

ABSTRACT
Purpose of Review: This article discusses the recent advances in the diagnosis and treatment of Alzheimer disease (AD).
Recent Findings: In recent years, significant advances have been made in the fields of genetics, neuroimaging, clinical diagnosis, and staging of AD. One of the most important recent advances in AD is our ability to visualize amyloid pathology in the living human brain. The newly revised criteria for diagnosis of AD dementia embrace the use for biomarkers as supportive evidence for the underlying pathology. Guidelines for the responsible use of amyloid positron emission tomography (PET) have been developed, and the clinical and economic implications of amyloid PET imaging are actively being explored.
Summary: Our improved understanding of the clinical onset, progression, neuroimaging, pathologic features, genetics, and other risk factors for AD impacts the approaches to clinical diagnosis and future therapeutic interventions.
INTRODUCTION
Alzheimer disease (AD) is a neurodegenerative disorder featuring gradually progressive cognitive and functional deficits as well as behavioral changes and is associated with accumulation of amyloid and tau depositions in the brain. Cognitive symptoms of AD most commonly include deficits in short-term memory, executive and visuospatial dysfunction, and praxis. Several rarer variants of AD with relative preservation of memory have been recognized. Clinical assessment, including cognitive testing, remains critical for the diagnosis and staging of AD, although recent advances in amyloid imaging and genetics show great promise for facilitating early and presymptomatic diagnosis of AD and its discrimination from other neurodegenerative disorders.
EPIDEMIOLOGY
AD is the most common neurodegenerative disorder and the sixth most common cause of death in the United States.1 Although there is increasing evidence that AD pathology starts depositing in the brain in midlife, the first clinical symptoms usually occur after the age of 65.2,3
AD prevalence is rapidly increasing in large part because the proportion of people 65 years and older is growing faster than any other age sector of the population worldwide. Between 1997 and 2050, the elderly population, defined as subjects 65 years of age and older, will increase from 63 to 137 million in the Americas, from 18 to 38 million in Africa, from 113 to 170 million in Europe, and from 172 to 435 million in Asia.4 One nationally representative US data set, the Aging, Demographics, and Memory Study (ADAMS), estimated that in the United States, 14% of people 71 years and older have dementia. AD dementia accounted for 70% of the dementia cases across the age spectrum in this cohort.5 In a subsequent publication, the ADAMS investigators reported that an additional 22% (or 5.4 million Americans) 71 years or older have cognitive impairment in the absence of overt dementia.6
Although age is the greatest risk factor for the development of AD, in and of itself, old age is not sufficient to cause AD. Other major risk factors include the presence of one or more apolipoprotein gene E4 alleles (APOE4), low educational and occupational attainment, family history of AD, moderate or severe traumatic brain injuries, and cardiovascular risk factors.
Gender modulates the prevalence of AD. Nearly two-thirds of all patients diagnosed with AD are women.7 According to ADAMS, 16% of women, but only 11% of men, live with dementia after 71 years of age.5 While it is true that women live longer than men, this alone does not explain the discrepancy. Genetic, hormonal, and societal factors (eg, lower education and occupational attainment among women currently in their 70s and 80s compared to men) likely play a significant role as well.
Racial disparities in AD prevalence have also been reported. Older African Americans and Hispanics have a higher prevalence of AD relative to older Caucasians in part because of lower education levels and higher prevalence of cardiovascular comorbidities,8–10 although other genetic and societal factors likely play a role as well.
CLINICAL PRESENTATION
Recognition of the clinical features and presenting symptoms of AD remains essential for the diagnosis and management of patients, even as biomarker tests such as amyloid positron emission tomography (PET) imaging that detect the underlying neuropathology of AD are increasingly available for patient care. Ascertainment of symptoms of cognitive decline, behavioral symptoms, functional decline, and cognitive testing remain the cornerstones to the clinical diagnosis and staging of patients with AD.
Cognitive Decline
Memory impairment is the most pervasive feature of AD. Although nonmemory cognitive deficits (eg, aphasia, executive dysfunction, apathy, or personality change) can manifest early and even be the presenting feature, in general, memory decline is considered the leading symptom. Early in the disease course, recent episodic memories are most affected, while memories from the distant past are usually spared. As the disease progresses, all aspects of episodic memory become affected. In contrast to episodic memory, working memory and semantic memory are preserved until later in the disease course.
Language disturbance, especially word-finding difficulties, is a common early symptom in AD but is generally mild. Subtle decline in visuospatial skills likewise occurs in the mild dementia stages and progresses throughout the course of the disease. Executive dysfunction, on the other hand, begins even earlier—in the predementia stages—and, similar to all other cognitive domains, worsens over the disease course.
Neuropsychiatric Symptoms
Patients with AD exhibit a variety of neuropsychiatric symptoms. Behavioral symptoms, once manifest, tend to worsen over the course of the disease; however, these symptoms often fluctuate and are not consistently present at each visit. Attention to these treatable components of excess morbidity is important as they have a profound impact on caregiver burden and are the leading cause of institutionalization.11
The earliest AD-associated neuropsychiatric symptoms are apathy, anxiety, and irritability. The latter two symptoms are often provoked in situations that the patient finds challenging. Mild to moderate depressive symptoms are also frequently present early on. Disturbances of appetite and sleep, disinhibition, and alterations in perception (hallucinations) or thought (delusions) commonly occur in the later stages of dementia. In addition to the classic neuropsychiatric behaviors, anosognosia (ie, lack of insight) often manifests early on and poses another difficult management problem.
Of all AD neuropsychiatric comorbidities, irritability, agitation, sundowning, psychosis, and diminished insight often present the most pressing therapeutic needs due to the psychological and physical strain they place on the family and caregivers.
Neurologic Examination
Outside of the mental status examination, findings on the neurologic examination are often normal in patients with AD. Parkinsonian symptoms can emerge in the later stages, but if these symptoms are present early in the course of the disease (eg, within 1 year of onset of cognitive problems) and especially when accompanied by cognitive fluctuations and early-onset psychosis, a diagnosis of dementia with Lewy bodies should be considered. Later in the disease course, pathologic reflexes such as grasp, root, and suck reflexes may be found. Patients become increasingly impaired in the moderate and severe stages and are ultimately mute, incontinent, and bedridden at the end stages of disease. At this stage, multiple complications arise such as risk of aspiration with unsafe swallowing, malnutrition, immobility with associated risks for bedsores, deep venous thrombosis, and infections. Often, these complications are the direct cause of death in patients with AD.
ATYPICAL ALZHEIMER DISEASE VARIANTS
In addition to the classic AD presentation, several less common AD variants should be recognized, which include frontal variant of AD, posterior cortical atrophy, and logopenic variant primary progressive aphasia due to AD.
Frontal variant of AD is characterized by substantial behavioral or personality changes that are out of proportion to the observed short-term memory loss. These patients are often profoundly impatient, irritable, impulsive, and disinhibited. On formal testing, they invariably show significant executive disturbances.12
Posterior cortical atrophy presents with visuospatial dysfunction often in the form of partial or full Balint syndrome (simultanagnosia, ocular apraxia, and ocular ataxia), partial or full Gerstmann syndrome (acalculia, agraphia, right/left disorientation, and finger agnosia), apperceptive visual agnosia, and environmental disorientation. Patients often develop constructional, dressing, and ideomotor apraxia early on in the presence of relatively preserved memory and insight.
Finally, the occasional patient with AD may also present with early progressive language involvement, most often in the form of logopenic aphasia with pronounced anomic deficits and impaired repetition but preserved grammar and syntax.
DIAGNOSTIC CRITERIA
Currently, the diagnostic standard for dementia is the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5).13 DSM-5 recognizes two cognitive syndromes: major neurocognitive impairment and mild neurocognitive impairment. The diagnosis of major neurocognitive impairment requires objective cognitive decline that is severe enough to interfere with activities of daily living and is not caused by delirium or another neurologic, medical, or psychiatric disorder. Patients with mild neurocognitive impairment have milder cognitive decline that does not yet deprive them of the ability to lead an independent lifestyle and perform complex daily activities such as managing finances or driving a car.
It should be noted that the DSM-5 introduces a major change in terms of diagnostic criteria for cognitive disorders. The criteria no longer require the presence of memory impairment for the diagnosis of neurodegenerative dementia to be established, as was the case in all previous DSM editions. DSM-5 thus recognizes that for some dementing disorders such as vascular and frontotemporal dementia, for instance, memory impairment is not an early symptom and may never manifest (Table 3-1).
Table 3-1.
Summary of Diagnostic Criteria for Mild and Major Neurocognitive Disordera

Another set of diagnostic criteria spanning all three major stages of AD (ie, the preclinical, the prodromal, and the overt dementia stages) were recently developed by the National Institute on Aging (NIA) and the Alzheimer’s Association (AA).14–16 Similarly to the DSM-5 criteria, the NIA-AA criteria for dementia of any cause no longer explicitly require memory impairment to be present, but rather, for the diagnosis of dementia to be established, call for documentation of impairment in two cognitive domains or one cognitive and one behavioral domain in addition to significant decline in day-to-day functioning (Table 3-2). For the first time, the NIA-AA criteria for probable Alzheimer dementia as a subtype of dementia recognized the diagnostic utility of disease biomarkers that have proven sensitivity, specificity, and pathologic validity (Table 3-2). At the present time, two types of biomarkers meet these criteria. Two neurodegenerative biomarkers—mesial temporal lobe atrophy on structural imaging (Figure 3-1) and posterior predominant hypometabolism with involvement of the posterior cingulate gyrus on fluorodeoxyglucose positron emission tomography (FDG-PET) (Figure 3-2)—have already received wide acceptance. However, neither of these are exclusively seen in AD dementia. Hippocampal atrophy occurs in normal aging,17,18 and both hippocampal atrophy and FDG-PET abnormalities occur in other dementing conditions.19,20 On the other hand, amyloid protein–based biomarkers such as a low CSF level of β-amyloid or a positive amyloid PET scan have been shown to be highly sensitive and specific in their ability to detect amyloid pathology in the brain.21,22
Table 3-2.
Summary of Diagnostic Criteria for Dementia Syndrome and Probable Alzheimer Disease from the National Institute on Aging and the Alzheimer’s Associationa

Figure 3-1.
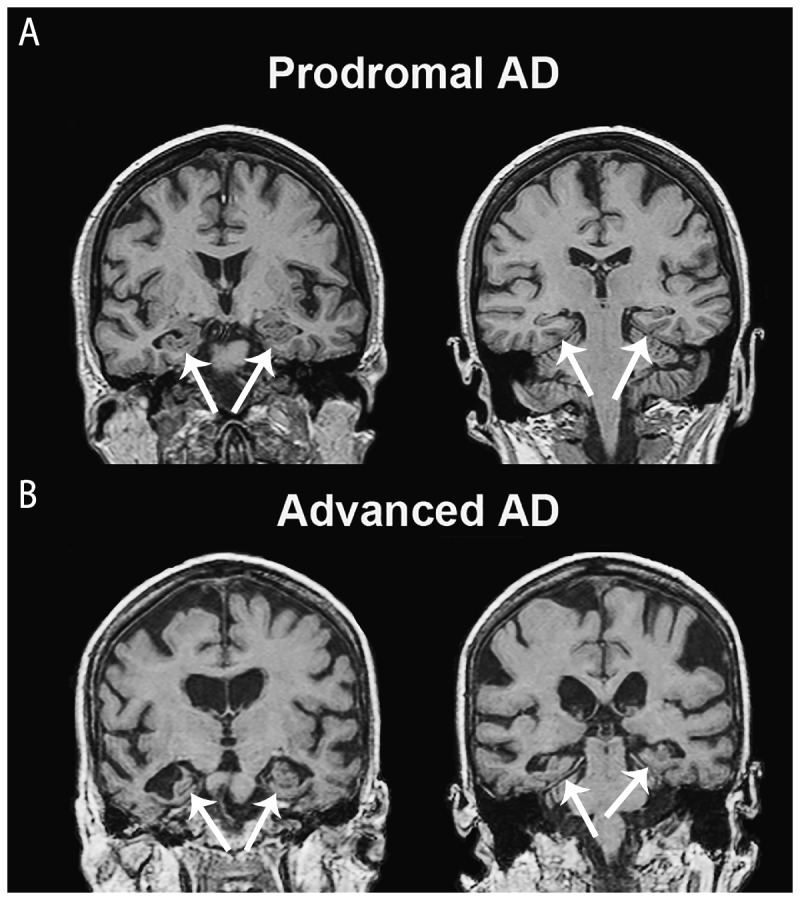
Coronal T1-weighted MRI slices with findings suggestive of Alzheimer disease (AD) pathology show mesial temporal atrophy (A, B, arrows) and, in the more advanced stages, global brain atrophy with pronounced ventricular enlargement.
Figure 3-3.
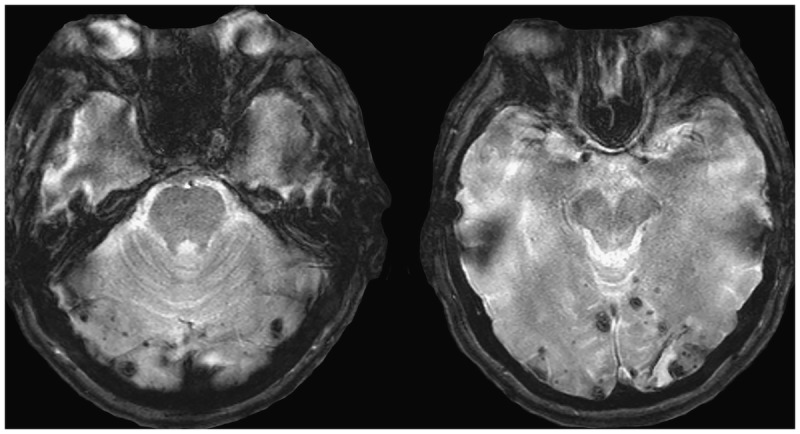
Cortico-subcortical microhemorrhages are often found on gradient echo MRI sequences and are suggestive of the presence of vascular amyloidosis.
Current Role of Biomarkers in Alzheimer Disease Diagnosis
In addition to supporting clinical diagnosis, biomarkers have historically played an important role in the workup of patients with dementia. The American Academy of Neurology (AAN) guidelines for the diagnostic evaluation of dementia require physicians to obtain a structural imaging scan in every patient with objective cognitive decline.23 This recommendation follows the review of the evidence presented in a Class II study showing that 5% of all patients with cognitive complaints harbored a causative nondegenerative lesion, such as a slow-growing brain neoplasm (most commonly of the frontal lobes), subdural hematoma, or normal pressure hydrocephalus.24 In addition to identifying a potentially treatable lesion, a brain CT or MRI scan can uncover ischemic changes that would prompt further workup and potential initiation of therapy aiming to reduce vascular risk factors or introduce behavioral modifications. MRI, with its improved resolution, allows better quantification of cerebral structures and better discrimination of normal from mildly affected patients with AD than is possible with CT. Some of the findings suggestive of AD pathology include mesial temporal atrophy25–27 and, in the more advanced stages, global brain atrophy with pronounced ventricular enlargement (Figure 3-1).28,29 Cortico-subcortical microhemorrhages are often found on gradient echo MRI sequences and are suggestive of the presence of vascular amyloidosis (Figure 3-3).
Figure 3-2.
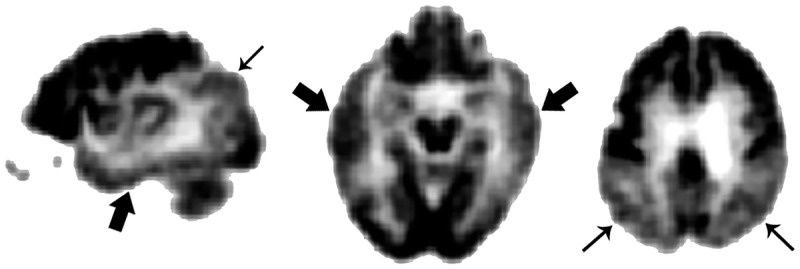
Temporal (thick arrows) and parietal (thin arrows) hypometabolism on fluorodeoxyglucose positron emission tomography (FDG-PET) is often seen in patients with Alzheimer disease dementia.
Functional brain imaging using single-photon emission computed tomography (SPECT) and PET technologies can be used to identify AD-specific patterns such as temporoparietal hypoperfusion/hypometabolism in patients with AD (Figure 3-2). More recently, arterial spin-labeling MRI sequences have been shown to capture perfusion abnormalities.30 Validation studies of SPECT and PET have generally shown high sensitivity yet relatively lower specificity, hence the increased risk for false-positive diagnoses.31,32 Currently, FDG-PET and SPECT use is only covered by Medicare for differentiating AD from frontotemporal dementia.
The recent discovery and validation of amyloid PET imaging has the potential to change the approach to clinical diagnosis of AD. This type of imaging allows the detection of moderate to severe amyloid deposition in the brain (Figure 3-4). Despite its well-documented sensitivity and specificity,22 this technology has not yet entered routine clinical practice, in part because insurance companies do not provide coverage for it. The major drawbacks among insurance carriers are: (1) the unproven economic benefit of using this relatively expensive PET technology for diagnostic purposes while disease-modifying therapies are lacking and (2) the potential risk for it to be used in cognitively normal individuals. Clearly, the latter scenario (ie, detecting AD in the latent stages in the form of asymptomatic brain amyloidosis) could inflict an insurmountable psychological burden given the absence of effective therapeutic approaches to delay disease onset or modify its course. In response to these concerns, a group of imaging and dementia experts convened to establish a set of recommendations of which patients should be imaged. These suggestions are the Appropriate Use Criteria for amyloid PET imaging,33,34 which recommend employing amyloid PET imaging in three specific clinical scenarios: (1) patients with amnestic mild cognitive impairment (ie, mild neurocognitive disorder), (2) patients in the dementia stages with suspected atypical AD or etiologically mixed presentation, and (3) those with early disease onset (younger than 65 years of age). At present, the impact of amyloid PET imaging on clinical practice and health outcomes is not yet known. Given the lack of disease-modifying drugs for AD, the rationale for the use of amyloid PET imaging in clinical practice includes: (1) helping the practitioner to select appropriate treatments and avoid unnecessary interventions and to aid in accurate diagnosis, (2) improving diagnostic accuracy, (3) advising patients and families on the clinical course and prognosis, and (4) educating patients and families on community services and resources for medical, financial, and legal planing.34 Case 3-1 and Case 3-2 describe exemplary scenarios.
Figure 3-4.
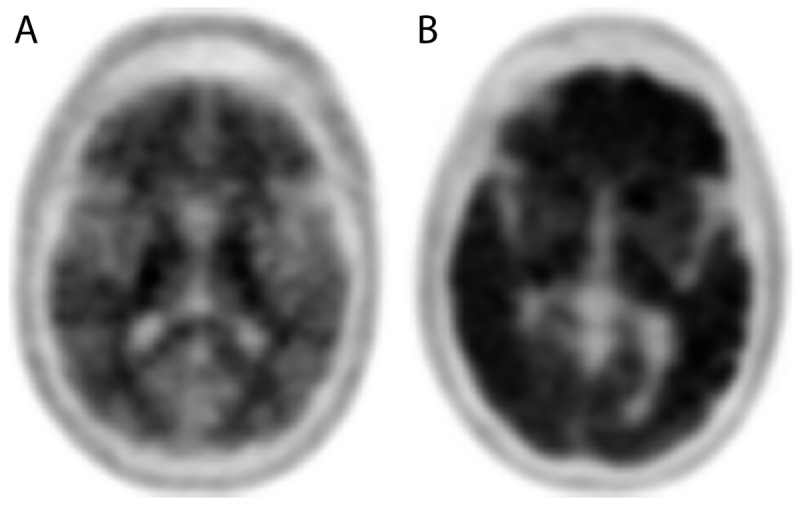
Amyloid positron emission tomography (PET) imaging allows the detection of amyloid pathology of the brain. A negative scan (A) shows only nonspecific white matter binding and indicates no to sparse amyloid plaque deposition, while a positive amyloid PET scan (B) shows significant uptake in multiple cortical areas and indicates the presence of moderate to severe amyloid pathology.
The Appropriate Use Criteria also define the inappropriate uses of amyloid PET imaging. Patients who should not be scanned include those lacking objective cognitive decline. Scanning solely on the basis of family history or APOE4 status was likewise determined to be inappropriate. Furthermore, amyloid PET scans cannot be used for determining dementia severity, as this is not feasible with this technology.
Another important suggestion of the Appropriate Use Criteria for amyloid PET imaging is the recommendation that the responsibility for determining patients’ eligibility (ie, appropriateness for imaging) should lie with medical professionals with significant expertise in evaluating and treating patients with dementia (defined as 25% or greater proportion of clinical practice devoted to cognitive disorders of the elderly).33 Last but not least, the committee recommended that amyloid PET scans are only appropriate if the scan results are expected to alter clinical management (see Case 3-1 and Case 3-2 for examples).
CSF amyloid-β (Aβ) and tau protein levels are the most established AD fluid biomarkers. Pathologic Aβ deposition in the brain tissue begins early in the disease course and is associated with decline in CSF Aβ.35 CSF total and phosphorylated tau levels rise in AD secondary to neurodegeneration of tau-containing neurons.36–39 CSF tau changes occur later in the disease course and are associated with cognitive decline.40–43 CSF Aβ and tau measurements are covered by most insurance companies in the United States as part of the workup for AD. Despite this, they are not routinely used due to relative invasiveness of the lumbar puncture procedure and the risk for side effects such as back discomfort, headache, and, in rare cases, iatrogenic meningitis. However, CSF Aβ and tau testing can be quite useful in diagnostically challenging cases as well as in those with early disease onset or an unusual clinical course. CSF Aβ level is highly correlated with the presence of amyloid deposition in the cortex.44 Thus, CSF Aβ levels can be used in lieu of an amyloid PET scan as a reliable proxy measure of the presence of brain amyloidosis.
Case 3-1
A 78-year-old man with 12 years of education presented to the memory disorder clinic reporting progressive memory difficulties for the past 1 to 2 years. His memory deficits were most noticeable when he recalled recent events as opposed to events from the distant past. He had on two occasions experienced mild spatial confusion when walking his dog but, in both instances, he was eventually able to find his way. He had no problems with attention, language skills, or judgment. He operated all appliances at home and safely drove a car. His family noticed mild anxiety in challenging situations and questioned whether he was a little depressed, as he had become quieter. The patient had no significant comorbidities and no family history of dementia. He had been prescribed memantine 10 mg once a day by an outside physician.
The patient’s cognitive evaluation revealed a Mini-Mental State Examination (MMSE) score of 25 out of 30, poor recollection of recent events, lower than expected performance on category fluency (number of animals produced in 1 minute), poor encoding and retention of verbal and nonverbal information, mild anxiety, and minimal depression. His attention, visuospatial skills, and abstract thinking were preserved. Formal neuropsychological testing confirmed the above deficits and further clarified his memory profile as most consistent with retrieval memory loss, as he showed significant improvement on cued recall (ie, the recognition portion of the memory tests). Review of an outside MRI revealed age-appropriate atrophy.
The patient was given a diagnosis of mild neurocognitive disorder by Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5)13 criteria (ie, mild cognitive impairment) most likely due to a neurodegenerative etiology. Although the leading consideration was Alzheimer disease (AD), the observed significant improvement on cued recall suggestive of retrieval rather than encoding deficits was considered atypical. An amyloid positron emission tomography (PET) scan was ordered and revealed diffuse amyloid tracer uptake in the cerebral cortical gray matter with complete loss of the gray-white matter differentiation consistent with the presence of moderate to severe amyloid pathology.
A diagnosis of mild neurocognitive disorder due to AD was made. The patient was prescribed donepezil. He experienced significant gastrointestinal side effects as the dose was increased from 5 mg/d to 10 mg/d. A slower escalation regimen (introducing an additional titration step of 7.5 mg/d for 1 month before introducing 10 mg/d was effective. Two years later, his MMSE score had declined to 19/30. His family reported difficulties with banking, shopping, navigation, and cooking. As his presentation was now consistent with moderate stage AD, he was started on memantine and the dosage was optimized to 10 mg twice a day.
Comment. This patient presented with atypical amnestic mild cognitive impairment that met appropriate use criteria33,34 for amyloid PET imaging. The diagnostic uncertainty stemmed from the patient’s pronounced memory retrieval as opposed to encoding deficit. The amyloid PET scan was beneficial in assisting the physician’s therapeutic decisions, specifically the potential symptomatic benefit of a cholinesterase inhibitor, and counseling the patient and family on the patient’s prognosis.
Case 3-2
A 59-year-old woman with 12 years of education presented to the memory disorder clinic because of memory difficulties for the past 1 to 2 years. Her husband reported that for the past few months they had the same conversation multiple times a day. She misplaced items more often. Her recall of distant memories and visuospatial function were preserved. She was also becoming more distractible. She managed their household without a problem and safely operated a motor vehicle. On a few occurrences, she reportedly paid some bills twice while being on time with the rest. She was still able to manage her appointments and medications with occasional reminders. She had become quieter in social situations and more reluctant to go out. She was no longer interested in volunteering at their church.
On bedside cognitive testing the patient scored 22 out of 30 on the Mini-Mental State Examination (MMSE). She showed relatively preserved encoding, impaired delayed recall, and endorsed multiple false-positive items on recognition testing. Her language and visuospatial skills were preserved. She made a few errors on frontal executive tasks. On formal neuropsychological testing, additional deficits in category fluency and nonverbal memory were noted.
The patient’s MRI revealed mild global atrophy with medial temporal lobe predominance and hippocampal atrophy. An amyloid PET scan was performed given her early age of onset and showed diffusely increased tracer uptake throughout the cortical cerebral gray matter with loss of the normal gray-white matter contrast consistent with the presence of moderate to severe amyloid pathology. A diagnosis of mild neurocognitive disorder (ie, mild cognitive impairment) due to early-onset AD was made. The patient was started on donepezil.
Comment. This patient presented with early-onset prodromal AD. The patient met the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5)13 criteria for mild neurocognitive disorder (ie, mild cognitive impairment) and met the appropriate use criteria for amyloid PET imaging by virtue of early disease onset. The amyloid PET scan was considered beneficial in assisting the physician’s therapeutic decisions and counseling of the patient.
GENETICS OF ALZHEIMER DISEASE
Although the APOE4 gene variant is the most established genetic risk factor for sporadic AD, screening for APOE4 is not recommended on a routine basis. While it has been estimated that one copy of this genetic variant increases the odds for developing AD threefold and two copies increase the odds 15-fold,45 a large multicenter study demonstrated that the presence of the APOE4 allele increased the positive predictive value of diagnosing AD by only 4% over diagnoses made on clinical grounds alone (from 90% to 94%).46 The APOE4 genotype should be considered a risk factor that is neither sufficient nor necessary for disease development.
More recently, several large genome-wide association studies have uncovered and successfully replicated another 20 risk/protective genetic variants.47 Relative to APOE4, these variants explain only a small fraction of the genetic heritability of sporadic AD where each risk variant conveys a relative risk of 1.1 to 1.3.47 The vast majority of these risk variants take part in processes implicated in the pathogenesis of AD such as amyloid metabolism, inflammation, oxidative stress, and energy metabolism. The search for other gene variants that may affect risk of AD is ongoing.
Family history of sporadic AD is a well-established risk factor. Individuals who have a first-degree relative diagnosed with AD are more likely to develop the disease than those who do not.48 Genetic makeup is not the only risk; shared environmental and lifestyle factors likely also play a role.
Several rare autosomal dominant variants of AD have been described. The first symptoms usually affect individuals in their 30s and 40s. These families show multiple affected individuals across generations. These autosomal dominant AD variants have been traced to genetic mutations in the amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes. Mutations in all three genes increase the levels of β-amyloid. Early-onset autosomal dominant AD accounts for less than 2% of all AD cases.
ALZHEIMER DISEASE PATHOLOGY
AD is thought to result from overproduction and impaired clearance of β-amyloid. Downstream events are tau hyperphosphorylation and neuronal toxicity. The primary pathologic features of AD are brain atrophy from regional neuronal and synaptic loss, extracellular β-amyloid deposition in the form of neuritic plaques, and intraneuronal tau protein deposition in the form of intraneuronal neurofibrillary tangles. β-Amyloid also deposits in the cerebral blood vessels. Cerebral amyloid angiopathy ranges in severity from small amounts of amyloid to major deposits that distort the artery architecture and cause cortical microinfarcts, microaneurysms, and cerebral microhemorrhages or macrohemorrhages (multiple microhemorhages in the occipital lobes of a patient with AD are shown in Figure 3-3).
Amyloid deposition is thought to begin 20 years prior to development of clinical symptoms.2 Longitudinal studies of cognitively normal individuals who are amyloid-positive are presently ongoing and are focusing on ascertainment of the risk for future development of dementia among these individuals. Neurofibrillary tangles are not exclusive to AD and can be found in other conditions, such as dementia pugilistica and chronic traumatic encephalopathy, prion disease, and in normal aging. Neurofibrillary tangle burden and neuronal loss show a robust association with global cognitive impairment.49,50
TREATMENT
The clinical management of patients with AD dementia should accomplish several important tasks. Patients need to receive diagnostic and prognostic counseling as well as appropriate medications in an optimized regimen. Patients’ coexisting behavioral and non-neurologic conditions need to be optimally managed. Vital importance should be placed on coordinating care among physicians, nurse practitioners, and social workers, and instituting appropriate oversight and safety precautions when patients have functional impairment and poor judgment. It is important to encourage patients to participate in social activities, adult day care centers and exercise programs, as well as to encourage both patients and caregivers to take part in support groups.
Acetylcholinesterase Inhibitors
Two classes of medications have been approved for AD: the acetylcholinesterase inhibitors (AChEIs) and the N-methyl-D-aspartate (NMDA) receptor antagonist memantine. Treatment with AChEIs should be considered in patients with mild to moderate AD per the AAN practice guidelines for dementia.51 Three agents are currently approved and marketed in the United States: donepezil, rivastigmine, and galantamine. All three have been shown to be effective in double-blind placebo-controlled trials, showing some benefit on cognitive measures including memory and concentration as well as global and functional outcome measures; however, their therapeutic cognitive and functional effects seem to be modest in size and purely symptomatic. Gastrointestinal side effects, more commonly seen during the dose escalation phase of treatment, occur with all three agents. Bradycardia and heart block may occur, especially in patients with underlying cardiac conduction deficits or in those individuals taking medications that cause PR interval prolongation such as beta-blockers. If one agent causes intolerable side effects, another AChEI should be tried.
Glutamate Receptor Modulators
Memantine is a low to moderate affinity NMDA receptor antagonist that is used as an add-on to ongoing AChEI therapy. A recent meta-analysis of nine trials with a combined sample size of 2433 revealed that memantine had a beneficial effect on cognition, behavior, activities of daily living, and global function.52 Memantine is approved by the US Food and Drug Administration (FDA) for the moderate to severe AD stages (Mini-Mental State Examination [MMSE] score of 5 to 15) in the United States. The main side effects of confusion and dizziness occur only rarely.
The timing of initiation of cholinesterase inhibitors and memantine therapy is an area of some controversy.53 Cholinesterase inhibitors and memantine are frequently prescribed off-label for mild cognitive impairment in the United States.54 Recent meta-analyses have not demonstrated a benefit of cholinesterase inhibitors in mild cognitive impairment, although a benefit for subgroups of patients remain undetermined.55,56 A separate meta-analysis suggests there is not a benefit of memantine in patients with mild AD.57 While the 2001 AAN practice parameter for treatment of dementia is currently under revision, 2011 guidelines from the National Institute for Health and Clinical Excellence now recommend memantine as an option for managing moderate AD for people who cannot take AChEIs and as an option for managing severe AD.58 In contrast, Canadian Consensus guidelines on dementia from 2013 state that, while combination therapy of a cholinesterase inhibitor and memantine is rational and appears to be safe, there is insufficient evidence to support or refute use of memantine in moderate to severe AD.59
Medications for Behavioral Symptoms
The first line of treatment for behavioral symptoms of AD are nonpharmacologic techniques. A quiet, familiar environment with labels on doors and sufficient lighting in all rooms is important to reduce disorientation. Aggressive behavior should always be addressed with positive and clear language to reassure and distract the patient.
Depressive symptoms are treated with selective serotonin reuptake inhibitors (SSRIs) due to their low propensity to cause anticholinergic effects. SSRIs may also ease anxiety, irritability, or other nonspecific symptoms that may accompany depression. The SSRI citalopram may be useful for agitation.
Agitation or disruptive behavior may require a neuroleptic for optimal therapeutic response. The newer “atypical” antipsychotic medications (quetiapine, risperidone, olanzapine) are often used in low doses with careful titration. Typical and atypical antipsychotic agents, however, carry a black box warning label due to an association with increased cardiovascular morbidity and mortality (higher for the typical compared to atypical antipsychotics) and cerebrovascular adverse events in the elderly with dementia-related psychosis.60–62 In addition, these medications have additional adverse effects: anticholinergic adverse events and orthostatic and metabolic disturbances. Traditional neuroleptics are more likely to produce extrapyramidal symptoms, which may worsen cognitive function. All antipsychotics, typical as well as atypical, when used in older adults with dementia, are associated with risk for death. This risk is quite comparable among atypical and typical antipsychotics. It is a black box warning for all antipsychotics as a class when used in older adults with dementia.62 Thus, judicious use of antipsychotics with frequent reassessment of the therapeutic need is appropriate.
Future Therapies
The majority of currently ongoing clinical trials are focused on interventions that directly influence the pathologic cascade in AD. One active immunization trial in humans was interrupted due to the development of neuroinflammation in a subset of the subjects. There was remarkable clearance of amyloid in the cortex in multiple subjects from that trial who died,63,64 suggesting that amyloid deposits can indeed be removed. Current major efforts are focused on passive immunization (ie, antibody infusion) as well as interference with β-amyloid and tau production and polymerization.
CONCLUSION
AD is an irreversible neurodegenerative disorder that affects over 5 million Americans. Although we are now able to diagnose AD in the early and even in the presymptomatic stages, we are still lacking preventative or disease-modifying medications that can alter its course. The effect of AD on individual patients and their families and caregivers is devastating. As the number of patients with AD climbs, families, caretakers, the medical system, and society as a whole will have to bear the burden of this disease unless we find a cure.
KEY POINTS
Alzheimer disease prevalence is rapidly increasing.
Although age is the greatest risk factor for the development of Alzheimer disease, old age is not sufficient, in and of itself, to cause Alzheimer disease.
Memory impairment is the most pervasive feature of Alzheimer disease.
Neuropsychiatric comorbidities often present the most pressing therapeutic needs due to the psychological and physical strain they place on the family and caregivers of patients with Alzheimer disease.
Several less common Alzheimer disease variants should be recognized, which include frontal variant of Alzheimer disease, posterior cortical atrophy, and logopenic variant primary progressive aphasia due to Alzheimer disease.
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria no longer require the presence of memory impairment for the diagnosis of neurodegenerative dementia to be established.
The National Institute on Aging and Alzheimer’s Association criteria for probable Alzheimer dementia recognize the diagnostic utility of disease biomarkers.
Amyloid positron emission tomography imaging allows the detection of moderate to severe amyloid deposition in the brain.
Amyloid positron emission tomography imaging is recommended for patients with amnestic mild cognitive impairment, patients in the dementia stages with suspected atypical Alzheimer disease or etiologically mixed presentation, and those with early disease onset.
Amyloid positron emission tomography scans are only appropriate if the scan results are expected to alter clinical management.
The APOE4 genotype should be considered a risk factor that is neither sufficient nor necessary for Alzheimer disease development.
The search for other gene variants that may affect risk of Alzheimer disease is ongoing.
Amyloid deposition is thought to begin 20 years prior to development of clinical symptoms.
Vital importance should be placed on coordinating care among physicians, nurse practitioners, and social workers, and instituting appropriate oversight and safety precautions when patients have functional impairment and poor judgment.
Treatment with acetylcholinesterase inhibitors should be considered in patients with mild to moderate Alzheimer disease per the American Academy of Neurology practice guidelines for dementia.
Memantine is US Food and Drug Administration approved for the moderate to severe stages of Alzheimer disease.
The first line of treatment for behavioral and neuropsychiatric symptoms of Alzheimer disease are nonpharmacologic modalities.
ACKNOWLEDGMENTs
The author is supported through grants from the National Institute on Aging (NIA R01 AG040770, NIA K02 AG048240, and NIA P30 AG010133).
REFERENCES
- 1.Alzheimer’s Association. 2015. Alzheimer’s disease facts and figures. Alzheimers Dement 2015; 11(3): 322– 384. [DOI] [PubMed] [Google Scholar]
- 2. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol 2013; 12(4): 357– 367. doi:10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 3. Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 2010; 9(1): 119– 128. doi:10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization. World atlas of ageing. Kobe, Japan: World Health Organization, Centre for Health Development, 1998. [Google Scholar]
- 5. Plassman BL, Langa KM, Fisher GG, et al. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 2007; 29(1–2): 125– 132. doi:10.1159/000109998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Plassman BL, Langa KM, Fisher GG, et al. Prevalence of cognitive impairment without dementia in the United States. Ann Intern Med 2008; 148(6): 427– 434. doi:10.7326/0003-4819-148-6-200803180-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003; 60(8): 1119– 1122. doi:10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 8. Gurland BJ, Wilder DE, Lantigua R, et al. Rates of dementia in three ethnoracial groups. Int J Geriatr Psychiatry 1999; 14(6): 481– 493. doi:10.1002/(SICI)1099-1166(199906)14:6<481::AID-GPS959>3.0.CO;2-5. [PubMed] [Google Scholar]
- 9. Haan MN, Mungas DM, Gonzalez HM, et al. Prevalence of dementia in older latinos: the influence of type 2 diabetes mellitus, stroke and genetic factors. J Am Geriatr Soc 2003; 51(2): 169– 177. doi:10.1046/j.1532-5415.2003.51054.x. [DOI] [PubMed] [Google Scholar]
- 10. Manly JJ, Mayeux R. Ethnic differences in dementia and Alzheimer disease. In: Anderson NB, Bulatao RA, Cohen B, eds. Critical perspectives on racial and ethnic differences in health in late life. Washington, DC: National Academies Press, 2004: 95– 142. [PubMed] [Google Scholar]
- 11. Kaufer DI, Cummings JL, Christine D, et al. Assessing the impact of neuropsychiatric symptoms in Alzheimer’s disease: the Neuropsychiatric Inventory Caregiver Distress Scale. J Am Geriatr Soc 1998; 46(2): 210– 215. doi:10.1111/j.1532-5415.1998.tb02542.x. [DOI] [PubMed] [Google Scholar]
- 12. Johnson JK, Head E, Kim R, et al. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol 1999; 56(10): 1233– 1239. doi:10.1001/archneur.56.10.1233. [DOI] [PubMed] [Google Scholar]
- 13.American Psychiatric Association. Diagnostic and statistical manual of mental disorders, fifth edition Washington, DC: American Psychiatric Publishing, 2013. [Google Scholar]
- 14. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7(3): 270– 279. doi:10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7(3): 263– 269. doi:10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7(3): 280– 292. doi:10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Apostolova LG, Green AE, Babakchanian S, et al. Hippocampal atrophy and ventricular enlargement in normal aging, mild cognitive impairment (MCI), and Alzheimer disease. Alzheimer Dis Assoc Disord 2012; 26(1): 17– 27. doi:10.1097/WAD.0b013e3182163b62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Leon MJ, George AE, Golomb J, et al. Frequency of hippocampal formation atrophy in normal aging and Alzheimer’s disease. Neurobiol Aging 1997; 18(1): 1– 11. doi:10.1016/S0197-4580(96)00213-8. [DOI] [PubMed] [Google Scholar]
- 19. Cerami C, Della Rosa PA, Magnani G, et al. Brain metabolic maps in Mild Cognitive Impairment predict heterogeneity of progression to dementia. Neuroimage Clin 2014; 7: 187– 194. doi:10.1016/j.nicl.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zarow C, Weiner MW, Ellis WG, Chui HC. Prevalence, laterality, and comorbidity of hippocampal sclerosis in an autopsy sample. Brain Behav 2012; 2(4): 435– 442. doi:10.1002/brb3.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Faull M, Ching SY, Jarmolowicz AI, et al. Comparison of two methods for the analysis of CSF Aβ and tau in the diagnosis of Alzheimer’s disease. Am J Neurodegener Dis 2014; 3(3): 143– 151. [PMC free article] [PubMed] [Google Scholar]
- 22. Beach TG, Schneider JA, Sue LI, et al. Theoretical impact of Florbetapir (18F) amyloid imaging on diagnosis of alzheimer dementia and detection of preclinical cortical amyloid. J Neuropathol Exp Neurol 2014; 73(10): 948– 953. doi:10.1097/NEN.0000000000000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Knopman DS, DeKosky ST, Cummings JL, et al. Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001; 56(9): 1143– 1153. doi:10.1212/WNL.56.9.1143. [DOI] [PubMed] [Google Scholar]
- 24. Chui H, Zhang Q. Evaluation of dementia: a systematic study of the usefulness of the American Academy of Neurology’s practice parameters. Neurology 1997; 49(4): 925– 935. doi:10.1212/WNL.49.4.925. [DOI] [PubMed] [Google Scholar]
- 25. Apostolova LG, Dutton RA, Dinov ID, et al. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Arch Neurol 2006; 63(5): 693– 699. doi:10.1001/archneur.63.5.693. [DOI] [PubMed] [Google Scholar]
- 26. Apostolova LG, Thompson PM, Green AE, et al. 3D comparison of low, intermediate, and advanced hippocampal atrophy in MCI. Hum Brain Mapp 2010; 31(5): 786– 797. doi:10.1002/hbm.20905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jack CR, Jr, Shiung MM, Gunter JL, et al. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology 2004; 62(2): 591– 600. doi:10.1212/01.WNL.0000110315.26026.EF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Apostolova LG, Steiner CA, Akopyan GG, et al. Three-dimensional gray matter atrophy mapping in mild cognitive impairment and mild Alzheimer disease. Arch Neurol 2007; 64(10): 1489– 1495. doi:10.1001/archneur.64.10.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thompson PM, Hayashi KM, de Zubicaray G, et al. Dynamics of gray matter loss in Alzheimer’s disease. J Neurosci 2003; 23(3): 994– 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kilroy E, Apostolova L, Liu C, et al. Reliability of two-dimensional and three-dimensional pseudo-continuous arterial spin labeling perfusion MRI in elderly populations: comparison with 15O-water positron emission tomography. J Magn Reson Imaging 2014; 39(4): 931– 939. doi:10.1002/jmri.24246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Silverman DH. Brain 18F-FDG PET in the diagnosis of neurodegenerative dementias: comparison with perfusion SPECT and with clinical evaluations lacking nuclear imaging. J Nucl Med 2004; 45(4): 594– 607. [PubMed] [Google Scholar]
- 32. Silverman DH, Truong CT, Kim SK, et al. Prognostic value of regional cerebral metabolism in patients undergoing dementia evaluation: comparison to a quantifying parameter of subsequent cognitive performance and to prognostic assessment without PET. Mol Genet Metab 2003; 80(3): 350– 355. doi:10.1016/S1096-7192(03)00139-2. [DOI] [PubMed] [Google Scholar]
- 33. Johnson KA, Minoshima S, Bohnen NI, et al. Update on appropriate use criteria for amyloid PET imaging: dementia experts, mild cognitive impairment, and education. Amyloid Imaging Task Force of the Alzheimer’s Association and Society for Nuclear Medicine and Molecular Imaging. Alzheimers Dement 2013; 9(4): e106– e109. doi:10.1016/j.jalz.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 34. Johnson KA, Minoshima S, Bohnen NI, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement 2013; 9(1): e- 1–16. doi:10.2967/jnumed.113.120618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol 2003; 2(10): 605– 613. [DOI] [PubMed] [Google Scholar]
- 36. Andreasen N, Minthon L, Davidsson P, et al. Evaluation of CSF-tau and CSF-Abeta42 as diagnostic markers for Alzheimer disease in clinical practice. Arch Neurol 2001; 58(3): 373– 379. [DOI] [PubMed] [Google Scholar]
- 37. Blennow K, Wallin A, Agren H, et al. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol 1995; 26(3): 231– 245. doi:10.1016/S1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- 38. Clark CM, Xie S, Chittams J, et al. Cerebrospinal fluid tau and beta-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol 2003; 60(12): 1696– 1702. doi:10.1001/archneur.60.12.1696. [DOI] [PubMed] [Google Scholar]
- 39. Galasko D, Chang L, Motter R, et al. High cerebrospinal fluid tau and low amyloid beta42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol 1998; 55(7): 937– 945. doi:10.1001/archneur.55.7.937. [DOI] [PubMed] [Google Scholar]
- 40. Buerger K, Ewers M, Andreasen N, et al. Phosphorylated tau predicts rate of cognitive decline in MCI subjects: a comparative CSF study. Neurology 2005; 65(9): 1502– 1503. doi:10.1212/01.wnl.0000183284.92920.f2. [DOI] [PubMed] [Google Scholar]
- 41. Buerger K, Teipel SJ, Zinkowski R, et al. CSF tau protein phosphorylated at threonine 231 correlates with cognitive decline in MCI subjects. Neurology 2002; 59(4): 627– 629. doi:10.1212/WNL.59.4.627. [DOI] [PubMed] [Google Scholar]
- 42. Riemenschneider M, Lautenschlager N, Wagenpfeil S, et al. Cerebrospinal fluid tau and beta-amyloid 42 proteins identify Alzheimer disease in subjects with mild cognitive impairment. Arch Neurol 2002; 59(11): 1729– 1734. doi:10.1001/archneur.59.11.1729. [DOI] [PubMed] [Google Scholar]
- 43. Wallin AK, Blennow K, Andreasen N, Minthon L. CSF biomarkers for Alzheimer’s Disease: levels of beta-amyloid, tau, phosphorylated tau relate to clinical symptoms and survival. Dement Geriatr Cogn Disord 2006; 21(3): 131– 138. doi:10.1159/000090631. [DOI] [PubMed] [Google Scholar]
- 44. Weigand SD, Vemuri P, Wiste HJ, et al. Transforming cerebrospinal fluid Aβ42 measures into calculated Pittsburgh Compound B units of brain Aβ amyloid. Alzheimers Dement 2011; 7(2): 133– 141. doi:10.1016/j.jalz.2010.08.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997; 278(16): 1349– 1356. doi:10.1001/jama.1997.03550160069041. [PubMed] [Google Scholar]
- 46. Mayeux R, Saunders AM, Shea S, et al. Utility of the apolipoprotein E genotype in the diagnosis of Alzheimer’s disease. Alzheimer’s Disease Centers Consortium on Apolipoprotein E and Alzheimer’s Disease. N Engl J Med 1998; 338(8): 506– 511. doi:10.1056/NEJM199802193380804. [DOI] [PubMed] [Google Scholar]
- 47. Chouraki V, Seshadri S. Genetics of Alzheimer’s disease. Adv Genet 2014; 87: 245– 294. doi:10.1016/B978-0-12-800149-3.00005-6. [DOI] [PubMed] [Google Scholar]
- 48. Green RC, Cupples LA, Go R, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA 2002; 287(3): 329– 336. doi:10.1001/jama.287.3.329. [DOI] [PubMed] [Google Scholar]
- 49. Sabbagh MN, Cooper K, DeLange J, et al. Functional, global and cognitive decline correlates to accumulation of Alzheimer’s pathology in MCI and AD. Curr Alzheimer Res 2010; 7(4): 280– 286. doi:10.2174/156720510791162340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Apostolova LG, Zarow C, Biado K, et al. Relationship between hippocampal atrophy and neuropathology markers: a 7T MRI validation study of the EADC-ADNI Harmonized Hippocampal Segmentation Protocol. Alzheimers Dement 2015; 11(2): 139– 150. doi:10.1016/j.jalz.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Doody RS, Stevens JC, Beck C, et al. Practice parameter: management of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001; 56(9): 1154– 1166. doi:10.1212/WNL.56.9.1154. [DOI] [PubMed] [Google Scholar]
- 52. Matsunaga S, Kishi T, Iwata N. Memantine monotherapy for Alzheimer’s disease: a systematic review and meta-analysis. PLoS One 2015; 10(4): e0123289 doi:10.1371/journal.pone.0123289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ehret MJ, Chamberlin KW. Current practices in the treatment of Alzheimer disease: Where is the evidence after the phase III trials? Clin Ther 2015; 37(8): 1604– 1616. doi:10.1016/j.clinthera.2015.05.510. [DOI] [PubMed] [Google Scholar]
- 54. Roberts JS, Karlawish JH, Uhlmann WR, et al. Mild cognitive impairment in clinical care: a survey of American Academy of Neurology members. Neurology 2010; 75(5): 425– 431. doi:10.1212/WNL.0b013e3181eb5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kelley BJ. Treatment of mild cognitive impairment. Curr Treat Options Neurol 2015; 17(9): 372 doi: 10.1007/s11940-015-0372-3. [DOI] [PubMed] [Google Scholar]
- 56. Russ TC, Morling JR. Cholinesterase inhibitors for mild cognitive impairment. Cochrane Database Syst Rev 2012; 9: CD009132 doi:10.1002/14651858.CD009132.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schneider LS, Dagerman KS, Higgins JP, McShane R. Lack of evidence for the efficacy of memantine in mild Alzheimer disease. Arch Neurol 2011; 68(8): 991– 998. doi:10.1001/archneurol.2011.60. [DOI] [PubMed] [Google Scholar]
- 58.National Institute for Health and Clinical Excellence. Donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease. NICE technology appraisal guidance (TA217). www.nice.org.uk/guidance/ta217. Updated March 2011. Accessed February 5, 2016. [Google Scholar]
- 59. Gauthier S, Patterson C, Chertkow H, et al. Recommendations of the 4th Canadian consensus conference on the diagnosis and treatment of dementia (CCCDTD4). Can Geriatr J 2012; 15(4): 120– 126. doi:10.5770/cgj.15.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schneider LS, Tariot PN, Dagerman KS, et al. CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med 2006; 355(15): 1525– 1538. [DOI] [PubMed] [Google Scholar]
- 61. Steinberg M, Lyketsos CG. Atypical antipsychotic use in patients with dementia: Managing safety concerns. Am J Psychiatry 2012; 169(9): 900– 906. doi:10.1176/appi.ajp.2012.12030342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schneeweiss S, Setoguchi S, Brookhart A, et al. Risk of death associated with the use of conventional versus atypical antipsychotic drugs among elderly patients. CMAJ 2007; 176(5): 627– 632. doi:10.1503/cmaj.061250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Masliah E, Hansen L, Adame A, et al. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 2005; 64(1): 129– 131. doi:10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- 64. Nicoll JA, Wilkinson D, Holmes C, et al. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 2003; 9(4): 448– 452. doi:10.1038/nm840. [DOI] [PubMed] [Google Scholar]