

Abstract
Key points
The transient receptor potential vanilloid type 1 (TRPV1) receptor is a polymodal molecular integrator in the pain pathway expressed in Aδ‐ and C‐fibre nociceptors and is responsible for the thermal hyperalgesia associated with inflammatory pain.
Noradrenaline strongly inhibited the activity of TRPV1 channels in dorsal root ganglia neurons. The effect of noradrenaline was reproduced by clonidine and antagonized by yohimbine, consistent with contribution of α2 adrenergic receptors.
The inhibitory effect of noradrenaline on TRPV1 channels was dependent on calcium influx and linked to calcium/calmodulin‐dependent protein kinase II.
In spinal cord slices, clonidine reduced the frequency of capsaicin‐induced miniature EPSCs in the presence of tetrodotoxin and ω‐conotoxin‐MVIIC, consistent with inhibition of presynaptic TRPV1 channels by α2 adrenergic receptors.
We suggest that modulation of presynaptic TRPV1 channels in nociceptive neurons by descending noradrenergic inputs may constitute a mechanism for noradrenaline to modulate incoming noxious stimuli in the dorsal horn of the spinal cord.
Abstract
The transient receptor potential vanilloid type 1 (TRPV1) receptor is a well‐known contributor to nociceptor excitability. To address whether noradrenaline can down‐regulate TRPV1 channel activity in nociceptors and reduce their synaptic transmission, the effects of noradrenaline and clonidine were tested on the capsaicin‐activated current recorded from acutely dissociated small diameter (<27 μm) dorsal root ganglia (DRG) neurons and on miniature (m)EPSCs recorded from large lamina I neurons in horizontal spinal cord slices. Noradrenaline or clonidine inhibited the capsaicin‐activated current by ∼60%, and the effect was reversed by yohimbine, confirming that it was mediated by activation of α2 adrenergic receptors. Similarly, clonidine reduced the frequency of capsaicin‐induced mEPSCs by ∼60%. Inhibition of capsaicin‐activated current by noradrenaline was mediated by GTP binding proteins, and was highly dependent on calcium influx. The inhibitory effect of noradrenaline on the capsaicin‐activated current was not affected either by blocking the activity of protein kinase A with H89, or by blocking the activity of protein kinase C with bisindolylmaleimide II. In contrast, when the calcium/calmodulin‐dependent protein kinase II (CaMKII) was blocked with KN‐93, the inhibitory effect of noradrenaline on the capsaicin‐activated current was greatly reduced, suggesting that activation of adrenergic receptors in DRG neurons is preferentially linked to CaMKII activity. We suggest that modulation of TRPV1 channels by noradrenaline in nociceptive neurons is a mechanism whereby noradrenaline may suppress incoming noxious stimuli at the primary synaptic afferents in the dorsal horn of the spinal cord.
Keywords: dorsal root ganglia neurons, noradrenaline, TRPV1 channels
Key points
The transient receptor potential vanilloid type 1 (TRPV1) receptor is a polymodal molecular integrator in the pain pathway expressed in Aδ‐ and C‐fibre nociceptors and is responsible for the thermal hyperalgesia associated with inflammatory pain.
Noradrenaline strongly inhibited the activity of TRPV1 channels in dorsal root ganglia neurons. The effect of noradrenaline was reproduced by clonidine and antagonized by yohimbine, consistent with contribution of α2 adrenergic receptors.
The inhibitory effect of noradrenaline on TRPV1 channels was dependent on calcium influx and linked to calcium/calmodulin‐dependent protein kinase II.
In spinal cord slices, clonidine reduced the frequency of capsaicin‐induced miniature EPSCs in the presence of tetrodotoxin and ω‐conotoxin‐MVIIC, consistent with inhibition of presynaptic TRPV1 channels by α2 adrenergic receptors.
We suggest that modulation of presynaptic TRPV1 channels in nociceptive neurons by descending noradrenergic inputs may constitute a mechanism for noradrenaline to modulate incoming noxious stimuli in the dorsal horn of the spinal cord.
Abbreviations
- BIM
bisindolylmaleimide II
- CaMKII
calcium/calmodulin‐dependent protein kinase II
- CMF
Ca2+‐, Mg2+‐free
- DRG
dorsal root ganglia
- GPCR
G‐protein‐coupled receptor
- mEPSCs
miniature EPSCs
- PKA
protein kinase A
- PKC
protein kinase C
- PLC
phospholipase C
- TRPV1
transient receptor potential vanilloid type 1
Introduction
Nociceptive signals relayed to the dorsal horn of the spinal cord by nociceptors are subject to modulation by supraspinal centres (Fields et al. 1991; Millan, 2002; Ossipov et al. 2014). Behavioural studies support the anti‐nociceptive effects of noradrenaline mediated by spinal α2 adrenergic receptors (Millan, 1992; Takano & Yaksh, 1992, 1993; Millan et al. 1994). Potentiation of descending noradrenergic inputs may be particularly significant during sustained nociceptive signals associated with nerve injury or inflammation (Cho et al. 1995; Tsuruoka & Willis, 1996; Green et al. 1998; Martin et al. 1999; Stone et al. 1999; Xu et al. 1999).
Noradrenergic fibres from A5, A6 and A7 regions provide the main source of spinal noradrenaline (Clark & Proudfit, 1991 b, a; Kwiat & Basbaum, 1992; Sluka & Westlund, 1992), and establish axo‐somatic and axo‐dendritic synaptic contacts with projection neurons and interneurons (Hagihira et al. 1990; Nicholas et al. 1993; Rosin et al. 1993; Stone et al. 1998; Shi et al. 1999; Riedl et al. 2009), supporting postsynaptic effects of noradrenaline. However, noradrenergic fibres, similar to other monoaminergic inputs, also possess varicosities with non‐synaptic terminations (Hagihira et al. 1990; Ridet et al. 1992, 1993), suggesting the possibility of volume transmission and presynaptic effects. Possible presynaptic effects of noradrenaline on primary sensory neurons are supported by expression of adrenergic receptors both in the cell body and in central terminals of dorsal root ganglia (DRG) neurons (Nicholas et al. 1993; Cho et al. 1997; Gold et al. 1997; Stone et al. 1998; Birder & Perl, 1999). Consistent with this pattern of expression, noradrenaline modulates calcium channels (Dunlap & Fischbach, 1981; Marchetti et al. 1986; Bean, 1989), ATP‐sensitive channels (Maruo et al. 2006), sodium channels (Khasar et al. 1999; Oda et al. 2007) and TRPM8 channels (Bavencoffe et al. 2010) in DRG neurons.
The transient receptor potential vanilloid type 1 (TRPV1) receptor is a polymodal molecular integrator in the pain pathway expressed in Aδ‐ and C‐fibre nociceptors (Szallasi et al. 1995; Caterina et al. 1997; Cavanaugh et al. 2011), and can be activated by vanilloids (capsaicin and resineferatoxin) (Caterina et al. 1997), endogenous lipids (anandamide) (Zygmunt et al. 1999), protons (Ahern et al. 2005; Dhaka et al. 2009), polyamines (Ahern et al. 2006) and noxious heat (Caterina et al. 1997). Pharmacological and genetic studies support the contribution of TRPV1 channels to the development of different pain conditions, including thermal hyperalgesia associated with inflammatory pain (Caterina et al. 2000; Davis et al. 2000), bone cancer pain (Ghilardi et al. 2005), migraine (Akerman et al. 2004), irritable bowel syndrome (Jones et al. 2005) and arthritis (Szabo et al. 2005).
TRPV1 channels are expressed on the soma, peripheral and central terminals of nociceptors which make synaptic contacts with second‐order neurons in the dorsal horn of the spinal cord (Holzer, 1991; Winter et al. 1993; Szallasi et al. 1995; Caterina et al. 1997; Tominaga et al. 1998; Guo et al. 1999; Hwang et al. 2004). Activation of presynaptic TRPV1 channels by endovanilloids or capsaicin triggers the release of peptides and modulates glutamatergic transmission in the dorsal horn of the spinal cord by increasing the frequency of miniature (m)EPSCs and by inhibiting evoked EPSCs (Yang et al. 1998, 1999; Tognetto et al. 2001; Nakatsuka et al. 2002; Baccei et al. 2003; Labrakakis & MacDermott, 2003; Tong & MacDermott, 2006; Medvedeva et al. 2008). The presynaptic localization of TRPV1 channels on the central terminals of nociceptors suggests that they are well positioned to interact with neuromodulators released in the dorsal horn of the spinal cord by descending fibres (Kim et al. 2014; Chakraborty et al. 2016), and point to a possible mechanism by which supraspinal centres may modulate the excitability of nociceptors.
Here, using acutely dissociated DRG neurons and horizontal spinal cord slices in vitro, we show that noradrenaline down‐regulates the activity of presynaptic TRPV1 channels expressed in nociceptors, and reduces the frequency of capsaicin‐induced mEPSCs recorded from large lamina I neurons in the dorsal horn of the spinal cord. Pharmacological studies indicate that the effect of noradrenaline is mediated by activation of α2 adrenergic receptors, is dependent on calcium influx and is coupled to calcium/calmodulin‐dependent protein kinase II (CaMKII).
Methods
Animals
Sprague–Dawley rats (both male and female) of postnatal days 18–28 were used in this study. This postnatal age was chosen because the postnatal development of dorsal horn sensory processing is mostly complete (Fitzgerald, 2005), although afferent synaptic input is further refined into adulthood (Park et al. 1999; Nakatsuka et al. 2000). Furthermore, in horizontal spinal cord slices, it is possible to record from large lamina I neurons at these ages, which becomes increasingly difficult with heavier laminar myelination at older postnatal ages. All procedures were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by Stony Brook University Institutional Animal Care and Use Committee.
Dissociated DRG neurons
Rats were deeply anaesthetized with isoflurane and decapitated. Both thoracic and lumbar segments of the spinal cord were removed and placed in a cold Ca2+‐, Mg2+‐free (CMF) Hank's solution containing (in mm): 137 NaCl, 5.3 KCl, 0.33 Na2HPO4, 0.44 KH2PO4, 5 Hepes and 5.5 glucose (pH 7.4 with NaOH). The bone surrounding the spinal cord was removed and DRG were exposed and pulled out. After removing the roots, ganglia were chopped in half and incubated for 20 min at 34°C in CMF Hank's solution containing 20 U ml−1 papain (Worthington Biochemical, Lakewood, NJ, USA) and 5 mm dl‐cysteine. Ganglia were then treated for 20 min at 34°C with 3 mg ml−1 collagenase (Type I, Sigma‐Aldrich, St. Louis, MO, USA) and 4 mg ml−1 Dispase II (Boehringer Mannheim, Indianapolis, IN, USA) in CMF Hank's solution. Ganglia were then washed with Leibovitz's L‐15 medium (Invitrogen, San Diego, CA, USA) supplemented with 10% fetal calf serum and 5 mm Hepes. Individual cells were dispersed by mechanical trituration using fire‐polished Pasteur pipettes with decreasing bore size and plated on glass coverslips treated with 30 μg ml−1 poly‐d‐lysine. Cells were incubated in the supplemented L‐15 solution at 34°C (in 5% CO2) for 2 h, and then stored at room temperature in Neurobasal medium (Gibco) and used over the next 4–6 h. This protocol yields spherical cell bodies without neurites. The cells can be lifted from the cover slip after establishing the whole‐cell configuration in order to facilitate rapid solution changes using flow pipes. Small DRG neurons (diameters <27 μm) sensitive to capsaicin were chosen for recording. Small DRG neurons were initially selected by measuring the diameter from images captured to a computer by a CCD camera (Oly‐150, Olympus Imaging America Inc., Center Valley, PA, USA) using a video acquisition card (dP dPict Imaging, Inc., Indianapolis, IN, USA). A more accurate measurement of cell diameter was obtained from measurements of whole‐cell capacitance assuming a membrane capacitance of 1 μF cm−2 and spherical shape. Cell capacitance was measured by integrating the average of 10 current responses to a –5 mV step from −80 mV filtered at 10 kHz and acquired at 50 kHz. DRG neurons were first tested for capsaicin sensitivity, and only those responding to capsaicin (75% of those tested), corresponding to a subset of nociceptors (Cardenas et al. 1995; Caterina et al. 1997; Petruska et al. 2000), were used for the experiments.
Horizontal spinal cord slices
Rats were deeply anaesthetized with isoflurane and decapitated. After decapitation, the ventral aspect of the vertebral column was exposed and immersed in ice‐cold dissecting solution (in mm): 87 NaCl, 2.5 KCl, 1.25 NaH2PO4.H2O, 26 NaHCO3, 6 MgCl2, 0.5 CaCl2, 20 glucose, 77 sucrose, 1 kynurenic acid, oxygenated with 95/5% O2/CO2. The lumbar part of the spinal cord was exposed and carefully removed with L4 and L5 dorsal roots attached. Horizontal slices were made manually. First, the spinal cord was cut in half with a razor blade through the parasagittal plane to produce a hemisected spinal cord. With a second cut, about 45 degree to the parasagittal plane, the ventral part of the hemisected cord was removed such that the result was a horizontal slice (400–500 μm thick) with the L4–L5 dorsal roots attached. Horizontal slices were then immersed in oxygenated recovery solution (same as dissecting solution, but without kynurenic acid) at 35°C and allowed to recover for 1 h. After 1 h the slices were transferred to a storage solution (same as recovery solution, but at room temperature) and kept for the next 4–5 h. Horizontal spinal cord slices, in contrast to coronal or parasagittal slices, offer the unique advantage that afferent fibres and large lamina I neurons in the dorsal horn of the spinal cord are preserved virtually intact. In horizontal slices, large lamina I neurons were visualized through the white matter using an infrared light‐emitting diode (IR‐LED) illumination (Safronov et al. 2007; Szucs et al. 2009; Li et al. 2015) and a CCD video camera (Oly 150, Olympus). The IR‐LED is attached to a 40× water immersion objective mounted on an upright microscope (BX51WI, Olympus). Large lamina I neurons (cross‐sectional soma area > 220 μm2), consistent with projection neurons (Al Ghamdi et al. 2009), located 20–25 μm below the white matter were selected for recording.
Electrophysiology in isolated DRG neurons
Whole‐cell recordings were made with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA). Patch pipettes were pulled from borosilicate glass (A‐M Systems, Sequim, WA, USA) using a Sutter P97 puller (Sutter Instrument, Novato, CA, USA). The resistance of the patch pipette was 1.3–1.8 MΩ when filled with the standard internal CsCl‐based solution. The shank of the patch pipette was wrapped with parafilm to reduce pipette capacitance. In whole‐cell mode, the capacity current was reduced by using the amplifier circuitry. To reduce voltage errors, 70–80% of series resistance compensation was applied. After the whole cell configuration was established, the cell was lifted up and placed in front of an array of quartz fibre flow pipes (320 μm internal diameter) containing the test solutions. Solutions were changed in ∼1 s by moving the cell from one pipe to another. All experiments with isolated DRGs were done at room temperature (22 ± 1°C), with the exception of those in Fig. 6 that were carried out at 35 ± 1°C. Solutions were heated with a temperature controller (Warner TC‐344B, Warner Instruments, Hamden, CT, USA). In voltage clamp experiments a Cs‐based internal solution was used to block outward currents through potassium channels. This solution contained (in mm): 125 CsCl, 10 NaCl, 2 MgCl2, 10 EGTA, 10 Hepes, 14 Tris‐creatine phosphate, 4 Mg‐ATP and 0.3 Na‐GTP (pH 7.2 with CsOH). The standard external solution was a modified Tyrode's solution containing (in mm): 151 NaCl, 2 CaCl2, 1 MgCl2, 2.5 KCl, 10 Hepes and 13 glucose (pH 7.4 with NaOH).
Figure 6. Effect of clonidine on the capsaicin‐activated current at physiological temperature.
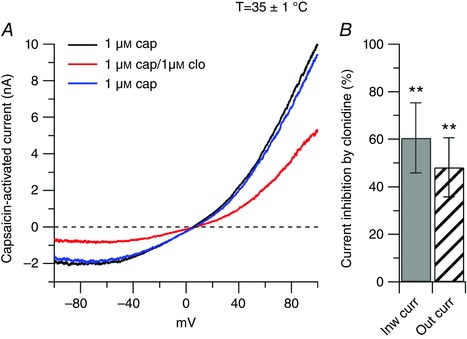
A, representative capsaicin‐activated currents recorded at physiological temperature (35 ± 1°C) in control (black trace), in the presence of 1 μm clonidine (clo, red trace) and upon washing out clonidine (blue trace). B, in collected results, 1 μm clonidine, applied on top of capsaicin, inhibited the inward current by 61 ± 15% (n = 8), ** P < 0.01, paired t test, and the outward current by 48 ± 12% (n = 8), ** P < 0.01, paired t test.
Voltage clamp protocols for isolated DRG neurons
TRPV1 current was determined as the capsaicin‐activated current by subtracting currents before and after application of 1 μm capsaicin. The I–V relationship for the TRPV1 current was determined using voltage ramps (1.6 mV ms−1). Using fast ramp commands rather than obtaining the time courses of inward/outwards currents activated by puffs of capsaicin offers two main advantages. First, it is possible to study the I–V relationship of the capsaicin‐activated current on a broad range of voltages (from −100 to +100 mV) during a single sweep. Second, ramps are delivered in the same sweep from −100 to +100 mV and then from +100 to −100 mV (Fig. 1 A, inset), with the ‘down ramp’ preceded by 50–200 ms at +100 mV. With this procedure, currents carried by voltage‐dependent sodium and calcium channels are largely inactivated before the ‘down ramp’, which allows more precise determination of TRPV1 currents by eliminating overlapping voltage‐dependent sodium and calcium currents (Puopolo et al. 2013; Chakraborty et al. 2016). I–V relationships for TRPV1 channels were therefore determined by using the ‘down ramp’. TRPV1 channels show some degree of desensitization upon repeated or sustained application of the agonist capsaicin (Caterina et al. 1997). To allow reliable comparison between capsaicin‐activated currents before and after drug treatment, the effect of drugs on the capsaicin‐activated current was measured as soon as the capsaicin‐activated current reached a stable value (usually 15–20 s, Fig. 1 B). The typical experiment consisted of 20 s of perfusion with capsaicin, followed by 20 s of the test drug in combination with capsaicin, and 20 s of washout in capsaicin.
Figure 1. Inhibition of capsaicin‐activated current by noradrenaline.
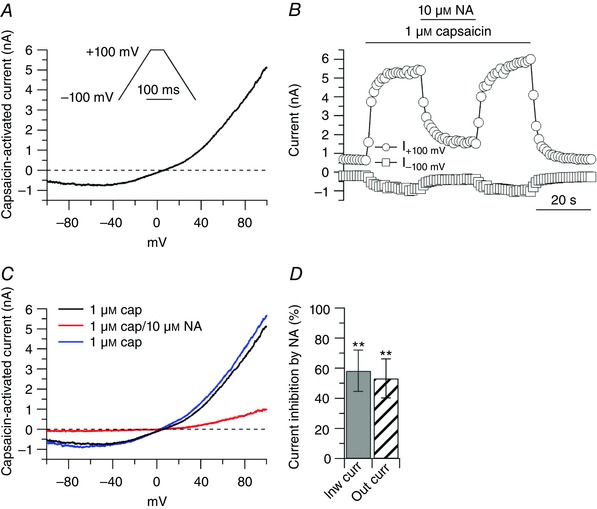
A, ionic currents through TRPV1 channels were determined by subtracting currents before and after application of 1 μm capsaicin. Representative capsaicin‐activated current recorded during the ‘down ramp’. Inset: voltage clamp protocol. Ramps (1.6 mV ms−1) were delivered in the same sweep from −100 to + 100 mV and from + 100 to −100 mV with the ‘down ramp’ preceded by 50–200 ms at + 100 mV to inactivate voltage‐dependent sodium and calcium channels. B, effects of 10 μm noradrenaline (NA) on capsaicin‐activated current. Inward and outward currents were measured at −100 and +100 mV, respectively. C, capsaicin‐activated currents from the cell in B: 1 μm capsaicin (black trace), 10 μm noradrenaline on top of capsaicin (red trace), 1 μm capsaicin upon washing out noradrenaline (blue trace). D, in collected results, 10 μm noradrenaline, applied on top of capsaicin, inhibited the inward current by 58 ± 13% (n = 6, ** P < 0.01, paired t test), and the outward current by 53 ± 13% (n = 6, ** P < 0.01, paired t test), with respect to control.
Electrophysiology in spinal cord slices
Whole‐cell voltage clamp recordings were made with a Multiclamp 700B amplifier (Molecular Devices). Patch pipettes were pulled from borosilicate glass (WPI, Sarasota, FL, USA) using a Sutter P97 puller (Sutter Instrument). The resistance of the patch pipette was 1.3–1.8 MΩ when filled with the standard internal Cs‐methanesulfonate‐based solution. The shank of the patch pipette was wrapped with parafilm to reduce pipette capacitance. In whole‐cell mode, the capacity current was further reduced by using the amplifier circuitry. To reduce voltage errors, 40–50% of series resistance compensation was applied. EPSCs were elicited by electrical stimulation of the dorsal root (L4 or L5) attached to the spinal cord slice. The dorsal root was included in a glass suction electrode and electrically stimulated at 0.016 Hz with 500 μA of constant current (100 μs duration) delivered with a constant current stimulus isolation unit (ISO‐flex, AMPI, Jerusalem, Israel). Spontaneous mEPSCs and evoked EPSCs were recorded in voltage clamp at a holding potential of −70 mV. For evoked EPSCs (Fig. 10), the external solution was (in mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4.H2O, 20 glucose, 0.01 bicuculline and 0.005 strychnine, oxygenated with 95/5% O2/CO2; the internal solution was (in mm): 125 caesium methanesulfonate, 10 NaCl, 2 MgCl2, 14 phosphocreatine, 4 Mg‐ATP, 0.3 GTP, 10 EGTA, 10 Hepes and 5 mm QX‐314 (pH 7.2 with CsOH). For mEPSCs, the external solution was (in mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4.H2O, 20 glucose, 0.01 bicuculline, 0.005 strychnine, 300 nm tetrodotoxin (Fig. 9) and 200 nm ω‐conotoxin‐MVIIC (Figs 11 and 12), oxygenated with 95/5% O2/CO2; the internal solution was (in mm): 125 caesium methanesulfonate, 10 NaCl, 2 MgCl2, 14 phosphocreatine, 4 Mg‐ATP, 0.6 GDP‐β‐S, 10 EGTA, 10 Hepes and 5 mm QX‐314 (pH 7.2 with CsOH). Recordings of evoked EPSCs and mEPSCs were made at 35 ± 1°C by heating the solutions with a temperature controller (Warner TC‐344B, Warner Instruments).
Figure 10. Contribution of N‐ and P/Q‐type calcium channels to neurotransmitter release from primary afferent fibres to lamina I neurons.
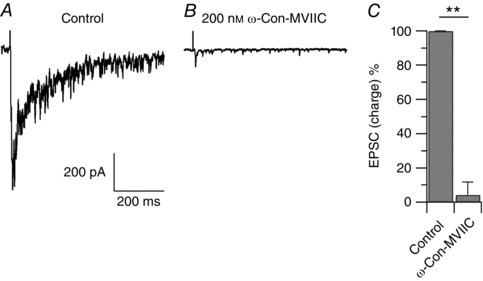
A, evoked EPSC recorded from a large lamina I neuron in a horizontal spinal cord slice. The EPSC was elicited by electrical stimulation of the L4 dorsal root attached to the spinal cord slice with 500 μA of constant current (100 μs duration). Shown is the average EPSC of three consecutive electrical stimulations at 0.016 Hz. B, 200 nm ω‐conotoxin‐MVIIC reduced the EPSC by 97%. C, in collected results, 200 nm ω‐conotoxin‐MVIIC reduced the EPSC by 94 ± 7% (n = 9), ** P < 0.01, paired t test.
Figure 9. Effect of clonidine on capsaicin‐induced mEPSCs.
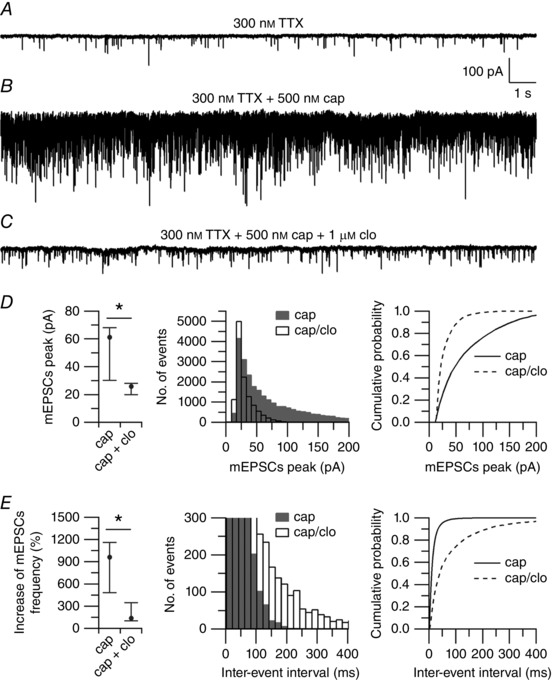
A, spontaneous mEPSCs recorded from a large lamina I neuron in a horizontal spinal cord slice. mEPSCS were recorded at a holding potential (V h) = −70 mV in the presence of 300 nm TTX, and with an intracellular solution in which Na‐GTP (0.3 mm) was replaced by the non‐hydrolysable analogue GDP‐β‐S (0.6 mm). B, capsaicin (500 nm) increased the median frequency of mEPSCs from 12 to 116 Hz. C, clonidine (1 μm), applied on top of capsaicin, reduced the median frequency of mEPSCs to 43 Hz. D, collected results showing the effects of clonidine on the peak of mEPSCs. Left: values are reported as median, first quartile (25th percentile), and third quartile (75th percentile). Capsaicin (cap): median = 61.2, 25th percentile = 30.1, 75th percentile = 68.1. Capsaicin + clonidine (cap + clo): median = 25.9, 25th percentile = 19.9, 75th percentile = 28.1 (n = 7). * P < 0.05, Wilcoxon matched‐pairs test. Middle: distribution of mEPSC peaks recorded in 500 nm capsaicin (shaded bars) and after application of 1 μm clonidine (open bars) on top of capsaicin (n = 7). Right: cumulative probabilities of mEPSC peaks recorded in 500 nm capsaicin (solid line) and after application of 1 μm clonidine (dashed line) on top of capsaicin (n = 7). **** P < 0.0001, Kolmogorov–Smirnov test. E, collected results showing the effects of clonidine on the frequency of mEPSCs. Left: values are reported as median, first quartile (25th percentile) and third quartile (75th percentile). Capsaicin (cap): median = 963%, 25th percentile = 483%, 75th percentile = 1160%. Capsaicin + clonidine (cap + clo): median = 137%, 25th percentile = 101%, 75th percentile = 346% (n = 7). * P < 0.05, Wilcoxon matched‐pairs test. Middle: distribution of inter‐event intervals of mEPSCs recorded in 500 nm capsaicin (shaded bars) and after application of 1 μm clonidine (open bars) on top of capsaicin (n = 7). Right: cumulative probabilities of inter‐event intervals of mEPSCs recorded in 500 nm capsaicin (solid line) and after application of 1 μm clonidine (dashed line) on top of capsaicin (n = 7). **** P < 0.0001, Kolmogorov–Smirnov test.
Figure 11. Effect of clonidine on capsaicin‐induced mEPSCs in the presence of calcium channel blockers.
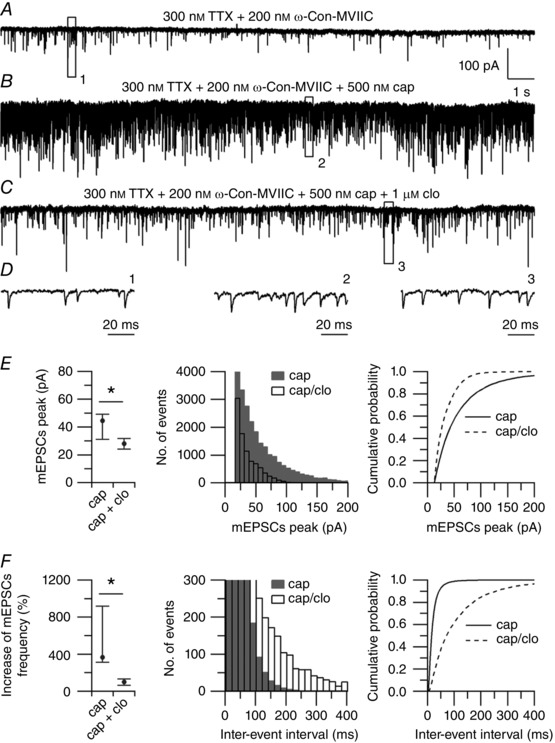
A, spontaneous mEPSCs recorded from a large lamina I neuron in a horizontal spinal cord slice. mEPSCS were recorded at a holding potential (V h) = −70 mV in the presence of 300 nm TTX + 200 nm ω‐conotoxin‐MVIIC, and with an intracellular solution in which Na‐GTP (0.3 mm) was replaced by the non‐hydrolysable analogue GDP‐β‐S (0.6 mm). B, capsaicin (500 nm) increased the median frequency of mEPSCs from 4 to 55 Hz. C, clonidine (1 μM), applied on top of capsaicin, reduced the median frequency of mEPSCs to 9 Hz. D, representative small events recorded in 300 nm TTX + 200 nm ω‐Con‐MVIIC (left, from box 1), after application of 500 nm capsaicin (middle, from box 2) and after application of 1 μm clonidine on top of capsaicin (right, from box 3). E, collected results showing the effects of clonidine on the peak of mEPSCs. Left: values are reported as median, first quartile (25th percentile) and third quartile (75th percentile). Capsaicin (cap): median = 44.5, 25th percentile = 31.1, 75th percentile = 49.1. Capsaicin + clonidine (cap + clo): median = 28.1, 25th percentile = 24.1, 75th percentile = 31.6 (n = 7). * P < 0.05, Wilcoxon matched‐pairs test. Middle: distribution of mEPSC peaks recorded in 500 nm capsaicin (shaded bars) and after application of 1 μm clonidine (open bars) on top of capsaicin (n = 7). Right: cumulative probabilities of mEPSC peaks recorded in 500 nm capsaicin (solid line) and after application of 1 μm clonidine (dashed line) on top of capsaicin (n = 7). **** P < 0.0001, Kolmogorov–Smirnov test. F, collected results showing the effects of clonidine on the frequency of mEPSCs. Left: values are reported as median, first quartile (25th percentile) and third quartile (75th percentile). Capsaicin (cap): median = 367%, 25th percentile = 315%, 75th percentile = 918%. Capsaicin + clonidine (cap + clo): median = 102%, 25th percentile = 65%, 75th percentile = 132% (n = 7). * P < 0.05, Wilcoxon matched‐pairs test. Middle: distribution of inter‐event intervals of mEPSCs recorded in 500 nm capsaicin (shaded bars) and after application of 1 μm clonidine (opne bars) on top of capsaicin (n = 7). Right: cumulative probabilities of inter‐event intervals of mEPSCs recorded in 500 nm capsaicin (solid line) and after application of 1 μm clonidine (dashed line) on top of capsaicin (n = 7). **** P < 0.0001, Kolmogorov–Smirnov test.
Figure 12. Effect of clonidine on capsaicin‐induced mEPSCs in the presence of CaMKII blockers.
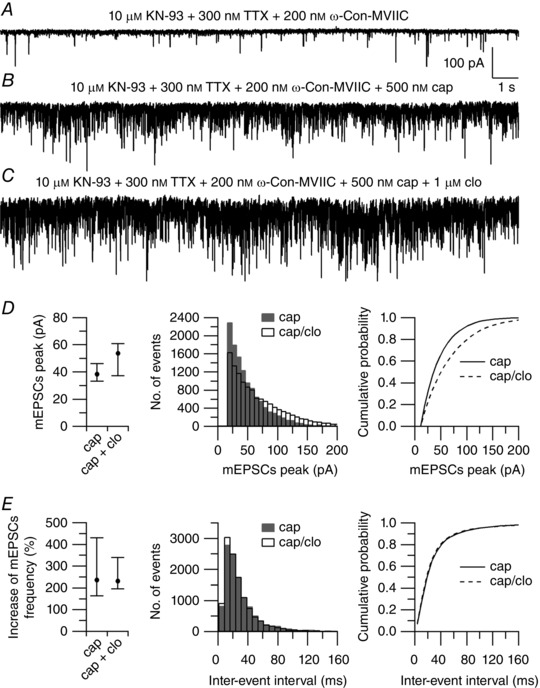
A, spontaneous mEPSCs recorded from a large lamina I neuron in a horizontal spinal cord slice pre‐incubated in 10 μm KN‐93 for 45 min. mEPSCS were recorded at a holding potential (V h) = −70 mV in the presence of 300 nm TTX + 200 nm ω‐conotoxin‐MVIIC, and with an intracellular solution in which Na‐GTP (0.3 mm) was replaced by the non‐hydrolysable analogue GDP‐β‐S (0.6 mm). B, capsaicin (500 nm) increased the median frequency of mEPSCs from 10 to 40 Hz. C, clonidine (1 μm), applied on top of capsaicin, reduced the median frequency of mEPSCs to 33 Hz. D, collected results showing the effects of clonidine on the peak of mEPSCs. Left: values are reported as median, first quartile (25th percentile) and third quartile (75th percentile). Capsaicin (cap): median = 38.5 pA, 25th percentile = 33.4 pA, 75th percentile = 46.2 pA. Capsaicin + clonidine (cap + clo): median = 53.8 pA, 25th percentile = 37.5 pA, 75th percentile = 60.9 pA (n = 7). P = 0.078, Wilcoxon matched‐pairs test. Middle: distribution of mEPSC peaks recorded in 500 nm capsaicin (shaded bars) and after application of 1 μm clonidine (open bars) on top of capsaicin (n = 7). Right: cumulative probabilities of mEPSC peaks recorded in 500 nm capsaicin (solid line) and after application of 1 μm clonidine (dashed line) on top of capsaicin (n = 7). **** P < 0.0001, Kolmogorov–Smirnov test. E, collected results showing the effects of clonidine on the frequency of mEPSCs. Left: values are reported as median, first quartile (25th percentile) and third quartile (75th percentile). Capsaicin (cap): median = 236%, 25th percentile = 163%, 75th percentile = 430%. Capsaicin + clonidine (cap + clo): median = 232%, 25th percentile = 196%, 75th percentile = 339% (n = 7). P = 0.687, Wilcoxon matched‐pairs test. Middle: distribution of inter‐event intervals of mEPSCs recorded in 500 nm capsaicin (shaded bars) and after application of 1 μm clonidine (open bars) on top of capsaicin (n = 7). Right: cumulative probabilities of inter‐event intervals of mEPSCs recorded in 500 nm capsaicin (solid line) and after application of 1 μm clonidine (dashed line) on top of capsaicin (n = 7). **P < 0.01, Kolmogorov–Smirnov test.
Data acquisition and analysis
Currents and voltages were controlled and sampled using a Digidata 1440A interface and pCLAMP 10.4 software (Molecular Devices). For isolated DRG neurons, current or voltage signals were filtered at 10 kHz (3 dB, 4‐pole Bessel) and digitized at 50 kHz. Capsaicin‐activated inward and outward currents were measured at −100 and +100 mV, respectively, during the “down ramp”. Spontaneous and evoked EPSCs were filtered at 1 kHz (3 dB, 4‐pole Bessel) and digitized at 50 kHz. Analysis was performed using Clampfit 10.4 and Igor Pro (version 6.2; WaveMetrics, Lake Oswego, OR, USA) using DataAccess (Bruxton, Seattle, WA) to import pCLAMP files into Igor. For mEPSCs analysis, the threshold was set at twice the average noise. Frequency and peak of mEPSCs were determined during a 1 min period in each condition. Reported voltages for spontaneous and evoked EPSCs were corrected for junction potential (−8 mV) between the internal solution and the extracellular solution measured using a flowing 3 m KCl electrode (Neher, 1992). Data are reported as mean ± SD.
Statistics
Statistical differences between data sets were analysed using Student's t test, or one‐way ANOVA, or Wilcoxon matched‐pairs test, or Kolmogorov–Smirnov test. Differences were considered significant at * P < 0.05.
Results
Ionic currents through TRPV1 channels were activated by challenging small DRG neurons with 1 μm capsaicin, and isolated by subtracting currents before and after application of capsaicin (Fig. 1 A). The capsaicin‐activated current showed the typical outward rectification and reversal potential close to 0 mV, as expected for a TRPV1 current (Caterina et al. 1997). To test whether activation of adrenergic receptors could modulate currents through TRPV1 channels, the effect of noradrenaline was tested on the capsaicin‐activated current. When applied on top of capsaicin, 10 μm noradrenaline caused substantial inhibition of the capsaicin‐activated current (Fig. 1 B and C), suggesting strong modulation of TRPV1 channels by adrenergic receptors. The effects of noradrenaline reached a maximum in 15–20 s and reversed completely with a similar time course. Noradrenaline inhibited the inward and outward currents (measured at −100 and + 100 mV, respectively) to a similar extent (Fig. 1 D), in a dose‐dependent manner. In collected results (Fig. 2), the capsaicin‐activated inward current was reduced by 43 ± 11% (n = 7) by 300 nm noradrenaline, by 55 ± 14 % (n = 17) by 1 μm noradrenaline, with little or no additional effects at 3 μm (reduction by 54 ± 17%, n = 9) and 10 μm (reduction by 58 ± 14%, n = 6).
Figure 2. Dose‐dependent inhibition of capsaicin‐activated current by noradrenaline.
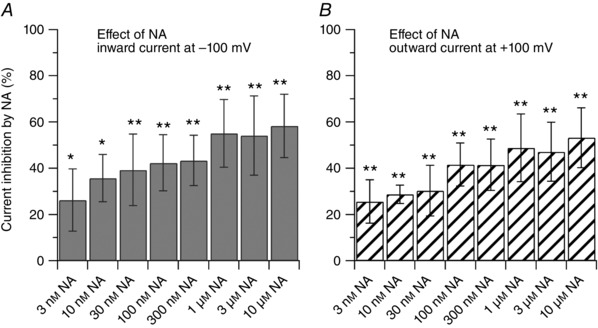
Dose‐dependent inhibition of capsaicin‐activated currents by noradrenaline showing similar effects on inward and outward currents. Each bar represents an independent experiment. For each concentration, statistical significance was assessed with a paired t test by comparing the effect of the drug to its own control. The IC50 was determined using a Log inhibitor versus normalized response equation: y = 100/(1 + 10^((x‐LogIC50))). A, the inward current was reduced by 26 ± 13% (n = 8, * P < 0.05) by 3 nm; by 36 ± 10% (n = 6, * P < 0.05) by 10 nm; by 39 ± 15% (n = 11, ** P < 0.01) by 30 nm; by 42 ± 12% (n = 8, ** P < 0.01) by 100 nm; by 43 ± 11% (n = 7, ** P < 0.01) by 300 nm; by 55 ± 14% (n = 17, ** P < 0.01) by 1 μm; by 54 ± 17% (n = 9, ** P < 0.01) by 3 μm; by 58 ± 13% (n = 6, ** P < 0.01) by 10 μm noradrenaline. The IC50 for the inward current was estimated to be 7.1 nm (2.1–22.7 nm, 95% confidence interval). B, the outward current was reduced by 26 ± 9% (n = 8, ** P < 0.01) by 3 nm; by 29 ± 4% (n = 6, ** P < 0.01) by 10 nm; by 30 ± 11% (n = 11, ** P < 0.01) by 30 nm; by 42 ± 9% (n = 8, ** P < 0.01) by 100 nm; by 42 ± 11% (n = 7, ** P < 0.01) by 300 nm; by 49 ± 15% (n = 17, ** P < 0.01) by 1 μm; by 47 ± 13% (n = 9, ** P < 0.01) by 3 μm; by 53 ± 13% (n = 6, ** P < 0.01) by 10 μm noradrenaline. The IC50 for the outward current was estimated to be 9.6 nm (2.5–34.2 nm, 95% confidence interval).
Our standard intracellular solution contained 0.3 mm GTP (see Methods). To confirm that the effect of noradrenaline was mediated by G‐protein‐coupled receptors (GPCRs) and dependent on GTP, we tested an intracellular solution in which GTP was replaced by the non‐hydrolysable analogue GDP‐β‐S. Replacing the intracellular GTP with GDP‐β‐S caused a small reduction in the current density of the capsaicin‐activated current. The inward current was reduced from 34 ± 20 to 25 ± 26 pA pF−1, and the outward current was reduced from 267 ± 120 to 204 ± 212 pA pF−1 (n = 10). The inhibitory effect of 1 μm noradrenaline on the capsaicin‐activated inward current was reduced from 55 ± 14 % (n = 17, with intracellular GTP) to 20 ± 10 % (n = 10, with intracellular GDP‐β‐S) (Fig. 3). Taken together, these results are consistent with coupling between adrenergic receptors and TRPV1 channels that is dependent on GTP acting via G‐proteins.
Figure 3. The effect of noradrenaline on the capsaicin‐activated current is mediated by G proteins.
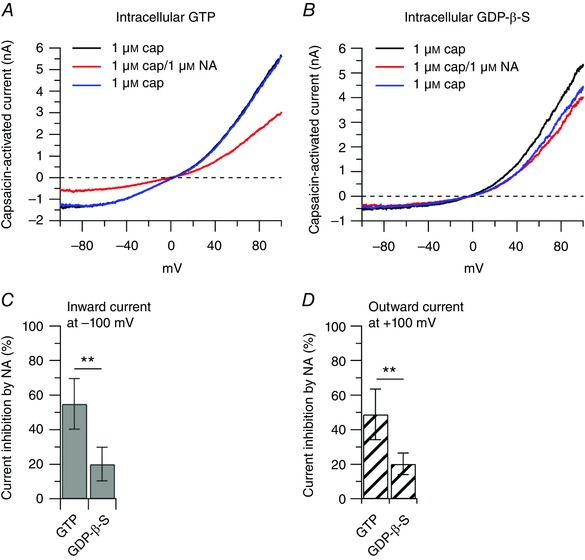
Collected results show reduction of the effect of 1 μm noradrenaline on the capsaicin‐activated inward and outward currents when the usual 0.3 mm GTP was replaced by 0.6 mm of the non‐hydrolysable analogue GDP‐β‐S. A, representative capsaicin‐activated currents recorded with 0.3 mm GTP in the intracellular solution: 1 μm capsaicin (black trace), after application of 1 μm noradrenaline on top of capsaicin (red trace) and upon removal of noradrenaline (blue trace). B, representative capsaicin‐activated currents recorded with 0.6 mm GDP‐β‐S in the intracellular solution: 1 μm capsaicin (black trace), after application of 1 μm noradrenaline on top of capsaicin (red trace) and upon removal of noradrenaline (blue trace). C, the inward current was reduced by 55 ± 14% (n = 17) with GTP included in the intracellular solution, and by 20 ± 10% when GTP was replaced by GDP‐β‐S (n = 10), unpaired t test, ** P < 0.01. D, the outward current was reduced by 49 ± 15% (n = 17) with GTP included in the intracellular solution, and by 20 ± 6% when GTP was replaced by GDP‐β‐S (n = 10), unpaired t test, ** P < 0.01.
The next step was to identify which adrenergic receptors mediate the effect of noradrenaline. The α2 adrenergic receptor agonist clonidine potently inhibited the capsaicin‐activated current similar to noradrenaline, in a dose‐dependent manner (Fig. 4). The capsaicin‐activated inward current was reduced by 4 ± 2% by 100 pm clonidine (n = 6), by 16 ± 6% by 300 pm clonidine (n = 7), by 54 ± 10% (n = 6) by 1 nm clonidine, with little or no additional effects at 3 nm (reduction by 58 ± 14 %, n = 6), 300 nm (reduction 54 ± 17 %, n = 7) and 1 μm (reduction by 63 ± 4 %, n = 7). The potency and saturation of the effects of clonidine suggest mediation by a receptor rather than a direct blocking effect on the channel. Mediation by a receptor was further tested by applying clonidine in combination with yohimbine, a selective antagonist at α2 adrenergic receptors. In this series of experiments, the inhibitory effect of 1 nm clonidine on the capsaicin‐activated inward current was reduced from 54 ± 10% (n = 6) when used alone to 8 ± 6% (n = 7) when applied in combination with 1 nm yohimbine (Fig. 5).
Figure 4. Dose‐dependent inhibition of capsaicin‐activated current by clonidine.
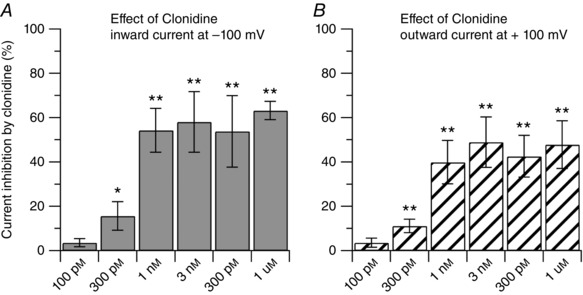
Dose‐dependent inhibition of capsaicin‐activated currents by the α2 agonist clonidine, showing similar effects on inward and outward currents. Each bar represents an independent experiment. For each concentration, statistical significance was assessed with a paired t test by comparing the effect of the drug to its own control. The IC50 was determined using a Log inhibitor versus normalized response equation: y = 100/(1 + 10^((x − LogIC50))). A, the inward current was reduced by 4 ± 2% (n = 6, P = 0.116) by 100 pm; by 16 ± 6% (n = 7, * P < 0.05) by 300 pm; by 54 ± 10% (n = 6, ** P < 0.01) by 1 nm; by 58 ± 14% (n = 6, ** P < 0.01) by 3 nm; by 54 ± 16% (n = 10, ** P < 0.01) by 300 nm; by 63 ± 4% (n = 7, ** P < 0.01) by 1 μm clonidine. The IC50 for the inward current was estimated to be 487.7 pm (209.9–1130 pm, 95% confidence interval). B, the outward current was reduced by 4 ± 2% (n = 6, P = 0.068) by 100 pm; by 11 ± 3% (n = 7, ** P < 0.01) by 300 pm; by 40 ± 10% (n = 6, ** P < 0.01) by 1 nm; by 49 ± 11% (n = 6, ** P < 0.01) by 3 nm; by 43 ± 9% (n = 10, ** P < 0.01) by 300 nm; by 48 ± 11% (n = 7, ** P < 0.01) by 1 μm clonidine. The IC50 for the outward current was estimated to be 470.1 pm (199.6–1089 pm, 95% confidence interval).
Figure 5. The effect of clonidine is reversed by the α2 antagonist yohimbine.
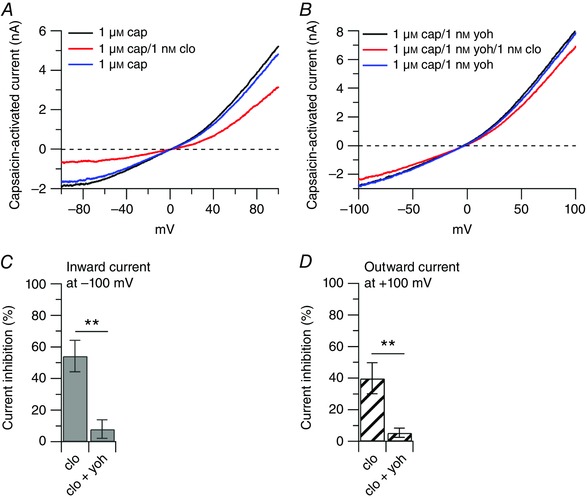
Collected results show the effect of yohimbine (α2 antagonist) on the capsaicin‐activated inward and outward currents. A, representative capsaicin‐activated currents recorded in the presence of 1 μm capsaicin (cap, black trace), after application of 1 nm clonidine (clo) on top of capsaicin (red trace), and upon removal of clonidine (blue trace). B, representative capsaicin‐activated currents recorded in the presence of 1 μm capsaicin + 1 nm yohimbine (yoh, black trace), after application of 1 nm clonidine on top of capsaicin and yohimbine (red trace), and upon removal of clonidine (blue trace). C, the inward current was reduced by 54 ± 10% (n = 6) by 1 nm clonidine and by 8 ± 5% by 1 nm clonidine + 1 nm yohimbine (n = 7), ** P < 0.01, unpaired t test. D, the outward current was reduced by 40 ± 10% (n = 6) by 1 nm clonidine and by 5 ± 3% by 1 nm clonidine + 1 nm yohimbine (n = 7), ** P < 0.01, unpaired t test.
A change in temperature can affect the response of TRPV1 channels to agonists (Neelands et al. 2008), its modulation by protons and the open probability (Neelands et al. 2010; Jara‐Oseguera et al. 2016), and the voltage dependence of activation (Voets et al. 2004). We therefore determined whether the inhibitory effect of clonidine on the capsaicin‐activated current observed at room temperature was unchanged at a more physiological temperature. For this, we tested the effect of 1 μm clonidine on the capsaicin‐activated current at 35 ± 1°C. As shown in Fig. 6, 1μm clonidine inhibited the inward current by 56 ± 19% and the outward current by 48 ± 12% (n = 8), which were not different from the inhibitory effects observed at room temperature.
Protein phosphorylation by protein kinase A (PKA), protein kinase C (PKC) and CaMKII has been shown to play prominent roles in the modulation of TRPV1 channels (Vellani et al. 2001; Bhave et al. 2002; Jung et al. 2004; Bangaru et al. 2015). Therefore, we tested whether PKA, PKC or CaMKII activity was linked to inhibition of TRPV1 channels by adrenergic receptor activation in DRG neurons. When PKA activity was blocked by pre‐incubating DRG neurons with 1 μm H89 for 30 min, the inhibitory effect of 1 μm noradrenaline on the capsaicin‐activated current remained unchanged: 1 μm noradrenaline inhibited the capsaicin‐activated inward current by 55 ± 10% (n = 9) in control and by 51 ± 13% (n = 11) in H89 (Fig. 7 A). The inhibitory effect of 1 μm noradrenaline on the capsaicin‐activated current remained unchanged also when PKC activity was blocked by pre‐incubating DRG neurons with 1 μm bisindolylmaleimide II (BIM) for 30 min: 1 μm noradrenaline inhibited the capsaicin‐activated inward current by 59 ± 10% (n = 7) in control and by 63 ± 4% (n = 7) in BMI (Fig. 7 C). Results were very similar when inhibitors of PKA or PKC were directly included in the patch pipette. When 1 μm H89 was included in the patch pipette, noradrenaline inhibited the inward current by 47 ± 10% and the outward current by 50 ± 10% (n = 6). When 1 μm BIM was included in the patch pipette, noradrenaline inhibited the inward current by 44 ± 14% and the outward current by 43 ± 9% (n = 6). In contrast, when CaMKII activity was blocked by including 10 μm KN‐93 in the patch pipette, the inhibitory effect of 1 μm noradrenaline on the capsaicin‐activated current was significantly affected: 1 μm noradrenaline inhibited the capsaicin‐activated inward current by 55 ± 14% (n = 17) in control and by 11 ± 9% (n = 12) with intracellular KN‐93 (Fig. 7 E). When KN‐93 was replaced by the inactive analogue KN‐92 (10 μm), 1 μm noradrenaline inhibited the capsaicin‐activated inward current by 52 ± 14% (n = 8) (Fig. 7 E). Taken together, these results suggest that activation of adrenergic receptors is linked to CaMKII activity, and point to a calcium‐dependent mechanism.
Figure 7. Intracellular pathways linked to α2 adrenergic receptors activation.
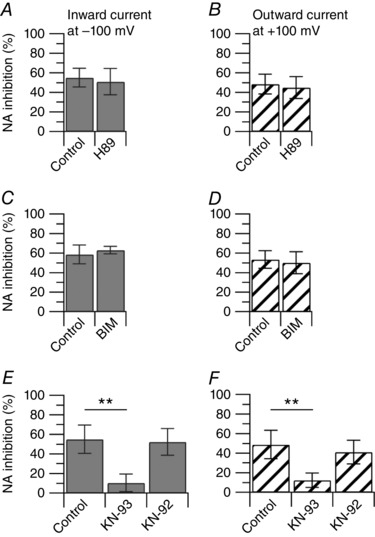
Collected results show the effect of 1 μm noradrenaline on the capsaicin‐activated inward and outward currents with manipulation of intracellular protein kinases. A, the inward current was reduced by 55 ± 10% (n = 9) in 1 μm noradrenaline alone, and by 51 ± 13% (n = 11) following incubation (30 min) with 1 μm H89, P = 0.420, unpaired t test. B, the outward current was reduced by 48 ± 10% (n = 9) in 1 μm noradrenaline alone, and by 45 ± 11% (n = 11) following incubation with 1 μm H89 (30 min), P = 0.474, unpaired t test. C, the inward current was reduced by 59 ± 10% (n = 7) in 1 μm noradrenaline alone, and by 63 ± 4% (n = 7) following incubation with 1 μm BIM (30 min), P = 0.362, unpaired t test. D, the outward current was reduced by 53 ± 9% (n = 7) in 1 μm noradrenaline alone, and by 50 ± 9% (n = 7) following incubation with 1 μm BIM (30 min), P = 0.609, unpaired t test. E, the inward current was reduced by 55 ± 14% (n = 17) in 1 μm noradrenaline alone, by 11 ± 9% (n = 12) with intracellular 10 μm KN‐93 and by 52 ± 14% (n = 8) with intracellular 10 μm KN‐92. Control versus KN‐93, ** P < 0.01; control versus KN‐92, P = 0.845, one way ANOVA followed by Dunnett's post hoc comparison test. F, the outward current was reduced by 49 ± 15% (n = 17) by 1 μm noradrenaline alone, by 12 ± 7% (n = 12) with intracellular 10 μm KN‐93 and by 41 ± 12% (n = 8) with intracellular 10 μm KN‐92. Control versus KN‐93, ** P < 0.01; control versus KN‐92, P = 0.364, one‐way ANOVA followed by Dunnett's post hoc comparison test.
A rise in cytoplasmic Ca2+ to activate CaMKII may reflect influx of external Ca2+, release of Ca2+ from internal stores or both. To test these possibilities, we carried out a series of experiments to manipulate the external concentration of calcium, to manipulate the calcium buffer capacity of the intracellular solution and to deplete the internal calcium stores (Fig. 8 A). When the external Ca2+ (2 mm) was replaced by an equimolar concentration of Mg2+, such that the external solution contained a final concentration of 0 mm Ca2+ and 3 mm Mg2+, using 10 mm EGTA as the intracellular calcium chelator, the inhibitory effect of 1 μm noradrenaline on the capsaicin‐activated inward current was reduced from 55 ± 14% (n = 17) in 2 mm Ca2+ to 15 ± 10% (n = 12) in 0 mm Ca2+, suggesting a strong contribution of calcium influx. The residual inhibitory effect of noradrenaline could reflect the contribution of calcium released from internal stores, or a calcium‐independent effect. To test these possibilities, first, the slow calcium buffer EGTA (10 mm) used in our experimental condition was replaced by 10 mm of the fast calcium buffer BAPTA. The inhibitory effect of 1 μm noradrenaline on the capsaicin‐activated inward current was reduced to 18 ± 15% (n = 6) in 2 mm Ca2+ external solution with BAPTA internally. Second, the external solution with 0 mm Ca2+ and 3 mm Mg2+ was tested with an internal solution containing 10 mm BAPTA. The inhibitory effect of 1 μm noradrenaline on the capsaicin‐activated inward current was reduced to 8 ± 3% (n = 10) in 0 mm Ca2+ external solution and internal BAPTA. Third, the 0 mm Ca2+ external solution with 10 mm internal EGTA was tested after depleting the internal calcium stores by pre‐incubating the cells with 1 μm thapsigargin, an inhibitor of the sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA), for 15 min. The inhibitory effect of 1 μm noradrenaline on the capsaicin‐activated inward current was reduced to 4 ± 3% (n = 6) under these conditions, consistent with a contribution of Ca2+ released from internal stores.
Figure 8. The effects of noradrenaline on the capsaicin‐activated current are dependent on calcium.
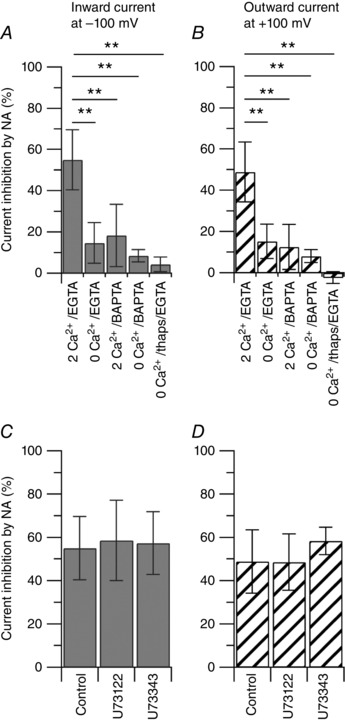
Collected results show the effect of 1 μm noradrenaline on the capsaicin‐activated inward and outward currents with manipulation of external and/or internal calcium. The effect of 1 μm noradrenaline was determined with the following conditions: (1) 2 mm Ca2+ in the external solution and 10 mm EGTA in the internal solution (2 Ca2+/EGTA); (2) 2 mm Ca2+ in the external solution was replaced by 2 mm Mg2+ (0 Ca2+) and tested with 10 mm EGTA in the internal solution (0 Ca2+/EGTA); (3) 2 mm Ca2+ in the external solution and 10 mm BAPTA in the internal solution (2 Ca2+/BAPTA); (4) 2 mm Ca2+ in the external solution was replaced by 2 mm Mg2+ (0 Ca2+) and tested with 10 mm BAPTA in the internal solution (0 Ca2+/BAPTA); (5) 2 mm Ca2+ in the external solution was replaced by 2 mm Mg2+ (0 Ca2+) and tested with 10 mm EGTA in the internal solution after depleting the internal calcium stores by pre‐incubating the cells with 1 μm thapsigargin for 15 min (0 Ca2+/thaps/EGTA). A, the inward current was reduced by 55 ± 14% (n = 17) in 2 Ca2+/EGTA; by 15 ± 10% (n = 12) in 0 Ca2+/EGTA; by 18 ± 15% (n = 6) in 2 Ca2+/BAPTA; by 8 ± 3% (n = 10) in 0 Ca2+/BAPTA; by 4 ± 3% (n = 6) in 0 Ca2+/thaps/EGTA. 2 Ca2+/EGTA versus 0 Ca2+/EGTA, 2 Ca2+/BAPTA, 0 Ca2+/BAPTA and 0 Ca2+/thaps/EGTA, ** P < 0.01, one‐way ANOVA followed by Dunnett's post hoc comparison test. B, the outward current was reduced by 49 ± 15% (n = 17) in 2 Ca2+/EGTA; by 15 ± 8% (n = 12) in 0 Ca2+/EGTA; by 13 ± 11% (n = 6) in 2 Ca2+/BAPTA; by 8 ± 3% (n = 10) in 0 Ca2+/BAPTA; by −2 ± 3% (n = 6) in 0 Ca2+/thaps/EGTA. 2 Ca2+/EGTA versus 0 Ca2+/EGTA, 2 Ca2+/BAPTA, 0 Ca2+/BAPTA and 0 Ca2+/thaps/EGTA, ** P < 0.01, one‐way ANOVA followed by Dunnett's post hoc comparison test. C, the capsaicin‐activated inward current was reduced by 55 ± 14% (n = 17) in 1 μm noradrenaline alone, by 59 ± 19% (n = 6) following incubation with 5 μm U73122 (PLC inhibitor) and by 57 ± 14% (n = 6) following incubation with 5 μm U73343 (inactive control). Control versus U73122, P = 0.862; control versus U73343, P = 0.953, one‐way ANOVA followed by Dunnett's post hoc comparison test. D, the capsaicin‐activated outward current was reduced by 49 ± 15% (n = 17) in 1 μm noradrenaline alone, by 49 ± 13% (n = 6) following incubation with 5 μm U73122 and by 58 ± 6% (n = 6) following incubation with 5 μm U73343. Control versus U73122, P = 0.998; control versus U73343, P = 0.352, one‐way ANOVA followed by Dunnett's post hoc comparison test.
Release of Ca2+ from internal stores may be triggered by activation of phospholipase C (PLC) and a subsequent increase in inositol 1,4,5‐trisphosphate (InsP3). When DRG neurons were pre‐incubated with 5 μm U73122 (a non‐selective inhibitor of PLC) or with 5 μm U73343 (inactive control) for 30 min, the inhibitory effects of 1 μm noradrenaline on the capsaicin‐activated inward current were 58 ± 18% (n = 6) and 57 ± 14% (n = 5), respectively, and were not different from those observed in control (55 ± 14%, n = 17), arguing against a contribution by U73122‐sensitive PLC. Taken together, the data suggest that the inhibitory effects of noradrenaline on the capsaicin‐activated current are strongly Ca2+‐dependent, mediated by CaMKII, and that both calcium influx and calcium released from internal stores contribute to the rise in cytoplasmic calcium activating CaMKII.
Activation of presynaptic TRPV1 channels by endovanilloids or capsaicin has been shown to trigger the release of peptides and modulate glutamatergic transmission in the dorsal horn of the spinal cord (Yang et al. 1998, 1999; Tognetto et al. 2001; Nakatsuka et al. 2002; Baccei et al. 2003; Labrakakis & MacDermott, 2003; Tong & MacDermott, 2006; Medvedeva et al. 2008). Based on these observations, our data predict that activation of presynaptic α2 adrenergic receptors could interact with presynaptic TRPV1 channels to modulate glutamatergic transmission between nociceptors and second‐order neurons in the dorsal horn of the spinal cord. To test this possibility, we recorded mEPSCs from large lamina I neurons in horizontal spinal cord slices. To restrict the effects of adrenergic agonists to presynaptic targets, mEPSCs were recorded in the presence of 300 nm TTX, and with an intracellular solution in which Na‐GTP (0.3 mm) was replaced by the non‐hydrolysable analogue GDP‐β‐S (0.6 mm) to inhibit GPCRs signalling in the postsynaptic neuron. Capsaicin (500 nm) increased the median frequency of mEPSCs by 963%, consistent with activation of presynaptic TRPV1 channels. Clonidine (1 μm), applied on top of capsaicin, strongly reduced the increased median frequency of mEPSCs to 137% (n = 7, Fig. 9 E), suggesting that activation of presynaptic α2 adrenergic receptors down‐regulates the capsaicin‐induced neurotransmitter release from nociceptors to lamina I neurons. Although clonidine also reduced the median of mEPSCs peak (Fig. 9 D, left panel), analysis of the distribution of events (Fig. 9, middle panel) indicated that the reduction of mEPSCs peak was restricted to events of larger size (probably reflecting multiple vesicles release), while smaller events (probably reflecting single vesicle release) remained largely unchanged (see also Fig. 11 E, middle panel), supporting our interpretation that the main effects of clonidine are on capsaicin‐induced presynaptic events and not inhibition of synaptic vesicle release per se or changes in postsynaptic excitability.
Neurotransmitter release from Aδ‐ and C‐fibre nociceptors to lamina I neurons is dependent on presynaptic N‐ and P/Q‐type calcium channels (Bao et al. 1998; Heinke et al. 2004), and noradrenaline is well known to inhibit presynaptic calcium channels and neurotransmitter release (Dunlap & Fischbach, 1981; Bean, 1989; Lipscombe et al. 1989; Pollo et al. 1992; Boehm & Huck, 1996; Ikeda, 1996; Li & Horn, 2008). These observations raise the possibility that the reduced frequency of capsaicin‐induced mEPSCs observed with clonidine (Fig. 9) could be mediated also by inhibition of calcium channels upon activation of presynaptic α2 adrenergic receptors. To exclude this possibility, the effects of clonidine on capsaicin‐induced mEPSCs recorded from large lamina I neurons were determined in the presence of ω‐conotoxin‐MVIIC, a selective blocker of both N‐ and P/Q‐type calcium channels (Hillyard et al. 1992; McDonough et al. 1996). Initial control experiments were carried out to confirm the contribution of presynaptic N‐ and P/Q‐type calcium channels to neurotransmitter release from primary afferent fibres to lamina I neurons. Evoked EPSCs, elicited by electrical stimulation of the dorsal root (L4 or L5) and recorded from large lamina I neurons in horizontal spinal cord slices, were reduced by 94 ± 7% (n = 9) by 200 nm ω‐conotoxin‐MVIIC (Fig. 10), consistent with a major contribution of N‐ and P/Q‐type calcium channels. Then, similar to the experiments in Fig. 9, mEPSCs were isolated in the presence of 300 nm TTX and 200 nm ω‐conotoxin‐MVIIC, and with GDP‐β‐S (0.6 mm) in the intracellular solution to block effects mediated by GPCRs in the postsynaptic neuron. The median frequency of mEPSCs recorded in TTX (15.2 Hz) was not affected by application of 200 nm ω‐conotoxin‐MVIIC on top of TTX (14.8 Hz). Application of 500 nm capsaicin on top of ω‐conotoxin‐MVIIC increased the median frequency of mEPSCs by 367%, and subsequent application of 1 μm clonidine on top of capsaicin reduced the increased median frequency of mEPSCs to 102% (n = 7, Fig. 11). Finally, we tested whether blocking the activity of CaMKII would reduce or eliminate the effects of clonidine on the frequency of mEPSCs. For these series of experiments, spinal cord slices were pre‐incubated with 10 μm KN‐93 for 30–45 min. Application of 500 nm capsaicin on top of TTX and ω‐conotoxin‐MVIIC increased the median frequency of mEPSCs by 236%, and subsequent application of 1 μm clonidine on top of capsaicin had little additional effects (232%, Fig. 12, n = 7), confirming that the inhibitory effect of clonidine requires the activity of CaMKII as seen in isolated DRG neurons. Taken together, the data suggest that activation of presynaptic α2 adrenergic receptors reduces the frequency of capsaicin‐induced mEPSCs, and that the effect is mediated by direct inhibition of presynaptic TRPV1 channels.
Discussion
The data presented here provide evidence that noradrenaline and clonidine strongly down‐regulate the activity of presynaptic TRPV1 channels expressed in DRG neurons through α2 adrenergic receptor activation. Only small DRG neurons (diameter of 23.8 ± 2.5 μm and cell capacitance of 18.1 ± 3.7 pF, n = 219) sensitive to capsaicin (75% of those tested, 255/339) were included in the study, comprising a population of probable nociceptors (Cardenas et al. 1995; Caterina et al. 1997; Petruska et al. 2000; Ho & O'Leary, 2011).
The inhibitory effect of noradrenaline on the capsaicin‐activated current was reduced by replacing GTP with GDP‐β‐S (a non‐hydrolysable analogue of GDP) in the intracellular solution (Fig. 3), confirming that the effects of noradrenaline require activation of GPCRs as expected for adrenergic receptors (Ruffolo et al. 1993; Saunders & Limbird, 1999). The lack of complete abolition of the response by GDP‐β‐S may reflect the difficulty in completely dialysing out GTP.
Of the three isoforms of α2 adrenergic receptors cloned in humans and rat (α2A, α2B, α2C) (Bylund et al. 1994), DRG neurons express mainly the α2A and α2C isoforms (Nicholas et al. 1993; Cho et al. 1997; Stone et al. 1998; Birder & Perl, 1999), although expression of the α2B isoform has been reported (Gold et al. 1997). Consistent with these observations, clonidine (α2 agonist), at saturating concentrations of 0.3–1 μm, fully mimicked the inhibitory effects of noradrenaline on the capsaicin‐activated current, and the effects were strongly antagonized by yohimbine (α2 antagonist), supporting the conclusion that noradrenaline down‐regulates the activity of TRPV1 channels by activation of α2 adrenergic receptors. Although available pharmacological agents do not discriminate between different α2 adrenergic receptor subtypes (Giovannoni et al. 2009), it has been shown that degeneration of capsaicin‐sensitive DRG neurons after neonatal capsaicin treatment in rats strongly decreases the expression of the α2A, but not α2C, isoform in primary afferent fibres (Stone et al. 1998), consistent with preferential expression of the α2A isoform in capsaicin‐sensitive DRG neurons. Because our study was limited only to capsaicin‐sensitive DRG neurons, it is likely that the effects of noradrenaline and clonidine reported here are mediated mainly by activation of α2A adrenergic receptors.
The inhibitory effects of noradrenaline and clonidine observed in acutely isolated DRG neurons, a preparation in which postsynaptic targets are removed, demonstrate that the machinery for noradrenaline inhibition of TRPV1 channels is present in DRG cell bodies. The inhibitory effects of clonidine on the capsaicin‐induced mEPSCs suggest that the elements for α2 adrenergic receptor inhibition of TRPV1 currents are also operational at presynaptic terminals. This conclusion is supported by several other observations. (1) TRPV1 channels are expressed not only on the cell body and peripheral terminals of nociceptors, but also on the central terminal which make synaptic contacts with second‐order neurons in the dorsal horn of the spinal cord (Holzer, 1991; Winter et al. 1993; Szallasi et al. 1995; Tominaga et al. 1998; Guo et al. 1999; Hwang et al. 2004). (2) Expression of α2 adrenergic receptors in the dorsal horn of the spinal cord co‐localizes with substance P (Stone et al. 1998), consistent with expression of α2 adrenergic receptors on the central terminals of nociceptors. (3) In spinal cord slices, mEPSCs were recorded from lamina I neurons in the presence of TTX and ω‐conotoxin‐MVIIC, and with an intracellular solution in which GTP was replaced by the non‐hydrolysable analogue GDP‐β‐S to block GPCRs in the postsynaptic lamina I neuron, arguing against a postsynaptic effect of clonidine. (4) The inhibitory effects of clonidine on capsaicin‐induced mEPSCs reported here parallel the inhibitory effects mediated by α2A adrenergic receptors on the capsaicin‐induced glutamate release in spinal cord synaptosomes (Li & Eisenach, 2001). (5) Although clonidine reduced the median peak of mEPSCs, the reduction was restricted to larger events, without any apparent reduction of the sizes of smaller events (Figs 9 D, 11 D and 11 E), suggesting no effect of clonidine on quantal release of transmitter, or suppression of postsynaptic excitability. All these observations point to a presynaptic effect of noradrenaline and clonidine mediated by adrenergic receptors expressed on the central terminals of nociceptors.
Protein phosphorylation plays a major role in the modulation of TRPV1 channels. Several serine and threonine residues have been reported to be phosphorylated by PKC (Tominaga et al. 2001; Numazaki et al. 2002; Bhave et al. 2003; Premkumar et al. 2004), or by PKA (De Petrocellis et al. 2001; Bhave et al. 2002; Rathee et al. 2002; Vlachova et al. 2003; Mohapatra & Nau, 2005; Jeske et al. 2006), or by CaMKII (Jung et al. 2004; Price et al. 2005; Hucho et al. 2012). Phosphorylation by PKC sensitizes TRPV1 channels by increasing the channel open probability (Vellani et al. 2001), while phosphorylation by PKA may reverse the desensitization of TRPV1 channels induced by prolonged or repeated application of the agonist (Bhave et al. 2002). Thus, stimulation of PKC or PKA would probably produce an increase of the capsaicin‐activated current. However, our data show that noradrenaline or clonidine consistently inhibited the capsaicin‐activated current, arguing against a direct involvement of PKC or PKA. In contrast, the inhibitory effect of noradrenaline on the capsaicin‐activated current was reduced by ∼50% when the CaMKII activity was blocked, suggesting that CaMKII activity is involved in the modulation of the capsaicin‐activated current by noradrenaline. We have previously shown in DRG neurons that CaMKII activity is required also for the modulation of TRPV1 channels upon activation of D1/D5 dopamine receptors (Chakraborty et al. 2016), raising the possibility for a convergent pathway involved in the modulation of TRPV1 channels by different catecholamines. Phosphorylation/dephosphorylation of TRPV1 channels by CaMKII/calcineurin have been suggested to modulate vanilloids binding to TRPV1 channels (Jung et al. 2004). Thus, an intriguing possibility is that activation of α2 adrenergic receptors may modulate the function of TRPV1 channels by affecting the binding of vanilloids to the channel. Because Ca2+ signalling regulates both CaMKII and calcineurin, different mechanisms have been proposed to explain the fine balance between phosphorylation and dephosphorylation, including different activation kinetics, different sensitivities to Ca2+ and calmodulin, or cross‐talk between these two enzymes–signalling pathways (Hashimoto et al. 1988; Klee, 1991; Tian & Karin, 1999; Tominaga & Tominaga, 2005; Stratton et al. 2013; Simon et al. 2015). Future experiments are required to fully elucidate the interaction between α2 adrenergic receptors and the CaMKII/calcineurin pathway.
Our results show that the inhibitory effect of noradrenaline on the capsaicin‐activated current is highly dependent on calcium. Removal of Ca2+ from the external solution strongly reduced the inhibitory effects of noradrenaline on the capsaicin‐activated current, suggesting a major contribution of Ca2+ influx for the rise in cytoplasmic Ca2+. Under physiological conditions, activation of voltage‐dependent calcium channels during action potential firing in nociceptors probably provides a major source of Ca2+ influx (Carbone & Lux, 1984; Scroggs & Fox, 1991, 1992; Blair & Bean, 2002; Bell et al. 2004; Castiglioni et al. 2006; Gemes et al. 2010), and will contribute to a rise in cytoplasmic Ca2+ necessary to engage downstream targets such as CaMKII. Activation of TRPV1 channels with their high permeability to Ca2+ (P Ca 2+/P Na + = ∼10) (Caterina et al. 1997) also results in influx of Ca2+. Removal of external Ca2+ reduced but did not eliminate noradrenaline inhibition of TRPV1 currents, suggesting an additional contribution from Ca2+ released from intracellular stores, consistent with further reduction of the noradrenaline effect when stores were depleted by thapsigargin treatment (Olah et al. 2001; Wong et al. 2014).
Calcium influx is suggested to play a prominent role in the desensitization of TRPV1 channels upon repeated or sustained stimulation with the agonist (Cholewinski et al. 1993; Docherty et al. 1996; Liu & Simon, 1996; Koplas et al. 1997; Jung et al. 2004; Rosenbaum et al. 2004), which predicts a reduced response of nociceptors to repeated or sustained noxious stimuli. However, in our experimental conditions, in which we used a single application of capsaicin, inhibition of capsaicin‐activated current by noradrenaline was consistently accompanied by rapid and complete recovery of the current upon washing out noradrenaline (Fig. 1), suggesting that the down‐regulation of TRPV1 channels by noradrenaline occurs via a mechanism different from Ca2+‐induced desensitization.
Our experiments in spinal cord slices show that clonidine can powerfully reduce the frequency of mEPSCs induced by capsaicin in the presence of ω‐conotoxin‐MVIIC (Fig. 11), a condition in which N‐ and P/Q‐type calcium channels are blocked (Fig. 10). The simplest interpretation is that under these conditions activation of presynaptic TRPV1 channels produces calcium entry into presynaptic terminals that promotes release of synaptic vesicles. This is consistent with a previous report showing a contribution of TRPV1 channels to a rise in cytoplasmic Ca2+ and neurotransmitter release in co‐cultures of DRG neurons and dorsal horn neurons (Medvedeva et al. 2008). The effect of α2 adrenergic receptor activation to reduce the capsaicin‐induced mEPSCs is then consistent with the inhibition of presynaptic TRPV1 channels like that seen in DRG cell bodies.
The inhibition of presynaptic TRPV1 channels by noradrenaline could contribute to overall inhibition of pain signaling by noradrenaline released by descending adrenergic fibres, although it remains to be determined under what circumstances presynaptic TRPV1 channels are activated. An intriguing possibility is that noradrenaline inhibition of TRPV1 signalling could also occur in peripheral terminals of primary nociceptors, but further work will be required to test this possibility.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
Conception and design of the experiments: M.P, M.R., M.K. Collection, analysis and interpretation of data: S.C., V.E., M.P, M.R., M.K. Drafting the article: S.C., M.P, M.R., M.K. All authors have approved the final version of the manuscript
Funding
This work was supported by internal funds from the Department of Anesthesiology, Stony Brook Medicine, Stony Brook, NY, to M.P.
Translational perspective.
Neurotransmitters released from supraspinal centres can strongly modulate pain signals in the dorsal horn of the spinal cord. TRPV1 channels play a critical role in setting the excitability of nociceptors and their synaptic transmission to second‐order neurons in dorsal horn of the spinal cord. Up‐regulation of TRPV1 channels is involved in the development of thermal hyperalgesia associated with inflammatory pain. This work shows that noradrenaline potently down‐regulates the activity of TRPV1 channels in nociceptors and inhibits the release of neurotransmitter from nociceptors to large lamina I neurons in the dorsal horn of the spinal cord. Potentiation of the descending noradrenergic system could play a prominent role in the setting of injury to reduce neurotransmitter release from nociceptors, thus inhibiting incoming noxious stimuli to the dorsal horn of the spinal cord.
Acknowledgements
We thank Dr Bruce Bean for helpful comments on the manuscript and Yong Lu for technical assistance.
Linked articles This article is highlighted by a Perspective by Carbone. To read this Perspective, visit http://dx.doi.org/10.1113/JP274103.
References
- Ahern GP, Brooks IM, Miyares RL & Wang XB (2005). Extracellular cations sensitize and gate capsaicin receptor TRPV1 modulating pain signaling. J Neurosci 25, 5109–5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern GP, Wang X & Miyares RL (2006). Polyamines are potent ligands for the capsaicin receptor TRPV1. J Biol Chem 281, 8991–8995. [DOI] [PubMed] [Google Scholar]
- Akerman S, Kaube H & Goadsby PJ (2004). Anandamide acts as a vasodilator of dural blood vessels in vivo by activating TRPV1 receptors. Br J Pharmacol 142, 1354–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Ghamdi KS, Polgar E & Todd AJ (2009). Soma size distinguishes projection neurons from neurokinin 1 receptor‐expressing interneurons in lamina I of the rat lumbar spinal dorsal horn. Neuroscience 164, 1794–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccei ML, Bardoni R & Fitzgerald M (2003). Development of nociceptive synaptic inputs to the neonatal rat dorsal horn: glutamate release by capsaicin and menthol. J Physiol 549, 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangaru ML, Meng J, Kaiser DJ, Yu H, Fischer G, Hogan QH & Hudmon A (2015). Differential expression of CaMKII isoforms and overall kinase activity in rat dorsal root ganglia after injury. Neuroscience 300, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J, Li JJ & Perl ER (1998). Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J Neurosci 18, 8740–8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavencoffe A, Gkika D, Kondratskyi A, Beck B, Borowiec AS, Bidaux G, Busserolles J, Eschalier A, Shuba Y, Skryma R & Prevarskaya N (2010). The transient receptor potential channel TRPM8 is inhibited via the α2A adrenoreceptor signaling pathway. J Biol Chem 285, 9410–9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP (1989). Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 340, 153–156. [DOI] [PubMed] [Google Scholar]
- Bell TJ, Thaler C, Castiglioni AJ, Helton TD & Lipscombe D (2004). Cell‐specific alternative splicing increases calcium channel current density in the pain pathway. Neuron 41, 127–138. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS & Gereau RWt (2003). Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1). Proc Natl Acad Sci USA 100, 12480–12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS & Gereau RWt (2002). cAMP‐dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 35, 721–731. [DOI] [PubMed] [Google Scholar]
- Birder LA & Perl ER (1999). Expression of α2‐adrenergic receptors in rat primary afferent neurones after peripheral nerve injury or inflammation. J Physiol 515, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair NT & Bean BP (2002). Roles of tetrodotoxin (TTX)‐sensitive Na+ current, TTX‐resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci 22, 10277–10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S & Huck S (1996). Inhibition of N‐type calcium channels: the only mechanism by which presynaptic α2‐autoreceptors control sympathetic transmitter release. Eur J Neurosci 8, 1924–1931. [DOI] [PubMed] [Google Scholar]
- Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, Molinoff PB, Ruffolo RR, Jr & Trendelenburg U (1994). International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev 46, 121–136. [PubMed] [Google Scholar]
- Carbone E & Lux HD (1984). A low voltage‐activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature 310, 501–502. [DOI] [PubMed] [Google Scholar]
- Cardenas CG, Del Mar LP & Scroggs RS (1995). Variation in serotonergic inhibition of calcium channel currents in four types of rat sensory neurons differentiated by membrane properties. J Neurophysiol 74, 1870–1879. [DOI] [PubMed] [Google Scholar]
- Castiglioni AJ, Raingo J & Lipscombe D (2006). Alternative splicing in the C‐terminus of CaV2.2 controls expression and gating of N‐type calcium channels. J Physiol 576, 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen‐Zeitz KR, Koltzenburg M, Basbaum AI & Julius D (2000). Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD & Julius D (1997). The capsaicin receptor: a heat‐activated ion channel in the pain pathway. Nature 389, 816–824. [DOI] [PubMed] [Google Scholar]
- Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R, O'Donnell D, Nicoll RA, Shah NM, Julius D & Basbaum AI (2011). Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J Neurosci 31, 5067–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Rebecchi M, Kaczocha M & Puopolo M (2016). Dopamine modulation of transient receptor potential vanilloid type 1 (TRPV1) receptor in dorsal root ganglia neurons. J Physiol 594, 1627–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HJ, Kim DS, Lee NH, Kim JK, Lee KM, Han KS, Kang YN & Kim KJ (1997). Changes in the α2‐adrenergic receptor subtypes gene expression in rat dorsal root ganglion in an experimental model of neuropathic pain. Neuroreport 8, 3119–3122. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Lee HS, Bae MA & Joo K (1995). Chronic arthritis increases tyrosine hydroxylase mRNA levels in the pontine noradrenergic cell groups. Brain Res 695, 96–99. [DOI] [PubMed] [Google Scholar]
- Cholewinski A, Burgess GM & Bevan S (1993). The role of calcium in capsaicin‐induced desensitization in rat cultured dorsal root ganglion neurons. Neuroscience 55, 1015–1023. [DOI] [PubMed] [Google Scholar]
- Clark FM & Proudfit HK (1991. a). The projection of locus coeruleus neurons to the spinal cord in the rat determined by anterograde tracing combined with immunocytochemistry. Brain Res 538, 231–245. [DOI] [PubMed] [Google Scholar]
- Clark FM & Proudfit HK (1991. b). The projection of noradrenergic neurons in the A7 catecholamine cell group to the spinal cord in the rat demonstrated by anterograde tracing combined with immunocytochemistry. Brain Res 547, 279–288. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A & Sheardown SA (2000). Vanilloid receptor‐1 is essential for inflammatory thermal hyperalgesia. Nature 405, 183–187. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Harrison S, Bisogno T, Tognetto M, Brandi I, Smith GD, Creminon C, Davis JB, Geppetti P & Di Marzo V (2001). The vanilloid receptor (VR1)‐mediated effects of anandamide are potently enhanced by the cAMP‐dependent protein kinase. J Neurochem 77, 1660–1663. [DOI] [PubMed] [Google Scholar]
- Dhaka A, Uzzell V, Dubin AE, Mathur J, Petrus M, Bandell M & Patapoutian A (2009). TRPV1 is activated by both acidic and basic pH. J Neurosci 29, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty RJ, Yeats JC, Bevan S & Boddeke HW (1996). Inhibition of calcineurin inhibits the desensitization of capsaicin‐evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflugers Arch 431, 828–837. [DOI] [PubMed] [Google Scholar]
- Dunlap K & Fischbach GD (1981). Neurotransmitters decrease the calcium conductance activated by depolarization of embryonic chick sensory neurones. J Physiol 317, 519–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM & Mason P (1991). Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci 14, 219–245. [DOI] [PubMed] [Google Scholar]
- Fitzgerald M (2005). The development of nociceptive circuits. Nat Rev Neurosci 6, 507–520. [DOI] [PubMed] [Google Scholar]
- Gemes G, Rigaud M, Koopmeiners AS, Poroli MJ, Zoga V & Hogan QH (2010). Calcium signaling in intact dorsal root ganglia: new observations and the effect of injury. Anesthesiology 113, 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghilardi JR, Rohrich H, Lindsay TH, Sevcik MA, Schwei MJ, Kubota K, Halvorson KG, Poblete J, Chaplan SR, Dubin AE, Carruthers NI, Swanson D, Kuskowski M, Flores CM, Julius D & Mantyh PW (2005). Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J Neurosci 25, 3126–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni MP, Ghelardini C, Vergelli C & Dal Piaz V (2009). α2‐Agonists as analgesic agents. Med Res Rev 29, 339–368. [DOI] [PubMed] [Google Scholar]
- Gold MS, Dastmalchi S & Levine JD (1997). α2‐Adrenergic receptor subtypes in rat dorsal root and superior cervical ganglion neurons. Pain 69, 179–190. [DOI] [PubMed] [Google Scholar]
- Green GM, Lyons L & Dickenson AH (1998). α2‐Adrenoceptor antagonists enhance responses of dorsal horn neurones to formalin induced inflammation. Eur J Pharmacol 347, 201–204. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X & Elde R (1999). Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci 11, 946–958. [DOI] [PubMed] [Google Scholar]
- Hagihira S, Senba E, Yoshida S, Tohyama M & Yoshiya I (1990). Fine structure of noradrenergic terminals and their synapses in the rat spinal dorsal horn: an immunohistochemical study. Brain Res 526, 73–80. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, King MM & Soderling TR (1988). Regulatory interactions of calmodulin‐binding proteins: phosphorylation of calcineurin by autophosphorylated Ca2+/calmodulin‐dependent protein kinase II. Proc Natl Acad Sci USA. 85, 7001–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinke B, Balzer E & Sandkuhler J (2004). Pre‐ and postsynaptic contributions of voltage‐dependent Ca2+ channels to nociceptive transmission in rat spinal lamina I neurons. Eur J Neurosci 19, 103–111. [DOI] [PubMed] [Google Scholar]
- Hillyard DR, Monje VD, Mintz IM, Bean BP, Nadasdi L, Ramachandran J, Miljanich G, Azimi‐Zoonooz A, McIntosh JM, Cruz LJ & et al (1992). A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron 9, 69–77. [DOI] [PubMed] [Google Scholar]
- Ho C & O'Leary ME (2011). Single‐cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol Cell Neurosci 46, 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P (1991). Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol Rev 43, 143–201. [PubMed] [Google Scholar]
- Hucho T, Suckow V, Joseph EK, Kuhn J, Schmoranzer J, Dina OA, Chen X, Karst M, Bernateck M, Levine JD & Ropers HH (2012). Ca++/CaMKII switches nociceptor‐sensitizing stimuli into desensitizing stimuli. J Neurochem 123, 589–601. [DOI] [PubMed] [Google Scholar]
- Hwang SJ, Burette A, Rustioni A & Valtschanoff JG (2004). Vanilloid receptor VR1‐positive primary afferents are glutamatergic and contact spinal neurons that co‐express neurokinin receptor NK1 and glutamate receptors. J Neurocytol 33, 321–329. [DOI] [PubMed] [Google Scholar]
- Ikeda SR (1996). Voltage‐dependent modulation of N‐type calcium channels by G‐protein β γ subunits. Nature 380, 255–258. [DOI] [PubMed] [Google Scholar]
- Jara‐Oseguera A, Bae C & Swartz KJ (2016). An external sodium ion binding site controls allosteric gating in TRPV1 channels. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeske NA, Patwardhan AM, Gamper N, Price TJ, Akopian AN & Hargreaves KM (2006). Cannabinoid WIN 55,212‐2 regulates TRPV1 phosphorylation in sensory neurons. J Biol Chem 281, 32879–32890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RC, 3rd , Xu L & Gebhart GF (2005). The mechanosensitivity of mouse colon afferent fibers and their sensitization by inflammatory mediators require transient receptor potential vanilloid 1 and acid‐sensing ion channel 3. J Neurosci 25, 10981–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H & Oh U (2004). Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin‐dependent kinase II regulates its vanilloid binding. J Biol Chem 279, 7048–7054. [DOI] [PubMed] [Google Scholar]
- Khasar SG, McCarter G & Levine JD (1999). Epinephrine produces a β‐adrenergic receptor‐mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. J Neurophysiol 81, 1104–1112. [DOI] [PubMed] [Google Scholar]
- Kim YS, Chu Y, Han L, Li M, Li Z, Lavinka PC, Sun S, Tang Z, Park K, Caterina MJ, Ren K, Dubner R, Wei F & Dong X (2014). Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain. Neuron 81, 873–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee CB (1991). Concerted regulation of protein phosphorylation and dephosphorylation by calmodulin. Neurochem Res 16, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Koplas PA, Rosenberg RL & Oxford GS (1997). The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci 17, 3525–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiat GC & Basbaum AI (1992). The origin of brainstem noradrenergic and serotonergic projections to the spinal cord dorsal horn in the rat. Somatosens Mot Res 9, 157–173. [DOI] [PubMed] [Google Scholar]
- Labrakakis C & MacDermott AB (2003). Neurokinin receptor 1‐expressing spinal cord neurons in lamina I and III/IV of postnatal rats receive inputs from capsaicin sensitive fibers. Neurosci Lett 352, 121–124. [DOI] [PubMed] [Google Scholar]
- Li C & Horn JP (2008). Differential inhibition of Ca2+ channels by α2‐adrenoceptors in three functional subclasses of rat sympathetic neurons. J Neurophysiol 100, 3055–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Kritzer E, Ford NC, Arbabi S & Baccei ML (2015). Connectivity of pacemaker neurons in the neonatal rat superficial dorsal horn. J Comp Neurol 523, 1038–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X & Eisenach JC (2001). α2A‐adrenoceptor stimulation reduces capsaicin‐induced glutamate release from spinal cord synaptosomes. J Pharmacol Exp Ther 299, 939–944. [PubMed] [Google Scholar]
- Lipscombe D, Kongsamut S & Tsien RW (1989). α‐Adrenergic inhibition of sympathetic neurotransmitter release mediated by modulation of N‐type calcium‐channel gating. Nature 340, 639–642. [DOI] [PubMed] [Google Scholar]
- Liu L & Simon SA (1996). Capsaicin‐induced currents with distinct desensitization and Ca2+ dependence in rat trigeminal ganglion cells. J Neurophysiol 75, 1503–1514. [DOI] [PubMed] [Google Scholar]
- Marchetti C, Carbone E & Lux HD (1986). Effects of dopamine and noradrenaline on Ca channels of cultured sensory and sympathetic neurons of chick. Pflugers Arch 406, 104–111. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Gupta NK, Loo CM, Rohde DS & Basbaum AI (1999). Differential effects of neurotoxic destruction of descending noradrenergic pathways on acute and persistent nociceptive processing. Pain 80, 57–65. [DOI] [PubMed] [Google Scholar]
- Maruo K, Yamamoto H, Yamamoto S, Nagata T, Fujikawa H, Kanno T, Yaguchi T, Maruo S, Yoshiya S & Nishizaki T (2006). Modulation of P2X receptors via adrenergic pathways in rat dorsal root ganglion neurons after sciatic nerve injury. Pain 120, 106–112. [DOI] [PubMed] [Google Scholar]
- McDonough SI, Swartz KJ, Mintz IM, Boland LM & Bean BP (1996). Inhibition of calcium channels in rat central and peripheral neurons by ω‐conotoxin MVIIC. J Neurosci 16, 2612–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva YV, Kim MS & Usachev YM (2008). Mechanisms of prolonged presynaptic Ca2+ signaling and glutamate release induced by TRPV1 activation in rat sensory neurons. J Neurosci 28, 5295–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ (1992). Evidence that an alpha 2A‐adrenoceptor subtype mediates antinociception in mice. Eur J Pharmacol 215, 355–356. [DOI] [PubMed] [Google Scholar]
- Millan MJ (2002). Descending control of pain. Prog Neurobiol 66, 355–474. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Bervoets K, Rivet JM, Widdowson P, Renouard A, Le Marouille‐Girardon S & Gobert A (1994). Multiple alpha‐2 adrenergic receptor subtypes. II. Evidence for a role of rat R alpha‐2A adrenergic receptors in the control of nociception, motor behavior and hippocampal synthesis of noradrenaline. J Pharmacol Exp Ther 270, 958–972. [PubMed] [Google Scholar]
- Mohapatra DP & Nau C (2005). Regulation of Ca2+‐dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP‐dependent protein kinase. J Biol Chem 280, 13424–13432. [DOI] [PubMed] [Google Scholar]
- Nakatsuka T, Ataka T, Kumamoto E, Tamaki T & Yoshimura M (2000). Alteration in synaptic inputs through C‐afferent fibers to substantia gelatinosa neurons of the rat spinal dorsal horn during postnatal development. Neuroscience 99, 549–556. [DOI] [PubMed] [Google Scholar]
- Nakatsuka T, Furue H, Yoshimura M & Gu JG (2002). Activation of central terminal vanilloid receptor‐1 receptors and αβ‐methylene‐ATP‐sensitive P2X receptors reveals a converged synaptic activity onto the deep dorsal horn neurons of the spinal cord. J Neurosci 22, 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelands TR, Jarvis MF, Faltynek CR & Surowy CS (2008). Elevated temperatures alter TRPV1 agonist‐evoked excitability of dorsal root ganglion neurons. Inflamm Res 57, 404–409. [DOI] [PubMed] [Google Scholar]
- Neelands TR, Zhang XF, McDonald H & Puttfarcken P (2010). Differential effects of temperature on acid‐activated currents mediated by TRPV1 and ASIC channels in rat dorsal root ganglion neurons. Brain Res 1329, 55–66. [DOI] [PubMed] [Google Scholar]
- Neher E (1992). Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol 207, 123–131. [DOI] [PubMed] [Google Scholar]
- Nicholas AP, Pieribone V & Hokfelt T (1993). Distributions of mRNAs for alpha‐2 adrenergic receptor subtypes in rat brain: an in situ hybridization study. J Comp Neurol 328, 575–594. [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H & Tominaga M (2002). Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cε and identification of two target serine residues. J Biol Chem 277, 13375–13378. [DOI] [PubMed] [Google Scholar]
- Oda A, Iida H, Tanahashi S, Osawa Y, Yamaguchi S & Dohi S (2007). Effects of alpha2‐adrenoceptor agonists on tetrodotoxin‐resistant Na+ channels in rat dorsal root ganglion neurons. Eur J Anaesthesiol 24, 934–941. [DOI] [PubMed] [Google Scholar]
- Olah Z, Szabo T, Karai L, Hough C, Fields RD, Caudle RM, Blumberg PM & Iadarola MJ (2001). Ligand‐induced dynamic membrane changes and cell deletion conferred by vanilloid receptor 1. J Biol Chem 276, 11021–11030. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Morimura K & Porreca F (2014). Descending pain modulation and chronification of pain. Curr Opin Support Palliat Care 8, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Nakatsuka T, Nagata K, Higashi H & Yoshimura M (1999). Reorganization of the primary afferent termination in the rat spinal dorsal horn during post‐natal development. Brain Res Dev Brain Res 113, 29–36. [DOI] [PubMed] [Google Scholar]
- Petruska JC, Napaporn J, Johnson RD, Gu JG & Cooper BY (2000). Subclassified acutely dissociated cells of rat DRG: histochemistry and patterns of capsaicin‐, proton‐, and ATP‐activated currents. J Neurophysiol 84, 2365–2379. [DOI] [PubMed] [Google Scholar]
- Pollo A, Lovallo M, Sher E & Carbone E (1992). Voltage‐dependent noradrenergic modulation of omega‐conotoxin‐sensitive Ca2+ channels in human neuroblastoma IMR32 cells. Pflugers Arch 422, 75–83. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Qi ZH, Van Buren J & Raisinghani M (2004). Enhancement of potency and efficacy of NADA by PKC‐mediated phosphorylation of vanilloid receptor. J Neurophysiol 91, 1442–1449. [DOI] [PubMed] [Google Scholar]
- Price TJ, Jeske NA, Flores CM & Hargreaves KM (2005). Pharmacological interactions between calcium/calmodulin‐dependent kinase II alpha and TRPV1 receptors in rat trigeminal sensory neurons. Neurosci Lett 389, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puopolo M, Binshtok AM, Yao GL, Oh SB, Woolf CJ & Bean BP (2013). Permeation and block of TRPV1 channels by the cationic lidocaine derivative QX‐314. J Neurophysiol 109, 1704–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathee PK, Distler C, Obreja O, Neuhuber W, Wang GK, Wang SY, Nau C & Kress M (2002). PKA/AKAP/VR‐1 module: a common link of Gs‐mediated signaling to thermal hyperalgesia. J Neurosci 22, 4740–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridet JL, Rajaofetra N, Teilhac JR, Geffard M & Privat A (1993). Evidence for nonsynaptic serotonergic and noradrenergic innervation of the rat dorsal horn and possible involvement of neuron–glia interactions. Neuroscience 52, 143–157. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Sandillon F, Rajaofetra N, Geffard M & Privat A (1992). Spinal dopaminergic system of the rat: light and electron microscopic study using an antiserum against dopamine, with particular emphasis on synaptic incidence. Brain Res 598, 233–241. [DOI] [PubMed] [Google Scholar]
- Riedl MS, Schnell SA, Overland AC, Chabot‐Dore AJ, Taylor AM, Ribeiro‐da‐Silva A, Elde RP, Wilcox GL & Stone LS (2009). Coexpression of α2A‐adrenergic and δ‐opioid receptors in substance P‐containing terminals in rat dorsal horn. J Comp Neurol 513, 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum T, Gordon‐Shaag A, Munari M & Gordon SE (2004). Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J Gen Physiol 123, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin DL, Zeng D, Stornetta RL, Norton FR, Riley T, Okusa MD, Guyenet PG & Lynch KR (1993). Immunohistochemical localization of α2A‐adrenergic receptors in catecholaminergic and other brainstem neurons in the rat. Neuroscience 56, 139–155. [DOI] [PubMed] [Google Scholar]
- Ruffolo RR, Jr , Nichols AJ, Stadel JM & Hieble JP (1993). Pharmacologic and therapeutic applications of α2‐adrenoceptor subtypes. Annu Rev Pharmacol Toxicol 33, 243–279. [DOI] [PubMed] [Google Scholar]
- Safronov BV, Pinto V & Derkach VA (2007). High‐resolution single‐cell imaging for functional studies in the whole brain and spinal cord and thick tissue blocks using light‐emitting diode illumination. J Neurosci Methods 164, 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders C & Limbird LE (1999). Localization and trafficking of α2‐adrenergic receptor subtypes in cells and tissues. Pharmacol Ther 84, 193–205. [DOI] [PubMed] [Google Scholar]
- Scroggs RS & Fox AP (1991). Distribution of dihydropyridine and ω‐conotoxin‐sensitive calcium currents in acutely isolated rat and frog sensory neuron somata: diameter‐dependent L channel expression in frog. J Neurosci 11, 1334–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggs RS & Fox AP (1992). Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J Physiol 445, 639–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi TJ, Winzer‐Serhan U, Leslie F & Hokfelt T (1999). Distribution of α2‐adrenoceptor mRNAs in the rat lumbar spinal cord in normal and axotomized rats. Neuroreport 10, 2835–2839. [DOI] [PubMed] [Google Scholar]
- Simon B, Huart AS & Wilmanns M (2015). Molecular mechanisms of protein kinase regulation by calcium/calmodulin. Bioorg Med Chem 23, 2749–2760. [DOI] [PubMed] [Google Scholar]
- Sluka KA & Westlund KN (1992). Spinal projections of the locus coeruleus and the nucleus subcoeruleus in the Harlan and the Sasco Sprague‐Dawley rat. Brain Res 579, 67–73. [DOI] [PubMed] [Google Scholar]
- Stone LS, Broberger C, Vulchanova L, Wilcox GL, Hokfelt T, Riedl MS & Elde R (1998). Differential distribution of α2A and α2C adrenergic receptor immunoreactivity in the rat spinal cord. J Neurosci 18, 5928–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone LS, Vulchanova L, Riedl MS, Wang J, Williams FG, Wilcox GL & Elde R (1999). Effects of peripheral nerve injury on alpha‐2A and alpha‐2C adrenergic receptor immunoreactivity in the rat spinal cord. Neuroscience 93, 1399–1407. [DOI] [PubMed] [Google Scholar]
- Stratton MM, Chao LH, Schulman H & Kuriyan J (2013). Structural studies on the regulation of Ca2+/calmodulin dependent protein kinase II. Curr Opin Struct Biol 23, 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo A, Helyes Z, Sandor K, Bite A, Pinter E, Nemeth J, Banvolgyi A, Bolcskei K, Elekes K & Szolcsanyi J (2005). Role of transient receptor potential vanilloid 1 receptors in adjuvant‐induced chronic arthritis: in vivo study using gene‐deficient mice. J Pharmacol Exp Ther 314, 111–119. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Nilsson S, Farkas‐Szallasi T, Blumberg PM, Hokfelt T & Lundberg JM (1995). Vanilloid (capsaicin) receptors in the rat: distribution in the brain, regional differences in the spinal cord, axonal transport to the periphery, and depletion by systemic vanilloid treatment. Brain Res 703, 175–183. [DOI] [PubMed] [Google Scholar]
- Szucs P, Pinto V & Safronov BV (2009). Advanced technique of infrared LED imaging of unstained cells and intracellular structures in isolated spinal cord, brainstem, ganglia and cerebellum. J Neurosci Methods 177, 369–380. [DOI] [PubMed] [Google Scholar]
- Takano Y & Yaksh TL (1992). Characterization of the pharmacology of intrathecally administered alpha‐2 agonists and antagonists in rats. J Neurosci Methods 261, 764–772. [PubMed] [Google Scholar]
- Takano Y & Yaksh TL (1993). Chronic spinal infusion of dexmedetomidine, ST‐91 and clonidine: spinal alpha 2 adrenoceptor subtypes and intrinsic activity. J Pharmacol Exp Ther 264, 327–335. [PubMed] [Google Scholar]
- Tian J & Karin M (1999). Stimulation of Elk1 transcriptional activity by mitogen‐activated protein kinases is negatively regulated by protein phosphatase 2B (calcineurin). J Biol Chem 274, 15173–15180. [DOI] [PubMed] [Google Scholar]
- Tognetto M, Amadesi S, Harrison S, Creminon C, Trevisani M, Carreras M, Matera M, Geppetti P & Bianchi A (2001). Anandamide excites central terminals of dorsal root ganglion neurons via vanilloid receptor‐1 activation. J Neurosci 21, 1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI & Julius D (1998). The cloned capsaicin receptor integrates multiple pain‐producing stimuli. Neuron 21, 531–543. [DOI] [PubMed] [Google Scholar]
- Tominaga M & Tominaga T (2005). Structure and function of TRPV1. Pflugers Arch 451, 143–150. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Wada M & Masu M (2001). Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP‐evoked pain and hyperalgesia. Proc Natl Acad Sci U S A 98, 6951–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong CK & MacDermott AB (2006). Both Ca2+‐permeable and ‐impermeable AMPA receptors contribute to primary synaptic drive onto rat dorsal horn neurons. Nat Rev Neurosci 575, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruoka M & Willis WD (1996). Descending modulation from the region of the locus coeruleus on nociceptive sensitivity in a rat model of inflammatory hyperalgesia. Brain Res 743, 86–92. [DOI] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB & McNaughton PA (2001). Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol 534, 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachova V, Teisinger J, Susankova K, Lyfenko A, Ettrich R & Vyklicky L (2003). Functional role of C‐terminal cytoplasmic tail of rat vanilloid receptor 1. J Neurosci 23, 1340–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Droogmans G, Wissenbach U, Janssens A, Flockerzi V & Nilius B (2004). The principle of temperature‐dependent gating in cold‐ and heat‐sensitive TRP channels. Nature 430, 748–754. [DOI] [PubMed] [Google Scholar]
- Winter J, Walpole CS, Bevan S & James IF (1993). Characterization of resiniferatoxin binding sites on sensory neurons: co‐regulation of resiniferatoxin binding and capsaicin sensitivity in adult rat dorsal root ganglia. Neuroscience 57, 747–757. [DOI] [PubMed] [Google Scholar]
- Wong CO, Chen K, Lin YQ, Chao Y, Duraine L, Lu Z, Yoon WH, Sullivan JM, Broadhead GT, Sumner CJ, Lloyd TE, Macleod GT, Bellen HJ & Venkatachalam K (2014). A TRPV channel in Drosophila motor neurons regulates presynaptic resting Ca2+ levels, synapse growth, and synaptic transmission. Neuron 84, 764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Kontinen VK & Kalso E (1999). Endogenous noradrenergic tone controls symptoms of allodynia in the spinal nerve ligation model of neuropathic pain. Eur J Pharmacol 366, 41–45. [DOI] [PubMed] [Google Scholar]
- Yang K, Kumamoto E, Furue H, Li YQ & Yoshimura M (1999). Action of capsaicin on dorsal root‐evoked synaptic transmission to substantia gelatinosa neurons in adult rat spinal cord slices. Brain Res 830, 268–273. [DOI] [PubMed] [Google Scholar]
- Yang K, Kumamoto E, Furue H & Yoshimura M (1998). Capsaicin facilitates excitatory but not inhibitory synaptic transmission in substantia gelatinosa of the rat spinal cord. Neurosci Lett 255, 135–138. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D & Hogestatt ED (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400, 452–457. [DOI] [PubMed] [Google Scholar]