGMP
- DPCPX
8‐cyclopentyl‐1,3‐dipropylxanthine
- Epac
exchange protein directly activated by cAMP
- NECA
5ʹ‐N‐ethylcarboxamidoadenosine
- NMDG
N‐methyl‐d‐glucamine
- PKA
protein kinase A
- STED
stimulated emission depletion
- TEER
transendothelial electrical resistance
Introduction
Gap junctions are intercellular channels that connect the cytoplasm of adjacent cells. They provide the possibility to exchange ions and small molecules (< 1.5 kDa) such as metabolites or second messengers between neighbouring cells. Gap junctions consist of two hemi‐channels, the so‐called connexons, each comprising six protein subunits, the connexins (Cxs). So far, 21 genes for different Cx isoforms have been identified in humans (Söhl & Willecke 2004; Beyer & Berthoud 2009). They are expressed in a tissue‐specific manner and are named according to their molecular weight (e.g. Cx43 has a molecular weight of 43 kDa; Söhl & Willecke 2004). Of these isoforms Cx37, Cx40, Cx43 and to a lesser extent Cx45 are commonly found in cells of the vascular wall (Haefliger et al. 2004; Johnstone et al. 2009; Figueroa & Duling 2009). They form gap junctions that mainly participate in the regulation of blood pressure and blood flow (Haefliger et al. 2004; Johnstone et al. 2009; Figueroa & Duling 2009).
In cerebral endothelial cells of different species, the expression of Cx37, Cx40, Cx43 and Cx45 has been shown (Nagasawa et al. 2006; Avila et al. 2011; Bock et al. 2012; Kaneko et al. 2015). At the functional level, Cxs were found to be closely associated with tight junction proteins in porcine blood–brain barrier (BBB) endothelial cells. It was also observed that chemical agents that close gap junctions such as 18β‐glycyrrhetinic acid affected the barrier function of these cells (Nagasawa et al. 2006). The association of Cxs with the endothelial tight junctions, which constitute the morphological basis of the barrier function of the BBB, suggests possible regulatory interactions as well as a co‐regulation of gap junction coupling and tight junctions by signalling mechanisms in BBB endothelial cells.
One of the signalling mechanisms involved in the physiology of the BBB is adenosine‐stimulated signalling. Adenosine is a metabolite ubiquitously present in tissues (Fredholm 2007; Blackburn et al. 2009). As an extracellular mediator, adenosine is produced by rapid (< 1ms) conversion of released adenine nucleotides like ATP or ADP through a series of ectonucleotidases such as CD73 and CD39 (Fredholm 2007; Eltzschig 2009).
Under pathophysiological conditions such as ischaemia, hypoxia or inflammation, external adenosine achieves high (100‐fold) and sustained concentrations, stimulating various cells in large portions of organs such as the brain (Sands & Palmer 2005; Fredholm 2007; Eltzschig 2009; Blackburn et al. 2009; Melani et al. 2014). Local increases of external adenosine by transient adenosine nucleotide release have also been observed in the brain under non‐pathophysiological conditions (Nguyen et al. 2014; Nguyen & Venton 2015) and can occur in the vasculature as well, especially in response to changed fluid flow (Bodin et al. 1991; Bodin & Burnstock 2001). As an external mediator, locally produced adenosine can affect the cells in an autocrine or paracrine manner by binding to the adenosine receptor subtypes A1, A2A, A2B or A3. These receptors are G protein‐coupled receptors which regulate the synthesis of cAMP in the cells and have a plethora of actions on the cells depending on the cell‐specific signalling regulated by cAMP. A1 and A3 subtypes inhibit the synthesis of cAMP via Gi proteins, while A2A and A2B subtypes stimulate the production of cAMP via Gs proteins (Fredholm 2007; Eltzschig 2009).
In endothelial cells of the BBB, the expression of A1, A2A and A2B adenosine receptor subtypes was shown at mRNA and protein levels (Schaddelee et al. 2003; Mills et al. 2011; Carman et al. 2011). In animal experiments, adenosine‐dependent signalling was demonstrated as a regulator of the BBB (Carman et al. 2011; Gao et al. 2014). However, the specific contribution of the participating cells (blood cells, endothelial cells, pericytes, glial cells or neurons) was not determined in those experiments and contradictory results have been published. While many authors claimed that adenosine signalling affected endothelial barrier function, Cheng et al. (2016) have shown that the classical adenosine receptor agonist 5′‐N‐ethylcarboxamidoadenosine (NECA) could stimulate cerebral extravasation of fluorescein and dextran without directly affecting the BBB (Cheng et al. 2016). The authors argued that NECA impaired renal function, resulting in retention of the tracer in the blood. This led to an increased concentration of the tracer in brain tissue (Cheng et al. 2016). Since adenosine receptor‐dependent signalling in the BBB was proposed as a potential target for pharmacological therapies of pathophysiological conditions such as hypoxia, ischaemia or stroke (Blackburn et al. 2009; Melani et al. 2014), or for controlled opening of the BBB to deliver drugs into the CNS (Carman et al. 2011; Gao et al. 2014; Kim & Bynoe 2015; reviewed by Bynoe et al. 2015), it is of interest to decipher the physiology of this signalling in the different cells of the BBB. Using the human cerebral microvascular endothelial cell line hCMEC/D3, which has been proposed as a suitable in vitro model for BBB endothelial cells (Weksler et al. 2005, 2013), we show in the present study that adenosine receptor signalling enhances gap junction coupling. We identify cyclic nucleotide‐gated (CNG) channels as the physiological link which allows an integration of the adenosine receptors and gap junction communication in a signalling unit in the BBB endothelial cells.
Methods
Materials
2‐PAA, MRS1754, SCH58261, forskolin, 8‐Br‐cAMP, 8‐Br‐cGMP, dibutyryl‐cGMP (db‐cGMP) and Lucifer Yellow were purchased from Sigma‐Aldrich (Taufkirchen, Germany). BAPTA AM was obtained from Santa Cruz Biotechnology (Heidelberg, Germany). EGTA AM was purchased from Biomol (Hamburg, Germany). CGS21680 was from Merck Millipore (Darmstadt, Germany). l‐cis‐diltiazem was from Abcam (Cambridge, UK). 8‐Cyclopentyl‐1,3‐dipropylxanthine (DPCPX), KT5720 and SQ22536 were from Enzo Life Sciences (Lörrach, Germany). Rp‐cAMPS and 8‐pCPT‐2ʹ‐O‐Me‐cAMP were from Biolog Life Science Institute (Bremen, Germany).
2‐PAA was dissolved in a mixture (1:1) of ethanol and buffer solution (121 mm NaCl, 5.4 mm KCl, 25 mm Hepes, 0.8 mm MgCl2, 5.5 mm glucose, 6 mm NaHCO3, 1.8 mm CaCl2, pH 7.4) while MRS1754, SCH58261, CGS21680, BAPTA AM, EGTA AM, KT5720 and 8‐pCPT‐2ʹ‐O‐Me‐cAMP were dissolved in DMSO. All inhibitors were added 20–30 min prior to addition of 2‐PAA. The vehicles DMSO or ethanol were added to control cells in all experiments at maximal concentrations of 0.2% or 0.3%, respectively. At these concentrations the vehicles by themselves did not affect the cells in any experiment.
Cell culture
The human cerebral endothelial cell line hCMEC/D3 was cultured as described before (Weksler et al. 2005) in EBM‐2 medium (Lonza, Basel, Switzerland) supplemented with 5% fetal calf serum (Biochrom, Berlin, Germany), 1 mg ml−1 penicillin, 0.1 mg ml−1 streptomycin (Biochrom), 1.4 μm hydrocortisone, 5 μg ml−1 ascorbic acid, 10 mm Hepes, 1 ng ml−1 bFGF (Sigma‐Aldrich), and chemically defined lipid concentrate (Thermo Fisher Scientific, Darmstadt, Germany) diluted 1:100. The cells were maintained in a cell culture incubator in a humidified atmosphere with 5% CO2 at 37 °C. The cell culture media was renewed every two to three days. Cells up to passage 36 were used for experiments.
Scrape loading/dye transfer
hCMEC/D3 cells were seeded at a density of 3 × 105 cells per coverslip on glass coverslips coated with collagen I (Trevigen, Gaithersburg, MD, USA). Cells were allowed to form a monolayer for 48 h before 2‐PAA was added to the cell culture medium for the indicated time periods. Scrape loading/dye transfer experiments were performed as described previously (Begandt et al. 2010). The cell monolayers were washed with Ca2+‐free buffer (121 mm NaCl, 5.4 mm KCl, 25 mm Hepes, 0.8 mm MgCl2, 5.5 mm glucose, 6 mm NaHCO3, 1 mm EGTA, pH 7.4) and then incubated in 0.25% Lucifer Yellow in Ca2+‐free buffer for 2 min. The monolayers were scraped with a razor blade and incubated for another 5 min to allow dye uptake by the injured cells and lateral dye diffusion in the cell monolayer. The cells were then washed twice, first with Ca2+‐free and then with Ca2+‐containing buffer before fixing with 4% formaldehyde in phosphate buffered saline (PBS, 137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, pH 7.4) for 10 min. The cells were stored in PBS at 4 °C prior to microscopic analysis.
Four images along the scrape with a size of 1273 μm x 1273 μm were acquired from each coverslip with a confocal Nikon Eclipse TE2000‐E laser scanning microscope (Nikon GmbH, Düsseldorf, Germany) and the software EZ‐C1 (Nikon GmbH). A home‐made ImageJ (http://imagej.nih.gov/ij) plugin and Matlab‐ or Octave‐based software was used to calculate the lateral diffusion distance of Lucifer Yellow within the monolayer (Begandt et al. 2010). For the graphical presentation of the data the dye diffusion distances were normalized to those found in vehicle‐treated cells. All experiments were repeated at least five times using at least three different cell passages.
To evaluate the time dependency we used the non‐linear curve fit assistant of Origin 7 (OriginLab) to fit the measured dye diffusion distances at different time points for 2‐PAA concentrations of 1 μm, 10 μm, 20 μm and 50 μm to an exponential asymptotic curve defined by the equation:
In this equation, b represents the relative dye diffusion distance measured at the time point 0 h and a represents the asymptotic value of the dye diffusion distance that would be achieved by 2‐PAA treatment for an infinite time. From the asymptote a, the half‐maximal increase in the dye diffusion distances was calculated as (a − b)/2 + 100 and the corresponding time point was considered as t ½. To analyse the concentration dependency, we used the non‐linear curve fit assistant of Origin 7 to fit the dye diffusion distances obtained after treatment with 2‐PAA concentrations ranging from 0.01 μm to 100 μm for 1 h to a sigmoidal Boltzmann function given by the equation:
In the equation, A 1 represents the diffusion distance obtained for 0.01 μm 2‐PAA and A 2 is the theoretical maximal diffusion distance calculated from the data. We estimated the half‐maximal increase as the value (A 1 + A 2)/2 (which corresponds to x = x 0) and the corresponding concentration was estimated as EC50 concentration for 2‐PAA.
Knock‐down of the A2B adenosine receptor subtype
For siRNA‐mediated knock‐down of the A2B adenosine receptor subtype, 2.5 × 105 cells were seeded on collagen I‐coated coverslips and grown for 24 h to a confluence of about 60%. The cell culture media was exchanged to Opti‐MEM (Thermo Fisher Scientific). Two different anti‐A2B adenosine receptor siRNAs (Qiagen, Hilden, Germany, SI02662982 and SI02662975) and Silencer Select Negative Control No. 2 siRNA (Thermo Fisher Scientific) were diluted in JetPrime dilution buffer (Polyplus transfection, Illkirch, France). Per well 1.5 μl JetPrime transfection reagent (Polyplus transfection) were added. The transfection mix was incubated for 10 min at room temperature before addition to the cells to a final siRNA concentration of 50 nm per well. After 6–7 h the transfection medium was exchanged for normal cell culture medium and the cells were cultivated for further 48 h before scrape loading/dye transfer experiments and quantification of A2B adenosine receptor mRNA was performed.
RT‐PCR
The RNeasy Mini kit (Qiagen) or the PeqGOLD Total RNA kit (Peqlab, Erlangen, Germany) were used for total RNA isolation according to the manufacturer's protocols. Briefly, cells were washed with PBS containing 1 mg ml−1 EDTA. Lysis buffer was then added and the cell lysate was collected in a new reaction tube. The RNA was eluted from the spin columns with 40–50 μl RNase‐free water. The RNA concentration was measured with a Nanodrop2000c spectrophotometer (Peqlab).
After removal of genomic DNA with DNaseI (Thermo Fisher Scientific), 500 ng of total RNA were reverse transcribed into cDNA in a 20 μl reaction mixture containing 0.2 μg of random hexamer primers, 10 μm of each dNTP, 1 × reaction buffer, 1 U RiboLock RNase inhibitor and 1 U MML‐V reverse transcriptase (Thermo Fisher Scientific). A first incubation phase at 25 °C for 5 min was followed by a second incubation phase at 37 °C for 1 h before the reaction was stopped at 70 °C for 5 min. Alternatively the Maxima First Strand cDNA synthesis kit for RT‐qPCR with dsDNase (Thermo Fisher Scientific) was used. Up to 5 μg of total RNA were incubated with 1 μl dsDNAse and 1 × dsDNAse buffer for 30 min at 37 °C. Then 1 × reaction buffer and 2 μl enzyme mix were added in a final volume of 20 μl. The reaction was carried out for 10 min at 25 °C followed by 30 min at 65 °C and 5 min at 85 °C. Complete removal of genomic DNA was confirmed before reverse transcription for all samples by PCR analysis using the DNase‐treated RNA as template.
The primer pairs for gene expression analysis of Cx isoforms, adenosine receptors and CNG channel subtypes are given in Table 1. The correct amplification of all genes was verified by sequencing of the amplificates. PCR was performed in a 25 μl reaction mixture containing 62.5 ng cDNA, 0.2 μm of each primer and 1 × GoTaq G2 Green Mastermix (Promega, Mannheim, Germany) or KAPA ReadyMix with dye (Kapa Biosystems, Wilmington, MA, USA) or OneTaq Quick‐Load Master Mix with standard buffer (New England Biolabs, Ipswich, MA, USA) in a peqSTAR Universal 2× gradient PCR cycler (Peqlab). The PCR reaction started with an initial denaturation at 95 °C for 3 min followed by 35 cycles of denaturation at 95 °C for 10 s, annealing of primers at 60 °C for 30 s and elongation at 72 °C for 45 s and a final elongation at 72 °C for 5 min. Each PCR was run with an appropriate positive and negative control for each primer pair. The PCR products were separated in 2% agarose gels stained with GelRed (Biotium, Hayward, CA, USA).
Table 1.
List of all primer pairs used for gene expression analyses and quantitative real time PCR
| Target gene | Primer sequence 5ʹ‐3ʹ | Amplicon size (bp) | |
|---|---|---|---|
| gapdh | For | GGGGAGCCAAAAGGGTCATCATCT | 701 |
| Rev | TGTGCTCTTGCTGGGGCTGGTG | ||
| adora1 | For | ATCCCTCTCCGGTACAAGATG | 550 |
| Rev | TCTGGATGCGGAAGGCATAG | ||
| adora2A | For | ATCGCAGTGGGTGTGCTCGC | 455 |
| Rev | GCCGCCAGGAAGATCCGCAAA | ||
| adora2B | For | GTGCCACCAACAACTGCACAGAAC | 518 |
| Rev | CTGACCATTCCCACTCTTGACATC | ||
| adora3 | For | GTGCTTCCAGCTCTGCTCCCA | 518 |
| Rev | GCCAGTGGGCCTAGCTCTCG | ||
| cx30 | For | GCTACCTGCTGCTGAAAGTG | 326 |
| Rev | CGTTGTGTATGAATGGAGCA | ||
| cx32 | For | CCTGGAGGAGGTGAAGAGGC | 250 |
| Rev | GCCAGAGGCAGCTAGCATGA | ||
| cx36 | For | CTGCCCAGTCTTTGTCTGCT | 119 |
| Rev | CAACAGGATCCTCCCGATCAT | ||
| cx37 | For | GGACCATGGAGCCCGTGTTTGTGT | 443 |
| Rev | AGAGCTGCTGGGACGACTTGGGGG | ||
| cx40 | For | CCGTATGCTCGTGCTGGGCA | 465 |
| Rev | CTGCGGCAGACATGCAGGGT | ||
| cx43 | For | AGCAAAAGAGTGGTGCCCAGG | 494 |
| Rev | TGATGTAGGTTCGCAGCAACCCC | ||
| cx45 | For | GGCTTCCAAGTCCACCCGTTTTAT | 453 |
| Rev | ATCCAAGCGTTCCTGAGCCATCC | ||
| cngA1 | For | TCAAGGAAGGCAAACTCGCT | 511 |
| Rev | GAGTCGATGGGCCCACTTTC | ||
| cngA2 | For | AAAATGGGCAATCGACGCAC | 436 |
| Rev | CAATGCTGGAGAGGCAGTTG | ||
| cngA3 | For | GCTACTTCGGGGAGATCAGC | 483 |
| Rev | CCCTGTGAAGCAGGAGACAG | ||
| cngA4 | For | CAGAGACCCGCACAGCTTAC | 447 |
| Rev | AGTCAATAACTCGCCGCTCC | ||
| cngB1 | For | CCCCTCGGAAGACCAAGATG | 451 |
| Rev | TTCTGCTCCAGCCACAGAAG | ||
| cngB3 | For | TGAGCTAAGGAAACACTACAGGAC | 524 |
| Rev | TGGTGTCATCCATGCAGGC | ||
| gapdh * | For | GCTCATTTCCTGGTATGAC | 274 |
| Rev | ACAGGGTACTTTATTGATGGT | ||
| actB * | For | ATATGAGATGCGTTGTTACAG | 257 |
| Rev | CAAAAGCCTTCATACATCTC | ||
| cx37 * | For | CCCAGCAGCTCTGCTTCTAA | 274 |
| Rev | TGCTGACTCTGTCTGTGCTC | ||
| cx40 * | For | AAATGCCCCACCTTGGTGAT | 265 |
| Rev | GGTTCGAGAGAGGACAACAG | ||
| cx43 * | For | AAGCTCTGTGCTCCAAGTTAC | 254 |
| Rev | GTTTGCCTAAGGCGCTCCA | ||
| cx45 * | For | CTGGACAACAGGGCATACCA | 269 |
| Rev | GGAACACCCAGAAGCGTACA | ||
| gapdh * | For | TTGAGGTCAATGAAGGGGTC | 117 |
| Rev | GAAGGTGAAGGTCGGAGTCA | ||
| actB * | For | CCTTGCACATGCCGGAG | 112 |
| Rev | GCACAGAGCCTCGCCTT | ||
| adora2B * | For | TTCTGGCCGTGGCAGTC | 100 |
| Rev | AGGACAGCAATGACCCCT |
*Primers for real time PCR.
Real time PCR was used to quantify the expression of Cx isoforms after 2‐PAA treatment and to analyse the knock‐down of the A2B adenosine receptor subtype after siRNA transfection. The glyceraldehyde 3‐phosphate dehydrogenase (gapdh) and beta‐actin (actB) genes were used as housekeeping genes for normalization. For real time PCR the KAPA SYBR FAST Universal mastermix (Kapa Biosystems) was used and PCR was performed in a peqSTAR 96Q real time PCR cycler (Peqlab). Real time PCR was carried out with an initial denaturation at 95 °C for 3 min followed by 40 cycles of denaturation at 95 °C for 15 s and annealing of primers and elongation at 55 to 65 °C, depending on the primers, for 30 s. Amplification of target sequences was confirmed by melting curve analysis and gel electrophoresis. The ∆∆ct method of the cycler software peqSTAR 96Q (Peqlab) was used for quantification of the relative mRNA amounts. At least three experiments with three different cell passages were performed for each treatment and each gene expression study.
Western blot
For isolation of total protein, cells grown in 60 mm diameter cell culture plates were washed twice with ice‐cold PBS and removed from the culture plate surface with a cell scraper in the presence of 1 ml PBS. The cells were centrifuged for 4 min at 900 × g at 4 °C. The cell pellet was resuspended in 15 μl RIPA buffer (25 mm Tris HCl, pH 7.6, 150 mm NaCl, 1% nonidet P‐40, 1% sodium desoxycholate, 0.1% SDS, freshly added 1% phosphatase inhibitor mix II (Serva, Heidelberg, Germany), 0.5% protease inhibitor cocktail (Roche, Waiblingen, Germany), 1.5 mm PMSF) and kept for 15 min on ice before centrifugation for 15 min at 14,000 × g at 4 °C. The protein concentration in the supernatant was determined with a Bradford assay (Sigma‐Aldrich) using bovine serum albumin (BSA) as standard. The protein solution was mixed with 1 × Laemmli buffer (13 mm Tris HCl, 2% glycerol, 0.4% SDS, 0.002% Bromophenol Blue, 10 mm DTT, pH 6.8) and heated at 70 °C for 10 min. Aliquots of 30 μg of protein per lane were separated in a 5% SDS‐polyacrylamide stacking gel and a 8% or 12% separation gel. The proteins were transferred onto a nitrocellulose membrane using a semi‐dry blotting system (transfer buffer: 25 mm Tris HCl, pH 8.3, 192 mm glycine, 0.1% SDS, 20% methanol). Afterwards, the membranes were blocked in 5% non‐fat dry milk powder in TBS (50 mm Tris HCl, 75 mm NaCl, pH 7.4) containing 0.1% Tween 20 (TBS‐T) for 2 h at room temperature. Anti‐β‐tubulin antibody for the loading control (Sigma‐Aldrich, T4026) was diluted 1:7500, anti‐CNGA2 antibody (Alomone Labs, Jerusalem, Israel, APC‐045) was diluted 1:750 and anti‐Cx37 antibody (Abcam, ab58918) was diluted 1:700 in TBS‐T and applied to the membranes at 4 °C overnight. After washing, the secondary anti‐rabbit and the secondary anti‐mouse antibody (each diluted 1:10,000 in TBS‐T, Sigma‐Aldrich, A9169 and A9044) were each applied for 1 h at room temperature. The detection was carried out with SuperSignal West chemiluminescent substrate (Thermo Fisher Scientific) and imaged with a CCD camera imaging system (Intas Science Imaging, Göttingen, Germany). The presence of CNGA2 and Cx37 protein was confirmed in at least five different cell passages.
Measurement of intracellular cAMP concentration
Approximately 4.5 × 105 hCMEC/D3 cells per well were seeded in a 24 multiwell plate and grown for 48 h until confluent. Measurement of cAMP levels was performed using the cAMP‐Screen Chemiluminescent Immunoassay System (Thermo Fisher Scientific) according to the manufacturer's instructions with slight modifications as described below. 100 μl of lysis buffer were added per well to the cells and incubated for 30 min at 37 °C with gentle agitation. 90 μl of lysed cell suspension were added to each well of the supplied ELISA 96 multiwell plate. 30 μl of the diluted cAMP‐AP conjugate and 60 μl of the anti‐cAMP antibody were added per well, followed by an incubation for 1 h at 37 °C with gentle agitation. Afterwards the wells were washed three times with 200 μl wash buffer before addition of 100 μl chemiluminescent substrate and incubation for 30 min at room temperature. Luminometric measurement was performed with a Varioskan Flash plate reader (Thermo Fisher Scientific) with a measurement time of 1 s per well. Defined cAMP concentrations served as standard. Chemiluminescence values of treated cell samples were normalized to those obtained from vehicle‐treated cell samples. The results are given as the mean ± SEM from at least six different cell passages.
Ca2+ imaging
The evaluation of changes of the intracellular Ca2+ concentration was performed by ratiometric Ca2+ imaging with Fura‐2 (Merck Millipore, Darmstadt, Germany) as described previously (Bintig et al. 2009). Cells grown on coverslips were loaded with 2 μm Fura‐2 AM dissolved in buffer (140 mm NaCl, 5 mm KCl, 10 mm HEPES, 1 mm MgCl2, 10 mm glucose, 2 mm CaCl2, pH 7.4) for 20 min at room temperature. The coverslips were transferred to a perfusion chamber mounted on an inverted microscope (Zeiss, Oberkochen, Germany) and perfused with buffer to remove external Fura‐2. Fura‐2 in single cells was excited at 340 and 380 nm using a monochromator polychrome II (T.I.L.L Photonics GmbH, Planegg, Germany) equipped with a 75 W XBO xenon lamp. Fluorescence emission at 510 nm was recorded with a digital CCD camera (C4742‐95, Hamamatsu Photonics Deutschland GmbH, Herrsching am Ammersee, Germany) controlled by Aquacosmos software (Hamamatsu Photonics GmbH). The Ca2+ concentration was accessed as the ratio of the fluorescence intensities elicited by excitation at 340 nm and 380 nm (R = F 340/F 380). At the beginning of the measurement, the cells were perfused with 0.3% ethanol‐containing buffer as vehicle control. 2‐PAA‐containing buffer was applied 1–2 min later, and the data were recorded for the following 20 min. For control experiments, the cells were continuously superfused with the vehicle‐containing buffer. For all cells, the ratio R measured during the first 1–2 min was averaged to generate a value R 0 and the standard deviation (SD) was calculated. A Ca2+ increase in a cell was assumed if the ratio R of the respective cell achieved a value higher than R 0 + 2 x SD over a period of at least 5 s within 20 min. The percentage of cells with an increased Ca2+ signal is given as the mean ± SEM for five different cell passages with at least 20 cells measured per passage.
Electrophysiology
CNG channel opening was analysed using the whole‐cell patch‐clamp technique. hCMEC/D3 cells were settled on coverslips placed in a perfusion chamber containing buffer (140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm glucose, 10 mm Hepes, pH 7.4). The perfusion chamber was mounted on a Ti‐E inverted fluorescence microscope (Nikon GmbH) equipped with a CCD Orca‐Flash 4 camera (Hamamtsu Photonics Deutschland GmbH) and the software NIS‐Elements AR (Nikon GmbH). A whole‐cell configuration was established using an EPC 10 USB Double (HEKA Elektronik Dr Schulze GmbH, Lambrecht/Pfalz, Germany) coupled to PulseMaster software (HEKA Elektronik Dr Schulze GmbH). The patch pipette filling solution was composed of 125 mm potassium gluconate, 15 mm CsCl, 0.2 mm CaCl2, 2.5 mm MgATP, 2 mm Na2ATP, 5 mm EGTA, 5.5 mm glucose, 10 mm Hepes (pH 7.25). The cells were clamped at −60 mV. Membrane currents were elicited by voltage pulses between −150 and 100 mV for 250 ms in 10 mV steps in the presence of buffer with 0.3% ethanol, or 20 μm 2‐PAA, or buffer containing 2‐PAA and 100 μm l‐cis‐diltiazem or l‐cis‐diltiazem alone. The results were obtained from two to four different cell passages measuring at least nine cells for each treatment.
Immunofluorescence
For immunostaining 7.5 × 104 cells were seeded on collagen I‐coated coverslips (diameter 10 mm) and grown for 48 h to a confluence of more than 70%. The cells were washed with PBS and fixed with 4% formaldehyde in PBS for 20 min at 4 °C. Blocking was performed with 0.5% BSA in PBS for 30 min at 37 °C. The primary antibodies anti‐Cx37 (1:100, Sigma‐Aldrich, SAB2100920 or Abcam, ab58918), anti‐Cx40 (1:100, Sigma‐Aldrich, SAB1304973), anti‐Cx43 (1:4000, Sigma‐Aldrich, C6219) and anti‐Cx45 (1:50, Sigma‐Aldrich, AV36631) were diluted as indicated in PBS containing 0.3% Triton X‐100 and 0.5% BSA (only for anti‐Cx40 antibody) and added to the cells overnight at 4 °C. The secondary fluorescein‐conjugated anti‐rabbit antibody (Merck Millipore, 401314) was diluted 1:100 in PBS containing 0.3% Triton X‐100 and added to the cells with 2 μm DAPI (Sigma‐Aldrich) for 1 h at 37 °C. The cells were washed with PBS and stored at 4 °C.
Immunostaining was imaged using the Eclipse TE2000‐E inverse confocal laser scanning microscope (Nikon GmbH) with a ×60 water immersion objective and the software EZ‐C1 (Nikon GmbH). Six sections of 212 μm x 212 μm were recorded of each coverslip. Cx40 and Cx43 were visible as green ‘dots’ either within the cytoplasm of the cells or as ‘plaques’ in the cell membranes between adjacent cells. For quantification, the number of cells containing Cx43 as ‘plaques’ in the cell membrane was counted in blinded samples. The percentage of vehicle‐treated cells containing membrane‐localized Cx43 was compared to the percentage of 2‐PAA‐treated cells containing Cx43 in the cell membrane. The experiments were repeated 12 times with at least three individual cell passages.
For evaluation of the Cx43 gap junction plaque size, additional super‐resolution stimulated emission depletion (STED) microscopy was applied. Staining was performed as described above, but without nuclear staining. The secondary goat anti‐rabbit AlexaFluor488‐conjugated antibody was diluted 1:1000. The STED set‐up (Urban et al. 2011) included a pulsed‐laser diode (Toptica Photonics, Graefelfing, Germany) with excitation at 488 nm and 100 ps pulses. The pulses for the STED beam were delivered by a Ti:Sapphire laser (MaiTai; Spectra‐Physics, Darmstadt, Germany) operating at 80 MHz and emitting light pulses at 795 nm that were converted to 595 nm by an optical parametric oscillator (APE, Berlin, Germany) and stretched to 400 ps by dispersion. The power of the STED beam was 20–30 mW in the back focal aperture of the objective lens. The STED focal doughnut was created with a vortex phase plate (RPC Photonics, Rochester, NY, USA) introduced into the path of the expanded STED beam. The STED and excitation pulses were synchronized via external triggering of the laser diode. Both beams were overlapped using custom‐made dichroic mirrors and focused into the 1.2 NA objective lens (PL APO, CORR CS, 63, water; Leica, Wetzlar, Germany). The fluorescence was collected by the same lens, separated by a dichroic mirror, filtered with a 535/50 band‐pass filter and imaged onto a multimode optical fibre connected to a single‐photon avalanche photodiode (PerkinElmer, Waltham, MA, USA). Images were recorded with resonant mirror scanning (15 kHz, SC‐30; EOPC, Glendale, NY, USA) along the x‐axis and piezo‐stage scanning (P‐733; Physik Instrumente, Karlsruhe, Germany) along the y‐axis.
Membrane areas between adjacent cells with Cx43 gap junction plaques were selected from confocal imaging and magnified with STED imaging. Areas of 18 μm x 18 μm were recorded with STED imaging. The plaque sizes in STED and corresponding confocal images were determined using the particle analyser tool of ImageJ after conversion into a 16‐bit image, applying the auto threshold MaxEntropy and selection of the plaque areas in the corresponding confocal images. For statistical analysis of plaque areas and the number of distinguishable plaques, linear mixed effects models (Pinheiro & Bates 2000) were used. Total area of plaques per image and number of distinguishable plaques per cell were log‐transformed before analysis. The model for comparing microscopy methods involved treatments, methods and their interaction as fixed effects and random effects for replicates, replicate‐method‐interaction and individual images. For the comparison of treatments, microscopy methods were analysed separately; the mixed models included treatments as fixed effects and random effects for replications, and treatment‐replication interaction. After model fitting, all pairwise comparisons of least square means of microscopy methods and treatments were tested using Kenward‐Roger degrees of freedom. Analysis was performed in R‐3.2.2 (https://www.R-project.org/) using add‐on packages lsmeans 2.20–23 and lme4 1.1–10. 4.
Statistical analysis
Statistical analysis was performed with a Student's paired two‐sided t test. Cells from the same passage treated at the same time were considered as pairs. All treatments were compared to the corresponding vehicle controls and treatments with inhibitors were additionally compared to samples treated with 2‐PAA only. Results with a P value < 0.05 were considered to be statistically significant. The exact P values are stated in the respective Results section.
Results
The aim of the present report was to study how stimulation of the adenosine receptors affects gap junction coupling of endothelial cells of the BBB. We first tested for the expression of the different adenosine receptor isoforms as well as of the vascular Cx isoforms in the hCMEC/D3 cells, which are an accepted in vitro model for human BBB endothelial cells (Weksler et al. 2005, 2013).
RT‐PCR showed that the adenosine receptors A1, A2A and A2B were expressed in hCMEC/D3 cells at the mRNA level (Fig. 1 A). The hCMEC/D3 cells also expressed Cx37, Cx40, Cx43 and Cx45 mRNA (Fig. 1 B). The Cx isoforms Cx30, Cx32 and Cx36 that are expressed in other cerebral cell types (Eugenin et al. 2012) were not found in hCMEC/D3 cells (Fig. 1 B). Cxs form gap junction channels which assemble in gap junction plaques that are easily recognized in immunocytochemistry experiments. Correspondingly, we found gap junction plaques composed of Cx40 and Cx43, while Cx37 and Cx45 were not observed in the membrane areas between contacting cells. For Cx37 and Cx45, a diffuse staining in the intracellular space was observed (Fig. 1 C).
Figure 1. Expression of adenosine receptor subtypes and Cx isoforms in hCMEC/D3 cells.
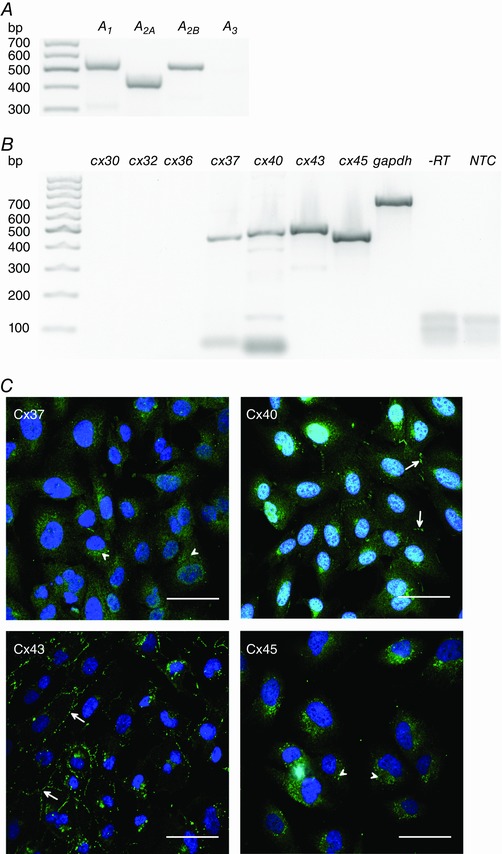
The mRNA of the adenosine receptor subtypes A1, A2A and A2B (A) and of the Cx isoforms Cx37, Cx40, Cx43 and Cx45 (B) was detected with RT‐PCR. Cx30, Cx32 and Cx36 were absent in hCMEC/D3 cells. ‐RT is the reverse transcription control with gapdh primers to confirm the absence of genomic DNA contamination. NTC is the negative control without template exemplarily shown for gapdh primers. C, immunofluorescence images of the different Cx isoforms in hCMEC/D3 cells (green). While Cx37 and Cx45 showed a diffuse intracellular staining (arrowheads), Cx40 and Cx43 staining showed gap junction plaques between neighbouring cells (arrows). Nuclei were counterstained with DAPI (blue). Scale bars represent 50 μm. [Color figure can be viewed at wileyonlinelibrary.com]
To activate adenosine receptors, we applied 2‐phenylaminoadenosine (2‐PAA), a non‐hydrolysable agonist for A2A and A2B adenosine receptors (Bruns et al. 1986; Choksi et al. 1997; Wilson & Batra 2002), to the hCMEC/D3 cells. With scrape loading/dye transfer assays, we analysed the gap junction coupling by measuring the dye diffusion distance in cell monolayers treated with 2‐PAA (20 μm) for 15 min to 6 h (Fig. 2 A). We found that compared to vehicle‐treated cells, the dye diffusion distance increased rapidly within the first 45 min of 2‐PAA application and reached an asymptotic maximum of approximately 135% after a 2‐PAA treatment period of 1 h (Fig. 2 B; Student's paired t test compared to vehicle‐treated cells: P = 4.5 × 10−3). For concentrations above 1 μm, the kinetic of the 2‐PAA‐induced increase in gap junction coupling was not concentration dependent as shown by the times for the half‐maximal increase (t ½) of approximatively 39 min calculated by fitting the dye diffusion distances for 1 μm, 10 μm, 20 μm and 50 μm 2‐PAA applied for 15 min to 6 h to an exponential asymptotic function (Table 2). With respect to the concentration dependency, we applied 2‐PAA at various concentrations between 0.01 μm and 100 μm for 1 h (Fig. 2 C). The dye diffusion distance in the cells increased beginning with 0.01 μm 2‐PAA (107%, Student's paired t test compared to vehicle‐treated cells: P = 5.3 × 10−2) and achieved a maximal increase of 130–140% at 20–50 μm (Student's paired t test compared to vehicle‐treated cells: P < 2.2 × 10−3). An EC50 of 1.9 μm was evaluated with a curve fit to a sigmoidal Boltzmann function (Fig. 2 C).
Figure 2. Enhancement of gap junction coupling by 2‐PAA.
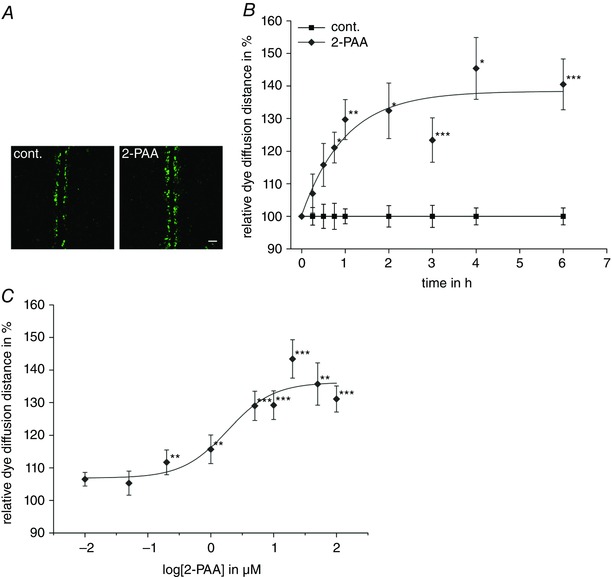
A, representative micrographs of scrape loading/dye transfer experiments in hCMEC/D3 cells treated with the vehicle (cont., 0.3% ethanol) or 2‐PAA (20 μm) for 1 h. Scale bar represents 100 μm. B, the time‐dependent increase in the dye diffusion distance induced by 2‐PAA (20 μm) as found by scrape loading/dye transfer assays relative to the vehicle control (cont., 0.3% ethanol). C, the concentration dependency of 2‐PAA on the increased dye diffusion distance. The data points represent the relative dye diffusion distance achieved in cell monolayers after application of 2‐PAA for 1 h. All results were analysed using Student's t test. *Significant differences to the vehicle control: * P < 0.05, ** P < 0.01, *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
Table 2.
The time for the half‐maximal increase in the dye diffusion distance for 2‐PAA concentrations ranging from 1 μm to 50 μm
| Concentration of 2‐PAA (μm) | Maximal relative dye diffusion distance (%) | Time for the half‐maximal dye diffusion increase (min) |
|---|---|---|
| 1 | 111.9 | 46 |
| 10 | 125.5 | 40 |
| 20 | 138.4 | 40 |
| 50 | 151.0 | 31 |
The maximal relative dye diffusion distance and the time for the half‐maximal increase were estimated by fitting the data points for the corresponding 2‐PAA concentration to a single exponential equation (Fig. 2 B).
The increase in gap junction coupling was not related to a change in the expression of the Cxs since quantitative real time PCR showed that the quantity of the mRNA for Cx37, Cx40, Cx43 and Cx45 was not significantly changed by an application of 2‐PAA for 1 or 6 h (data not shown). At the morphological level, the 2‐PAA‐related enhancement of the gap junction coupling correlated to an increase in cells with gap junction plaques composed of Cx43 (Fig. 3 A). It is noteworthy that the other Cx isoforms were not affected. We observed 73% of cells with Cx43 gap junction plaques in control conditions and found an increase to 80% of cells with Cx43 gap junction plaques after 2‐PAA treatment for 1 h (Fig. 3 B; Student's paired t test compared to vehicle‐treated cells: P = 1.6 × 10−2, n = 12). Evaluation of the STED microscopy showed that compared to the corresponding confocal microscopic images, in STED microscopic images the mean area of the plaques per image was reduced to one‐third (P < 1.0 × 10−4) but the total amount of individually distinguishable particles increased 10‐fold (P < 1.0 × 10−4, Fig. 3 C). The different treatments were compared for each microscopic method individually. A significant increase in the total plaque area per image was found for 2‐PAA‐treated cells both in the confocal images (123% of the vehicle control, P = 4.9 × 10−2) and in the images generated with STED microscopy (144% compared to ethanol‐treated cells, P = 4.5 × 10−2).
Figure 3. 2‐PAA induced the formation of gap junction plaques between hCMEC/D3 cells.
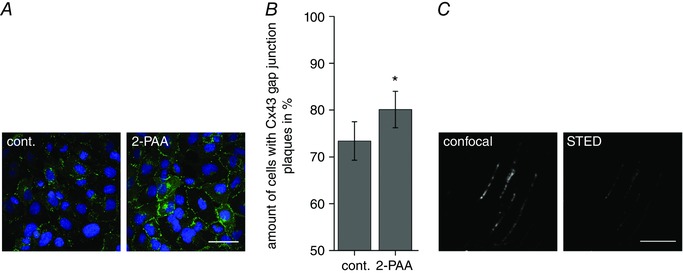
A, representative microscopic images of Cx43 (green) immunofluorescence in hCMEC/D3 cells after 20 μm 2‐PAA treatment for 1 h. Nuclei were counterstained with DAPI (blue). Scale bar represents 50 μm. B, the amount of hCMEC/D3 cells with Cx43 gap junction plaques was significantly increased after 2‐PAA treatment (20 μm) for 1 h compared to the vehicle control (cont., 0.3% ethanol). The results were analysed using Student's t test. *Significant differences to the vehicle control: * P < 0.05. C, exemplary confocal and STED images with increased resolution of individual Cx43 gap junction plaques. Scale bar represents 5 μm. [Color figure can be viewed at wileyonlinelibrary.com]
In recent publications we showed that the adenosine transporter inhibitor dipyridamole increased the gap junction coupling in aortic endothelial cells by activating adenosine receptors and subsequently the cAMP–PKA pathway (Begandt et al. 2010, 2013 b). Therefore, we analysed whether the same pathway is activated in the BBB endothelial cells. We found that 2‐PAA (20 μm) stimulated the synthesis of cAMP to 124% compared to the vehicle (Fig. 4 A; Student's paired t test compared to vehicle‐treated cells: P = 8.5 × 10−4). The adenylyl cyclase inhibitor SQ22536 (400 μm) applied concomitantly with 2‐PAA suppressed the increase in the intracellular cAMP concentration evoked by 2‐PAA alone with a relative cAMP concentration of 91% compared to the vehicle control (Fig. 4 A; Student's paired t test compared to 2‐PAA‐treated cells: P = 2.4 × 10−3, n = 7). Consequently, we found that SQ22536 attenuated the 2‐PAA‐induced enhancement of the dye diffusion distance in scrape loading/dye transfer experiments (Fig. 4 B; relative dye diffusion distance of 109% for 2‐PAA with SQ22536 compared to 121% for 2‐PAA alone, Student's paired t test compared to 2‐PAA‐treated cells: P = 2.6 × 10−2, n = 7). Additionally, a general increase in the intracellular cAMP concentration either by activation of adenylyl cyclase using forskolin (0.5 μm) or by application of the membrane permeable cAMP analogue 8‐Br‐cAMP (1 mm) significantly increased the gap junction coupling (Fig. 4 B; relative diffusion distance of 123% for forskolin and 129% for 8‐Br‐cAMP, Student's paired t test compared to vehicle‐treated cells: P = 4.6 × 10−8, n = 23 for forskolin and P = 3.3 × 10−2, n = 7 for 8‐Br‐cAMP), suggesting a cAMP‐dependent mechanism might also be induced by 2‐PAA.
Figure 4. The pharmacology of the 2‐PAA‐related increase in gap junction coupling.
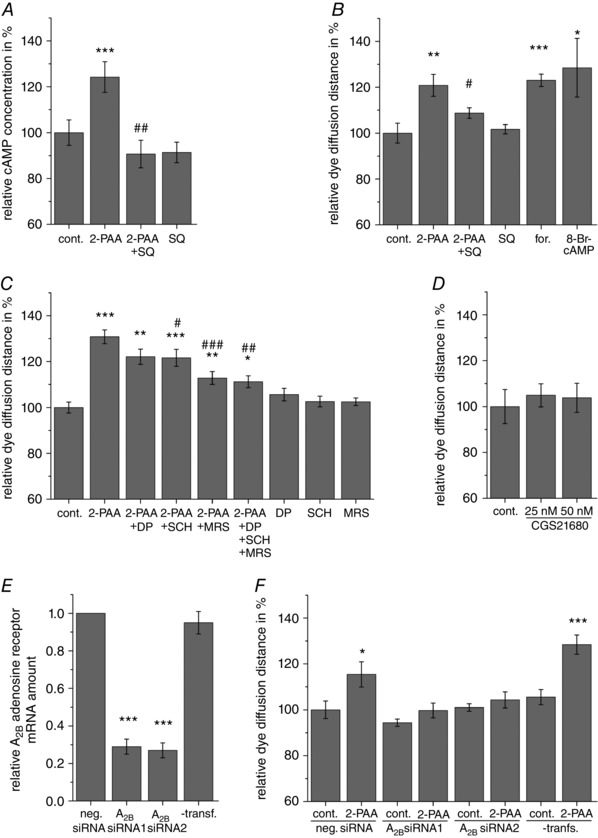
A, 2‐PAA (20 μm) increased the intracellular cAMP concentration. This increase could be blocked by the adenylyl cyclase inhibitor SQ22536 (SQ, 400 μm). B, the increase in the dye diffusion distance in scrape loading/dye transfer assays was significantly attenuated by the adenylyl cyclase inhibitor SQ22536 (SQ, 400 μm). The adenylyl cyclase activator forskolin (for., 0.5 μm) and the cAMP analogue 8‐Br‐cAMP (1 mm) significantly increased the dye diffusion distance within 1 h similar to 2‐PAA. C, the increased dye diffusion distance by 2‐PAA (20 μm, 1 h) was not significantly affected by the A1 adenosine receptor subtype antagonist DPCPX (DP, 25 nm) and only slightly affected by the A2A adenosine receptor subtype antagonist SCH58261 (SCH, 0.5 μm). The A2B adenosine receptor subtype antagonist MRS1754 (MRS, 0.5 μm) alone or together with DPCPX and SCH58261 nearly completely blocked the 2‐PAA‐induced increase in the dye diffusion distance. D, the A2A adenosine receptor subtype‐specific agonist CGS21680 did not change the dye diffusion distance. E, transfection of the hCMEC/D3 cells with two different anti‐A2B adenosine receptor siRNAs (A2B siRNA) significantly decreased the mRNA amount of the A2B adenosine receptor after 48 h compared to transfection with negative control (neg.) siRNA. Cells that were not treated with transfection reagent (‐transf.) served as control. F, the transfection with both anti‐A2B adenosine receptor siRNAs (A2B siRNA) attenuated the increase in the dye diffusion distance after 2‐PAA (20 μm) application compared to cells transfected with negative control (neg.) siRNA. The result of cells that were not treated with transfection reagent (‐transf.) is shown as control. The relative dye diffusion distances were normalized to vehicle‐treated cells transfected with negative control siRNA. All results were analysed using Student's t test. *Significant differences to the vehicle control: * P < 0.05, ** P < 0.01, *** P < 0.001; #significant differences to 2‐PAA: # P < 0.05, ## P < 0.01, ### P < 0.001.
As agonist of adenosine receptors, 2‐PAA predominantly targets A2A and A2B adenosine receptor subtypes (Bruns et al. 1986; Choksi et al. 1997; Wilson & Batra 2002). However, since 2‐PAA is an adenosine derivate, an action on the A1 adenosine receptor subtype, which was also expressed in hCMEC/D3 cells, cannot be completely excluded. The classical pathway of the A1 adenosine receptor subtype is activation of Gi, thereby inhibiting the adenylyl cyclase. This pathway seemed unlikely since we found that 2‐PAA increased cAMP in the cells and the adenylyl cyclase inhibitor SQ22536 antagonized the 2‐PAA‐related increase in the dye diffusion distance (Fig. 4 A and B). A second pathway of A1 adenosine receptor activation is related to cGMP accumulation (Kurtz et al. 1988; Serpa et al. 2014; Pinto et al. 2016). Therefore we analysed whether A1 adenosine receptor‐ and cGMP‐dependent pathways may be involved in the 2‐PAA‐induced enhancement of the gap junction coupling. We found that DPCPX (25 nm), an A1 adenosine receptor subtype antagonist, had no effect on the 2‐PAA‐induced increase in the dye diffusion distance (Fig. 4 C; relative dye diffusion distance of 122% for 2‐PAA with DPCPX compared to 131% for 2‐PAA alone, Student's paired t test compared to 2‐PAA‐treated cells: P = 0.11, n = 9). Moreover, the membrane permeable cGMP analogues db‐cGMP (1 mm) and 8‐Br‐cGMP (1 mm) did not influence the dye diffusion distance (relative dye diffusion distance of 96% for both db‐cGMP and 8‐Br‐cGMP, Student's paired t test compared to vehicle‐treated cells: P = 0.36 for db‐cGMP and 0.57 for 8‐Br‐cGMP, n = 7, respectively).
With respect to the A2A and A2B adenosine receptor subtypes, MRS1754 (0.5 μm), a specific antagonist of the A2B adenosine receptor subtype, almost completely antagonized the effect of 20 μm 2‐PAA on the gap junction coupling (Fig. 4 C; relative dye diffusion distance of 113% for 2‐PAA with MRS1754 compared to 131% for 2‐PAA alone, Student's paired t test compared to 2‐PAA‐treated cells: P = 1.2 × 10−5, n = 15). In contrast, the A2A adenosine receptor subtype antagonist SCH58261 (0.5 μm) only had a slight inhibitory effect on the increased dye diffusion distance after 2‐PAA application (Fig. 4 C; relative diffusion distance of 122% for 2‐PAA with SCH58261 compared to 131% for 2‐PAA alone, Student's paired t test compared to 2‐PAA‐treated cells: P = 2.5 × 10−2, n = 15). The combination of all three adenosine receptor inhibitors with 2‐PAA led to the same result as the combination of 2‐PAA with only MRS1754 (Fig. 4 C; relative dye diffusion distance of 111% for 2‐PAA with all three inhibitors compared to 131% for 2‐PAA alone, Student's paired t test compared to 2‐PAA‐treated cells: P = 8.3 × 10−3, n = 9). Additionally, the specific A2A adenosine receptor agonist CGS21680 (25 nm and 50 nm) did not affect the gap junction coupling of hCMEC/D3 cells with maximal dye diffusion distances of 105% compared to the vehicle control (Fig. 4 D; Student's paired t test compared to vehicle‐treated cells: P > 0.38, n = 5). The siRNA‐mediated knock‐down of the A2B adenosine receptor subtype also confirmed the importance of this receptor subtype in the observed 2‐PAA‐related enhancement of gap junction coupling. Transfection of the hCMEC/D3 cells with siRNA specific for A2B adenosine receptor mRNA led to significant decreases in A2B adenosine receptor mRNA amounts of 29% and 27% for siRNA1 and siRNA2, respectively, compared to cells transfected with negative control siRNA after 48 h (Fig. 4 E; Student's paired t test compared to negative control siRNA‐transfected cells: P = 5.7 × 10−7for siRNA1 and P = 1.8 × 10−7 for siRNA2, n = 8). Scrape loading/dye transfer experiments showed that 2‐PAA (20 μm, 1 h) enhanced the dye diffusion distance in cells transfected with negative control siRNA (Fig. 4 F; relative dye diffusion distance of 115% for 2‐PAA, Student's paired t test compared to vehicle‐treated cells: P = 2.8 × 10−2, n = 8). In cells that were transfected with anti‐A2B adenosine receptor siRNAs the effect of 2‐PAA on the dye diffusion distance was significantly reduced (Fig. 4 F; relative dye diffusion distances of 106% and 103% for siRNA1 and siRNA2, respectively, for 2‐PAA treatment compared to the respective vehicle‐treated cells, Student's paired t test compared to vehicle‐treated cells: P = 0.20 for siRNA1 and P = 0.41 for siRNA2, n = 8). The observed reduced increase in the dye diffusion distance induced by 2‐PAA in cells transfected with negative control siRNA as compared to non‐transfected cells (Fig. 4 F) seems to reflect a general stress induced in the cells by the transfection.
As second messenger, cAMP can activate the protein kinase A (PKA; Tasken & Aandahl 2004), the exchange protein directly activated by cAMP (Epac; Banerjee & Cheng 2015), and cyclic nucleotide‐gated (CNG) channels (Kaupp & Seifert 2002). We used pharmacological approaches in combination with patch‐clamp and Ca2+ measurements to evaluate the involvement of the different cAMP‐dependent pathways in the 2‐PAA‐induced enhancement of the gap junction coupling in the hCMEC/D3 cells. We found that the PKA inhibitors Rp‐cAMPS (200 μm) and KT15720 (1 μm) did not affect the 2‐PAA‐related enhancement of the gap junction coupling in the cerebral microvascular endothelial cells hCMEC/D3 (Fig. 5 A; relative dye diffusion distances of 132% for 2‐PAA alone and 133% with Rp‐cAMPS and 123% with KT5720, respectively, Student's paired t test compared to 2‐PAA‐treated cells: P = 0.20, n = 17 for 2‐PAA with Rp‐cAMPS and P = 0.95, n = 9 for 2‐PAA with KT5720). Furthermore, the specific Epac activator 8‐pCPT‐2′‐O‐Me‐cAMP (100 μm) did not induce an increase in the gap junction coupling in the hCMEC/D3 cells with a dye diffusion distance of 98% compared to the vehicle control (Fig. 5 A; Student's paired t test compared to vehicle‐treated cells: P = 0.65, n = 5). We therefore hypothesized that the 2‐PAA‐related enhancement of the gap junction coupling in these cells was related to an activation of CNG channels. Accordingly, we found that the CNG channel inhibitor l‐cis‐diltiazem (100 μm) suppressed the 2‐PAA‐related enhancement of the gap junction coupling (Fig. 5 B; relative dye diffusion distance of 102% for 2‐PAA with l‐ cis‐diltiazem compared to 125% for 2‐PAA alone, Student's paired t test compared to 2‐PAA‐treated cells: P = 1.8 × 10−2, n = 7). We therefore analysed the expression and activation of CNG channels in hCMEC/D3 cells.
Figure 5. The signalling mechanism induced by 2‐PAA.

A, the increase in the dye diffusion distance induced by 2‐PAA (20 μm, 1 h) was not blocked by the protein kinase A inhibitors Rp‐cAMPS (Rp, 200 μm) and KT5720 (KT, 1 μm). Additionally, the activator of the exchange protein directly activated by cAMP, 8‐pCPT‐O‐Me‐cAMP (100 μm) did not affect the dye diffusion distance. B, the cyclic nucleotide‐gated (CNG) channel inhibitor l‐cis‐diltiazem (L‐cis‐dil., 100 μm) could prevent the increase in the dye diffusion distance induced by 2‐PAA (20 μm, 1 h) relative to the vehicle control (cont., 0.3% ethanol) in scrape loading/dye transfer assays. C, RT‐PCR showed that the CNG channel subunits A1, A2 and B1 were expressed in hCMEC/D3 cells. D, furthermore, the cAMP‐sensitive subunit CNGA2 was confirmed to be expressed at protein level with β‐tubulin (β‐Tb) serving as loading control. E, in whole‐cell patch‐clamp experiments, 2‐PAA (20 μm) increased the current measured in hCMEC/D3 cells. This increased current was completely abolished by simultaneous application of the CNG channel blocker l‐cis‐diltiazem (L‐cis‐dil., 100 μm) with 2‐PAA. All results were analysed using Student's t test. *Significant differences to the vehicle control: * P < 0.05, ** P < 0.01, *** P < 0.001; #significant differences to 2‐PAA: # P < 0.05.
There are six different subunits known to form CNG channels, A1, A2 and A3 and the three modulatory subunits A4, B1 and B3 (Kaupp & Seifert 2002). RT‐PCR experiments showed that the hCMEC/D3 cells expressed the A1, the A2 and the B1 subunits of the CNG channels (Fig. 5 C). Western blotting confirmed the presence of the particularly cAMP‐sensitive A2 subunit (Kaupp & Seifert 2002; Cheng et al. 2008) in the cells (Fig. 5 D). Correspondingly, the patch‐clamp technique showed that the application of 20 μm 2‐PAA increased a whole‐cell current registered in the cells (Fig. 5 E; Student's paired t test compared to vehicle‐treated cells: P < 0.08, n = 9). The increase in the current was abolished by simultaneous application of the CNG channel blocker l‐ cis‐diltiazem (100 μm; Fig. 5 E; Student's paired t test compared to vehicle‐treated cells: P > 0.25, n = 15).
CNG channels are rather unselective channels permeable to cations. Beside Na+ influx and K+ efflux, the CNG channels also allow Ca2+ entry into the cells. Accordingly, Ca2+ imaging experiments with Fura‐2 showed an increased Ca2+ signal in more cells treated with 2‐PAA (61%) compared to cells treated with the vehicle (30%; Fig. 6 A; Student's paired t test compared to vehicle‐treated cells: P = 1.7 × 10−2, n = 5). The increase in the Ca2+ signal was slow and rapid transient Ca2+ spikes were rarely observed, suggesting that the fractional Ca2+ current was reduced, as the CNG channels are permeable to both Ca2+ and Na+ (Kaupp & Seifert 2002). We therefore replaced the external Na+ by 5′‐N‐ethylcarboxamidoadenosine (NMDG) and repeated the Ca2+ imaging. In the absence of external Na+, 2‐PAA more readily induced rapid transient Ca2+ spikes (Fig. 6 B). To analyse whether Ca2+ was necessary for the 2‐PAA‐related enhancement of the gap junction coupling, 2‐PAA (20 μm) was applied after preloading the cells with BAPTA (10 μm) as chelator of intracellular Ca2+ or in the absence of extracellular Ca2+. Under these conditions, the 2‐PAA‐related enhancement of the dye diffusion distance was strongly reduced (Fig. 6 C; relative dye diffusion distances of 109% for 2‐PAA with BAPTA and 113% for 2‐PAA without extracellular Ca2+ compared to 126% for 2‐PAA alone, Student's paired t test compared to 2‐PAA‐treated cells: P = 2.1 × 10−3, n = 12 for 2‐PAA with BAPTA and Student's paired t test compared to 2‐PAA‐treated cells with extracellular Ca2+: P = 5.1 × 10−3, n = 17 for 2‐PAA in absence of extracellular Ca2+).
Figure 6. The 2‐PAA‐induced increase in gap junction coupling was Ca2+ dependent.
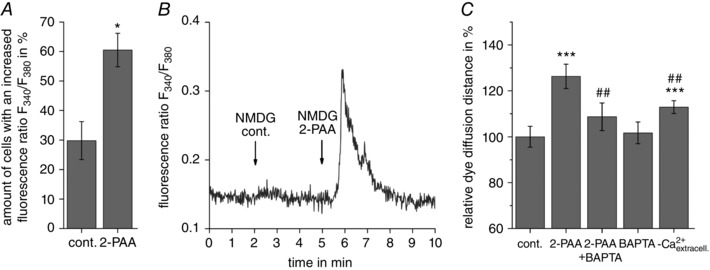
A, application of 2‐PAA (20 μm) increased the intracellular Ca2+ signal (fluorescence ratio F 340/F 380). The columns show the results obtained by counting the amount of cells with an increased [Ca2+]i after treatment with the vehicle control (cont., 0.3% ethanol) or 2‐PAA (20 μm) in presence of 140 mm external Na+. B, a representative rapid transient Ca2+ signal induced by application of 2‐PAA (20 μm) when external Na+ was replaced by NMDG. C, the 2‐PAA‐induced increase in the dye diffusion distance was abolished when 2‐PAA (20 μm, 1 h) was applied on cells preloaded with the Ca2+ chelator BAPTA (10 μm) or in absence of external Ca2+ (‐ Ca2+ extracell.). All results were analysed using Student's t test. *Significant differences to the vehicle control: * P < 0.05: *** P < 0.001; #significant differences to 2‐PAA: ## P < 0.01.
Collectively, the data presented in this report identify CNG channels as a previously unrecognized physiological link between adenosine receptors and gap junction coupling in human cerebral microvascular endothelial cells. Specifically, we show that following activation of predominantly A2B adenosine receptors in hCMEC/D3 cells, the second messenger cAMP leads to the opening of CNG channels, allowing a Ca2+ influx that evokes the increase in gap junction coupling.
Discussion
By allowing the exchange of ions and metabolites between cells, the gap junction coupling permits the formation of physiological units in tissues. A prerequisite for a regulation of these units is the physiological integration of the gap junction coupling in the general signalling pathways of the cells within a tissue. In the present report we used hCMEC/D3 cells as an accepted in vitro model for human BBB endothelial cells (Weksler et al. 2005, 2013) and found that adenosine receptor‐dependent signalling regulated the gap junction coupling in these cells via activation of CNG channels (Figs 2 and 5).
In accordance with other studies on BBB endothelial cells (Nagasawa et al. 2006; Avila et al. 2011; Bock et al. 2012; Kaneko et al. 2015), RT‐PCR analysis revealed that Cx37, Cx40, Cx43 and Cx45 were expressed in hCMEC/D3 cells (Fig. 1 B). Transcripts of three other isoforms, Cx30, Cx32 and Cx36, that are known to be expressed in other cerebral cell types (Eugenin et al. 2012) were not found in hCMEC/D3 cells (Fig. 1 B). Immunofluorescence staining showed that in hCMEC/D3 cells Cx40 and Cx43 formed gap junction plaques while Cx37 and Cx45 were solely stained in the intracellular space (Fig. 1 C). The structural and physiological significance of the absence of Cx37 and Cx45 in gap junction plaques is not clear. It is possible that Cx37 and Cx45 are not inserted into the membrane of the cells. The expression of Cxs independently of gap junction channel formation has for example been proposed to modulate cell migration (Kameritsch et al. 2012) or act as tumour suppressors (McLachlan et al. 2006). Alternatively Cx37 and Cx45 could form gap junction hemichannels or gap junction channels which are not clustered like the Cx43 and Cx40 plaques and are therefore not easily recognizable with fluorescence microscopy (Nakagawa et al. 2011). In endothelial cells, particularly those derived from the BBB, the existence and functional role of hemichannels has been proposed (Bock et al. 2012; Kaneko et al. 2015). These hemichannels are permeable to metabolites such as ATP (Stout et al. 2002; Kang et al. 2008) and pathophysiological conditions like hypoxia or ischaemia are known to increase their open probability (Davidson et al. 2012; Kaneko et al. 2015). Furthermore, the presence of non‐clustered gap junction channels containing Cx37 and Cx45 is possible as it is known that the different Cxs form gap junction channels with variable dynamics (Thomas et al. 2005; Stout et al. 2015).
Considering gap junction coupling, we found that in hCMEC/D3 cells the adenosine receptor agonist 2‐PAA enhanced the gap junction coupling (Fig. 2), which achieved its maximum within 45–60 min (Fig. 2 B). At the morphological level, the 2‐PAA‐induced increase in gap junction coupling was only paralleled by an increased formation of Cx43 gap junction plaques as well as increased Cx43 gap junction plaque size between adjacent cells as revealed by quantitative evaluation of confocal and STED microscopic images (Fig. 3). Although no effect of 2‐PAA on the localization of Cx37, Cx45 or Cx40 was discernible (data not shown), the increased presence of Cx43 in the membrane in response to 2‐PAA treatment suggests that the increase in gap junction coupling may primarily be related to an increased formation of gap junction channels between the cells. However, changes in the biophysical properties of the gap junction channels such as an increase in single gap junction channel conductance or the open probability of the channels may be secondarily involved as well. An increase in gap junction channels can be achieved by enhanced gene expression, Cx synthesis, less connexon degradation or enhanced connexon trafficking to the membrane. The increase in gap junction coupling can also be achieved by the insertion of new connexons into the membrane or a reduced removal of the gap junctions from the membrane. The synthesis and trafficking, as well as the removal, of gap junction channels are processes which necessitate a time too long (Jordan et al. 1999; Falk et al. 2014) to explain the observed rapid 2‐PAA‐induced increase in gap junction coupling within 30 min (Fig. 2 B), which is therefore more compatible with 2‐PAA stimulated formation of gap junction channels in the membrane.
Regarding the kinetics of the effect of adenosine receptor stimulation on the physiological function of the BBB in animal models, an increase in the BBB permeability was visible within 15–30 min (Carman et al. 2011). However, while the animal model reflects systemic reactions integrating all cells in a tissue, the specific contribution of the different cell types, e.g. endothelial cells, is very difficult to estimate. Other authors have analysed how adenosine receptor stimulation affected the BBB function using isolated microvascular endothelial cells. It was found that the A2A adenosine receptor‐specific agonist regadenoson (lexiscan) and the non‐specific adenosine receptor agonist NECA decreased the transendothelial electrical resistance (TEER) of the cells within 5–15 min of application (Gao et al. 2014; Kim & Bynoe 2015), which is faster than the effect of 2‐PAA on the gap junction coupling described in the present report (Fig. 2 B). The pharmacology shows that the decrease of the TEER was mainly due to the stimulation of the A2A adenosine receptors (Gao et al. 2014; Kim & Bynoe 2015). 2‐PAA, which was used in the present report, stimulates both A2A and A2B adenosine receptor subtypes (Bruns et al. 1986; Choksi et al. 1997; Wilson & Batra 2002). Accordingly, we found that 2‐PAA increased the cellular cAMP concentration and that the adenylyl cyclase inhibitor SQ22536 suppressed the increased cAMP synthesis (Fig. 4 A). SQ22536 also reduced the 2‐PAA‐related increase in gap junction coupling (Fig. 4 B), which showed that cAMP was necessary for the 2‐PAA‐induced increase in gap junction coupling. It is noteworthy that SQ22536 did not completely suppress the 2‐PAA‐induced increase in gap junction coupling, suggesting that other mechanisms could be involved in the 2‐PAA‐induced enhancement of gap junction coupling in the cells. Although 2‐PAA is described as an agonist for A2A and A2B adenosine receptors (Bruns et al. 1986; Choksi et al. 1997; Wilson & Batra 2002) and we found that 2‐PAA stimulation induced cAMP synthesis, as an adenosine derivative 2‐PAA might be able to bind to A1 adenosine receptors, which are linked to Gi or to the synthesis of cGMP (Kurtz et al. 1988; Eltzschig 2009; Serpa et al. 2014; Pinto et al. 2016). The following findings, however, argue against the involvement of the A1 adenosine receptor in the action of 2‐PAA on the gap junction coupling. First, we found that 2‐PAA induced a cAMP increase (Fig. 4 A), which is incompatible with activation of A1 adenosine receptors, which, via Gi, preferentially inhibits adenylyl cyclase (Fredholm 2007; Eltzschig 2009). Secondly, the A1 adenosine receptor antagonist DPCPX did not influence the 2‐PAA‐related enhancement of the gap junction coupling (Fig. 4 C), and additionally cGMP analogues were not able to mimic the effect of 2‐PAA on the gap junction coupling. Hence, an action of 2‐PAA that involves binding to A1 adenosine receptors and activation of cGMP synthesis seems very unlikely. The results therefore suggest that 2‐PAA acted solely by binding to Gs coupled receptors, which activated adenylyl cyclase to synthesize cAMP. Nine different adenylyl cyclase isoforms can be expressed in cells (Seifert et al. 2012) and none of the widely used adenylyl cyclase inhibitors is able to inhibit all isoforms equally well (Seifert et al. 2012). Therefore we assume that even in the presence of SQ22536, 2‐PAA could still induce cAMP synthesis to some extent, which could affect the gap junction coupling in a statistically non‐significant manner as observed in this report. Regarding the A2A and A2B adenosine receptor subtypes which are both linked to Gs and stimulation of adenylyl cyclase, the A2A adenosine receptor antagonist SCH58261 and the A2B adenosine receptor antagonist MRS1754, as well as the A2A adenosine receptor‐specific agonist CGS21680, revealed that the 2‐PAA‐related increase in gap junction coupling was mainly dependent on stimulation of the A2B adenosine receptor subtype and not the A2A adenosine receptor subtype (Fig. 4 C and D). The predominant role of the A2B adenosine receptor subtype in the regulation of gap junction coupling in cerebral endothelial cells presented here is also sustained by the transient knock‐down of the A2B adenosine receptor subtype using siRNA. We found that when expression of the A2B adenosine receptor subtype was decreased, 2‐PAA could no longer significantly increase the gap junction coupling in hCMEC/D3 cells (Fig. 4 F). It is noteworthy that neither the pharmacological inhibition nor the siRNA‐dependent knock‐down of the A2B adenosine receptor could completely suppress the effect of 2‐PAA. With siRNA we achieved a knock‐down of mRNA levels of over 70% (Fig. 4 E) and with the pharmacological inhibitors our aim was to avoid high inhibitor concentrations, which are not specific anymore and might induce unintended side‐effects on the cells. We therefore assume that under our experimental conditions, a reduced activation of adenosine receptors by 2‐PAA was still possible, thereby inducing an increase in gap junction coupling. This increase was, however, not statistically significant. Moreover, an involvement of the A2A adenosine receptor subtype to a low extent cannot completely be ruled out. However, our results obtained using hCMEC/D3 cells as model suggest that the regulation of gap junction coupling in cerebral microvascular cells is mainly related to A2B adenosine receptor subtypes.
Activation of the A2B adenosine receptor subtype and subsequent upregulation of its expression in endothelial cells has been proposed as a mechanism for preventing vascular leakage (Eltzschig et al. 2003; Eckle et al. 2008) and inflammatory signalling (Yang et al. 2006; Blackburn et al. 2009). Additionally, it was found that activation of the A2B adenosine receptor subtype stimulated the expression of vascular endothelial growth factor and angiogenesis in microvascular endothelial cells (Feoktistov et al. 2002; Du et al. 2015). Our findings identify gap junction coupling as another target of the A2B adenosine receptor‐related signalling in endothelial cells of the microvascular system. Whether and how gap junction coupling is integrated in, for example, the anti‐inflammatory or angiogenic properties of A2B adenosine receptor stimulation is an exciting topic for future research; to this end the present report clearly shows the physiological link between A2B adenosine receptor activation and cAMP‐dependent regulation of gap junction coupling.
The enhancement of gap junction coupling in different cells by cAMP‐ and PKA‐dependent pathways is an accepted mechanism (Mehta et al. 1992; Atkinson et al. 1995; Paulson et al. 2000; Lampe & Lau 2004). A general increase in the cAMP concentration by application of forskolin or 8‐Br‐cAMP also increased the gap junction coupling in hCMEC/D3 cells (Fig. 4 B). Recently, we showed that the adenosine transporter inhibitor dipyridamole induced an enhancement of gap junction coupling in aortic endothelial cells as well as in aortic smooth muscle cells through activation of adenosine receptors, an effect that was suppressed in presence of PKA inhibitors (Begandt et al. 2010, 2013 a,b). In the present report the PKA inhibitors Rp‐cAMPS and KT5720 were not able to suppress the 2‐PAA‐related enhancement of gap junction coupling in hCMEC/D3 cells (Fig. 5 A). Additionally, an activator of Epac, another effector of cAMP, also failed to affect the gap junction coupling (Fig. 5 A), suggesting that another cAMP‐dependent mechanism is activated in these BBB endothelial cells.
CNG channels constitute a third target for cAMP. RT‐PCR experiments showed that the CNG channel subunits A1, A2 and B1 were expressed in hCMEC/D3 cells (Fig. 5 C). Western blotting confirmed that the especially cAMP‐sensitive A2 subunit of CNG channels (Kaupp & Seifert 2002; Cheng et al. 2008) is expressed in hCMEC/D3 cells (Fig. 5 D). Electrophysiological patch‐clamp experiments showed that 2‐PAA increased the current in the cells (Fig. 5 E) without significantly affecting the membrane potential. However, the CNG channel inhibitor l‐ cis‐diltiazem antagonized the 2‐PAA‐related current changes (Fig. 5 E), suggesting that 2‐PAA induced the opening of CNG channels. It could be expected that BBB endothelial cells might express CNG channels since expression and a physiological role of CNG channels in various endothelial systems has been shown previously (Zhang et al. 2002; Cheng et al. 2003; Cheng et al. 2008). Moreover, it was shown that an adenosine receptor‐related increase in cAMP enhanced the membrane current and intracellular Ca2+ concentration in endothelial cells and that these effects were both blocked by CNG channel inhibitors (Cheng et al. 2008). Interestingly, in those experiments it was also the A2B adenosine receptor subtype that led to the CNG channel activation (Cheng et al. 2008), which is in accordance with our results.
With respect to gap junction coupling of hCMEC/D3 cells, l‐ cis‐diltiazem suppressed the 2‐PAA‐related enhancement of gap junction coupling as shown by scrape loading/dye transfer experiments (Fig. 5 B). This indicates that activation of CNG channels was required for the 2‐PAA‐induced enhancement of gap junction coupling. Interestingly, l‐ cis‐diltiazem also attenuated the increase in gap junction coupling induced by the adenylyl cyclase activator forskolin (data not shown), indicating that cAMP‐dependent activation of CNG channels was crucial for the cAMP‐mediated effect on the gap junction coupling. As cation permeable channels, the CNG channels can affect the membrane potential of the cells and allow a Ca2+ influx into the cells (Kaupp & Seifert 2002). Since we did not observe a significant change in the membrane potential of the cells when we applied 2‐PAA in the whole‐cell patch‐clamp experiments (Fig. 5 E), we concluded that the 2‐PAA‐related increase in gap junction coupling necessitated a Ca2+ influx. The finding that the 2‐PAA‐related increase in gap junction coupling was reduced if 2‐PAA was applied to cells preloaded with the internal Ca2+ chelator BAPTA or if 2‐PAA was applied in the absence of external Ca2+ (Fig. 6 C) support this conclusion. Preloading the cells with BAPTA also attenuated the forskolin‐induced increase in gap junction coupling (data not shown), indicating the importance of Ca2+ in the action mechanism of cAMP on the gap junction coupling. Additionally, Ca2+ imaging showed that 2‐PAA led to a slow and moderate increase in the intracellular Ca2+ signal (Fig. 6 A). A rapid and transient increase in Ca2+ could, however, be observed if 2‐PAA was applied in the absence of external Na+ (Fig. 6 B). These results suggest that 2‐PAA induced an activation of CNG channels but that under normal extracellular Na+ concentrations the fractional Ca2+ current is reduced. We therefore assume that under normal conditions, the intracellular Ca2+ increase was mostly related to a locally restricted Ca2+ increase in the vicinity of the membrane rather than to a general influx of Ca2+ into the cytoplasmic space. This assumption is supported by comparison of the results obtained when 2‐PAA was applied to cells preloaded with the Ca2+ chelators EGTA or BAPTA. BAPTA antagonized the 2‐PAA‐induced increase in gap junction coupling, but EGTA did not (data not shown). This might be because BAPTA diffuses readily, coming into close proximity with the cell membrane (Rousset et al. 2004), and is therefore able to efficiently chelate Ca2+ that is located right underneath the cell membrane, while EGTA, due to its limited diffusion, mainly chelates the Ca2+ in the core of the cells and does not reach the membrane region (Rousset et al. 2004). Furthermore, BAPTA can effectively chelate Ca2+ derived from nano‐ and microdomains due to its faster Ca2+ binding rate constant (Parekh 2008; Fakler & Adelman 2008). Overall, the results suggest that 2‐PAA by binding to A2B adenosine receptors activates adenylyl cyclase which synthesises cAMP. cAMP in turn induces a locally restricted Ca2+ influx through CNG channels. This Ca2+ stimulates insertion of connexons in the membrane and leads to an increase in gap junction plaques between the cells, resulting in an increase in gap junction coupling.
To our knowledge, a physiological link between the A2B adenosine receptor, CNG channels and gap junction coupling as shown in this report (Fig. 7) has not been described before. The detailed mechanism by which the reinforcement of gap junction plaques and gap junction coupling in hCMEC/D3 cells is achieved by a Ca2+ influx through CNG channel activation is currently a matter of speculation. Protein kinase C (PKC) or Ca2+/calmodulin‐dependent protein kinase II (CaMKII)‐dependent pathways do not seem to play a role, since the PKC activator TPA did not enhance gap junction coupling in the hCMEC/D3 cells. Moreover, inhibitors of PKC (bisindolylmaleimide II) and of CaMKII (KN93) did not significantly affect the 2‐PAA‐related enhancement of gap junction coupling (data not shown). Considering the relatively fast increase in gap junction coupling and the observation of a parallel increased formation of gap junction plaques, we propose that the increased Ca2+ induced formation of new gap junction channels in the membrane in a secretion process of connexon containing vesicles.
Figure 7. Proposed mechanism of action of 2‐PAA to increase the gap junction coupling in hCMEC/D3 cells (continuous arrows).
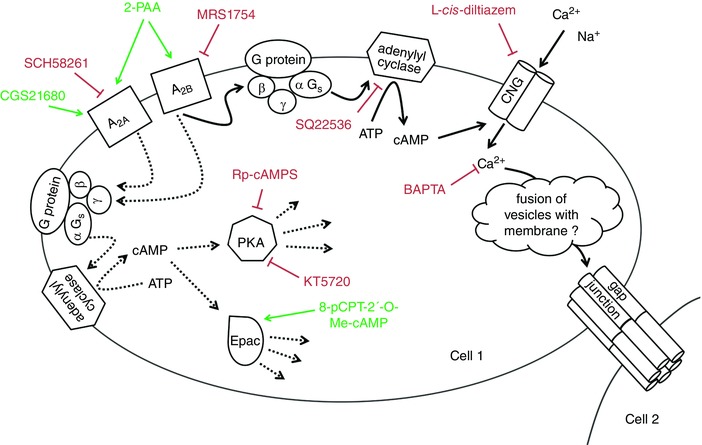
The stimulation of A2B adenosine receptors induces synthesis of cAMP which leads to a Ca2+ influx by opening CNG channels. We hypothesise that Ca2+ in turn induces the increase in gap junction plaques resulting in an enhancement of the gap junction coupling. The increase in the gap junction plaques is related by an as‐yet‐unidentified mechanism which is probably a fusion of connexon‐containing vesicles with the cell membrane. The green arrows indicate activation, the red bars indicate inhibition. The dotted arrows indicate other possible adenosine receptor‐dependent signalling mechanisms which were found not to be involved in the regulation of gap junction coupling in the cerebral microvascular endothelial cells in the present report. [Color figure can be viewed at wileyonlinelibrary.com]
Additional information
Competing interests
The authors declare no conflict of interests.
Author contributions
Experiments were performed in the laboratory of A.N. A.B., W.B., D.B. and A.N. contributed to the conception and design of the work as well as writing and revising the manuscript. A.B., W.B., A.K., I.G.S., C.G., F.S. and A.N. contributed to the acquisition, analysis, or interpretation of data for the work. B.W., I.R. and P.O.C. provided the hCMEC/D3 cells and revised the work critically. S.W.H. provided the STED microscopy and a secondary antibody and also revised the work critically. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The work was partly supported by Boehringer Ingelheim International GmbH and the DFG project “Elektrodenoptimierung für Neuroprothesen” (NG 4/10‐1).
Acknowledgements
The authors thank Nicolai Urban for help with the STED microscopy set‐up. The authors also thank Kristina Schmitt for her assistance with the experiments and Nadine Dilger for her help with the patch‐clamp data analysis.
Linked articles This article is highlighted by a Perspective by Sáez. To read this Perspective, visit http://dx.doi.org/10.1113/JP274027.
References
- Atkinson MM, Lampe PD, Lin HH, Kollander R, Li XR & Kiang DT (1995). Cyclic AMP modifies the cellular distribution of connexin43 and induces a persistent increase in the junctional permeability of mouse mammary tumor cells. J Cell Sci 108, 3079–3090. [DOI] [PubMed] [Google Scholar]
- Avila MA, Sell SL, Hawkins BE, Hellmich HL, Boone DR, Crookshanks JM, Prough DS & DeWitt DS (2011). Cerebrovascular connexin expression: effects of traumatic brain injury. J Neurotrauma 28, 1803–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee U & Cheng X (2015). Exchange protein directly activated by cAMP encoded by the mammalian rapgef3 gene: structure, function and therapeutics. Gene 570, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begandt D, Bader A, Dreyer L, Eisert N, Reeck T & Ngezahayo A (2013. a). Biphasic increase of gap junction coupling induced by dipyridamole in the rat aortic A‐10 vascular smooth muscle cell line. J Cell Commun Signal 7, 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begandt D, Bader A, Gerhard L, Lindner J, Dreyer L, Schlingmann B & Ngezahayo A (2013. b). Dipyridamole‐related enhancement of gap junction coupling in the GM‐7373 aortic endothelial cells correlates with an increase in the amount of connexin 43 mRNA and protein as well as gap junction plaques. J Bioenerg Biomembr 45, 409–419. [DOI] [PubMed] [Google Scholar]
- Begandt D, Bintig W, Oberheide K, Schlie S & Ngezahayo A (2010). Dipyridamole increases gap junction coupling in bovine GM‐7373 aortic endothelial cells by a cAMP‐protein kinase A dependent pathway. J Bioenerg Biomembr 42, 79–84. [DOI] [PubMed] [Google Scholar]
- Beyer EC & Berthoud VM (2009). The family of connexin genes In Connexins: A Guide, eds Harris AL. & Locke D, pp. 3–26. Springer, New York. [Google Scholar]
- Bintig W, Baumgart J, Walter WJ, Heisterkamp A, Lubatschowski H & Ngezahayo A (2009). Purinergic signalling in rat GFSHR‐17 granulosa cells: an in vitro model of granulosa cells in maturing follicles. J Bioenerg Biomembr 41, 85–94. [DOI] [PubMed] [Google Scholar]
- Blackburn MR, Vance CO, Morschl E & Wilson CN (2009). Adenosine receptors and inflammation. Handb Exp Pharmacol 193, 215–269. [DOI] [PubMed] [Google Scholar]
- Bock M de, Culot M, Wang N, da CA, Decrock E, Bol M, Bultynck G, Cecchelli R & Leybaert L (2012). Low extracellular Ca2+ conditions induce an increase in brain endothelial permeability that involves intercellular Ca2+ waves. Brain Res 1487, 78–87. [DOI] [PubMed] [Google Scholar]
- Bodin P, Bailey D & Burnstock G (1991). Increased flow‐induced ATP release from isolated vascular endothelial cells but not smooth muscle cells. Br J Pharmacol 103, 1203–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodin P & Burnstock G (2001). Evidence that release of adenosine triphosphate from endothelial cells during increased shear stress is vesicular. J Cardiovasc Pharmacol 38, 900–908. [DOI] [PubMed] [Google Scholar]
- Bruns RF, Lu GH & Pugsley TA (1986). Characterization of the A2 adenosine receptor labeled by [3H]NECA in rat striatal membranes. Mol Pharmacol 29, 331–346. [PubMed] [Google Scholar]
- Bynoe MS, Viret C, Yan A & Kim DG (2015). Adenosine receptor signaling: a key to opening the blood–brain door. Fluids Barriers CNS 12, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman AJ, Mills JH, Krenz A, Kim DG & Bynoe MS (2011). Adenosine receptor signaling modulates permeability of the blood–brain barrier. J Neurosci 31, 13272–13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CC, Yang YL, Liao KH & Lai TW (2016). Adenosine receptor agonist NECA increases cerebral extravasation of fluorescein and low molecular weight dextran independent of blood–brain barrier modulation. Sci Rep 6, 23882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Chan FL, Huang Y, Chan WY & Yao X (2003). Expression of olfactory‐type cyclic nucleotide‐gated channel (CNGA2) in vascular tissues. Histochem Cell Biol 120, 475–481. [DOI] [PubMed] [Google Scholar]
- Cheng KT, Leung YK, Shen B, Kwok YC, Wong CO, Kwan HY, Man YB, Ma X, Huang Y & Yao X (2008). CNGA2 channels mediate adenosine‐induced Ca2+ influx in vascular endothelial cells. Arterioscler Thromb Vasc Biol 28, 913–918. [DOI] [PubMed] [Google Scholar]
- Choksi NY, Hussain A & Booth RG (1997). 2‐Phenylaminoadenosine stimulates dopamine synthesis in rat forebrain in vitro and in vivo via adenosine A2 receptors. Brain Res 27, 151–155. [DOI] [PubMed] [Google Scholar]
- Davidson JO, Green CR, Nicholson LF, O'Carroll SJ, Fraser M, Bennet L & Gunn AJ (2012). Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann Neurol 71, 121–132. [DOI] [PubMed] [Google Scholar]
- Du X, Ou X, Song T, Zhang W, Cong F, Zhang S & Xiong Y (2015). Adenosine A2B receptor stimulates angiogenesis by inducing VEGF and eNOS in human microvascular endothelial cells. Exp Biol Med (Maywood) 240, 1472–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF & Eltzschig HK (2008). A2B adenosine receptor dampens hypoxia‐induced vascular leak. Blood 111, 2024–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK (2009). Adenosine: an old drug newly discovered. Anesthesiology 111, 904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC & Colgan SP (2003). Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med 198, 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Basilio D, Sáez JC, Orellana JA, Raine CS, Bukauskas F, Bennett MV & Berman JW (2012). The role of gap junction channels during physiologic and pathologic conditions of the human central nervous system. J Neuroimmune Pharmacol 7, 499–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakler B & Adelman JP (2008). Control of KCa channels by calcium nano/microdomains. Neuron 59, 873–881. [DOI] [PubMed] [Google Scholar]
- Falk MM, Kells RM & Berthoud VM (2014). Degradation of connexins and gap junctions. FEBS Lett 588, 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistov I, Goldstein AE, Ryzhov S, Zeng D, Belardinelli L, Voyno‐Yasenetskaya T & Biaggioni I (2002). Differential expression of adenosine receptors in human endothelial cells: role of A2B receptors in angiogenic factor regulation. Circ Res 90, 531–538. [DOI] [PubMed] [Google Scholar]
- Figueroa XF & Duling BR (2009). Gap junctions in the control of vascular function. Antioxid Redox Signal 11, 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB (2007). Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 14, 1315–1323. [DOI] [PubMed] [Google Scholar]
- Gao X, Qian J, Zheng S, Changyi Y, Zhang J, Ju S, Zhu J & Li C (2014). Overcoming the blood–brain barrier for delivering drugs into the brain by using adenosine receptor nanoagonist. ACS Nano 8, 3678–3689. [DOI] [PubMed] [Google Scholar]
- Haefliger JA, Nicod P & Meda P (2004). Contribution of connexins to the function of the vascular wall. Cardiovasc Res 62, 345–356. [DOI] [PubMed] [Google Scholar]
- Johnstone S, Isakson B & Locke D (2009). Biological and biophysical properties of vascular connexin channels. Int Rev Cell Mol Biol 278, 69–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K, Solan JL, Dominguez M, Sia M, Hand A, Lampe PD & Laird DW (1999). Trafficking, assembly, and function of a connexin43‐green fluorescent protein chimera in live mammalian cells. Mol Biol Cell 10, 2033–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameritsch P, Pogoda K & Pohl U (2012). Channel‐independent influence of connexin 43 on cell migration. Biochim Biophys Acta 1818, 1993–2001. [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Tachikawa M, Akaogi R, Fujimoto K, Ishibashi M, Uchida Y, Couraud PO, Ohtsuki S, Hosoya K & Terasaki T (2015). Contribution of pannexin 1 and connexin 43 hemichannels to extracellular calcium‐dependent transport dynamics in human blood–brain barrier endothelial cells. J Pharmacol Exp Ther 353, 192–200. [DOI] [PubMed] [Google Scholar]
- Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J & Nedergaard M (2008). Connexin 43 hemichannels are permeable to ATP. J Neurosci 28, 4702–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp UB & Seifert R (2002). Cyclic nucleotide‐gated ion channels. Physiol Rev 82, 769–824. [DOI] [PubMed] [Google Scholar]
- Kim DG & Bynoe MS (2015). A2A adenosine receptor regulates the human blood–brain barrier permeability. Mol Neurobiol 52, 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz A, Della Bruna R, Pfeilschifter J & Bauer C (1988). Role of cGMP as second messenger of adenosine in the inhibition of renin release. Kidney Int 33, 798–803. [DOI] [PubMed] [Google Scholar]
- Lampe PD & Lau AF (2004). The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol 36, 1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan E, Shao Q, Wang HL, Langlois S & Laird DW (2006). Connexins act as tumor suppressors in three‐dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res 66, 9886–9894. [DOI] [PubMed] [Google Scholar]
- Mehta PP, Yamamoto M & Rose B (1992). Transcription of the gene for the gap junctional protein connexin43 and expression of functional cell‐to‐cell channels are regulated by cAMP. Mol Biol Cell 3, 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melani A, Pugliese AM & Pedata F (2014). Adenosine receptors in cerebral ischemia. Int Rev Neurobiol 119, 309–348. [DOI] [PubMed] [Google Scholar]
- Mills JH, Alabanza L, Weksler BB, Couraud P‐O, Romero IA & Bynoe MS (2011). Human brain endothelial cells are responsive to adenosine receptor activation. Purinergic Signal 7, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa K, Chiba H, Fujita H, Kojima T, Saito T, Endo T & Sawada N (2006). Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J Cell Physiol 208, 123–132. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Gong XQ, Maeda S, Dong Y, Misumi Y, Tsukihara T & Bai D (2011). Asparagine 175 of connexin32 is a critical residue for docking and forming functional heterotypic gap junction channels with connexin26. J Biol Chem 286, 19672–19681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MD, Lee ST, Ross AE, Ryals M, Choudhry VI & Venton BJ (2014). Characterization of spontaneous, transient adenosine release in the caudate‐putamen and prefrontal cortex. PLoS One 9, e87165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MD & Venton BJ (2015). Fast‐scan cyclic voltammetry for the characterization of rapid adenosine release. Comput Struct Biotechnol J 13, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB (2008). Ca2+ microdomains near plasma membrane Ca2+ channels: impact on cell function. J Physiol 586, 3043–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson AF, Lampe PD, Meyer RA, TenBroek E, Atkinson MM, Walseth TF & Johnson RG (2000). Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J Cell Sci 113, 3037–3049. [DOI] [PubMed] [Google Scholar]
- Pinheiro JC & Bates DM (2000). Mixed‐effects models in S and S‐PLUS In Statistics and Computing, eds Chambers J, Eddy W, Härdle W, Sheather S. & Tierney L. Springer, New York. [Google Scholar]
- Pinto I, Serpa A, Sebastião AM & Cascalheira JF (2016). The role of cGMP on adenosine A1 receptor‐mediated inhibition of synaptic transmission at the hippocampus. Front Pharmacol 7, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset M, Cens T, Vanmau N & Charnet P (2004). Ca2+‐dependent interaction of BAPTA with phospholipids. FEBS Lett 576, 41–45. [DOI] [PubMed] [Google Scholar]
- Sands WA & Palmer TM (2005). Adenosine receptors and the control of endothelial cell function in inflammatory disease. Immunol Lett 101, 1–11. [DOI] [PubMed] [Google Scholar]
- Schaddelee MP, Voorwinden HL, van Tilburg EW, Pateman TJ, Ijzerman AP, Danhof M & Boer AG de (2003). Functional role of adenosine receptor subtypes in the regulation of blood–brain barrier permeability: possible implications for the design of synthetic adenosine derivatives. Eur J Pharm Sci 19, 13–22. [DOI] [PubMed] [Google Scholar]
- Seifert R, Lushington GH, Mou TC, Gille A & Sprang SR (2012). Inhibitors of membranous adenylyl cyclases. Trends Pharmacol Sci 33, 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpa A, Sebastião AM & Cascalheira JF (2014). Modulation of cGMP accumulation by adenosine A1 receptors at the hippocampus: influence of cGMP levels and gender. Eur J Pharmacol 5, 83–90. [DOI] [PubMed] [Google Scholar]
- Söhl G & Willecke K (2004). Gap junctions and the connexin protein family. Cardiovasc Res 62, 228–232. [DOI] [PubMed] [Google Scholar]
- Stout CE, Costantin JL, Naus CC & Charles AC (2002). Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem 277, 10482–10488. [DOI] [PubMed] [Google Scholar]
- Stout RF Jr, Snapp EL & Spray DC (2015). Connexin type and fluorescent protein fusion tag determine structural stability of gap junction plaques. J Biol Chem 290, 23497–23514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasken K & Aandahl EM (2004). Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev 84, 137–167. [DOI] [PubMed] [Google Scholar]
- Thomas T, Jordan K, Simek J, Shao Q, Jedeszko C, Walton P & Laird DW (2005). Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J Cell Sci 118, 4451–4462. [DOI] [PubMed] [Google Scholar]
- Urban NT, Willig KI, Hell SW & Nagerl UV (2011). STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys J 101, 1277–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksler B, Romero IA & Couraud PO (2013). The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksler BB, Subileau EA, Perrière N, Charneau P, Holloway K, Leveque M, Tricoire‐Leignel H, Nicotra A, Bourdoulous S, Turowski P, Male DK, Roux F, Greenwood J, Romero IA & Couraud PO (2005). Blood–brain barrier‐specific properties of a human adult brain endothelial cell line. FASEB J 19, 1872–1874. [DOI] [PubMed] [Google Scholar]
- Wilson CN & Batra VK (2002). Lipopolysaccharide binds to and activates A1 adenosine receptors on human pulmonary artery endothelial cells. J Endotoxin Res 8, 263–271. [DOI] [PubMed] [Google Scholar]
- Yang D, Zhang Y, Nguyen HG, Koupenova M, Chauhan AK, Makitalo M, Jones MR, St HC, Seldin DC, Toselli P, Lamperti E, Schreiber BM, Gavras H, Wagner DD & Ravid K (2006). The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest 116, 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Xia SL, Block ER & Patel JM (2002). NO upregulation of a cyclic nucleotide‐gated channel contributes to calcium elevation in endothelial cells. Am J Physiol Cell Physiol 283, C1080–C1089. [DOI] [PubMed] [Google Scholar]