

Abstract
Background:
Because the graphical presentation and analysis of motif distribution can provide insights for experimental hypothesis, PISMA aims at identifying motifs on DNA sequences, counting and showing them graphically. The motif length ranges from 2 to 10 bases, and the DNA sequences range up to 10 kb. The motif distribution is shown as a bar-code–like, as a gene-map–like, and as a transcript scheme.
Results:
We obtained graphical schemes of the CpG site distribution from 91 human papillomavirus genomes. Also, we present 2 analyses: one of DNA motifs associated with either methylation-resistant or methylation-sensitive CpG islands and another analysis of motifs associated with exosome RNA secretion.
Availability and Implementation:
PISMA is developed in Java; it is executable in any type of hardware and in diverse operating systems. PISMA is freely available to noncommercial users. The English version and the User Manual are provided in Supplementary Files 1 and 2, and a Spanish version is available at www.biomedicas.unam.mx/wp-content/software/pisma.zip and www.biomedicas.unam.mx/wp-content/pdf/manual/pisma.pdf.
Keywords: Software, graphical user interface, Java, DNA motif distribution methodology
Introduction
It seems that there is a trend toward looking for DNA-overrepresented sequence patterns, called DNA motifs, possessing potential biological functions. Normally, DNA motifs are fairly short (5-20 base pairs [bp] in length) and are known to be present in different genes or within a gene.1 DNA motifs can occur on both strands of the DNA. Some examples of DNA motifs are as follows: CpG methylation sites involved in transcription regulation,2 nonmethylated CpG-containing oligodeoxynucleotides able to stimulate immune responses,3 potential RNA secretory motifs which are oligonucleotide sequences associated with RNA in exosomes,4 and oligonucleotides differentially associated with genome regions such as exons and introns.5
Microsatellites are another example of interesting motifs. Microsatellites are mid-sized (a few hundred of primary repeat units or motifs) families of tandem repeat DNA sequences and have motifs typically 1 to 5 bp in length. Microsatellites are located in both coding and noncoding DNAs, as well as in regulatory regions, and may act as enhancers that regulate disease-relevant genes.6,7
Motif identification is an important step toward our understanding of gene regulatory networks and protein function. Many algorithms and computational tools have been designed to solve the motif-finding problem.8 Several of these tools are as follows: BioProspector, CompareProspector, and MDscan9; MEME10; info-gibbs11; BAMBI12; BEST13; GAME14; GALF-P15; and Hegma.16 However, most of them are devoted to looking for regulatory motif prediction in more than 1 nucleotide sequence, and none of them shows graphically both the global motif distribution and the individual location of each motif into the sequence, in an immediate way.
In our understanding, the graphical representation and analysis of DNA motif distributions are very useful as they can provide insights for experimental hypothesis. Following this idea, we developed PISMA, a computational tool that shows the distribution of user-selected motifs on a DNA sequence of up to 10 000 nucleotides. PISMA is a friendly graphical interface which gives a clear picture of motif distribution along the different DNA regions, such as promoters, open reading frames (ORFs) (or genes), exons, introns, and other user-selected regions. The name PISMA is the acronym of the original Spanish name of the project: Proyecto de Identificación de Sitios Metilables en ADN (Project for Identification of Methylation Sites). We wanted to maintain the name PISMA because this project brought us together as a team.
When compared with other motif-finding tools, the main difference in PISMA is the goal. Most of the tools are designed to identify functional motifs shared by several nucleotide and/or amino acid sequences. In this way, PISMA is devoted to looking for user-defined motifs—even without a functional meaning—in just 1 DNA sequence. In addition, PISMA offers the possibility of graphically showing the motif distribution on 2 user-defined schemes: as a gene-map–like and as a transcript scheme. None of the other tools are able to do this.
BioProspector, CompareProspector, and MDscan are together in a suite similar to the suite MEME. Both of the suites share the same goal: prediction of regulatory motifs. Accordingly, the main output from the first suite is a list of motifs, as position-specific probability matrices. Thus, it has no immediate direct visual insight of the motif distribution, such as that of PISMA. The MEME suite uses a nice graphical interface to show the outputs. However, it refers to more than 1 nucleotide or amino acid sequences to calculate the abundance of relative motifs. In addition, the MEME suite is able to present the motif distribution in each sequence; however, each motif position can be displayed as a temporary label. Info-gibbs and BAMBI (Bayesian Multiple-Instance Motif Discovery) are other motif finder tools, similar in their goals to the aforementioned tools. Info-gibbs efficiently optimizes motif discovery by directly focusing the search on the motif information content or the motif log-likelihood ratio, and BAMBI is based on a sequential Monte Carlo motif-identification algorithm. The Info-gibbs output is similar to that of the MEME and BAMBI tools, whose outputs are .txt files, and thus, all 3 are different to the graphical outputs of PISMA.
BEST is another motif-finding suite, similar to the previously described tools, which includes 4 programs: AlignACE, BioProspector, Consensus, and MEME. BEST compares similarly with PISMA to the above-described tools. However, BEST is limited to only running on Linux machines. PISMA can be run on any type of hardware and in diverse operative systems because it was developed in Java, an object-oriented programming language which is platform independent.
GAME and GALF-P tools were proposed to solve some algorithm failures from the above-described suites. The main difference is that GAME and GALF-P use genetic algorithm–based approaches to search for the motifs. However, the time and memory complexity of genetic algorithms are higher than for more specific algorithms. Currently, both GAME and GALF-P tools are no longer available via the Internet.
Finally, Hegma is a word-based motif discovery tool. It uses a DNA Gray code and equiprobable oligomers to be applicable to large-scale data, even if only a small fraction of the examined sequences contain the motifs. Thus, its purpose is also different from that of PISMA.
PISMA presents the motif distribution in 3 different contexts: as a bar-code–like, as a gene-map–like, and as a transcript scheme. To date, we have been unable to find computational tools that give a similar graphical representation as their result and at the same time allow locating different user-defined DNA motifs. The most similar tools to this are the CpG island searcher17 that shows the CpG motifs on a straight line and the CpG analyzer.18 In our view, both the CpG island searcher and the CpG analyzer are quite good tools; they are, however, complementary to PISMA. CpG island searcher is devoted to identifying CpG islands and shows the CpG island graphically only as bar-code–like graph. PISMA is able to show any user-defined motif distribution (up to 10 bases), not just CpG sites, and show them in different graphical formats; it does not identify CpG islands. The graphical interface CpG analyzer is closer to PISMA. It shows the CpG site distribution as a bar-code–like graph, and it also makes a list of positions for all of the cytosines in a CpG context, along a user-selected DNA sequence. Each position number is taken from the GenBank format for the DNA sequence. In addition, CpG analyzer is a very useful tool for DNA methylation studies because it can predict the resulting sequence from sodium bisulfite–treated DNA. Sodium bisulfite treatment switches any nonmethylated cytosine to thymine, whereas any methylated cytosine remains as cytosine. However, PISMA is able to show any user-defined motif distribution (not just the CpG motif) in several different graph formats, instead of giving as the result either a list of motif positions or coloring them on the sequence.
Another interesting computational tool is QGRS-H Predictor.19 However, this is specifically devoted to identify homologous quadruplex-forming G-rich sequence motifs in nucleotide sequences. As far as we know, no other computational tool is able to graphically locate any user-defined motif sites in any of the contexts that PISMA does.
Methods
Graphical interface description
PISMA is developed in Java. Java is a general-purpose and object-oriented platform-independent programming language. This means that applications developed using Java can be run on any hardware and software platform and are supported by every Java-compatible browser.
To achieve the goal of localization and identification of motifs ranging between 2 and 10 bases, in up to 10-kb DNA sequences, and in addition counting the number of motifs in defined regions, the first principal task of PISMA is the search of bases in a complete DNA sequence. The search function is described in Supplementary File 3.
PISMA is able to (1) count all the DNA motifs in the complete genomic sequence, (2) count all the DNA motifs in any user-defined regions, and (3) display the motif distribution in 3 different graphs: a bar-code–like, a gene-map–like, and a transcript scheme.
The graphical user interface (GUI) we developed is shown in Figure 1. Panel A shows the user-defined genomic regions in a gene-map–like view. Panel B shows the complete genomic sequence as a bar-code–like graph. Panel C shows the user-defined genomic regions as a transcript scheme. Panel D shows the plain text DNA sequence.
Figure 1.

PISMA graphical user interface. Panel A: Motif distribution on user-defined genomic regions, as a gene-map–like view. Panel B: Motif distribution on the complete genomic sequence, as a bar-code–like view. Panel C: Motif distribution on user-defined genomic regions, as a transcripts scheme. Panel D: Plain text DNA sequence.
The gene-map–like graph (Figure 1, panel A) is based on genome physical mapping. It shows graphically the physical distance between user-selected DNA fragments, according to the position given as number of bases along the 10 000-base-long sequence. The fragments can represent genes, promoter regions, and so on. In the bar-code–like graph (Figure 1, panel B), the selected motifs (2- to 10-base-long) are drawn as vertical lines on a horizontal line representing a genome fragment. It is possible to zoom in from 100%, where the whole genomic fragment is in the window, up to 800%, where it is possible to have a look of each single nucleotide position. The transcript scheme (Figure 1, panel C) is built based on the relative position of primary transcripts with respect to the gene-map–like graph. It could give a clear view of alternative transcription into a coding region, as well as show the spatial relationship between exons and introns.
This computational tool is directly installable from Supplementary File 1, and the User Manual is included in Supplementary File 2. Otherwise, a Spanish version of PISMA is also available at www.biomedicas.unam.mx/wp-content/software/pisma.zip and www.biomedicas.unam.mx/wp-content/pdf/manual/pisma.pdf.
Input and configuration
The input data are any DNA sequence up to 10 000 nucleotides included in a plain text file (.txt). As soon as the input file is chosen, the corresponding sequence text is shown on the right side of the “file” icon (Figure 1, panel D).
PISMA was primarily developed for locating CpG dinucleotides, but it can search for any other user-defined DNA motifs, from 2 to 10 nucleotides in the sequence. Each user-defined motif must be written in the small bottom left window in panel B (Figure 1) before the “Search” magnifier icon is pressed.
The Configuration tab in panel A of Figure 1 (illustrated in Figure 2A) allows the user to define selected regions from the nucleotide sequence, such as genes or ORFs. Once the regions have been defined, the gene map is presented by pressing the “Finish” key. This last key switches to a “Refresh” option in previously defined gene maps.
Figure 2.
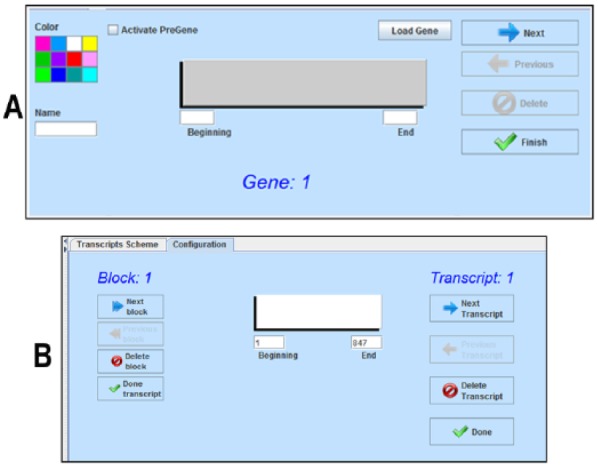
(A) Gene map configuration option allows selection of color, name, and length (start and end, nucleotides) for each user-selected region, such as open reading frame. (B) Transcript configuration option allows selection of start and end nucleotide numbers for each transcription region, as well as for each exor, called block.
The Configuration tab in panel C of Figure 1 (illustrated in Figure 2B) allows the user to define exons in transcript regions. The user can select the extent of every exon or “Block,” in each transcript.
In the same manner, it is possible to include as many transcripts as are experimentally defined.
Output
After the input sequence is processed according to the configured information, either the default output (CpG sites) or any user-selected motif is shown graphically on each panel.
It is possible to have the total number of motifs in any specific region by selecting the region with the cursor in any panel.
Panel A in Figure 1 shows a gene-map–like graph. Panel B shows the DNA sequence as a horizontal line, and a set of vertical lines represents the occurrence of the motifs. The 2 magnifier icons allow zooming of the motif location up to each base level, from 100× to 800×, as well as to show any individual position by typing the nucleotide number. It is also possible to save the images as PISMA files. Panel C shows a transcript scheme. The exons are shown as rectangle frames that include the genomic regions as colored in the gene-map–like view, and the introns are represented as lack of lines between each 2 exons. Panel D shows the DNA sequence being analyzed in sets of 100 nucleotides each line.
The camera icon facility in every panel allows saving each graphical result to help the user to correspond to all 3 graphs: the motif distribution on the genome, the gene-map–like, and the transcription-map–like. All 3 graphs have the genomic nucleotide line as scale.
As an example, Figure 3 shows all the graphs for human papillomavirus (HPV) type 1. The scheme allows quick visual identification of differences and similarities between regional motif distribution among ORFs.
Figure 3.

CpG site distribution, referred to gene map and transcript scheme in the HPV 1 genome. Genes are as follows: E6 = yellow, E7 = red, E1 = green, E2 = blue, E4 = fuchsia, E5 = pink, E5B = bright green, L2 = bluish green, and L1 = purple. Long control region and pre-genes = white.
Results
We show some graphical results from PISMA that allow expedite comparison between HPV gene maps and between CpG motif distributions. We have included both the 91 gene maps and the CpG site distributions for each HPV type, in Supplementary File 4. In addition, we present 2 preliminary analyses of motifs: (1) DNA motifs associated with either methylation-resistant CpG islands (MRM) or methylation-sensitive CpG islands (MSM) in the HPV E2/E4 ORFs and (2) motifs potentially specific for RNA secretion associated with exosomes in HPV genomes. Although this kind of analysis does not require a graphical visualization of the motifs, PISMA is able to identify and count them in a quick and accurate way.
Comparison of HPV gene maps in HPV genomes
The gene is the unit of inherited information in DNA and is defined as a nucleotide sequence that is used as a template for the RNA-copying process. Every gene contains the information to produce at least 1 protein. In most of the reported HPV genomes, there are around 7 ORFs that could be considered as viral genes. The early gene expression region includes 6 ORFs: E6 and E7 are the viral oncogenes, and E1, E2, E4, and E5 are genes encoding proteins involved in different viral functions, and the late gene expression region comprises 2 genes, L2 and L1, coding the capsid proteins.
The HPV genomes are variable in size which leads to a lack of correspondence between both ORF sizes and first codon position for each gene in every HPV type. This fact is evident when graphical HPV gene maps are shown together. Figure 4 shows the gene maps of the 5 most common virus types; HPV-2 is the most prevalent type in common warts, HPV-6 and HPV-11 are the most frequent virus types in mucosal papillomas, and HPV-16 and HPV-18 are the main causes of cervical cancer.20 We have included 91 HPV gene maps in Supplementary Data File 4.
Figure 4.

Gene maps of the 5 most common human papillomavirus (HPV) types. All genomic sequences start at the first ATG belonging to the E6 open reading frame. Genes are as follows: E6 = yellow, E7 = red, E1 =green: E2 = blue, E4 = fuchsia, E5 = pink, E5B = bright green, L2 = bluish green, and L1 = purple.
As usual, the genes located in the first line in each gene map start at the first nucleotide of the triplet, the genes located in the second line start at the second nucleotide of the triplet, and the genes located in the third line start at the third nucleotide of the triplet.
This graphical view shows that except for the 2 oncogenes (E6 in yellow and E7 in red), all other genes are differently distributed according to the nucleotide number in the triplet. In this comparison, it is easy to see that the oncogenes are the most conserved ORFs in size and location in these HPV genomes and that HPV-11 differs from the rest due to it having an extra gene, 5B.
Comparison of CpG site distributions in HPV genomes
Methylation is the only known covalent modification of DNA in eukaryotes and plays an important regulatory role in vertebrates by silencing specific genes during development and cell differentiation. Cytosine methylation occurs mainly within a CpG context, although methylation of cytosine in different contexts has also been described.21,22
In the genomes of higher eukaryotes, CpG dinucleotides are usually underrepresented, from one-third down to 5% of their expected frequency.23 Similar to their hosts, CpG dinucleotides are underrepresented in HPV genomes, although to a lesser extent, around 50%.24 Nevertheless, such underrepresentation is not homogeneous along the viral genomes; there are several CpG clusters or islands (CGI) in some genomic regions. In a previous study, we made an exhaustive analysis of CpG methylation sites and CpG islands among 92 HPV DNA genomes.24 Specifically, we found 1 CGI in every E4 ORF identified among the HPV genomes. We have included the distribution of CpG in 91 HPV genomes in Supplementary File 4. We have left out one of the genomes because it lacks the start position for the E6 ORF and could not be aligned with the other HPV genome sequences.
Figure 5 shows the graphical representations of CpG site distribution in the 5 most common HPV types. These look like bar codes and show, at first glance, obvious differences in CpG site regional densities. HPV-2 clearly shows the highest amount of CpG sites, followed in decreasing order by HPVs 18, 6, 11, and finally HPV-16. It is also clear from Figure 6 that there is a CGI in the E4 region on each of the 5 HPV genomes between nucleotides 3000 and 3300. A comparison between E4 CpG sites and complete viral genome CpG sites, in 68 HPV types with reported E4 gene, is shown in Figure 6.
Figure 5.

CpG site distribution on the 5 most common human papillomavirus (HPV) types as bar-code–like diagrams. All genomic sequences start at the first ATG belonging to the E6 open reading frame.
Figure 6.
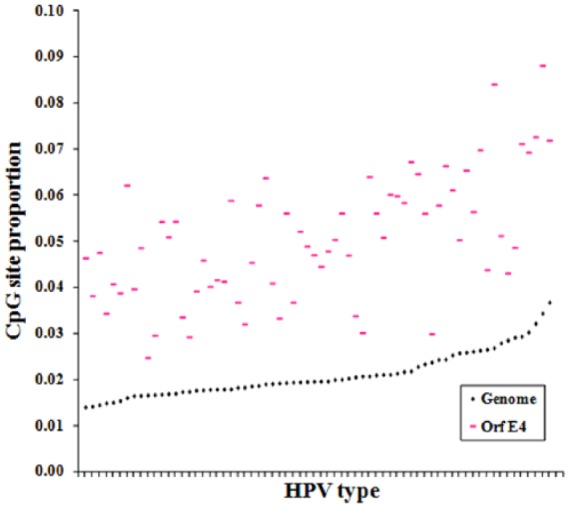
Relationship between CpG site proportion in E4 gene (pink) and human papillomavirus (HPV) whole genome (black), among 68 HPV types with reported E4 gene.
Preliminary analysis of DNA motifs associated with either MRM or MSM in the HPV ORF E4
Several DNA-related attributes which differ significantly between methylated and unmethylated CpG islands were identified.25 The analysis was done on the 132 CpG islands across the entire human chromosome 21. In all, 36 attributes are short DNA motifs. In total, 18 of those motifs were associated with the chromosome plus-strand pattern and the other 18 motifs were nonstrand specific. In our view, the distribution analysis of those motifs could provide insight into the methylation susceptibility from CpG islands in different types of HPVs. However, because the viral genomes hardly behave as chromosomal DNA, except when they are integrated in the cell genome, we choose motifs that were nonstrand specific for the analysis. So, using the computational tool PISMA, we explored all of the 18 nonstrand-specific DNA motifs associated with MRM as well as 2 of the 2 DNA motifs associated with MSM.
For this analysis, we used the CpG islands located in E2/E4 ORF from the different HPV types. This is the only HPV genome region where there is at least 1 CpG island. Table 1 shows a summary of the results obtained. We used motifs/kb as results because of the differences between the lengths of CpG islands. We also calculated the MRM/MSM rate to gain an insight about the magnitude of the differences.
Table 1.
DNA motifs associated with either methylation-resistant or methylation-sensitive CpG islands.
| HPV types | E2/E4 region | MRM (MOTIFS/KIB) | MRM (MOTIFS/KIB) | MRM/MSM rate |
|---|---|---|---|---|
| All | 93 | 53.7 | 20.3 | 2.65 |
| Cutaneous | 35 | 59.9 | 16.4 | 3.65 |
| Cutaneous/mucosal | 15 | 48.4 | 20.8 | 2.33 |
| Mucosal | 43 | 50.4 | 23.2 | 2.17 |
| Cutaneous | ||||
| Warts | 10 | 51.3 | 12.3 | 4.19 |
| Epidermoplasia | 25 | 63.4 | 18.1 | 3.51 |
| Mucosal | ||||
| LR-HPV | 22 | 48.0 | 25.8 | 1.86 |
| HR-HPV | 21 | 52.9 | 20.5 | 2.58 |
Abbreviations: HPV, human papillomavirus; HR, high risk; LR, low risk; MRM, methylation-resistant CpG islands; MSM, methylation-sensitive CpG islands.
Behind the taxonomy, there is a trend to classify the HPV types in function of their preferential tropism and the associated lesions. Because of their preferential tropism, HPVs are grouped as cutaneous, mucosal, and those without a clear preference, which are referred to as cutaneous/mucosal. The cutaneous HPV group includes both those HPVs associated with epidermodysplasia verruciformis (EV) and those HPVs associated with warts. On the other side, the mucosal HPV group is subgrouped based on their association with cervical cancer and other kinds of cancers as low-risk HPVs (LR-HPVs) and high-risk HPVs (HR-HPVs).
Previously, we found HR-HPV types that exhibit lesser and shorter CpG islands than any other HPV group.24 On the basis of this screening, the MRM/MSM average rates point to there being much more MRM than MSM, which could be a direct consequence of the motifs originally found by Bock et al25: 18 MRMs and 2 MSMs. Nevertheless, it points to the hypothesis about the HPV CpG islands in the E2/E4 region being resistant to methylation in all of the viral groups, however, by following an order as follows: cutaneous HPV CpG islands (3.65) being more resistant than in cutaneous/mucosal HPVs (2.33), and CpG islands in this group being more resistant than in mucosal HPV types (2.17).The comparison between MRM/MSM rates in the cutaneous subgroups points to a higher methylation resistance from the E2/E4 CpG islands in the HPV types associated with warts (4.19) than those associated with EV (3.51); similarly, in the mucosal HPVs, it seems that there is a higher methylation resistance in the islands from the HR-HPV subgroup (2.58) compared with the islands from the LR-HPV (1.86) subgroup. By looking at the average motifs/kb, there is an inverse relationship between MRM and MSM when all the 3 main groups are taken into account. The cutaneous HPV subgroups (59.9) have much more MRM per island than cutaneous/mucosal (48.4) and mucosal (50.4) HPVs, as well as cutaneous HPV subgroups (16.4) have lesser MSM per island than the 2 other HPV groups (cutaneous/mucosal: 20.8 and mucosal: 23.2). Islands from both cutaneous/mucosal and mucosal HPVs share MRM/kb and MSM/kb average values, which points to a higher similarity between their island resistance and a difference with that from the cutaneous HPVs. Into the mucosal HPVs, both subgroups, M-HR and M-LR, also show an inverse relationship between the MRM and MSM in their islands; in the HR subgroup (52.9), there are more MRMs than in the LR subgroup (48.0) as well as there are less MSMs in the HR subgroup (20.5) than in the LR subgroup (25.8). However, there is not an inverse relationship between the MRM and MSM in the islands from both cutaneous subgroups; HPV types associated with EV show more MRM (63.4) and MSM (18.1) than HPV types associated with warts (51.3 and 12.3, respectively).
Preliminary analysis of motifs potentially specific for RNA secretion associated with exosomes in HPV genomes
Exosomes are small (50-150 nm) membrane vesicles released from various cell types that have attracted a great interest because of their role in intercellular communication.26 These vesicles contain proteins, lipids and their bound carbohydrates, and RNA. In the recipient cells, the messenger RNA can be translated into protein,27 and the microRNAs are able to repress the expression of other genes.28 In fact, exosomes are being considered as promising tools for the delivery of therapeutic RNAs for the treatment of various conditions ranging from cancer to diabetes.29
Batagov et al4 identified specific sequence motifs from exosome-secreted RNAs (eRNAs) that potentially function as cis-acting elements targeting RNAs to exosomes. Three motifs (ACCAGCCU, CAGUGAGC, and UAAUCCCA) satisfy all criteria the authors had chosen for evaluation as potentially specific for RNA secretion, and the combination of these 3 motifs is preferred in eRNAs over single and double motif co-occurrence.
Using the computational tool PISMA, we explored these 3 RNA motifs, as DNA sequences, in the whole genome of 91 HPV types. Table 2 shows a summary of the results obtained. Interestingly, not considering the 2 HPV types with exosome-associated motifs in the long control region which is not transcribed, only 14 HPV types possess exosome-associated motifs: 6 with skin main tropism and 8 with mucosa main tropism (4 low risk and 4 high risk). Nevertheless, none possesses more than 1 motif in any ORF, which suggest that viral RNAs are not candidates for secretion via exosome.
Table 2.
RNA motifs potentially associated to RNA exosome secretion in HPV ORFs.
| HPV type | Main tropism | Motif sequence |
||
|---|---|---|---|---|
| ACCAGCCT | CAGTGAGC | TAATCCCA | ||
| 34 | Mucosa LR | E6 | ||
| 67 | Mucosa LR | E2 | ||
| 83 | Mucosa LR | L1 | ||
| 56 | Mucosa HR | L1 | ||
| 59 | Mucosa HR | L2 | ||
| 2a | Skin | E1 | ||
| 50 | Skin | L1 | ||
| 63 | Skin | L1 | ||
| cand85 | Mucosa LR | LCR | ||
| 7 | Skin | E1 | ||
| 4 | Skin | L2 | ||
| 47 | Skin | L2 | ||
| 40 | Mucosa LR | E1 | ||
| 16 | Mucosa HR | L2 | ||
| 53 | Mucosa HR | L2 | ||
| 11 | Mucosa LR | LCR | ||
Abbreviations: HPV, human papillomavirus; HR, high risk; LCR, long control region; LR, low risk; ORFs, open reading frames.
Discussion
PISMA is a tool with many advantages. It is developed in Java, a platform-independent tool, and its use is free of charge. We intended to design a friendly GUI, requiring minimal experience with software, and developed a user manual as short and simple as possible. Moreover, PISMA is highly versatile. It allows seeking of the distribution of any DNA motif ranging between 2 and 10 nucleotides. PISMA eases the motif regional distribution analysis in an up to 10 000-nucleotide sequence. The gene-map–like view, which usually helps to point out the relative positions of genes, is used by PISMA in such a way that it allows not only ORF selection but also several other genomic regions such as gene promoters and enhancers, and all fragments are user defined. The transcript-map–like view allows establishing graphical relationships between ORFs and the exon/intron regions. In our view, the main benefit of PISMA is the graphical approach on motif quantity and distribution. This type of analysis helps produce insights that can eventually become hypotheses on the functional relevance of motif distribution patterns. From the 91 HPV gene maps (Supplementary File 4), we can clearly see that most of the contiguous ORFs start in a different nucleotide of the triplet, even without overlapping between ORFs. There is a possible functional constriction affecting the viral genomes. The high density of CpG in a specific genomic region suggests that most viral types share a CpG island. It may be that this region is associated with gene expression control. In addition to the insight that can be generated by the graphical representation of motifs, this GUI can also be used simply to identify and count motifs in an expedited and confident way. This is shown by the following 2 examples of motif analysis: the analysis of CpG motifs in the HPV genomes, which suggests the only shared CpG islands in all HPVs, seems to be methylation resistant, and the analysis of motifs in RNA that suggests viral RNAs are no candidates to secretion via exosomes. As far as we know, there is no other tool which allows performing such kind of tasks this way.
The hypothesis derived using PISMA either from the graphical approach on motif distribution or from the identification and counting of motifs could be experimentally tested.
PISMA can be used to identify and present graphically other important motifs, such as those from microsatellites. Mutations in microsatellites within ORFs can affect the physical and chemical properties of proteins30 and they can rapidly change gene expression within promoters and other cis-regulatory regions,31 and microsatellites can be used to assess loss of heterozygosity.32
Regarding PISMA’s limitations, this tool is only directly applicable to DNA sequences of up to 10 000 bases, which means that it is limited in sequence size and nature. Nevertheless, these limitations could be overcome. It is possible to use PISMA—at least for motif distribution—with RNA sequences. To do so, it would be necessary to change U (uracil) for T (thymine), in the plain sequence text, using a word processor. It is also possible to use PISMA for large sequences, if these are broken down into smaller fractions of up to 10 000 nucleotides. In our opinion, the motif distribution along very large sequences may lead to loss of graphical resolution because of the usual size of monitors.
Supplementary Material
Acknowledgments
This work was performed mainly at 2 locations: Unidad de Investigación Biomédica en Cáncer, Instituto de Investigaciones Biomédicas, and Facultad de Ingeniería, both in the Universidad Nacional Autónoma de México. The authors thank Prof. Victor M Galvan for helpful discussions, Dr. Elizabeth Langley for critical review of the manuscript, Ing. Cristian Hernández-Escobedo who helped us to develop the search function included in the Supplementary File 3, Mr. Robert Paskin and Biolgist L Hermann Bojórquez-Galván for English editing, as well as the Article Reviewers because of their comments that helped us to improve its content.
Footnotes
Peer Review:Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1180 words, excluding any confidential comments to the academic editor.
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Dirección General de Asuntos del Personal Académico de la UNAM (grant number: PAPIIT-IN215411-3).
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article
Author Contributions: SCG and RAS conceived and designed the study, wrote the first draft of the manuscript, and jointly developed the structure and arguments for the paper. MAH, GDC, and RAS developed the software. MSB, SC, and SCG analyzed the data. GDC, MAH, MSB, and SC contributed to the writing of the manuscript and made critical revisions and approved the final version. RAS, MAH, GDC, MSB, SC, and SCG agreed with manuscript results and conclusions. All authors reviewed and approved the final manuscript.
Disclosures and Ethics: As a requirement of publication, authors have provided to the publisher signed confirmation of compliance with legal and ethical obligations including, but not limited to the, following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
References
- 1. Rombauts S, Dehais P, Van Montagu M, Rouze P. PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999;27:295–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doerfler W. In pursuit of the first recognized epigenetic signal—DNA methylation: a 1976 to 2008 synopsis. Epigenetics. 2008;3:125–133. [DOI] [PubMed] [Google Scholar]
- 3. Krieg AM. Lymphocyte activation by CpG dinucleotide motifs in prokaryotic DNA. Trends Microbiol. 1996;4:73–77. [DOI] [PubMed] [Google Scholar]
- 4. Batagov AO, Kuznetsov VA, Kurochkin IV. Identification of nucleotide patterns enriched in secreted RNAs as putative cis-acting elements targeting them to exosome nano-vesicles. BMC Genomics. 2011;12:S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hackenberg M, Rueda A, Carpena P, Bernaola-Galván P, Barturen G, Oliver JL. Clustering of DNA words and biological function: a proof of principle. J Theor Biol. 2012;297:127–136. [DOI] [PubMed] [Google Scholar]
- 6. Gymrek M, Willems T, Guilmatre A, et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat Genet. 2016;48:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grünewald TGP, Bernard V, Gilardi-Hebenstreit P, et al. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet. 2015;47:1073–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tompa M, Li N, Bailey TL, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol. 2005;23:137–144. [DOI] [PubMed] [Google Scholar]
- 9. Liu Y, Wei L, Batzoglou S, Brutlag DL, Liu JS, Liu XS. A suite of web-based programs to search for transcriptional regulatory motifs. Nucleic Acids Res. 2004;32:204–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bailey TL, Bodén M, Whitington T, Machanick P. The value of position-specific priors in motif discovery using MEME. BMC Bioinformatics. 2010;11:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Defrance M, van Helden J. Info-gibbs: a motif discovery algorithm that directly optimizes information content during sampling. Bioinformatics. 2009;25:2715–2722. [DOI] [PubMed] [Google Scholar]
- 12. Jajamovich GH, Wang X, Arkin AP, Samoilov MS. Bayesian multiple-instance motif discovery with BAMBI: inference of recombinase and transcription factor binding sites. Nucleic Acids Res. 2011;39:e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Che D, Jensen S, Cai L, Liu JS. BEST: binding-site estimation suite of tools. Bioinformatics. 2005;21:2909–2911. [DOI] [PubMed] [Google Scholar]
- 14. Wei Z, Jensen ST. GAME: detecting cis-regulatory elements using a genetic algorithm. Bioinformatics. 2006;22:1577–1584. [DOI] [PubMed] [Google Scholar]
- 15. Chan TM, Leung KS, Lee KH. TFBS identification based on genetic algorithm with combined representations and adaptive post-processing. Bioinformatics. 2008;24:341–349. [DOI] [PubMed] [Google Scholar]
- 16. Ichinose N, Yada T, Gotoh O. Large-scale motif discovery using DNA Gray code and equiprobable oligomers. Bioinformatics. 2012;28:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takai D, Jones PA. The CpG island searcher: a new WWW resource. In Silico Biol. 2003;3:235–240. [PubMed] [Google Scholar]
- 18. Xu YH, Manoharan HT, Pitot HC. CpG analyzer, a Windows-based utility program for investigation of DNA methylation. Biotechniques. 2005;39:656–662. [DOI] [PubMed] [Google Scholar]
- 19. Menendez C, Frees S, Bagga PS. QGRS-H Predictor: a web server for predicting homologous quadruplex forming G-rich sequence motifs in nucleotide sequences. Nucleic Acids Res. 2012;40:W96–W103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. [DOI] [PubMed] [Google Scholar]
- 21. Gowher H, Jeltsch A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J Mol Biol. 2001;309:1201–1208. [DOI] [PubMed] [Google Scholar]
- 22. Pinney SE. Mammalian non-CpG methylation: stem cells and beyond. Biology (Basel). 2014;1;3:739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schorderet DF, Gartler SM. Analysis of CpG suppression in methylated and nonmethylated species. Proc Natl Acad Sci U S A. 1992;89:957–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Galván SC, Martínez-Salazar M, Galván VM, et al. Analysis of CpG methylation sites and CGI among human papillomavirus DNA genomes. BMC Genomics. 2011;12:580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bock C, Paulsen M, Tierling S, Mikeska T, Lengauer T, Walter J. CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA structure. PLoS Genet. 2006;2:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. 2010;73:1907–1920. [DOI] [PubMed] [Google Scholar]
- 27. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. [DOI] [PubMed] [Google Scholar]
- 28. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A. 2010;107:6328–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tan A, De La Peña H, Seifalian AM. The application of exosomes as a nanoscale cancer vaccine. Int J Nanomedicine. 2010;5:889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fondon JW, III, Garner HR. Molecular origins of rapid and continuous morphological evolution. Proc Natl Acad Sci U S A. 2004;101:18058–18063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rockman MV, Wray GA. Abundant raw material for cis-regulatory evolution in humans. Mol Biol Evol. 2002;19:1991–2004. [DOI] [PubMed] [Google Scholar]
- 32. Migdalska-Sęk M, Karowicz-Bilińska A, Pastuszak-Lewandoska D, et al. Assessment of the frequency of genetic alterations (LOH/MSI) in patients with intraepithelial cervical lesions with HPV infection: a pilot study. Med Oncol. 2016;33:51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.