

Abstract
Key points
Heart failure with preserved ejection fraction (HFpEF) is associated with disordered breathing patterns, and sympatho‐vagal imbalance.
Although it is well accepted that altered peripheral chemoreflex control plays a role in the progression of heart failure with reduced ejection fraction (HFrEF), the pathophysiological mechanisms underlying deterioration of cardiac function in HFpEF are poorly understood.
We found that central chemoreflex is enhanced in HFpEF and neuronal activation is increased in pre‐sympathetic regions of the brainstem.
Our data showed that activation of the central chemoreflex pathway in HFpEF exacerbates diastolic dysfunction, worsens sympatho‐vagal imbalance and markedly increases the incidence of cardiac arrhythmias in rats with HFpEF.
Abstract
Heart failure (HF) patients with preserved ejection fraction (HFpEF) display irregular breathing, sympatho‐vagal imbalance, arrhythmias and diastolic dysfunction. It has been shown that tonic activation of the central and peripheral chemoreflex pathway plays a pivotal role in the pathophysiology of HF with reduced ejection fraction. In contrast, no studies to date have addressed chemoreflex function or its effect on cardiac function in HFpEF. Therefore, we tested whether peripheral and central chemoreflexes are hyperactive in HFpEF and if chemoreflex activation exacerbates cardiac dysfunction and autonomic imbalance. Sprague‐Dawley rats (n = 32) were subjected to sham or volume overload to induce HFpEF. Resting breathing variability, chemoreflex gain, cardiac function and sympatho‐vagal balance, and arrhythmia incidence were studied. HFpEF rats displayed [mean ± SD; chronic heart failure (CHF) vs. Sham, respectively] a marked increase in the incidence of apnoeas/hypopnoeas (20.2 ± 4.0 vs. 9.7 ± 2.6 events h−1), autonomic imbalance [0.6 ± 0.2 vs. 0.2 ± 0.1 low/high frequency heart rate variability (LF/HFHRV)] and cardiac arrhythmias (196.0 ± 239.9 vs. 19.8 ± 21.7 events h−1). Furthermore, HFpEF rats showed increase central chemoreflex sensitivity but not peripheral chemosensitivity. Accordingly, hypercapnic stimulation in HFpEF rats exacerbated increases in sympathetic outflow to the heart (229.6 ± 43.2% vs. 296.0 ± 43.9% LF/HFHRV, normoxia vs. hypercapnia, respectively), incidence of cardiac arrhythmias (196.0 ± 239.9 vs. 576.7 ± 472.9 events h−1) and diastolic dysfunction (0.008 ± 0.004 vs. 0.027 ± 0.027 mmHg μl−1). Importantly, the cardiovascular consequences of central chemoreflex activation were related to sympathoexcitation since these effects were abolished by propranolol. The present results show that the central chemoreflex is enhanced in HFpEF and that acute activation of central chemoreceptors leads to increases of cardiac sympathetic outflow, cardiac arrhythmogenesis and impairment in cardiac function in rats with HFpEF.
Keywords: autonomic imbalance, cardiac function, central chemoreflex, heart failure preserved ejection fraction, respiratory disorders
Key points
Heart failure with preserved ejection fraction (HFpEF) is associated with disordered breathing patterns, and sympatho‐vagal imbalance.
Although it is well accepted that altered peripheral chemoreflex control plays a role in the progression of heart failure with reduced ejection fraction (HFrEF), the pathophysiological mechanisms underlying deterioration of cardiac function in HFpEF are poorly understood.
We found that central chemoreflex is enhanced in HFpEF and neuronal activation is increased in pre‐sympathetic regions of the brainstem.
Our data showed that activation of the central chemoreflex pathway in HFpEF exacerbates diastolic dysfunction, worsens sympatho‐vagal imbalance and markedly increases the incidence of cardiac arrhythmias in rats with HFpEF.
Abbreviations
- ACF
aorto‐caval fistula
- AHI
apnoea/hypopnoea index
- CHF
chronic heart failure
- CO
cardiac output
- DHE
dihydroethidium
- EDPVR
end‐diastolic pressure–volume relationship
- EF
ejection fraction
- ESPVR
end‐systolic pressure–volume relationship
- HCVR
hypercapnic ventilatory response
- HF
heart failure
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- HR
heart rate
- HRV
heart rate variability
- HVR
hypoxic ventilatory response
- LV
left ventricle
- LVEDP
left ventricular end‐diastolic pressure
- LVEDV
left ventricular end‐diastolic volume
- LVESP
left ventricular end‐systolic pressure
- LVESV
left ventricular end‐systolic volume
- PSD
power spectral density
- PV
pressure–volume
- RR
respiratory frequency
- RTN
retrotrapezoid nucleus
- RVLM
rostral ventrolateral medulla
- SV
stroke volume
- VE
minute ventilation
- Vt
tidal volume
Introduction
Heart failure (HF) with preserved ejection fraction (HFpEF) is a highly prevalent form of HF affecting 47% of all HF patients (Owan et al. 2006). HFpEF is characterized by progressive cardiac diastolic function impairment (Oktay et al. 2013; Yancy et al. 2013), sleep disordered breathing (Oldenburg et al. 2007), increased sympathetic outflow, parasympathetic withdrawal and decreased heart rate variability (Kristen et al. 2002; Borlaug & Kass, 2006; Abassi et al. 2011; Andrade et al. 2015). Importantly, autonomic imbalance and sleep disordered breathing are related to the progression of HF (Florea & Cohn, 2014). Altered peripheral and central chemoreflex drive contributes to autonomic imbalance and sleep disordered breathing (Blain et al. 2010; Mansukhani et al. 2014) and has been proposed as a key component in the progression of HF. Chemoreflex activation in HF with reduced EF (HFrEF) is associated with increased sympathetic nerve activity and incidence of cardiac arrhythmias, both being inversely correlated with survival (Giannoni et al. 2009). In concordance with these observations in clinical populations, Del Rio et al. (2013) found that selective bilateral denervation of the carotid body, the main peripheral chemoreceptor, significantly decreased cardiac sympathetic drive and markedly improved survival of HFrEF rats. Therefore, alterations in the chemoreflex pathways may also contribute to the progression of HFpEF. Despite what is known about chemoreflex function in HFrEF, few studies have addressed the potential contribution of the chemoreflex to autonomic imbalance and disordered breathing patterns in HFpEF.
Existing evidence suggests that peripheral chemoreceptors are not involved in HFpEF pathophysiology but that central chemoreceptors may contribute to the onset and maintenance of autonomic imbalance and disordered breathing in HFpEF (Andrade et al. 2015; Toledo et al. 2016). Kristen et al. (2002) showed an increase in renal sympathetic nerve discharge following acute stimulation with hypercapnia in HFpEF rats. These results suggest that activation of central chemoreceptors in HFpEF may increase sympathetic outflow. However, there are no studies showing the effects of activation of the peripheral and/or central chemoreflex drive in cardiac function and/or cardiac sympatho‐vagal balance in HFpEF.
There are numerous regions in the brain that have been shown to exhibit CO2 sensitivity, although the retrotrapezoid nucleus (RTN) is currently considered one of the main central chemoreceptor areas (Lazarenko et al. 2009; Guyenet & Mulkey, 2010; Wang et al. 2013). In normal conditions, subtle changes in cerebrospinal fluid CO2 and/or pH induce RTN neuron depolarization leading to excitatory stimulation of respiratory and cardiovascular control centres in the brainstem (Guyenet et al. 2005, 2010; Guyenet, 2014). Importantly, it has been shown that RTN neurons project to pre‐sympathetic neurons of the rostral ventrolateral medulla (RVLM), a key area involved in autonomic regulation (Moreira et al. 2006; Rosin et al. 2006). We recently showed that chronic activation of the RVLM in HFrEF rats is critically dependent on enhanced afferent stimulation from the carotid body chemoreceptors and is associated with HF progression and mortality (Del Rio et al. 2013). To date, no studies have addressed the effects of changes in central chemoreflex drive on pre‐sympathetic RVLM neuron activation in HFpEF.
We hypothesized that acute activation of the chemoreflex drive in HFpEF, which may result from disordered breathing patterns, exacerbates cardiac dysfunction and cardiac autonomic imbalance and further increases the incidence of arrhythmia. In the present study we sought to determine whether HFpEF rats display respiratory abnormalities and altered peripheral and/or central chemoreflex function. Furthermore, we studied whether cardiac autonomic imbalance in HFpEF is associated with chronic activation of RVLM neurons. Finally, we tested whether acute activation of central and peripheral chemoreflex pathways alters cardiac sympatho‐vagal balance, increases arrhythmia incidence and exacerbates cardiac dysfunction in HFpEF.
Methods
Ethical approval
All animals were housed and handled according to the American Physiological Society and the National Institutes of Health guidelines for the Care and Use of Laboratory Animals and the Guía para el Cuidado y Uso de los Animales de Laboratorio from CONICYT. All experimental protocols were approved by the Universidad Autónoma de Chile and conformed to the principles and regulations as described by Grundy (2015). Efforts were made to limit the number of subjects and minimize animal suffering according to the principles of reduction, replacement and refinement in experimental design. Animals were given access to food (Prolab RMH 3000, LabDiet, St Louis, MO, USA) and water ad libitum.
Animals
Adult male Sprague‐Dawley rats (n = 32), weighing initially ∼250 g, were used in this study. All experiments were performed 8 weeks following chronic heart failure (CHF) induction. Rats were group housed (maximum three per cage). At the end of the appropriate experiments, all animals were humanely killed by an overdose of anaesthetic (sodium pentobarbital 100 mg kg−1 i.p.).
Rat model of CHF
CHF with preserved ejection fraction was produced in rats (n = 16) by volume overload by aorto‐caval fistula (ACF) using the needle technique, as described previously (Brodsky et al. 1998; Modesti et al. 2004; Ryan et al. 2007; Toischer et al. 2010; Abassi et al. 2011). Briefly, under anaesthesia (2% isoflurane/98%O2) a laparotomy was performed. The wall between the inferior vena cava and the abdominal aorta was grasped through a longitudinal incision made in the aorta, and a fistula was created between the two vessels using a 1.20 × 40 mm needle (BD Precision Glide, Franklin Lakes, NJ, USA). The opening in the aorta was then closed with tissue adhesive (Hystoacryl, Braun, USA). Following these manoeuvres, the abdomen was closed in layers. Sham‐operated rats (n = 16) were prepared in the same manner but did not undergo ACF. All experiments were performed at 8 weeks after ACF or sham surgery.
Resting breathing and ventilatory chemoreflex function
Resting breathing was evaluated by unrestrained whole body plethysmography in Sham (n = 6) and CHF rats (n = 6) (Del Rio et al. 2013). Apnoea episodes (cessation of breathing ≥3 breaths) and hypopneas [reductions ≥50% in tidal volume (V t)] were averaged during resting breathing, as previously described (Del Rio et al. 2013; Haack et al. 2014; Marcus et al. 2014a). Respiratory stability during resting breathing was illustrated by construction of Poincare plots and quantified by analysis of SD1 and SD2 of the inter‐breath interval variability (Haack et al. 2014; Marcus et al. 2014a). Tidal volume, respiratory frequency (RR) and minute ventilation (V E: V t × RR) were determined by unrestrained whole body plethysmography (Del Rio et al. 2013). Tidal volume was measured by temporarily (15–30 s) sealing the air ports and measuring the pressure changes in the sealed chamber using a Validyne (MP‐45) differential pressure transducer and amplifier connected to a PowerLab System (AD Instruments, Australia). Chamber pressure fluctuations were proportional to tidal volume. Resting breathing was recorded for 2 h while the rats breathed room air. Peripheral chemoreceptors were stimulated preferentially by allowing the rats to breathe hypoxic (10% O2/balance N2) gas for 2–5 min under isocapnic conditions. Hyperoxic hypercapnia (7% CO2/93% O2) gas challenges were given for 2–5 min. The hypoxic ventilatory response (HVR) was calculated by the slope between 21% and 10%. The hypercapnic ventilatory response (HCVR) was calculated by the slope between 0.03% and 7%. All recordings were made at an ambient temperature of 25 ± 2°C, as previously described (Del Rio et al. 2013; Haack et al. 2014; Marcus et al. 2014a).
Measurements of arterial blood gases during hypercapnic stimulation
The arterial blood gases were measured in conscious freely moving rats (n = 4, CHF; n = 4, Sham) using a blood gas analyser (i‐STAT1 Analyser). Under isoflurane (2%) rats were anaesthetized and a vascular access port was placed in the carotid artery. One week after surgery the animals were put in a plexiglass chamber and 100 μl was withdrawn during normoxia and hypercapnia. The volume of withdrawn blood was immediately replaced by an equal volume of sterile saline solution (0.9% NaCl).
Measurements of baseline haemodynamic and cardiac function
Arterial blood pressure and left ventricular haemodynamic parameters were measured with a conductance catheter in anaesthetized animals (n = 10, CHF; n = 10, Sham) as previously described (Pacher et al. 2008; Rommel et al. 2016). Rats were anaesthetized with α‐chloralose and urethane (40 and 800 mg kg−1, respectively), and were intubated with a 16‐g cannula. A pressure–volume (PV) conductance catheter (SPR‐869; Millar, USA) was placed into the right carotid artery and advanced to the left ventricle (LV) (Cingolani et al. 2004; Pacher et al. 2008; Rommel et al. 2016). We made a laparotomy to visualize the arteria‐venous fistula, and then isolated the cava vein to perform the vein occlusion for the measurement of cardiac function. After a 25 min equilibration period, baseline PV loops were recorded and LV haemodynamic parameters were calculated using 10–15 consecutive PV loops. Baseline haemodynamic PV loop parameters were: left ventricular end‐systolic volume (LVESV); left ventricular end‐diastolic volume (LVEDV); left ventricular end‐systolic pressure (LVESP); left ventricular end‐diastolic pressure (LVEDP); stroke volume (SV); cardiac output (CO) and ejection fraction (EF). Load‐dependent cardiac function parameters were determined by means of the calculation of dp/dt max and dp/dt min. Load‐independent cardiac function parameters were studied by calculating the end‐systolic PV relationship (ESPVR) and the end‐diastolic PV relationship (EDPVR). Briefly, transient occlusions of the inferior vena cava were performed to obtain ESPVR and EDPVR by fitting the data to a linear and an exponential function, respectively (Cingolani & Kass, 2011; Hamdani et al. 2013; Leite et al. 2015).We also calculated ESPVR and EDPVR during hypercapnia ( 7%) and hypoxia ( 10%) exposure to estimate the effects of central and peripheral chemoreflex activation on cardiac function, respectively. Volumes were calibrated using arterial blood using the cuvette calibration method (Pacher et al. 2008). We also performed saline injections (n = 4 per group) to fully calibrate the conductance catheter before propranolol administration (Pacher et al. 2008). Data analysis was performed using the PV loop module of the LabChart 7.0 software.
Sympatho‐vagal balance during hypercapnia
Heart rate variability (HRV) was analysed using LabChart pro 7.0 software in Sham (n = 4) and CHF rats (n = 4). We used dp/dt signals obtained from blood pressure recordings to calculate heart rate. Power spectral density (PSD) of HRV was obtained using a Fast Fourier Transform algorithm after Hann windowing with 50% overlap (Pagani et al. 1986; Marcus et al. 2014b). Analysis was performed in 10 min intervals obtained when the rats were in normoxia ( 21%) and hypercapnia ( 7%). Cut‐off frequencies were defined as: low frequency (LF), 0.04–0.6 Hz; and high frequency (HF), 0.6–2.4 Hz, as previously described (Ryan et al. 2007). In addition, propranolol chlorhydrate (1 mg kg−1, i.v.) and atropine sulphate (1 mg kg−1, i.v.) were used to fully determine sympathetic and parasympathetic tone to the heart, respectively (Del Rio et al. 2016). The cardiac chronotropic response was expressed as ΔHR (n = 6, CHF; n = 6, Sham).
Arrhythmia incidence
Heart rate was derived from blood pressure dp/dt waveforms. Irregular heartbeats were visually inspected and counted as previously described (n = 6, CHF; n = 6, Sham) (Aboukhalil et al. 2008; Del Rio et al. 2013). Arrhythmias were defined as premature or delayed beats with changes greater than 3 standard deviations (SD) from the mean beat‐to‐beat interval duration. We also evaluated arrhythmia incidence during hypercapnic stimulation ( 7%). The contribution of sympathetic drive on arrhythmia incidence was studied using intravenous infusion of propranolol (1 mg kg−1 i.v.). The arrhythmia index was expressed as events h–1.
Western blot
Chronic neuronal activation was assessed by immunoblot of Fos‐B (1:100 Santa Cruz Biotechnology, Dallas, TX, USA) in RVLM micropunches obtained from CHF (n = 6) rats and Sham rats (n = 6) as previously described (Haack et al. 2012; Rommel et al. 2016). Fos‐B expression is induced during chronic neuronal activation. Briefly, coronal sections (100 μm) were cut through the medulla at the level of the RVLM according to the atlas of Paxinos et al. (1980). Later, RVLMs were bilaterally punched using a diethylpyrocarbonate (DEPC)‐treated blunt 18‐gauge needle attached to a syringe as previously described (Palkovits, 1983). Micropunches containing the RVLM were removed and snap‐frozen on dry ice and stored at −80°C. Micropunches were lysed in RIPA buffer plus protease inhibitor cocktail (1%) followed by sonication. Total protein extract concentration was determined using a BCA protein assay kit (ThermoScientific, Waltham, MA, USA). Proteins were then separated by polyacrylamide gel electrophoresis (10%), transferred to PVDF membranes (Immobilon‐P, Millipore, Billerica, MA, USA) and probed with a rabbit polyclonal anti Fos‐B antibody (1:100, Santa Cruz Biotechnology) overnight at 4°C. Membranes were washed in Tris‐buffered saline with 0.1% Tween, and incubated for 2 h with a horseradish peroxidase‐conjugated goat anti‐rabbit secondary antibody (Thermo Fisher). Following stripping procedure (Restor Plus, Thermo Scientific), the membranes were probed with a monoclonal anti‐β‐actin antibody (1:2000) (Sigma‐Aldrich, St Louis, MO, USA). The relative amount of protein of interest was calculated as the ratio of intensity of the band relative to the intensity of β‐actin. No difference in the level of expression of β‐actin was found between groups.
Oxidative stress in pre‐sympathetic neurons from the RVLM
Superoxide levels in the RVLM were measured using dihydroethidium (DHE), a cell membrane‐permeable superoxide‐sensitive fluorescent dye, in Sham (n = 6) and CHF rats (n = 6) as described previously (Banes et al. 2005; Oliveira‐Sales et al. 2010). Snap‐frozen brains were sectioned on a cryostat at 20 μm thickness at −20°C and placed onto electrostatically charged microscope slides (Superfrost, VWR Scientific, Radnor, PA, USA). Brain sections containing the RVLM were incubated with DHE at 1 μm (Life Technologies Inc., Carlsbad, CA, USA) in PBS for 30 min at 37°C, and washed three times for 5 min in PBS. Next, slices were fixed with 4% paraformaldehyde for 10 min at room temperature, and washed twice for 5 min with PBS. After blocking for 60 min (5% gelatin from cold water fish skin, 0.5% Triton X‐100 in PBS), the sections were incubated for 1 h at room temperature with anti‐tyrosine hydroxylase monoclonal antibody (1:100, Millipore) diluted in the same blocking solution. Later, samples were rinsed three times in PBS for 5 min, and incubated with Alexa fluor 488 goat anti‐mouse (Invitrogen) diluted 1:250 in PBS for 1 h at room temperature. Finally, sections were rinsed three times in PBS for 5 min and mounted with DAPI fluorescent mounting media (Vector Shield, Vector Laboratories, Burlingame, CA, USA). Images were acquired with a high‐resolution fluorescence microscope (Leica). Slides were scanned under the same conditions for magnification, laser intensity, brightness, gain and pinhole size. Images were processed using the ImageJ software (NIH image).
Statistical analysis
Data were expressed as means ± SD. Student's t test was used to compare the differences between two groups. Differences among two or more conditions (i.e. treatments) were assessed with one‐ or two‐way ANOVA, followed by Sidak's post hoc comparisons. A P value of < 0.05 was considered statistically significant.
Results
Baseline physiological characteristics
Resting physiological variables of both Sham and CHF animals are summarized in Table 1. CHF rats showed overt signs of cardiac hypertrophy compared to Sham rats, as evidenced by a significant increase in the heart weight to body weight ratio (HW/BW) (3.9 ± 0.6 vs. 2.9 ± 0.3 mg g−1, CHF vs. Sham, respectively; P < 0.01). No significant changes in body weight were observed between groups (Table 1). LVEDP, LVEDV and LVESV were all significantly increased in CHF compared to the values obtained in Sham rats (Table 1). Importantly, LVEF values remained similar between CHF and sham conditions (75.9 ± 10.7 vs. 72.1 ± 7.2%, CHF vs. Sham, respectively; P > 0.05). Baseline heart rate, systolic blood pressure, diastolic blood pressure and pulse pressure were not significantly different in CHF rats compared to Sham rats (Table 1).
Table 1.
Baseline physiological variables
| Sham (n = 10) | CHF (n = 10) | |
|---|---|---|
| BW (g) | 498.0 ± 45.8 | 530.6 ± 42.3 |
| HW (g) | 1.4 ± 0.3 | 2.1 ± 0.3* |
| HW/BW (mg g−1) | 2.9 ± 0.3 | 3.9 ± 0.6* |
| Lung W/D (g g−1) | 4.2 ± 0.6 | 4.2 ± 0.3 |
| SBP (mmHg) | 101.1 ± 12.0 | 102.2 ± 12.9 |
| DBP (mmHg) | 68.8 ± 11.7 | 71.6 ± 16.1 |
| PP (mmHg) | 35.3 ± 10.7 | 30.9 ± 7.5 |
| MABP (mmHg) | 77.6 ± 11.7 | 82.1 ± 16.1 |
| HR (beats min−1) | 344.2 ± 37.6 | 318.3 ± 51.2 |
| LVESV (μl) | 84.6 ± 24.0 | 134.5 ± 40.7* |
| LVEDV (μl) | 245.1 ± 46.8 | 460.1 ± 75.8* |
| LVEDP (mmHg) | 3.8 ± 0.9 | 5.6 ± 0.3* |
| LVEF (%) | 72.1 ± 7.2 | 75.9 ± 10.7 |
Values are expressed as mean ± SD. BW, body weight; HW, heart weight; HW/BW, heart weight/body weight; Lung W/D, Lung wet/dry; SBP, systolic blood pressure; DBP, diastolic blood pressure; PP, pulse pressure; MABP, mean arterial blood pressure; HR, heart rate; LV, left ventricular; ESV, end‐systolic volume; EDV, end‐diastolic volume; EDP, end‐diastolic pressure; EF, ejection fraction. Unpaired t‐test. * vs. Sham, P < 0.01.
Respiratory disorders and chemoreflex function
Irregular resting breathing patterns were found in CHF rats compared to Sham rats. Figure 1 A and B illustrates the loss of ventilator stability and an increased incidence of apnoea in CHF rats. The apnoea/hypopnea index (AHI) was significantly increased in CHF compared to the Sham condition (20.2 ± 4.0 vs. 9.7 ± 2.6 events h−1, CHF vs. Sham, respectively; P < 0.01, Fig. 1 C). In addition, both the short‐term (SD1) and the long‐term (SD2) breath‐to‐breath interval variability increased twofold in the CHF group compared to Sham condition (Fig. 1 D and E). Activation of the peripheral chemoreflex with hypoxia ( 10%) resulted in a large increase in V E in Sham rats (Fig. 2 A–C). In contrast, CHF rats showed reduced HVR (Fig. 2 A–C). The peripheral chemoreflex gain in CHF was 2.1 ± 1.0 ΔV E/% compared to 6.1 ± 1.8 ΔV E/% obtained in Sham rats. Sham rats increased V E in response to hypercapnia, a measure of central chemoreflex sensitivity, and this response was significantly enhanced in CHF rats (Fig. 2 A, D and E). The central chemoreflex gain was higher in CHF rats compared to Sham rats (19.1 ± 3.1 ΔV E/% CHF vs. 13.9 ± 2.3 ΔV E/%, Sham; P < 0.01, Fig. 2 E).
Figure 1. Disordered breathing patterns in HFpEF.
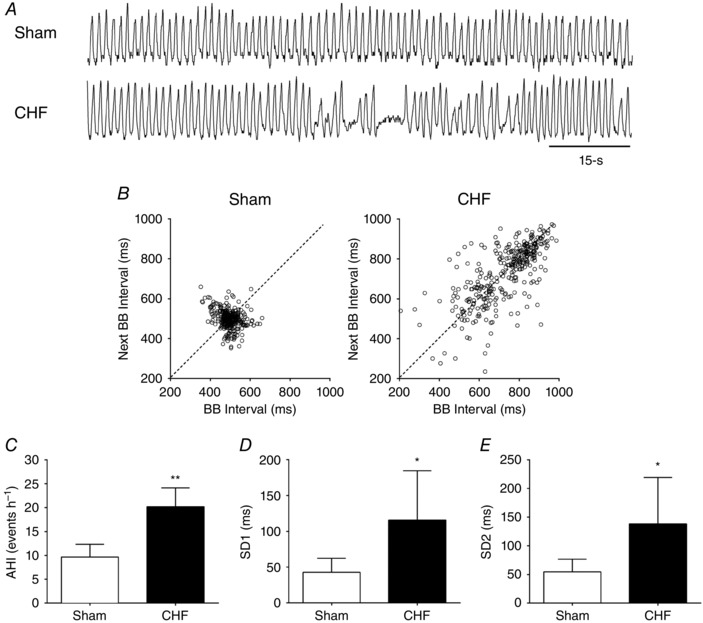
A, representative traces of tidal volume (V t) at rest in one Sham rat and one CHF rat. B, representative Poincaré plot displaying breath‐to‐breath (BB) interval variability in Sham vs. CHF condition. C, apnoea–hypopnea index (AHI) was increased in CHF rats vs. Sham rats. D and E, summary data showing short term (SD1) and long term (SD2) breathing interval variability in CHF vs. Sham condition. * P < 0.05; ** P < 0.01 vs. Sham; n = 6 rats.
Figure 2. Hypercapnic ventilatory responses (HCVR) but not the hypoxic ventilatory responses (HVR) are increased in HFpEF.
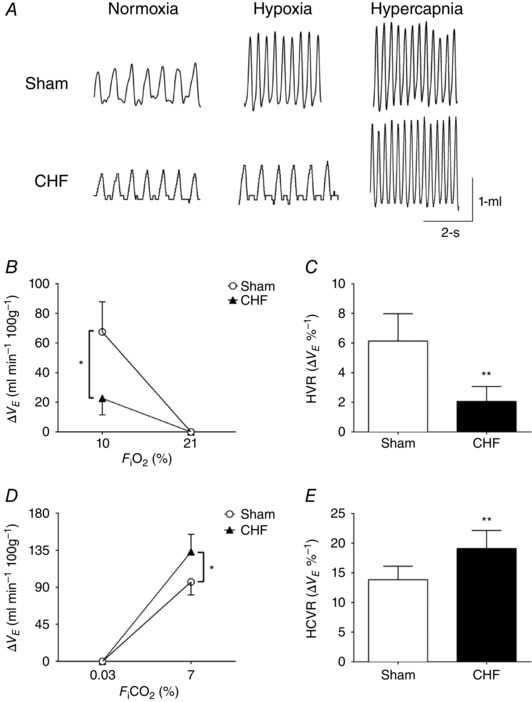
A, representative plethysmography recordings of tidal volume (V t) in normoxia ( 21%), hypoxia ( 10%) and hypercapnia ( 7%) in Sham and CHF rats. B and C, the magnitude and the gain of HVR was decreased in CHF rats compared to Sham rats. D and E, the magnitude and the gain of the HCVR was significantly increased in CHF rats compared to Sham rats. * P < 0.05; ** P < 0.01 vs. Sham condition, n = 6 rats.
RVLM neuronal activation and cardiac autonomic control
CHF animals showed greater Fos‐B protein expression in RVLM micropunches compared to Sham (252.0 ± 218.7 vs. 100.0 ± 15.2%, in CHF vs. Sham rats, respectively; P < 0.05, Fig. 3 B) indicating that neuronal activation in the RVLM is elevated in HFpEF. In addition, we observed that DHE staining in the RVLM of CHF animals was 7.5‐fold higher than that observed in Sham animals (12.3 ± 4.7 vs. 3.7 ± 2.0 AU, in CHF vs. Sham rats, respectively; P < 0.001, Fig. 3), suggesting that HFpEF is associated with increased superoxide in the RVLM. Importantly, DHE staining was found to co‐localize with tyrosine hydroxylase, a C1 neuronal marker in the RVLM from CHF rats (Fig. 3 A).
Figure 3. HFpEF rats displayed rostral ventrolateral medulla (RVLM) oxidative stress and chronic neuronal activation and sympatho‐vagal imbalance.
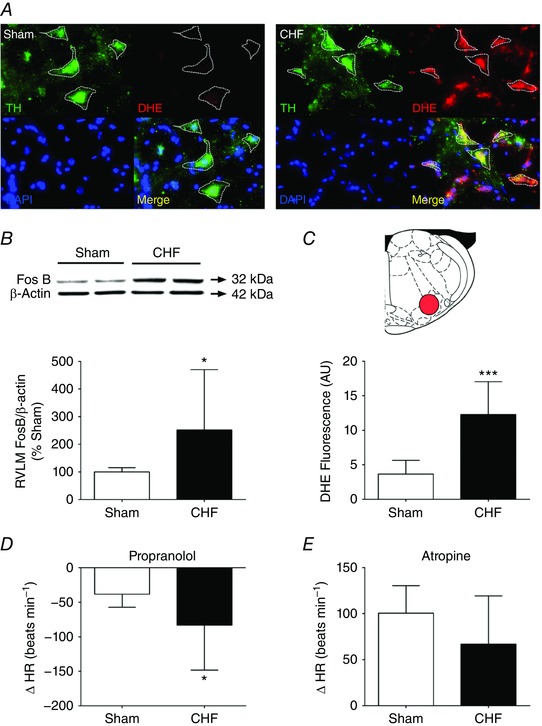
A, tyrosine hydroxylase (TH)‐positive C1 cells from the RVLM of CHF rats showed increases in dihydroethidium (DHE) staining compared to Sham rats. B, expression of Fos‐B on the RVLM. Note that CHF rats displayed increased Fos‐B expression in the RVLM compared to Sham rats. C, summary data showing ROS production in the RVLM from CHF and Sham rats. D, the bradycardic response (ΔHR) following propanolol was markedly higher in CHF compared to the Sham condition. E, the tachycardic response (ΔHR) following atropine was slightly lower in CHF compared to Sham rats. * P < 0.05; *** P < 0.001 vs. Sham condition, n = 6 rats. [Color figure can be viewed at wileyonlinelibrary.com]
Cardiac autonomic control was assessed by measuring the chronotropic responses following intravenous injection of propranolol and atropine. Perturbation of cardiac sympatho‐vagal balance was characterized by greater increases in sympathetic influence of heart rate in CHF vs. Sham rats (Fig. 3 D and E) (ΔHR to propanolol: −83.3 ± 65.0 beats min−1 CHF vs. −38.3 ± 18.2 beats min−1 Sham; P < 0.05).
Central chemoreflex activation and autonomic imbalance in CHF
Stimulation of central chemoreceptors with hypercapnia exacerbated cardiac sympatho‐vagal imbalance in CHF rats (Fig. 4). Using beat‐to‐beat analysis of HRV in the frequency domain we found that the LF band of HRV was increased in CHF rats following hypercapnia (179.8 ± 26.2% CHF + Normoxia vs. 212.5 ± 30.3% CHF + Hypercapnia; P < 0.05), without significant changes in HF band (Fig. 4 A–C). The LF/HFHRV ratio was significantly increased in CHF rats during hypercapnia (229.6 ± 43.2% CHF + Normoxia vs. 296.0 ± 43.9% CHF + Hypercapnia; P < 0.05, Fig. 4 A and D). Furthermore, we found that the deleterious effects of central chemoreflex activation on sympatho‐vagal balance in CHF rats was partly mediated by increased sympathetic outflow because intravenous propranolol attenuated the effect of hypercapnia on the LF band of HRV (41.9 ± 8.8 normalized units (n.u.) CHF + Hypercapnia vs. 10.5 ± 6.8 n.u. CHF + Hypercapnia + Propranolol; P < 0.05, Table 2). Accordingly, the effects of hypercapnia on the LF/HFHRV ratio was reversed after intravenous administration of propranolol (296.0 ± 43.9% CHF + Hypercapnia vs. 66.3 ± 23.1% CHF + Hypercapnia + Propranolol; P < 0.05, Table 2).
Figure 4. Acute central chemoreflex activation worsens cardiac sympatho‐vagal balance in HFpEF rats.
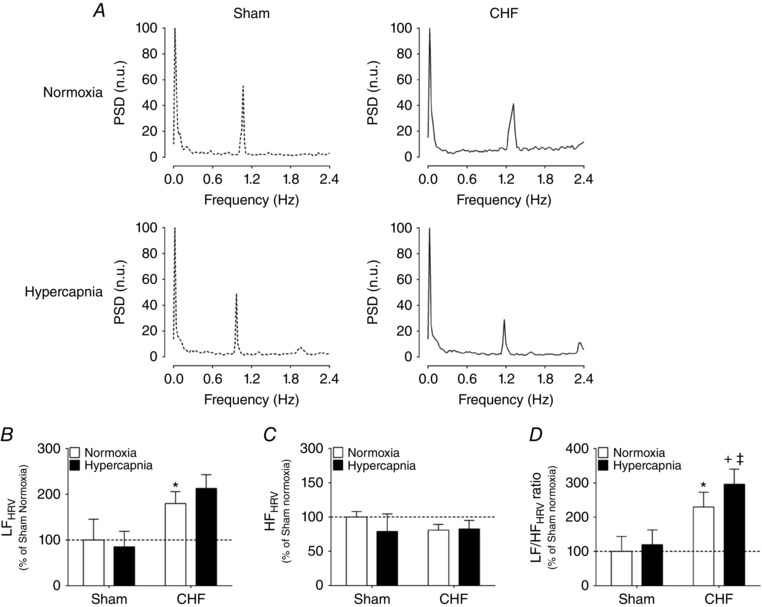
A, representative HRV spectra during normoxia ( 21%) and hypercapnia ( 7%) in one Sham rat and one CHF rat. B and C, hypercapnic stimulation increases the low‐frequency (LF) component (B) and decreases the high‐frequency (HF) component (C) of the HRV in CHF rats. D, summary data showing the effects of hypercapnia on the LF/HF ratio of HRV. * P < 0.01 vs. normoxia; + P < 0.05 vs. CHF + Hypercapnia, n = 4 rats.
Table 2.
Effect of sympathetic blockers during hypercapnia on heart rate variability
| Sham (n = 4) | CHF (n = 4) | |||||||
|---|---|---|---|---|---|---|---|---|
| Normoxia | Normoxia + Propranolol | Hypercapnia | Hypercapnia + Propranolol | Normoxia | Normoxia + Propranolol | Hypercapnia | Hypercapnia + Propranolol | |
| LF (n.u.) | 13.5 ± 10.6 | 13.4 ± 8.8 | 17.3 ± 5.4 | 18.7 ± 10.7 | 31.3 ± 9.7* | 19.7 ± 13.2 | 41.9 ± 8.8*, + | 10.5 ± 6.8α |
| HF (n.u.) | 70.1 ± 5.7 | 83.1 ± 8.9 | 58.7 ± 15.7 | 81.3 ± 10.7 | 56.2 ± 6.5* | 80.2 ± 13.0 | 57.5 ± 9.4 | 89.5 ± 6.8+ α |
| LF/HF ratio | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.3 ± 0.1 | 0.2 ± 0.1 | 0.6 ± 0.2* | 0.3 ± 0.2 | 0.8 ± 0.2*, + | 0.1 ± 0.1 α |
Values are expressed as mean ± SD. Normoxia, 21%; hypercapnia, 7%; Propranolol, 1 mg kg−1 i.v.; LF, low frequency; HF, high frequency; LF/HF ratio: low frequency to high frequency ratio. Two‐way ANOVA, followed by Sidak post hoc analysis. * vs. Sham + Normoxia, + vs. CHF + Normoxia, α vs. CHF + Hypercapnia, P < 0.05.
Cardiac function in CHF and chemoreflex activation
Breathing normoxic air, CHF rats displayed both systolic (ESPVR: 0.24 ± 0.1 mmHg μl−1 CHF vs. 0.48 ± 0.2 mmHg μl−1 Sham; P < 0.05) and diastolic cardiac function impairment compared to sham rats (EDPVR: 0.008 ± 0.004 mmHg μl−1 CHF vs. 0.005 ± 0.003 mmHg μl−1 Sham; P < 0.05, Fig. 5 A–C). To determine the effects of central and peripheral chemoreflex activation on cardiac function, rats were acutely exposed to hypercapnia and hypoxia, respectively. In Sham rats, neither exposure to hypercapnia nor exposure to hypoxia induced a significant change in ESPVR or EDPVR (Fig. 5, Tables 3 and 4). Similarly, exposure to hypoxia induced no significant changes in ESPVR and EDPVR in CHF rats (Table 4). In contrast, hypercapnia induced a marked impairment of diastolic cardiac function in CHF rats as evidenced by a threefold increase in EDPVR compared to normoxic conditions (0.027 ± 0.027 mmHg μl−1 CHF + Hypercapnia vs. 0.008 ± 0.004 mmHg μl−1 CHF + Normoxia; P < 0.05, Fig. 5 C). Systolic cardiac function did not deteriorate further following hypercapnic stimulation in CHF rats (ESPVR: 0.16 ± 0.11 mmHg μl−1 CHF + Hypercapnia vs. 0.29 ± 0.11 mmHg μl−1 CHF + Normoxia rats; P < 0.05, Fig. 5 B). Other left ventricular haemodynamic parameters were not altered by hypercapnia or by hypoxia in either Sham or the CHF condition (Tables 3 and 4, respectively). Importantly, arterial pH, , and HCO3 − were not different in normoxia and during hypercapnic stimulation between Sham and CHF rats (Table 5).
Figure 5. Diastolic function is impaired by acute central chemoreflex activation in HFpEF.
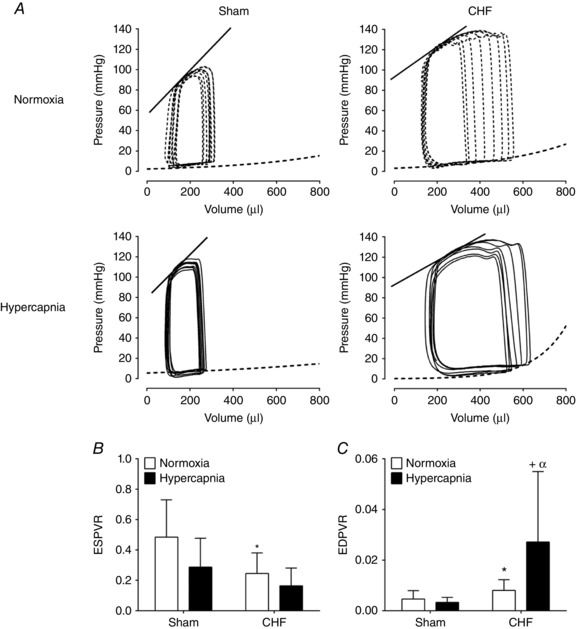
A, representative recording of intraventricular pressure–volume loops (PV‐loop) during normoxia ( 21%) and hypercapnia ( 7%) in one Sham rat and one CHF rat. The straight line represents the end‐systolic pressure–volume relationship (ESPVR) and segmented line represents the end‐diastolic pressure–volume relationship (EDPVR). B, systolic function (ESPVR) was significantly decreased in CHF vs. Sham. No effect of hypercapnia on ESPVR was found in the CHF group. C, diastolic function (EDPVR) was markedly impaired in CHF vs. Sham. Note that hypercapnia further exacerbates diastolic dysfunction in CHF rats. * P < 0.05 vs. Sham; + P < 0.05 vs. CHF; α P < 0.05 vs. Sham + Hypercapnia, n = 10.
Table 3.
Effect of hypercapnia on PV loops haemodynamic parameters
| Sham + Normoxia (n = 10) | Sham + Hypercapnia (n = 10) | CHF + Normoxia (n = 10) | CHF + Hypercapnia (n = 10) | |
|---|---|---|---|---|
| Global cardiac function | ||||
| HR (beats min−1) | 344.2 ± 23.1 | 353.6 ± 46.1 | 318.3 ± 51.0 | 314.1 ± 34.1 |
| CO (ml min−1) | 55.5 ± 13.3 | 52.4 ± 17.7 | 106.6 ± 39.2* | 83.2 ± 35.1* |
| LVEDP (mmHg) | 3.8 ± 0.9 | 5.0 ± 2.2 | 5.6 ± 0.3* | 5.7 ± 1.2* |
| Preload and afterload | ||||
| LVEDV (μl) | 245.1 ± 46.8 | 263.0 ± 22.7 | 460.1 ± 75.8* | 465.1 ± 48.3* |
| E a (mmHg μl−1) | 0.6 ± 0.3 | 0.8 ± 0.6 | 0.2 ± 0.0*, + | 0.4 ± 0.2+ |
| Systolic left ventricular function | ||||
| LVEF (%) | 72.1 ± 7.2 | 82.2 ± 8.5 | 75.9 ± 10.5 | 73.8 ± 22.7 |
| LVESV (μl) | 84.6 ± 24.0 | 81.2 ± 11.1 | 134.5 ± 40.8*, + | 195.2 ± 27.5*, + |
| dp/dt max (mmHg s−1) | 8260.0 ± 3307.7 | 8612.0 ± 3058.2 | 8057.0 ± 2585.7 | 7942 ± 2356.2 |
| Diastolic left ventricular function | ||||
| Active relaxation | ||||
| Tau (ms) | 9.6 ± 2.8 | 11.1 ± 2.8 | 8.9 ± 1.5 | 10.8 ± 2.2 |
| dp/dt min (mmHg s−1) | −6603.0 ± 1178.8 | −6429 ± 1653.5 | −5903.0 ± 2044.7 | −6429 ± 1748.4 |
Values are expressed as mean ± SD. HR, heart rate; CO, cardiac output; LV, left ventricular; EDP, end‐diastolic pressure; EDV, end‐diastolic volume; E a, arterial elastance; EF, ejection fraction; ESV, end‐systolic volume; Tau, time constant of relaxation. Two way ANOVA, followed by Sidak post hoc analysis. * vs. Sham + Normoxia, + vs. Sham + Hypercapnia, P < 0.01.
Table 4.
Effect of hypoxia on PV loops haemodynamic parameters
| Sham + Normoxia (n = 10) | Sham + Hypoxia (n = 10) | CHF + Normoxia (n = 10) | CHF + Hypoxia (n = 10) | |
|---|---|---|---|---|
| Global cardiac function | ||||
| HR (beats min−1) | 343.2 ± 34.4 | 343.2 ± 31.9 | 317.3 ± 44.7 | 341.0 ± 45.2 |
| CO (ml min−1) | 55.4 ± 14.5 | 56.8 ± 25.9 | 106.6 ± 39.2* | 98.1 ± 39.2 |
| LVEDP (mmHg) | 3.7 ± 0.9 | 4.8 ± 0.9 | 5.5 ± 0.1 | 5.8 ± 0.9* |
| Preload and after load | ||||
| LVEDV (μl) | 246.1 ± 43.6 | 230.2 ± 90.4 | 462.1 ± 76.5* | 398.9 ± 113.8* |
| E a (mmHg μl−1) | 0.6 ± 0.3 | 0.7 ± 0.3 | 0.2 ± 0.0*, + | 0.3 ± 0.3*, + |
| Systolic left ventricular function | ||||
| LVEF (%) | 73.1 ± 7.2 | 77.0 ± 12.9 | 77.9 ± 10.7 | 78.1 ± 9.4 |
| LVESV (μl) | 85.6 ± 24.0 | 68.3 ± 38.2 | 136.5 ± 40.7*, + | 130.7 ± 48.6*, + |
| dp/dt max (mmHg s−1) | 8263.0 ± 3310.9 | 7652.8 ± 1915.3 | 8056.0 ± 2,588.9 | 10 804.8 ± 4041.1 |
| ESPVR | 0.48 ± 0.2 | 0.26 ± 0.3 | 0.24 ± 0.1 | 0.17 ± 0.3 |
| Diastolic left ventricular function | ||||
| Active relaxation | ||||
| Tau (ms) | 9.5 ± 2.8 | 13.4 ± 6.3* | 8.8 ± 1.5 | 9.4 ± 1.8 |
| dp/dt min (mmHg s−1) | −6633.0 ± 1185.2 | −5292.3 ± 2298.3 | −5901.0 ± 2047.8 | −6247.7 ± 1652.9 |
| EDPVR | 5 × 10−3 ± 3 × 10−3 | 3 × 10−3 ± 3 × 10−3 | 8 × 10−3 ± 4 × 10−3 * | 5 × 10−3 ± 3 × 10−3 |
Values are expressed as mean ± SD. Normoxia, 21%; hypoxia, 10%; HR, heart rate; CO, cardiac output; LV, left ventricular; EDP, end‐diastolic pressure; EDV, end‐diastolic volume; E a, arterial elastance; EF, ejection fraction; ESV, end‐systolic volume; Tau, time constant of relaxation. Two‐way ANOVA, followed by Sidak post hoc analysis. * vs. Sham + Normoxia, + vs. Sham + Hypoxia, P < 0.01.
Table 5.
Arterial blood gases
| Sham + Normoxia (n = 4) | Sham + Hypercapnia (n = 4) | CHF + Normoxia (n = 4) | CHF + Hypercapnia (n = 4) | |
|---|---|---|---|---|
| pH | 7.47 ± 0.06 | 7.38 ± 0.06** | 7.49 ± 0.02 | 7.40 ± 0.04** |
| (mmHg) | 32.75 ± 4.44 | 50.20 ± 0.40** | 38.53 ± 1.92 | 48.85 ± 0.06** |
| (mmHg) | 82.00 ± 2.00 | 109.00 ± 2.00*** | 80.33 ± 0.66 | 99.50 ± 0.58*** |
| HCO3 − (mmol l−1) | 28.60 ± 0.20 | 29.65 ± 2.70 | 29.83 ± 1.42 | 31.05 ± 1.86 |
Values are expressed as mean ± SD. , partial pressure of CO2; , partial pressure of O2; HCO3 −, bicarbonate. Two‐way ANOVA, followed by Sidak post hoc analysis. ** vs. CHF + Normoxia, P < 0.01; *** vs. CHF + Normoxia, P < 0.001.
To determine if the effect of central chemoreflex activation on cardiac diastolic function in CHF was driven by sympathetic activation, we performed hypercapnic stimulation following sympathetic blockade with propranolol. Remarkably, the deleterious effect of hypercapnia on EDPVR in CHF rats was totally abolished by propranolol (Table 6). Taken together, these results suggest that central chemoreflex activation contributes to cardiac diastolic dysfunction in CHF by activation of the sympathetic nervous system.
Table 6.
Effect of sympathetic and parasympathetic blockade on cardiac function during hypercapnia
| Sham (n = 10) | CHF (n = 10) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Norm | Norm + Prop | Hyperc | Hyperc + Prop | Norm + Atro | Hyperc + Atro | Norm | Norm + Prop | Hyperc | Hyperc + Prop | Norm + Atro | Hyperc + Atro | |
| Systolic cardiac function | ||||||||||||
| ESPVR | 0.588 ± 0.275 | 0.378 ± 0.325 | 0.430 ± 0.284 | 0.159 ± 0.303 | 0.007* ± 0.003 | 0.039 ± 0.041 | 0.232* ± 0.129 | 0.002+ ± 0.003 | 0.151 ± 0.113 | 0.005+ ± 0.009 | 0.158 ± 0.113 | 0.113 ± 0.126 |
| Diastolic cardiac function | ||||||||||||
| EDPVR | 0.004 ± 0.003 | 0.003 ± 0.003 | 0.005 ± 0.003 | 0.005 ± 0.006 | 0.007 ± 0.003 | 0.004 ± 0.003 | 0.008* ± 0.003 | 0.002+ ± 0.003 | 0.025+ ± 0.025 | 0.001+ ± 0.000 | 0.007 ± 0.003 | 0.113+, α ± 0.072 |
Values are expressed as mean ± SD. Norm, normoxia ( 21%); Hyperc, hypercapnia ( 7%); Prop, propranolol 1 mg kg−1 (i.v.); Atro, atropine 1 mg kg−1 (i.v.); ESPVR, end‐systolic pressure–volume relationship; EDPVR, end‐diastolic pressure–volume relationship. Two‐way ANOVA, followed by Sidak post hoc analysis. * vs. Sham + Norm, + vs. CHF + Norm, α vs. CHF + Norm + Atro, P < 0.05.
Arrhythmia incidence and chemoreflex activation
Under normoxic conditions, the incidence of cardiac arrhythmias was significantly higher in CHF rats compared to Sham rats (196.0 ± 239.9 events h−1 CHF vs. 19.8 ± 21.7 events h−1 Sham; P < 0.05, Fig. 6 A and B). Stimulation of the central chemoreflex with hypercapnia increased arrhythmia incidence in both Sham and CHF rats. In Sham animals, hypercapnia induced an 8.5‐fold increase in arrhythmia incidence, which was comparable to the numbers of events displayed by CHF animals at rest (Fig. 6 A and B). In CHF rats, hypercapnia also induced a significant 2.5‐fold increase in the number of arrhythmic events compared to values obtained without hypercapnic stimulation (196.0 ± 239.9 events h−1 CHF + Normoxia vs. 576.7 ± 472.9 events h−1 CHF + Hypercapnia; P < 0.05, Fig. 6 A and B). Most of the arrhythmic events detected during hypercapnic stimulation in both sham and CHF animals were related to premature ventricular contractions. No detection of atrial fibrillation was found during hypercapnia in either sham or CHF rats. Importantly, the effects of hypercapnia on cardiac arrhythmias were abolished by propranolol treatment in both Sham and CHF rats (Fig. 6 A and B).
Figure 6. Central chemoreflex activation increases incidence of arrhythmia.
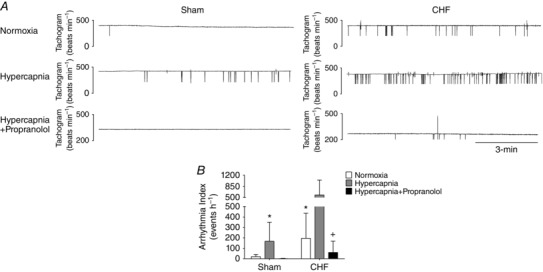
A, representative tachograms obtained from one Sham rat and one CHF rat before and after acute exposure to hypercapnia and prior to propranolol administration. The horizontal and vertical line represent 3 min and 200 beats min−1, respectively. B, arrhythmia incidence was markedly increased by hypercapnia in both Sham and CHF groups. Note that propranolol administration totally abolished the effects of hypercapnia on arrhythmia incidence in Sham and CHF rats. * P < 0.05 vs. Sham; + P < 0.05 vs. CHF + Hypercapnia, n = 6.
Discussion
Our results show that rats with volume overload‐induced CHF display: (i) preserved ejection fraction despite overt signs of cardiac hypertrophy, (ii) altered resting breathing patterns, (iii) increased central chemoreflex gain without alterations in peripheral chemoreflex gain, (iv) cardiac autonomic imbalance characterized by increased sympathetic outflow and (v) oxidative stress and evidence of chronic neuronal activation in the RVLM. Furthermore, we show for the first time that acute activation of the central chemoreflex with hypercapnia enhances sympathetic stimulation of the heart, exacerbates existing cardiac diastolic dysfunction and significantly increases arrhythmia incidence. Importantly, the effects of central chemoreflex activation on cardiac function and arrhythmia incidence were abolished by blockade of β‐adrenoreceptors. This suggests that stimulation of central chemoreceptors with hypercapnia affects cardiac function and arrhythmia incidence via increases in cardiac sympathetic outflow. Together our results indicate that acute central chemoreflex activation in the setting of HFpEF exacerbates autonomic and cardiac dysfunction and contributes to cardiac arrhythmogenesis.
Autonomic control and chemoreflex activation in heart failure
Sympatho‐vagal imbalance is a hallmark of heart failure (both HFrEF and HFpEF) that is positively correlated with disease progression (Kitzman et al. 2002; Florea & Cohn, 2014). Understanding the physiological mechanisms that contribute to this imbalance is important to developing future strategies intended to improve autonomic function in HF. In HFrEF, it has been shown that hyperreflexia rising from the carotid bodies is an important contributor to sympathatoexcitation (Ponikowski et al. 1997). In the present study we found that rats with HFpEF displayed no alterations in the peripheral chemoreflex response to hypoxia but an enhanced central chemoreflex response to hypercapnia. Acute stimulation of the central chemoreflex exacerbates cardiac dysfunction via activation of the sympathetic nervous system. Our findings strongly suggest that central chemoreflex and not peripheral chemoreflex activation is involved in sympatho‐vagal imbalance and/or cardiac dysfunction observed in HFpEF rats. However, we cannot rule out the possibility that the carotid bodies play a role in the early phase of CHF development in this model. Future studies should address the contribution of the carotid bodies to sympatho‐vagal imbalance and cardiac dysfunction in HFpEF.
RVLM pre‐sympathetic neurons and autonomic imbalance in heart failure
Previous work has shown that chronic activation of sympathetic neurons in the RVLM occurs in HFrEF and is associated with sympathoexcitation (Del Rio et al. 2013). In this study, we observed increased Fos‐B expression in the RVLM, indicating that chronic activation of these neurons is also a feature of HFpEF. Furthermore, we found that chronic activation of RVLM neurons occurs in tandem with marked increases in oxidative stress, which has previously been identified as a major contributor to activation of RVLM neurons and regulation of sympathetic nerve activity in HF (Lindley et al. 2004; Ding et al. 2009). Central chemoreceptor neurons from the RTN make glutamatergic contacts with RVLM neurons (Rosin et al. 2006). Also, it has been shown that increases in synaptic glutamate release lead to oxidative stress (Lafon‐Cazal et al. 1993; Vesce et al. 2004). Therefore, it is plausible that the enhanced RTN drive to the RVLM in HFpEF contributes to superoxide radical production and neuronal activation. Further studies should focus on the role of RTN on RVLM oxidative stress and hyper‐activation in HFpEF.
The RVLM is an important area of the brainstem that plays a major role in the control of sympathetic outflow. It has been shown that chemoreceptor fibres project to the RVLM and can regulate neuronal activity and sympathetic outflow to the heart (Moreira et al. 2006; Guyenet et al. 2010; Del Rio et al. 2013). In the present study we showed that activation of the central chemoreflex increases arrhythmia incidence and exacerbates cardiac dysfunction, an effect that was associated with increased sympathetic activation. One of the main central chemoreceptor areas in the brain is the RTN (Guyenet et al. 2010). It has been shown that RTN neurons project to pre‐sympathetic neurons of the RVLM (Rosin et al. 2006) suggesting a plausible role in the regulation of sympathetic outflow through a first‐order synapse with C1 neurons. Furthermore, it has been shown that activation of central chemoreceptors can trigger respiratory synchronous modulation of sympathetic nerve activity and subsequent respiratory–sympathetic coupling (Narkiewicz et al. 1999; Lazarenko et al. 2009). This is consistent with our finding of increased sympathetic drive in HFpEF rats following central chemoreflex activation. Interestingly, these effects were not observed in control animals. Future studies should assess the degree of entrainment of both respiratory and sympathetic activities in HFpEF.
Disordered breathing in heart failure
Human HF patients display disordered breathing patterns, with a high prevalence of central apnoeas and periodic breathing (Lorenzi‐Filho et al. 1999; Giannoni et al. 2008). It has been proposed that the hypoxemia/hypercapnia associated with disordered breathing in HF plays a pivotal role in the progression of the disease by repetitive stimulation of the sympathetic nervous system (Toledo et al. 2016). Interestingly, different experimental models of HF display similar degrees of disordered breathing, characterized by increased incidence of apnoea/hypopnea and breathing irregularity (Bitter et al. 2009; Herrscher et al. 2011; Chan & Lam, 2013). In this study, we showed for the first time that rats with HFpEF display significant increases in breath‐to‐breath interval variability and a marked increase in AHI compared to control rats. Furthermore, our data indicate that HFpEF rats have an exaggerated central chemoreflex response to acute hypercapnic stimulation. These results strongly suggest that repetitive stimulation of central chemoreceptors during the episodes of disordered breathing found in HFpEF (i.e. apnoeas, hypopneas, Cheyne–Stokes breathing) contribute to sympatho‐vagal imbalance and compromised cardiac function. Indeed, our results showed that hypercapnic stimulation exacerbates cardiac sympatho‐vagal imbalance. In agreement with this finding, Kristen et al. (2002) showed that the renal sympathetic nerve response to hypercapnia was increased in HFpEF rats. Taken together, these data suggest that central chemoreflex control of ventilation and sympathetic nerve activity are augmented in HFpEF.
Arrhythmogenesis in heart failure
Cardiac arrhythmias constitute one of the largest predictors of morbidity and mortality in HF (Esler, 1998; Giannoni et al. 2008). Recently, we showed that cardiac arrhythmogenesis is critically dependent on input from the carotid body in HFrEF (Del Rio et al. 2013; Marcus et al. 2014a). In this study we showed that acute activation of the central chemoreflex increased arrhythmia incidence. The effects of hypercapnia‐induced activation of central chemoreflex on cardiac arrhythmogenesis were related to augmented sympathetic activation. It is widely known that RVLM sympathetic neurons are modulated by several afferent inputs, one of them being projections from central chemoreceptors (Moreira et al. 2006; Rosin et al. 2006; Abbott et al. 2013). Therefore, we propose that in HFpEF, activation of the central chemoreflex by acute hypercapnic episodes directly activates RVLM sympathetic neurons resulting in increased sympathetic outflow to the heart and subsequent arrhythmogenesis. The role played by RVLM sympathetic neurons on cardiac arrhythmias following stimulation of the central chemoreflex in HFpEF merits further study.
Additional considerations
Several limitations are inherent in our study. Volume overload in rats results in increased EDPVR and EDP, both of which are considered diagnostic criteria for the recognition of human HFpEF (Yancy et al. 2013). However, we did not observe changes in end‐systolic elastance and LV filling times in our model. Furthermore, the increases in ventricular volumes reported in our model are not normally observed in all HFpEF patients. Therefore, it is important to note that our experimental model recapitulates some but not all of the pathophysiological components of the human HFpEF. Differences between our experimental model and human HFpEF may be related to several comorbidities associated with the progression of HFpEF (Edelmann et al. 2011; Yancy et al. 2013; Rosita et al. 2014). Indeed, high incidences of cardiovascular and non‐cardiovascular comorbidities have been found in HFpEF patients (Edelmann et al. 2011). Data from population‐based studies indicate a prevalence of coronary artery disease of 20–76%, diabetes mellitus of 13–70%, atrial fibrillation of 15–41% and hypertension of 25–88% in HFpEF (Rosita et al. 2014). Therefore, it is plausible that co‐morbidities may account for some of the differences between our model and human HFpEF. Future studies will be needed to identify the contribution of co‐morbidities on cardiac function in HFpEF.
Additional information
Competing interests
None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
Author contributions
C.T. and D.C.A contributed to the concept of the project, performed data collection and analysis, performed interpretation of the data and contributed to the preparation of the manuscript. C.L, A.A.A., H.S.D., V.A., M.M. and M.F. performed data collection and analysis, and contributed to the preparation of the manuscript. N.J.M. and H.D.S. performed interpretation of the data and contributed to the preparation of the manuscript. R.D.R. and H.S.D. contributed to the concept of the project and the experimental design. R.D.R. performed interpretation of the data and contributed to the preparation of the manuscript. All data collection was undertaken in the laboratory of R.D.R. All authors approved the final version of the manuscript.
Funding
This work was supported by FONDECYT 1140275 grant from the National Fund for Scientific and Technological Development of Chile.
Translational perspective
Tonic and episodic chemoreflex activation plays a pivotal role in sympatho‐vagal imbalance, respiratory disorders and cardiac dysfunction in HF, all of which strongly predict mortality in HF patients. We found that HFpEF rats have enhanced central chemoreflex drive, altered resting breathing patterns, chronic activation of RVLM neurons, cardiac sympatho‐vagal imbalance and increased arrhythmia incidence. Importantly, acute activation of the central chemoreflex resulted in additional impairment of cardiac function in HFpEF rats. Future studies should focus on delineating the signalling pathways associated with enhanced central chemoreflex sensitivity and chronic activation of RVLM neurons. Progress in identifying chemoreflex mechanisms that regulate cardiorespiratory function and how these mechanisms contribute to development of autonomic dysfunction and breathing disturbances will contribute to novel treatment strategies in HFpEF patients.
Acknowledgements
The authors thank Dr Julio Alcayaga for his technical support and Mr Fidel Flores for management of the animal facility at Universidad Autónoma de Chile.
References
- Abassi Z, Goltsman I, Karram T, Winaver J & Hoffman A (2011). Aortocaval fistula in rat: a unique model of volume‐overload congestive heart failure and cardiac hypertrophy. J Biomed Biotechnol 2011, 729497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott SB, DePuy SD, Nguyen T, Coates MB, Stornetta RL & Guyenet PG (2013). Selective optogenetic activation of rostral ventrolateral medullary catecholaminergic neurons produces cardiorespiratory stimulation in conscious mice. J Neurosci 33, 3164–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboukhalil A, Nielsen L, Saeed M, Mark RG & Clifford GD (2008). Reducing false alarm rates for critical arrhythmias using the arterial blood pressure waveform. J Biomed Inform 41, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade DC, Lucero C, Toledo C, Madrid C, Marcus NJ, Schultz HD & Del Rio R (2015). Relevance of the carotid body chemoreflex in the progression of heart failure. Biomed Res 2015, 467597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banes AK, Shaw SM, Tawfik A, Patel BP, Ogbi S, Fulton D & Marrero MB (2005). Activation of the JAK/STAT pathway in vascular smooth muscle by serotonin. Am J Physiol Cell Physiol 288, C805–812. [DOI] [PubMed] [Google Scholar]
- Bitter T, Faber L, Hering D, Langer C, Horstkotte D & Oldenburg O (2009). Sleep‐disordered breathing in heart failure with normal left ventricular ejection fraction. Eur J Heart Fail 11, 602–608. [DOI] [PubMed] [Google Scholar]
- Blain GM, Smith CA, Henderson KS & Dempsey JA (2010). Peripheral chemoreceptors determine the respiratory sensitivity of central chemoreceptors to CO2 . J Physiol 588, 2455–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlaug BA & Kass DA (2006). Mechanisms of diastolic dysfunction in heart failure. Trends Cardiovasc Med 16, 273–279. [DOI] [PubMed] [Google Scholar]
- Brodsky S, Gurbanov K, Abassi Z, Hoffman A, Ruffolo RR Jr, Feuerstein GZ & Winaver J (1998). Effects of eprosartan on renal function and cardiac hypertrophy in rats with experimental heart failure. Hypertension 32, 746–752. [DOI] [PubMed] [Google Scholar]
- Cingolani OH & Kass DA (2011). Pressure–volume relation analysis of mouse ventricular function. Am J Physiol Heart Circ Physiol 301, H2198–2206. [DOI] [PubMed] [Google Scholar]
- Cingolani OH, Yang XP, Liu YH, Villanueva M, Rhaleb NE & Carretero OA (2004). Reduction of cardiac fibrosis decreases systolic performance without affecting diastolic function in hypertensive rats. Hypertension 43, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MM & Lam CS (2013). How do patients with heart failure with preserved ejection fraction die? Eur J Heart Fail 15, 604–613. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Andrade DC, Lucero C, Arias P & Iturriaga R (2016). Carotid body ablation abrogates hypertension and autonomic alterations induced by intermittent hypoxia in rats. Hypertension 68, 436–445. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Marcus NJ & Schultz HD (2013). Carotid chemoreceptor ablation improves survival in heart failure: rescuing autonomic control of cardiorespiratory function. J Am Coll Cardiol 62, 2422–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Li YL, Zimmerman MC, Davisson RL & Schultz HD (2009). Role of CuZn superoxide dismutase on carotid body function in heart failure rabbits. Cardiovasc Res 81, 678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann F, Stahrenberg R, Gelbrich G,Durstewitz K, Angermann CE, Düngen H‐D, Scheffold T, Zugck C, Maisch B, Regitz‐Zagrosek V, Hasenfuß G, Pieske BM & Wachter R (2011). Contribution of comorbidities to functional impairment is higher in heart failure with preserved than with reduced ejection fraction. Clin Res Cardiol 100, 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler M (1998). High blood pressure management: potential benefits of I1 agents. J Hypertens Suppl 16, S19–24. [PubMed] [Google Scholar]
- Florea VG & Cohn JN (2014). The autonomic nervous system and heart failure. Circ Res 114, 1815–1826. [DOI] [PubMed] [Google Scholar]
- Giannoni A, Emdin M, Bramanti F, Iudice G, Francis DP, Barsotti A, Piepoli M & Passino C (2009). Combined increased chemosensitivity to hypoxia and hypercapnia as a prognosticator in heart failure. J Am Coll Cardiol 53, 1975–1980. [DOI] [PubMed] [Google Scholar]
- Giannoni A, Emdin M, Poletti R, Bramanti F, Prontera C, Piepoli M & Passino C (2008). Clinical significance of chemosensitivity in chronic heart failure: influence on neurohormonal derangement, Cheyne‐Stokes respiration and arrhythmias. Clin Sci 114, 489–497. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG (2014). Regulation of breathing and autonomic outflows by chemoreceptors. Compr Physiol 4, 1511–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG & Mulkey DK (2010). Retrotrapezoid nucleus and parafacial respiratory group. Respir Physiol Neurobiol 173, 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Mulkey DK, Stornetta RL & Bayliss DA (2005). Regulation of ventral surface chemoreceptors by the central respiratory pattern generator. J Neurosci 25, 8938–8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Abbott SB, Depuy SD, Fortuna MG & Kanbar R (2010). Central CO2 chemoreception and integrated neural mechanisms of cardiovascular and respiratory control. J Appl Phys 108, 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack KK, Engler CW, Papoutsi E, Pipinos II, Patel KP & Zucker IH (2012). Parallel changes in neuronal AT1R and GRK5 expression following exercise training in heart failure. Hypertension 60, 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack KK, Marcus NJ, Del Rio R, Zucker IH & Schultz HD (2014). Simvastatin treatment attenuates increased respiratory variability and apnea/hypopnea index in rats with chronic heart failure. Hypertension 63, 1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdani N, Franssen C, Lourenco A, Falcao‐Pires I, Fontoura D, Leite S, Plettig L, Lopez B, Ottenheijm CA, Becher PM, Gonzalez A, Tschope C, Diez J, Linke WA, Leite‐Moreira AF & Paulus WJ (2013). Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail 6, 1239–1249. [DOI] [PubMed] [Google Scholar]
- Herrscher TE, Akre H, Overland B, Sandvik L & Westheim AS (2011). High prevalence of sleep apnea in heart failure outpatients: even in patients with preserved systolic function. J Card Fail 17, 420–425. [DOI] [PubMed] [Google Scholar]
- Kitzman DW, Little WC, Brubaker PH, Anderson RT, Hundley WG, Marburger CT, Brosnihan B, Morgan TM & Stewart KP (2002). Pathophysiological characterization of isolated diastolic heart failure in comparison to systolic heart failure. JAMA 288, 2144–2150. [DOI] [PubMed] [Google Scholar]
- Kristen AV, Just A, Haass M & Seller H (2002). Central hypercapnic chemoreflex modulation of renal sympathetic nerve activity in experimental heart failure. Basic Res Cardiol 97, 177–186. [DOI] [PubMed] [Google Scholar]
- Lafon‐Cazal M, Pietri S, Culcasi M & Bockaert J (1993). NMDA‐dependent superoxide production and neurotoxicity. Nature 364, 535–537. [DOI] [PubMed] [Google Scholar]
- Lazarenko RM, Milner TA, Depuy SD, Stornetta RL, West GH, Kievits JA, Bayliss DA & Guyenet PG (2009). Acid sensitivity and ultrastructure of the retrotrapezoid nucleus in Phox2b‐EGFP transgenic mice. J Comp Neurol 517, 69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite S, Rodrigues S, Tavares‐Silva M, Oliveira‐Pinto J, Alaa M, Abdellatif M, Fontoura D, Falcao‐Pires I, Gillebert TC, Leite‐Moreira AF & Lourenco AP (2015). Afterload‐induced diastolic dysfunction contributes to high filling pressures in experimental heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol 309, H1648–1654. [DOI] [PubMed] [Google Scholar]
- Lorenzi‐Filho G, Rankin F, Bies I & Douglas Bradley T (1999). Effects of inhaled carbon dioxide and oxygen on cheyne‐stokes respiration in patients with heart failure. Am J Respir Crit Care Med 159, 1490–1498. [DOI] [PubMed] [Google Scholar]
- Lindley TE, Doobay MF, Sharma RV & Davisson RL (2004). Superoxide is involved in the central nervous system activation and sympathoexcitation of myocardial infarction‐induced heart failure. Circ Res 94, 402–409. [DOI] [PubMed] [Google Scholar]
- Mansukhani MP, Kara T, Caples SM & Somers VK (2014). Chemoreflexes, sleep apnea, and sympathetic dysregulation. Curr Hypertens Rep 16, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Del Rio R, Schultz EP, Xia XH & Schultz HD (2014a). Carotid body denervation improves autonomic and cardiac function and attenuates disordered breathing in congestive heart failure. J Physiol 592, 391–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Del Rio R & Schultz HD (2014b). Central role of carotid body chemoreceptors in disordered breathing and cardiorenal dysfunction in chronic heart failure. Front Physiol 5, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modesti PA, Vanni S, Bertolozzi I, Cecioni I, Lumachi C, Perna AM, Boddi M & Gensini GF (2004). Different growth factor activation in the right and left ventricles in experimental volume overload. Hypertension 43, 101–108. [DOI] [PubMed] [Google Scholar]
- Moreira TS, Takakura AC, Colombari E & Guyenet PG (2006). Central chemoreceptors and sympathetic vasomotor outflow. J Physiol 577, 369–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkiewicz K, Pesek CA, van de Borne PJ, Kato M & Somers VK (1999). Enhanced sympathetic and ventilatory responses to central chemoreflex activation in heart failure. Circulation 100, 262–267. [DOI] [PubMed] [Google Scholar]
- Oktay AA, Rich JD & Shah SJ (2013). The emerging epidemic of heart failure with preserved ejection fraction. Curr Heart Fail Rep 10, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburg O, Lamp B, Faber L, Teschler H, Horstkotte D & Topfer V (2007). Sleep‐disordered breathing in patients with symptomatic heart failure: a contemporary study of prevalence in and characteristics of 700 patients. Eur J Heart Fail 9, 251–257. [DOI] [PubMed] [Google Scholar]
- Oliveira‐Sales EB, Colombari DS, Davisson RL, Kasparov S, Hirata AE, Campos RR & Paton JF (2010). Kidney‐induced hypertension depends on superoxide signaling in the rostral ventrolateral medulla. Hypertension 56, 290–296. [DOI] [PubMed] [Google Scholar]
- Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL & Redfield MM (2006). Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 355, 251–259. [DOI] [PubMed] [Google Scholar]
- Pacher P, Nagayama T, Mukhopadhyay P, Batkai S & Kass DA (2008). Measurement of cardiac function using pressure–volume conductance catheter technique in mice and rats. Nat Protoc 3, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani M, Lombardi F, Guzzetti S, Rimoldi O, Furlan R, Pizzinelli P, Sandrone G, Malfatto G, Dell'Orto S & Piccaluga E (1986). Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho‐vagal interaction in man and conscious dog. Circ Res 59, 178–193. [DOI] [PubMed] [Google Scholar]
- Palkovits M (1983). Punch sampling biopsy technique. Methods Enzymol 103, 368–376. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson CR & Emson PC (1980). AChE‐stained horizontal sections of the rat brain in stereotaxic coordinates. J Neurosci Methods 3, 129–149. [DOI] [PubMed] [Google Scholar]
- Ponikowski P, Chua TP, Piepoli M, Ondusova D, Webb‐Peploe K, Harrington D, Anker SD, Volterrani M, Colombo R, Mazzuero G, Giordano A & Coats AJ (1997). Augmented peripheral chemosensitivity as a potential input to baroreflex impairment and autonomic imbalance in chronic heart failure. Circulation 96, 2586–2594. [DOI] [PubMed] [Google Scholar]
- Rommel KP, von Roeder M, Latuscynski K, Oberueck C, Blazek S, Fengler K, Besler C, Sandri M, Lucke C, Gutberlet M, Linke A, Schuler G & Lurz P (2016). Extracellular volume fraction for characterization of patients with heart failure and preserved ejection fraction. J Am Coll Cardiol 67, 1815–1825. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Chang DA & Guyenet PG (2006). Afferent and efferent connections of the rat retrotrapezoid nucleus. J Comp Neurol 499, 64–89. [DOI] [PubMed] [Google Scholar]
- Ryan TD, Rothstein EC, Aban I, Tallaj JA, Husain A, Lucchesi PA & Dell'Italia LJ (2007). Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J Am Coll Cardiol 49, 811–821. [DOI] [PubMed] [Google Scholar]
- Rosita Z, Borlaug BA, McNulty SE, Mohammed SF, Lewis GD, Semigran MJ, Deswal A, LeWinter M, Hernandez AF, Braunwald E, Redfield MM (2014). Impact of atrial fibrillation on exercise capacity in heart failure with preserved ejection fraction: clinical perspective. Circ Heart Fail 7, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toischer K, Rokita AG, Unsold B, Zhu W, Kararigas G, Sossalla S, Reuter SP, Becker A, Teucher N, Seidler T, Grebe C, Preuss L, Gupta SN, Schmidt K, Lehnart SE, Kruger M, Linke WA, Backs J, Regitz‐Zagrosek V, Schafer K, Field LJ, Maier LS & Hasenfuss G (2010). Differential cardiac remodeling in preload versus afterload. Circulation 122, 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo C, Andrade DC, Lucero C, Schultz HD, Marcus N, Retamal M, Madrid C & Del Rio R (2016). Contribution of peripheral and central chemoreceptors to sympatho‐excitation in heart failure. J Physiol 2016, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesce S, Kirk L & Nicholls DG (2004). Relationships between superoxide levels and delayed calcium deregulation in cultured cerebellar granule cells exposed continuously to glutamate. J Neurochem 90, 683–693. [DOI] [PubMed] [Google Scholar]
- Wang S, Shi Y, Shu S, Guyenet PG & Bayliss DA (2013). Phox2b‐expressing retrotrapezoid neurons are intrinsically responsive to H+ and CO2 . J Neurosci 33, 7756–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ & Wilkoff BL, American College of Cardiology F & American Heart Association Task Force on Practice G (2013). 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 62, e147–239. [DOI] [PubMed] [Google Scholar]