

ABSTRACT
Purpose of Review: This article provides an overview of the clinical features, neuropathologic findings, diagnostic criteria, and management of dementia with Lewy bodies (DLB) and Parkinson disease dementia (PDD), together known as the Lewy body dementias.
Recent Findings: DLB and PDD are common, clinically similar syndromes that share characteristic neuropathologic changes, including deposition of α-synuclein in Lewy bodies and neurites and loss of tegmental dopamine cell populations and basal forebrain cholinergic populations, often with a variable degree of coexisting Alzheimer pathology. The clinical constellations of DLB and PDD include progressive cognitive impairment associated with parkinsonism, visual hallucinations, and fluctuations of attention and wakefulness. Current clinical diagnostic criteria emphasize these features and also weigh evidence for dopamine cell loss measured with single-photon emission computed tomography (SPECT) imaging and for rapid eye movement (REM) sleep behavior disorder, a risk factor for the synucleinopathies. The timing of dementia relative to parkinsonism is the major clinical distinction between DLB and PDD, with dementia arising in the setting of well-established idiopathic Parkinson disease (after at least 1 year of motor symptoms) denoting PDD, while earlier cognitive impairment relative to parkinsonism denotes DLB. The distinction between these syndromes continues to be an active research question. Treatment for these illnesses remains symptomatic and relies on both pharmacologic and nonpharmacologic strategies.
Summary: DLB and PDD are important and common dementia syndromes that overlap in their clinical features, neuropathology, and management. They are believed to exist on a spectrum of Lewy body disease, and some controversy persists in their differentiation. Given the need to optimize cognition, extrapyramidal function, and psychiatric health, management can be complex and should be systematic.
INTRODUCTION
In 1912, Frederick Lewy first described the cytoplasmic inclusions now known as Lewy bodies in the substantia nigra in Parkinson disease (PD).1 Cortical Lewy bodies were first reported in association with dementia in 1961,2 but they were felt to be a relatively rare finding until the 1980s, when first ubiquitin and later α-synuclein immunostains made it easier to see them3 and demonstrated that Lewy bodies were a common neuropathologic finding in dementia, second only to Alzheimer disease (AD). Lewy body–related pathology is observed in dementia with Lewy bodies (DLB), idiopathic PD, and multiple system atrophy (MSA), and DLB and the dementia that arises in PD (ie, Parkinson disease dementia [PDD]) together comprise the Lewy body dementias. The clinical features of DLB and PDD are similar and include hallucinations, cognitive fluctuations, and dementia in the setting of the extrapyramidal motor impairments known as parkinsonism. The cognitive domains that are impacted in DLB and PDD overlap substantially, with prominent executive dysfunction and visual-spatial abnormalities and variable impairment in memory capacities.4 In DLB, dementia often heralds the onset of illness in advance of parkinsonian motor signs, but by consensus may follow their development up to 1 year from their onset.5 In contrast, a diagnosis of PDD is made when cognitive impairments develop in the setting of well-established PD.6
Despite the different temporal sequences of motor and cognitive deficits, PDD and DLB show remarkably convergent neuropathologic changes at autopsy. These changes include widespread limbic and cortical Lewy bodies7 and Lewy neurites composed of aggregates of α-synuclein that involve the brainstem as well as limbic and neocortical regions (referred to as Lewy body disease), loss of midbrain dopamine cells,8 and loss of cholinergic neurons in ventral forebrain nuclei.9 Neuritic plaques that contain amyloid and neurofibrillary tangles are found in the majority of cases of DLB and are common in PD.10 Current neuropathologic criteria of Lewy body disease weigh α-synuclein pathology against AD neurofibrillary tangle pathology to estimate the probability that Lewy body disease caused the clinical syndrome in life.5 It is notable that Lewy body disease at autopsy does not successfully predict whether patients had DLB or PDD syndromes in life. The overlap of clinical, neuropsychological, and neuropathologic features has led to the hypothesis that PDD and DLB may be different phenotypic expressions of the same underlying process.11,12 This hypothesis implies that future disease-modifying therapies will be effective in both diseases.
CLINICAL FEATURES AND DIAGNOSTIC EVALUATION OF DEMENTIA WITH LEWY BODIES
DLB is associated with a stereotyped set of clinical features.
Cognitive Symptoms
The typical patient with DLB presents with early dementia, often in association with visual hallucinations. Extrapyramidal motor symptoms and signs characteristic of PD often develop simultaneously or soon thereafter. Progressive cognitive decline begins early, typically after age 55. It is useful to identify the first cognitive domains impaired, as these can point toward DLB. Although short-term memory may be involved, cognitive domains other than memory are frequently affected as well, including attention, executive function, and visual-spatial skill. Patients may therefore report early difficulty multitasking at work or home and may start to lose the thread of conversations. In addition, patients may occasionally get lost while driving or grow increasingly dependent on global positioning system (GPS) devices. Short-term memory loss can be significant as well. While reminiscent of the impairment of hippocampal-dependent memory encoding seen in AD, in many patients with DLB, the impairment of short-term memory reflects instead a problem of retrieval of stored information, which can be improved with cues. Errors of memory encoding and retrieval can be differentiated on detailed cognitive testing (see the following section on diagnostic criteria for DLB). Over time, patients’ cognitive impairments progress and spread to involve other cognitive domains. When they are sufficiently severe to impair social or occupational function (impacting instrumental or basic activities of daily living), they reach criteria for a diagnosis of dementia.5
Neuropsychiatric Symptoms
Recurrent, complex visual hallucinations are common in patients with DLB, and their early presence in a dementia syndrome is diagnostically useful. These hallucinations are usually well formed and animate, and it is common for these hallucinations to include adults or small children, deceased family members, and small animals. Early in the course of the illness, hallucinations are usually unimodal, without sound, smell, or touch. They are frequently well-tolerated and emotionally neutral, but occasionally can be dysphoric or fear provoking. These hallucinations are distinct from visual illusions, in which an object is visually misinterpreted (eg, a corner lamp that is misinterpreted as a person). Such illusions are common early in the illness as well, particularly at night in dimly lit environments, but are nonspecific.
Delusions can also arise in patients with DLB, typically later in the course, and usually have a paranoid quality. Delusions of infidelity, house intruders, and theft are common, the latter often occurring as patients misplace items around the home. As cognition continues to deteriorate, patients may believe that their spouse or other caregiver has been replaced by an imposter, a phenomenon known as Capgras syndrome. One hypothesis for this phenomenon is the loss of valence (ie, emotional) associations for a memory, such that a familiar face, for example, loses its ability to retrieve emotional associations.
Fluctuations of Attention and Arousal
Attention and alertness may fluctuate, leading to episodes of staring and perturbed flow of ideas, or to frequent daytime drowsiness and naps during the day. These episodes can be hard to quantify and need to be disentangled from toxic metabolic processes such as medication side effects or infections. A recent fluctuations scale vetted for this purpose is the Dementia Cognitive Fluctuation Scale,13 which aggregates prior scales. The fluctuations screen requires a positive response to at least three of the following: (1) Does the patient’s inability to organize thoughts in a coherent way vary significantly over the course of the day? (2) Does the patient spend more than 1 hour sleeping during the waking day? (3) Is the patient drowsy and lethargic for more than 1 hour during the day, despite getting the usual amount of sleep the night before? (4) Is the patient difficult to arouse on a usual day? This approach had a sensitivity of 80% and a specificity of 76% in differentiating clinical syndromes of DLB and PDD from AD and vascular dementia, but has yet to be neuropathologically validated.
Motor Features of Parkinsonism
Parkinsonian motor signs often develop concurrently with or subsequent to these problems and are also diagnostically very useful. These motor signs are often symmetric, and bradykinesia and gait impairment are more common than rest tremor. However, the variance of the motor presentation is high. Some patients may present with a classic asymmetric pill-rolling tremor of PD while others may have no motor concerns yet will display clear extrapyramidal dysfunction on examination. In contrast to patients with PD, who have a sustained beneficial response to PD medications such as carbidopa/levodopa, patients with DLB often have a limited response to such medications. These patients nonetheless show reduced dopamine transporter (DAT) activity on single-photon emission computed tomography (SPECT) or positron emission tomography (PET) imaging, when performed. Generalized myoclonus can also occur in some patients with DLB.
Neuroleptic Sensitivity
In part as a result of dopamine cell loss, patients with DLB are particularly sensitive to neuroleptics. Such agents can trigger or exacerbate parkinsonism, as they can in PD, and this may be irreversible. In addition, neuroleptics have been associated with increased mortality, and patients with DLB are at increased risk for neuroleptic malignant syndrome. Neuroleptics can also affect cognition and impair attention and alertness. This issue of neuroleptic sensitivity is clinically important, as many patients will at some time be evaluated in an emergency department for psychosis or confusion, where haloperidol and other neuroleptics are dispensed liberally. As such, it is worthwhile to teach patients and their caregivers that patients with DLB are essentially “allergic” to haloperidol and other neuroleptics with significant D2 receptor antagonism.
Other Associated Symptoms
As in PD (see the following section on preclinical synucleinopathies), rapid eye movement (REM) sleep behavior disorder, loss of olfaction, and constipation are common and may antedate the illness by several years.14 Epidemiologic data suggest that these problems are risk factors for all of the synucleinopathies (PD, DLB, and MSA). In addition, many patients with DLB will report a chronic, high sensitivity to medications in general. It is unclear how or whether this relates to the underlying disease process.
Rapid Eye Movement Sleep Behavior Disorder
REM sleep behavior disorder refers to a syndrome in which the normal paralysis of REM sleep is impaired. As a result, patients’ bed partners may report that they act out their dreams with behaviors such as kicking, punching, and yelling. The observation that most REM sleep behavior disorder behaviors are violent suggests that the impairment of paralysis may be relative, with a reduction in threshold that is overcome by only the most emotionally salient dreams, perhaps on the basis of catecholamine or amygdala drive.
Autonomic Impairment
Autonomic impairment is common in DLB but is not as profound as in MSA. Constipation is common in both and can be problematic if not treated aggressively. Some patients also experience orthostatic hypotension and its complications, particularly syncope and falls. This is more common later in the course of DLB and can be exacerbated by medications. Denervation of cardiac sympathetic ganglia is widespread and can be appreciated using metaiodobenzylguanidine (MIBG) cardiac scans.15 In addition, some patients experience neurogenic urinary frequency or incontinence.
DIAGNOSTIC CRITERIA FOR DEMENTIA WITH LEWY BODIES
The consensus criteria for a clinical diagnosis of DLB reflect the clinical features described previously in this article (Table 4-1). Progressive cognitive decline to dementia is required, often involving attention, executive function, and visual-spatial skills. The core features of these criteria include the following: (1) recurrent visual hallucinations that are well formed and detailed; (2) fluctuations in attention and alertness; and (3) parkinsonian motor signs. Supportive features, also common in PD, include the presence of REM sleep behavior disorder, severe neuroleptic sensitivity, or low DAT uptake in the basal ganglia on SPECT or PET. A diagnosis of clinically probable DLB requires at least two out of three of the core features to be present or one core feature and one supportive feature. A diagnosis of clinically possible DLB requires only one of the three core features to be present.
Table 4-1.
Diagnostic Criteria for the Clinical Diagnosis of Dementia With Lewy Bodiesa

Although the specificity of these criteria for a diagnosis of DLB is high (estimated to range from 79% to 100%), the sensitivity can be low (12% to 88%), improving with the addition of the supportive features.16 These data suggest that we are missing patients with DLB in our clinics. Thus, further refinement of these criteria is needed.
As touched upon previously in this article, the clinical features of DLB can overlap with those of AD. For example, short-term memory loss can occur in both dementias. However, impairment of short-term memory is usually the dominant and earliest feature in AD, where it reflects an error of encoding, due to hippocampal-dependent impairment. In contrast, the pattern of memory loss is more variable in DLB, instead reflecting an error of retrieval in some patients. On cognitive testing, controlling for severity of dementia, patients with DLB are often more impaired than those with AD in tests of attention, executive function, and visual-spatial skills.5 However, late in the course of DLB and AD, the profiles of cognitive impairment may converge. Although hallucinations and parkinsonism can occur late in the course of AD, neither is common and pervasive in AD, and their early presence should point toward DLB. Fluctuations of awareness or attention are unusual in early AD except when due to a toxic-metabolic process, but daytime sleepiness often increases with increasing dementia severity.
It is useful to keep in mind that parkinsonism and cognitive impairment can also arise in the parkinsonian tauopathy syndromes, progressive supranuclear palsy (PSP), and corticobasal syndrome (CBS). The specific constellation of cognitive and motor impairments differentiates these clinical presentations from DLB and PDD (Case 4-1). These conditions are briefly discussed in the following sections. For a more comprehensive discussion of PSP and CBS, refer to the article “Frontotemporal Dementias” by Elizabeth C. Finger, MD, FRCPC,17 in this issue of Continuum.
Case 4–1
A 63-year-old man presented for a neurologic evaluation for progressive cognitive and motor symptoms. He then developed a shuffling gait, kyphosis, and a postural tremor, and noted increasing difficulty with buttons. He was involved in a minor motor vehicle accident 1 year later. Short-term memory loss was first noted at this time and was insidiously progressive. He developed nonthreatening visual hallucinations of small children, particularly in the evenings. His performance as a computer technician declined, prompting retirement. His wife took over his medications and the finances at that time. He was often somnolent and difficult to arouse during the day, regularly sleeping for at least 2 hours. He grew apathetic and less engaged in conversations. He gave up most of his household chores due to their cognitive rather than physical demands, but he still enjoyed driving around town on overlearned routes. His wife reported that he had been episodically acting out violent dreams over the last few years but otherwise slept well. He had stopped driving prior to this evaluation. On examination, he had a Montreal Cognitive Assessment (MoCA) score of 21, with 5 out of 5 errors of 5-minute recall, improving to 3 out of 5 errors with cues, and 1 error of concentration. Spatial testing showed markedly distorted figure copy and clock. Bradyphrenia was prominent. Praxis and language were normal. Vertical gaze was preserved. He had a masked facies, and tone was increased in the neck and arms. Fast repetitive movements were slow in the arms, where a symmetric postural tremor was noted. Gait was slow, with symmetric, reduced arm swing, narrowed stride, and en bloc turns. He was slightly unstable on the pull test. The patient’s blood testing was normal. MRI showed minimal global atrophy, and fluorodeoxyglucose positron emission tomography (FDG-PET) revealed temporal, parietal, and occipital hypometabolism. Cognitive testing confirmed the cognitive profile observed on examination. Given these results, he was diagnosed with probable dementia with Lewy bodies (DLB). Donepezil was started and was associated with significantly improved cognitive function, along with resolution of his hallucinations. Carbidopa/levodopa 25/100 times a day was subsequently initiated and associated with modest improvement of bradykinesia and gait. Physical therapy, occupational therapy, and a home safety evaluation were helpful. His possible rapid eye movement (REM) sleep behavior disorder was deemed mild and left untreated. Visual hallucinations returned after about 6 months as cognition began to again deteriorate but were nonthreatening and well tolerated.
Comment. This case illustrates an uncomplicated case of DLB. The constellation of early parkinsonism and hallucinations in the setting of cognitive impairment sufficiently severe to interfere with activities of daily living supports the clinical diagnosis of probable DLB. Both cognitive and motor impairments can impact driving safety in patients with DLB. There are no Lewy body dementia–specific guidelines for driving. Driving issues in patients with dementia are discussed further in the Patient Management Problem by Elizabeth C. Finger, MD,17 in this issue of Continuum.
CLINICAL FEATURES AND DIAGNOSTIC EVALUATION OF PARKINSON DISEASE DEMENTIA
Cognitive and neuropsychiatric impairments in PDD are common and are similar in quality to those of DLB.
Mild Cognitive Impairment and Dementia in Parkinson Disease
Contrary to Dr James Parkinson’s introduction to “An Essay on the Shaking Palsy,”18 the intellect is not “uninjured” in PD. Although patients with PD first come to medical attention because of characteristic motor signs, including rest tremor, rigidity, bradykinesia, and gait abnormality, specific cognitive impairments in executive function, visual-spatial skill, and even memory function in patients with PD are common and have been known for more than 40 years.19 PD hastens deterioration of these cognitive abilities over time, with the incidence and prevalence of cognitive impairments increasing with duration and severity of illness.20,21 In this sense, PD can be considered a risk factor for dementia. Formal criteria have been developed for mild cognitive impairment (MCI) in PD (Table 4-2).22,23 These criteria attempt to account for the contribution of motor impairment to functional decline and are now being validated.
Table 4-2.
Movement Disorder Society Task Force Guidelines for Diagnostic Criteria of Mild Cognitive Impairment in Parkinson Diseasea

Dementia in PD is common, with prevalence rates as high as 78%24 and incidence rates ranging from 3% to 10% per year, approximately threefold higher than in the normal population.25–27 Dementia in PD is associated with high morbidity and mortality, antedating death by approximately 4 years on average.28 The risk of developing cognitive impairment and dementia in PD has been associated with older age, greater severity of extrapyramidal motor impairment, and longer duration of illness.20,24,25 Additional risk factors have been identified as well, including male gender, atypical motor syndromes (notably the postural instability gait disorder (PIGD) variant, axial symmetrical parkinsonism, and akinetic dominant parkinsonism), and the early development of hallucinations (Table 4-3).24,27
Table 4-3.
Risk Factors for Cognitive Impairment in Parkinson Disease

The cognitive profile in PD dementia overlaps significantly with that observed in DLB.4 Patients typically demonstrate executive dysfunction and visual-spatial impairment. Caregivers will note new errors with the patient’s (usually complex) medication regimen and will often need to intervene. Errors with the finances can be significant. Attention is often impaired and may fluctuate. In fact, late in the course of PD in some patients, inattention may cycle as a peak-dose phenomenon, phase-locked to dopamine replacement dosing. Although naming is often impaired to a variable degree, frank language impairments are not present. As in DLB, free recall is often impaired but tends to improve with cues, suggesting an error of recall rather than encoding. Lastly, bradyphrenia (slowed thinking) may be significant.
Visual hallucinations are common in PDD. Like those of DLB, these are often animate and unimodal and only occasionally dysphoric or fear provoking. Delusions are less common but can arise as well. Both hallucinations and delusions can be precipitated or exacerbated by dopamine replacement medications, and dopamine agonists are particularly notorious. Reducing these agents or shifting from one class of agent to another (eg, from dopamine agonist to carbidopa/levodopa) can often dramatically improve these problems.
The differential diagnosis for cognitive impairment in PD includes toxic metabolic processes in general and PD medications specifically. Excessive dopamine replacement can worsen executive dysfunction and attention and precipitate or exacerbate hallucinations or delusions. Although this is true of all agents, dopamine agonists are notorious offenders, and amantadine can be problematic in some patients. Carbidopa/levodopa is best tolerated in this regard, but at sufficiently high dose, carbidopa/levodopa can also exacerbate cognitive impairments and precipitate psychosis. Of note, the central anticholinergic agent trihexyphenidyl, used to treat PD tremor, can be particularly deleterious to cognition.
DIAGNOSTIC CRITERIA FOR PARKINSON DISEASE DEMENTIA
Consensus criteria for PDD were developed in 2007 (Table 4-4).6,29 These criteria require cognitive impairments across multiple domains but emphasize that noncognitive features such as hallucinations are common. As described previously in the article, the clinical and neuropsychological features of DLB and PDD are similar. Indeed, it is the relative timing of dementia and parkinsonism that defines the clinical distinction between DLB and PDD. Controversy exists over how or whether to distinguish these syndromes.30
Table 4-4.
Consensus Criteria for a Clinical Diagnosis of Parkinson Disease Dementiaa

CLINICAL STUDIES
Patients with suspected DLB or PDD should receive a standard evaluation for cognitive impairment, including blood testing to exclude reversible contributions to cognitive impairment such as a thyroid disorder or vitamin B12 deficiency; a brain MRI scan; and detailed cognitive testing. The MRI scan is nondiagnostic in DLB and PDD, although some degree of global, symmetric atrophy may be appreciated.31 The finding of marked, disproportionate hippocampal atrophy would point toward AD, while focal cortical atrophy may point toward CBS (see the following section on differential diagnosis). Detailed cognitive testing (if well performed) can provide a valuable assessment of the patient’s function across multiple cognitive domains. This is often diagnostically useful and also provides a baseline for future comparisons. In patients with profound fluctuations of attention or arousal, an EEG can exclude seizure activity. In DLB, the EEG often shows diffuse slowing in the theta or delta range.
In a subset of patients with DLB, fluorodeoxyglucose positron emission tomography (FDG-PET) or cerebral blood flow SPECT can also be informative. These often show a characteristic pattern of symmetric hypometabolism involving not just the parietal and temporal regions, as seen in AD, but also the occipital lobes.31 However, in some patients, only the AD pattern of hypometabolism may be present. FDG-PET appears to be more sensitive than SPECT, with a sensitivity of 83% to 92% and a specificity of 67% to 93%.32 Clinical context is important in interpreting the FDG-PET scan, as occipital hypometabolism has also been described in cognitively normal PD,33 in PDD,33,34 and in posterior cortical atrophy.35
On SPECT or PET imaging, reduced DAT levels are also observed in DLB and PDD.31 Because this has high sensitivity (78% to 88%) and specificity (90% to 100%) to differentiate DLB from AD, reduction in DAT levels is one of the suggestive diagnostic features of DLB. However, it is important to note that DAT imaging is also abnormal in CBD and PSP.
CSF assessment is increasingly used in the workup of dementia patients, as the pattern of CSF amyloid-β (Aβ) and tau has high sensitivity and specificity for AD.36 Because of the frequent coexistence of Alzheimer pathology in DLB, however, the AD CSF pattern does not exclude DLB. Limited data exist for CSF evaluation in DLB, except as a research tool, where molecules such as α-synuclein are under study and have been found to be reduced in DLB (see the following section on trends for more information).
Differential diagnosis OF THE LEWY BODY DEMENTIAS
Few clinically useful biomarkers differentiate DLB and PD from MSA and the parkinsonian tauopathies PSP and CBD, and careful history and examination remain the method of choice. Although unusual, cognitive impairment and dementia have recently been described in MSA37,38 and can no longer be used as strong evidence against the diagnosis. The early and profound development of dysautonomia, in association with parkinsonism and/or cerebellar ataxia characterizes MSA39 and can help in its differentiation from DLB and PDD. When present, ataxia is a strong distinguishing feature of MSA. Conversely, the presence of visual hallucinations and fluctuations would argue in favor of DLB or PDD. Late in the course of MSA, cerebellar atrophy and the hot cross bun pons sign may be appreciated on MRI.
While it can be straightforward to differentiate Richardson syndrome, the most common clinical variant of PSP, from PD and DLB in advanced disease, differentiation can be challenging early in the disease. Executive dysfunction and extrapyramidal motor symptoms/signs characteristic of PD (parkinsonism) are common in both PSP and DLB (as well as in PDD). While many patients with DLB have symmetric or axial predominant parkinsonism, axial predominant features are the rule in Richardson syndrome, in association with a lordotic posture rather than the kyphotic posture common to DLB and PD. Other clinically useful features of Richardson syndrome include a specific impairment of vertical gaze, including downgaze, which is preserved in DLB and PD; a frequent facial expression of fear or surprise uncommon in DLB or PD; and a propensity for falls backward early in the course of the illness. While falls are not uncommon in DLB and PD, falls backward are unusual. Late in the course of PSP, midbrain atrophy may be appreciated on midsagittal T1 sequence MRI, revealing the brainstem hummingbird sign.
CBS refers to a classically asymmetric neurodegenerative syndrome of parkinsonism or dystonia, accompanied by asymmetric cortical signs, such as apraxia or cortical sensation loss. Differentiation from DLB and PD is made on the basis of the marked asymmetry and presence of both cortical and extrapyramidal features in CBS. Multiple neuropathologies can underlie the syndrome, with corticobasal degeneration (CBD) accounting for approximately 50% of cases. CBS is often associated with hypometabolism on FDG-PET around the central sulcus and the ipsilateral striatum. Late in the course, focal cortical atrophy may be appreciated in the primary motor and primary sensory cortices. For more information on PSP and CBS, refer to article “Frontotemporal Dementias” by Elizabeth C. Finger, MD, FRCPC,17 in this issue of Continuum.
In addition, hallucinations are uncommon in MSA, PSP, and CBS, as are fluctuations of attention and arousal. The presence of these problems should direct the clinician toward DLB and PDD. REM sleep behavior disorder has been described in both PSP and CBD but is more common in the synucleinopathies (including MSA). In contrast to PD, motor impairments in MSA, PSP, and CBD are rarely responsive to dopamine replacement (Case 4-2).
Case 4–2
A 65-year-old man developed shuffling of his feet and a left rest tremor, associated with impaired fine manual coordination. On examination, masked facies, hypophonia, and left predominant rest tremor, rigidity, and bradykinesia were noted. The patient’s gait was parkinsonian. He was started on ropinirole and had a marked improvement of function. He was diagnosed with idiopathic Parkinson disease (PD), and rasagiline was added. As his disease progressed, ropinirole was gradually increased. Physical, occupational, and speech therapy provided additional benefit. For mild depression and anxiety, he was treated with escitalopram. He subsequently developed rapid eye movement (REM) sleep behavior disorder, which was managed with lorazepam. Six years into his illness, he developed the hallucination of a stranger in the living room and began having trouble maintaining his complex medication regimen. On examination, he was markedly inattentive and encephalopathic and moderately dyskinetic. Ropinirole was replaced with carbidopa/levodopa, melatonin was subsequently substituted for lorazepam, and his wife took over his medication regimen. In this context, hallucinations, confusion, and dyskinesias markedly improved. He resumed his golf game and bridge, but he underperformed at both compared to baseline. Six months later, his hallucinations returned. Although attention was improved, he still demonstrated mild executive dysfunction and visual spatial impairment. A metabolic panel including thyroid-stimulating hormone (TSH) and vitamin B12 level was normal. Brain MRI showed mild generalized atrophy without evidence for medial temporal lobe atrophy. Donepezil was started and associated with robust improvement of cognition and reduced frequency of hallucinations. Over the next 3 years, the patient’s cognition gradually deteriorated. He gave up his hobbies and grew increasingly reliant on his wife to coordinate their plans. He required help to get dressed due to the cognitive demands of the task. He was diagnosed with Parkinson disease dementia (PDD). Memantine was added but was not found to be helpful and was ultimately discontinued.
One night, he developed escalating agitation and concern about intruders in the house. He was brought to an outside emergency department, where he was paranoid and anxious. Mental status testing was notable for disorientation to date, reduced short-term memory, and visual spatial impairment. Workup was negative for a toxic metabolic process. ECG confirmed a normal QTc. Haloperidol was considered by the emergency department staff but the neurology consultant intervened. After discussion with family about the risks and benefits of starting an atypical antipsychotic agent, quetiapine was started and slowly uptitrated, and his paranoia improved. He was discharged home with resolution of the delusions and with neurology follow-up.
Comment. This case illustrates the challenges of managing patients with advanced PD and the common manifestations of cognitive impairment and psychosis in this setting. The need to avoid central D2 receptor antagonists applies to both dementia with Lewy bodies and PD. Given their risks, use of atypical antipsychotics should be minimized.
GENETICS OF THE LEWY BODY DEMENTIAS
A number of genetic mutations have been associated with DLB and PDD, which are interesting clinically but also hold promise to elucidate fundamental mechanisms of disease. Some genetic errors appear to be dose dependent. For example, mutation or duplication of α-synuclein causes autosomal dominant PD, but triplication is often associated with both parkinsonism and dementia.40 Several other genes also confer risk for DLB and PDD. The most prominent of these is GBA, the gene encoding glucocerebrosidase.41,42 While double mutations of GBA cause Gaucher disease (which is autosomal recessive), single GBA mutations are associated with DLB as well as with a variant of PD that carries an increased risk of cognitive impairment. Not all PD-related genes confer such risk, however. For example, the LRRK2 mutation causes autosomal dominant PD without cognitive impairment.43 In addition, a small number of genes have been identified that carry risk for DLB but not PDD, including the apolipoprotein E (APOE) ɛ4 allele.44 Mutations in the MAPT gene, which have been associated with the tauopathies such as FTD with parkinsonism, and in the COMT gene, have also been variably observed in PDD.45 In patients with a strong family history, genetic counseling should be provided and genetic studies should be considered.
SYMPTOMATIC TREATMENT IN DEMENTIA WITH LEWY BODIES AND PARKINSON DISEASE DEMENTIA
This section discusses several therapeutic strategies for the problems that arise in DLB and PDD, and Table 4-546 provides a comprehensive list. However, few of these agents have been evaluated for their efficacy in clinical trials, and such studies remain an important need.
Table 4-5.
Symptomatic Treatments in Dementia With Lewy Bodies and Parkinson Disease Dementiaa
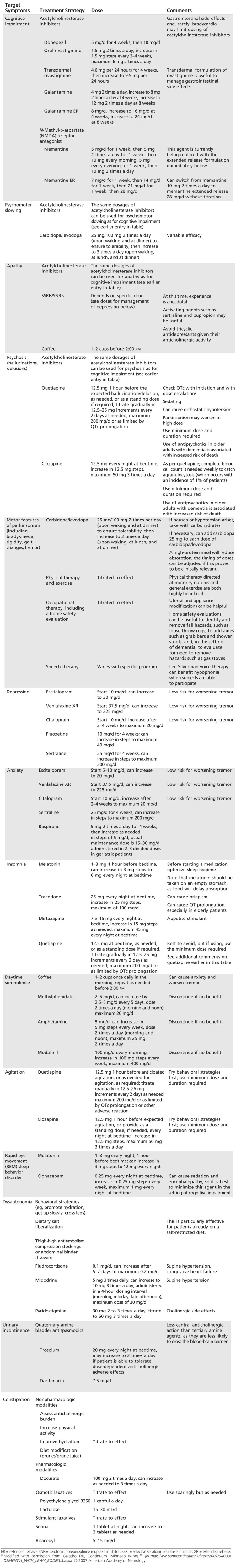
A useful first step is to streamline the medication list to remove possible offending agents and drug interactions (Table 4-6). In general, there is value in making single changes systematically and serially, starting at low dose, tackling the most severe problem first. This simple strategy accounts for the frequent sensitivity to medications observed in DLB and allows for straightforward interpretation of the effects of manipulations. In the process of treating multiple problems, patients are at risk for complications of polypharmacy, and agents should be selected cautiously.
Table 4-6.
Agents to Avoid in Patients With Dementia With Lewy Bodies and Parkinson Disease Dementia

Cognitive Impairment
The marked loss of acetylcholine neurons in DLB and PDD is the basis for the use of acetylcholinesterase inhibitors in these illnesses. In placebo-controlled clinical trials, donepezil and rivastigmine have been demonstrated to be effective in treating cognitive impairment in DLB and PDD, respectively.47 In some patients, the benefit can be marked and unambiguous and may be associated with improvement of hallucinations or delusions as well.
There is little evidence to suggest that acetylcholinesterase inhibitors differ in their efficacy. However, they do vary in their probability of common adverse reactions. Most of these are dose related, and nausea is particularly common. Because such side effects most often occur as a peak-dose phenomenon, they may resolve with transition to a transdermal formulation (rivastigmine transdermal system, for example), where the peak dose is reduced. Another important side effect to consider in the appropriate patient is bradycardia. Parkinsonism is not usually affected, although a minority may experience worsened tremor. A large multicenter clinical trial in 2010 suggested greater efficacy, and higher adverse reaction rate, for high-dose donepezil (23 mg/d) compared to standard dose (10 mg/d) in moderate to severe AD.48 This study has opened the door to higher dosing in DLB and PDD, and high-dose studies of acetylcholinesterase inhibitors in DLB and PDD are needed.
In small studies, memantine has also been found to be modestly effective in DLB and PDD.47 Larger studies have not yet been performed to confirm these results. Many patients note little subjective benefit from this agent, but a small subpopulation of patients may report significant improvement.
Many patients with PDD develop their cognitive impairments in the setting of a complex medication regimen tailored for patients with PD with moderate to severe motor disease. When necessary, the cautious withdrawal of trihexyphenidyl or dopamine agonists, transition to carbidopa/levodopa, and, if needed, general dose reduction of dopamine replacement, may improve cognition in these patients. In part due to the complexity of the regimen, medication errors are a common contributor to cognitive impairment or psychosis in this setting. Supervision of medications can be helpful in both DLB and PDD.
Psychosis
Medication review is also critical in managing psychosis. For example, in PDD, the cautious withdrawal of dopamine replacement agents, or transition to carbidopa/levodopa, may improve psychosis.
Hallucinations that are not aversive do not require medical treatment. Acetylcholinesterase inhibitors are the first line of defense for nonemergency visual hallucinations and delusions. They can be very effective for this purpose, and they lack the cardiac risk of the neuroleptics. The basis for their benefit is unclear but suggests that lack of acetylcholine receptor activation, possibly in the ventral visual stream, contributes to these psychotic features.
When atypical antipsychotic agents are needed, quetiapine and clozapine have been found to be least likely to exacerbate parkinsonism49 or to cause neuroleptic malignant syndrome. These agents should be started at a low dose and slowly titrated to the minimum dose required. Given the increased risk for death in patients with dementia treated with antipsychotic agents, primarily due to cardiac arrest, congestive heart failure, and pneumonia,50 and the frequent presence of dysautonomia in DLB, the QTc should be monitored with a baseline ECG and with follow-up ECGs for significant dose escalation, and the dose and duration of treatment should be minimized. Furthermore, the risks and benefits should be discussed frankly with the patient and caregiver. Clozapine is often less sedating than quetiapine. However, due to the low but significant risk for agranulocytosis, clozapine use requires weekly complete blood cell count monitoring.
Parkinsonism
In DLB, a trial of carbidopa/levodopa (25 mg/100 mg 2 or 3 times a day) can improve motor features in some patients without worsening cognition or psychosis. However, it tends to be much less effective in DLB than in idiopathic PD. Should cognition or hallucinations worsen, this agent can be reduced or discontinued, if necessary. Patients with DLB and PDD benefit from physical therapy, which can provide gait assistance and focus on particular motor impairments such as focal hand bradykinesia. Occupational therapy can be helpful as well, providing tools to help with feeding and other basic functions. A home safety evaluation is useful to guide caregivers, for example, in the removal of throw rugs and the addition of safety bars.
Rapid Eye Movement Sleep Behavior Disorder
REM sleep behavior disorder does not require medical treatment if the patient is not harming himself or his caregiver. If treatment is required, a number of nonpharmacologic steps may be useful. These include removing sharp objects from the sleep environment, adding soft bedding to the floor next to the patient, and using separate beds. Several medications can be effective in REM sleep behavior disorder. Benzodiazepines are particularly effective, but these carry the risk of exacerbating confusion. Melatonin can be effective as well and is usually well tolerated. Some patients with REM sleep behavior disorder have concomitant obstructive sleep apnea, and the use of positive airway pressure may resolve both obstructive sleep apnea and REM sleep behavior disorder.
TRENDS
Recent advances in our understanding of DLB and PD are likely to impact future diagnosis and management of these diseases. These advances include the concept of preclinical features, the search for diagnostic features of MCI predictive of future DLB, and efforts to determine the causes of dementia in these illnesses.
Preclinical Synucleinopathies
Several preclinical features antedate cognitive, motor, and neuropsychiatric impairments in DLB, PD, and MSA, including constipation, REM sleep behavior disorder, and olfactory loss. These features support the premise that the synucleinopathies can be identified at a preclinical stage, but they do not clearly differentiate between them. These impairments are attributed to ascending α-synuclein pathology, corresponding respectively with Lewy bodies identified in the enteric plexus, in brainstem sleep centers, and in the olfactory bulb, as suggested by cross-sectional neuropathologic studies.14,51 Efforts are now underway to improve screening to identify at-risk patients. Future neuroprotective strategies will likely take advantage of these and related preclinical features.
Mild Cognitive Impairment Preceding Dementia With Lewy Bodies
Patients with the clinical features of DLB but who remain independent for their instrumental and basic activities of daily living meet criteria for Lewy body spectrum MCI.14 The sensitivity and specificity for a diagnosis of Lewy body–MCI (LB-MCI) are likely to be lower than for DLB, in part due to milder manifestations of the core criteria. Ancillary testing has yet to be validated in LB-MCI. For example, the prevalence of occipital hypometabolism appears to be reduced in LB-MCI compared with DLB. In addition, the sensitivity of the DAT scan may be reduced when extrapyramidal symptoms are mild. Like preclinical DLB, LB-MCI is a useful construct for therapeutic clinical trials and for biomarker studies.
Mechanisms for Dementia and Disease-Modifying Treatment Trials
Multiple pathologic processes have been linked to cognitive impairment and psychosis in DLB and PDD, including α-synuclein deposition with secondary synapse impairment,7,52,53 amyloid burden,10,54 and dopamine55 and acetycholine9 cell loss (Table 4-7).52–60 The difference in the timing of cognitive and motor impairments in DLB and PDD likely reflects a difference in the temporal sequence of these pathologies. One possibility is that in DLB, cortical lesions, mostly β-amyloid, arise early, driving cognitive impairment. Then, α-synuclein pathology ascends from brainstem to cortex. In contrast, in PDD, cortical lesions arise late, and ascending α-synuclein pathology drives the clinical syndrome. Amyloid PET imaging in DLB and PDD supports this model, showing high amyloid burden in most cases of DLB, with more modest accumulation in PDD.54 Antibodies targeting β-amyloid have entered clinical trials in AD and MCI.61 Although the outcomes are uncertain, the strategy is applicable to DLB and possibly to PDD, where amyloid accumulation appears to contribute to certain clinical features, including the timing and rate of cognitive decline.54 A similar immune targeting approach is under development for α-synuclein. If successful, this strategy would be applicable to both DLB and PD, irrespective of cognitive impairment.
Table 4-7.
Putative Brain Substrates for Major Clinical Features of Dementia with Lewy Bodies and Parkinson Disease Dementia

CONCLUSION
DLB and PDD are clinically and neuropathologically similar illnesses distinguished on the basis of the relative timing of dementia and parkinsonism. The core features of these illnesses include dementia, parkinsonism, hallucinations, and fluctuations of attention or arousal. The deposition of α-synuclein is central to both of these illnesses. Additional neuropathologic changes such as dopamine and acetylcholine cell loss are likely secondary. Superimposed AD-associated neuropathologic changes are common in DLB and PDD and appear to be synergistic. Treatment strategies targeting specific clinical impairments in DLB and PDD need to be carefully selected to avoid worsening other domains of impairment. Disease-modifying therapies remain a major unmet need.
USEFUL WEBSITES
Lewy Body Dementia Association. The Lewy Body Dementia Association is a nonprofit organization that works to support individuals diagnosed with Lewy body dementia and raise awareness about the disease through scientific research.
National Parkinson Foundation. The National Parkinson Foundation supports the care of individuals with Parkinson disease through its commitment to expert research and education about the disease.
American Parkinson Disease Association. The American Parkinson Disease Association funds research and promotes patient care and education, as well as working to promote public awareness about the condition.
Michael J. Fox Foundation for Parkinson’s Research. The Michael J. Fox Foundation for Parkinson’s Research works toward the goal of curing Parkinson disease through funding research and developing improved therapies for individuals living with the disease.
KEY POINTS
Deposition of α-synuclein is the hallmark neuropathologic finding of the synucleinopathies, which include dementia with Lewy bodies, Parkinson disease, and multiple system atrophy. The Lewy body dementias include dementia with Lewy bodies and Parkinson disease dementia.
The Lewy body dementias are the second most common neurodegenerative dementia, after Alzheimer disease.
In dementia with Lewy bodies, α-synuclein pathology is observed beyond the brainstem in limbic and neocortical regions. In contrast, in Parkinson disease, α-synuclein pathology is first observed in the brainstem, in association with extrapyramidal impairment, and appears to spread with progression of disease to involve limbic and neocortical regions.
Amyloid deposition is common and variably present in dementia with Lewy bodies and Parkinson disease dementia.
Early dementia, visual hallucinations, fluctuations of attention and arousal, and the motor manifestations of parkinsonism characterize dementia with Lewy bodies.
Due to neuroleptic sensitivity in dementia with Lewy bodies, D2 receptor antagonists such as typical and most atypical neuroleptics are dangerous and contraindicated.
-
Rapid eye movement sleep behavior disorder, impairment of olfaction, chronic constipation, and neuroleptic sensitivity are common in dementia with Lewy bodies and Parkinson disease. These features may precede the development of typical clinical symptoms in these illnesses.
Clinical diagnostic criteria for dementia with Lewy bodies have better specificity than sensitivity.
Early anterograde amnesia is the sine qua non of Alzheimer disease. Hallucinations and parkinsonism can arise late in the course of Alzheimer disease. Early additional cognitive features and the early appearance of hallucinations, parkinsonism, and fluctuations of attention or arousal point to dementia with Lewy bodies.
Parkinson disease hastens cognitive decline and is a risk factor for dementia.
Parkinson disease medications can impair cognition. This is particularly true of trihexyphenidyl and the dopamine agonists but can be seen in all agents at sufficient dose.
The relative timing of dementia and parkinsonism defines the clinical distinction between dementia with Lewy bodies and Parkinson disease dementia.
Dementia with Lewy bodies and Parkinson disease dementia can be distinguished from multiple system atrophy, progressive supranuclear palsy, and corticobasal syndrome on the basis of their clinical features. However, no firm biomarkers have been developed that can predict a pathologic diagnosis.
It is generally advisable to make single changes in treatment systematically and serially, starting at low dose and tackling the most severe problem first. This simple strategy accounts for the frequent sensitivity to medications in dementia with Lewy bodies and allows for straightforward interpretation of the effects of manipulations.
The marked loss of acetylcholine neurons in dementia with Lewy bodies and Parkinson disease dementia likely underlies the efficacy of acetylcholinesterase inhibitors in these illnesses.
In Parkinson disease dementia, it is often useful to streamline the medication regimen in the service of cognition.
When psychosis in dementia with Lewy bodies or Parkinson disease dementia requires medical treatment, acetylcholinesterase inhibitors, quetiapine, and clozapine can be useful. However, the latter two agents require caution, given their risk of significant and severe adverse reactions.
Physical therapy, occupational therapy, and home safety evaluations are valuable treatments for motor impairments in dementia with Lewy bodies and Parkinson disease dementia.
Medical and nonmedical strategies exist to manage rapid eye movement sleep behavior disorder.
Given its prevalence, dementia with Lewy bodies is likely to be a common cause of mild cognitive impairment.
ACKNOWLEDGMENTS
The author thanks John Growdon, MD, for helpful comments on the manuscript.
REFERENCES
- 1. Lewy FH. Paralysis agitans. I. Pathologische Anatomie. Lewandowsky’s Handbuch der Neurologie, 3 Band: Spez Neurologie II. Berlin, Germany: Springer, 1912: 920– 933. [Google Scholar]
- 2. Okazaki H, Lipkin LE, Aronson SM. Diffuse intracytoplasmic ganglionic inclusions (Lewy type) associated with progressive dementia and quadriparesis in flexion. J Neuropathol Exp Neurol 1961; 20: 237– 244. [DOI] [PubMed] [Google Scholar]
- 3. Spillantini MG, Schmidt ML, Lee VM, et al. Alpha-synuclein in Lewy bodies. Nature 1997; 388(6645): 839– 840. [DOI] [PubMed] [Google Scholar]
- 4. Lippa CF, Duda JE, Grossman M, et al. ; DLB/PDD Working Group. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 2007; 68(11): 812– 819. doi:10.1212/01.wnl.0000256715.13907.d3. [DOI] [PubMed] [Google Scholar]
- 5. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005; 65(12): 1863– 1872. doi:10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 6. Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 2007; 22(12): 1689– 1707. doi:10.1002/mds.21507. [DOI] [PubMed] [Google Scholar]
- 7. Harding AJ, Halliday GM. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol 2001; 102(4): 355– 363. doi:10.1007/s004010100390. [DOI] [PubMed] [Google Scholar]
- 8. Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 1991; 114(pt 5): 2283– 2301. doi:10.1093/brain/114.5.2283 2283–2301. [DOI] [PubMed] [Google Scholar]
- 9. Tiraboschi P, Hansen LA, Alford M, et al. Cholinergic dysfunction in diseases with Lewy bodies. Neurology 2000; 54(2): 407– 411. doi:10.1212/WNL.54.2.407. [DOI] [PubMed] [Google Scholar]
- 10. Mattila PM, Röyttä M, Torikka H, et al. Cortical Lewy bodies and Alzheimer-type changes in patients with Parkinson’s disease. Acta Neuropathol 1998; 95(6): 576– 582. doi:10.1007/s004010050843. [DOI] [PubMed] [Google Scholar]
- 11. McKeith IG, Burn D. Spectrum of Parkinson’s disease, Parkinson’s dementia, and Lewy body dementia. In: DeKosky ST, ed. Neurologic clinics. Philadelphia, PA: WB Saunders, 2000: 865– 883. [DOI] [PubMed] [Google Scholar]
- 12. Aarsland D, Ballard CG, Halliday G. Are Parkinson’s disease with dementia and dementia with Lewy bodies the same entity? J Geriatr Psychiatry Neurol 2004; 17(3): 137– 145. doi:10.1177/0891988704267470. [DOI] [PubMed] [Google Scholar]
- 13. Lee DR, McKeith I, Mosimann U, et al. The dementia cognitive fluctuation scale, a new psychometric test for clinicians to identify cognitive fluctuations in people with dementia. Am J Geriatr Psychiatry 2014; 22(9): 926– 935. doi:10.1016/j.jagp.2013.01.072. [DOI] [PubMed] [Google Scholar]
- 14. Donaghy PC, O’Brien JT, Thomas AJ. Prodromal dementia with Lewy bodies. Psychol Med 2015: 45(2): 259− 268. doi:10.1017/S0033291714000816. [DOI] [PubMed] [Google Scholar]
- 15. Yoshita M, Arai H, Arai H, et al. Diagnostic accuracy of 123I-meta-iodobenzylguanidine myocardial scintigraphy in dementia with Lewy bodies: a multicenter study. PLoS One 2015; 10(3): e0120540 doi:10.1371/journal.pone.0120540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang Y, Halliday G. Can we clinically diagnose dementia with Lewy bodies yet? Transl Neurodegener 2013; 2(1): 4 doi:10.1186/2047-9158-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Finger EC. Frontotemporal dementias. Continuum (Minneap Minn) 2016; 22(2 Dementia): 464– 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parkinson J. An essay on the shaking palsy. England: Sherwood, Neely, and Jones, 1817. [Google Scholar]
- 19. Growdon JH, Corkin S, Rosen TJ. Distinctive aspects of cognitive dysfunction in Parkinson’s disease. Adv Neurol 1990; 53: 365– 376. [PubMed] [Google Scholar]
- 20. Locascio JJ, Corkin S, Growdon JH. Relation between clinical characteristics of Parkinson’s disease and cognitive decline. J Clin Exp Neuropsychol 2003; 25(1): 94– 109. doi:10.1076/jcen.25.1.94.13624. [DOI] [PubMed] [Google Scholar]
- 21. Mortimer JA, Pirozzolo FJ, Hansch EC, Webster DD. Relationship of motor symptoms to intellectual deficits in Parkinson disease. Neurology 1982; 32(2): 133– 137. doi:10.1212/WNL.32.2.133. [DOI] [PubMed] [Google Scholar]
- 22. Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson’s disease. Arch Neurol 1993; 50(2): 140– 148. doi:10.1001/archneur.1993.00540020018011. [DOI] [PubMed] [Google Scholar]
- 23. Litvan I, Goldman JG, Tröster AI, et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov Disord 2012; 27(3): 349– 356. doi:10.1002/mds.24893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aarsland D, Andersen K, Larsen JP, et al. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003; 60(3): 387– 392. doi:10.1001/archneur.60.3.387. [DOI] [PubMed] [Google Scholar]
- 25. Hughes TA, Ross HF, Musa S, et al. A 10-year study of the incidence of and factors predicting dementia in Parkinson’s disease. Neurology 2000; 54(8): 1596– 1602. doi:10.1212/WNL.54.8.1596. [DOI] [PubMed] [Google Scholar]
- 26. Mayeux R, Chen J, Mirabello E, et al. An estimate of the incidence of dementia in idiopathic Parkinson’s disease. Neurology 1990; 40(10): 1513– 1517. doi:10.1212/WNL.40.10.1513. [DOI] [PubMed] [Google Scholar]
- 27. Uc EY, McDermott MP, Marder KS, et al. ; Parkinson Study Group DATATOP Investigators. Incidence of and risk factors for cognitive impairment in an early Parkinson disease clinical trial cohort. Neurology 2009; 73(18): 1469– 1477. doi:10.1212/WNL.0b013e3181bf992f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kempster PA, O’Sullivan SS, Holton JL, et al. Relationships between age and late progression of Parkinson’s disease: a clinico-pathological study. Brain 2010; 133(pt 6): 1755– 1762. doi:10.1093/brain/awq059. [DOI] [PubMed] [Google Scholar]
- 29.American Psychiatric Association. Diagnostic and statistical manual of mental disorders, fifth edition Washington, DC: American Psychiatric Publishing, 2013. [Google Scholar]
- 30. Berg D, Postuma RB, Bloem B, et al. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov Disord 2014; 29(4): 454– 462. doi:10.1002/mds.25844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mak E, Su L, Williams GB, O’Brien JT. Neuroimaging characteristics of dementia with Lewy bodies. Alzheimers Res Ther 2014; 6(2): 18 doi:10.1186/alzrt248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davison CM, O’Brien JT. A comparison of FDG-PET and blood flow SPECT in the diagnosis of neurodegenerative dementias: a systematic review. Int J Geriatr Psychiatry 2014; 29(6): 551– 561. doi:10.1002/gps.4036. [DOI] [PubMed] [Google Scholar]
- 33. Jokinen P, Scheinin N, Aalto S, et al. [(11)C]PIB-, [(18)F]FDG-PET and MRI imaging in patients with Parkinson’s disease with and without dementia. Parkinsonism Relat Disord 2010; 16(10): 666– 670. doi:10.1016/j.parkreldis.2010.08.021. [DOI] [PubMed] [Google Scholar]
- 34. Bohnen NI, Koeppe RA, Minoshima S, et al. Cerebral glucose metabolic features of Parkinson disease and incident dementia: longitudinal study. J Nucl Med 2011; 52(6): 848– 855. doi:10.2967/jnumed.111.089946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Singh TD, Josephs KA, Machulda MM, et al. Clinical, FDG and amyloid PET imaging in posterior cortical atrophy. J Neurol 2015; 262(6): 1483– 1492. doi:10.1007/s00415-015-7732-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lleó A, Cavedo E, Parnetti L, et al. Cerebrospinal fluid biomarkers in trials for Alzheimer and Parkinson diseases. Nat Rev Neurol 2015; 11(1): 41– 55. doi:10.1038/nrneurol.2014.232. [DOI] [PubMed] [Google Scholar]
- 37. Kawai Y, Suenaga M, Takeda A, et al. Cognitive impairments in multiple system atrophy: MSA-C vs MSA-P. Neurology 2008; 70(16 pt 2): 1390– 1396. doi:10.1212/01.wnl.0000310413.04462.6a. [DOI] [PubMed] [Google Scholar]
- 38. Kitayama M, Wada-Isoe K, Irizawa Y, Nakashima K. Assessment of dementia in patients with multiple system atrophy. Eur J Neurol 2009; 16(5): 589– 594. doi:10.1111/j.1468–1331.2009.02544.x. [DOI] [PubMed] [Google Scholar]
- 39. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71(9): 670– 676. doi:10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ibáñez P, Lesage S, Janin S, et al. ; French Parkinson’s Disease Genetics Study Group. Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol 2009; 66(1): 102– 108. doi:10.1001/archneurol.2008.555. [DOI] [PubMed] [Google Scholar]
- 41. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 2009; 361(17): 1651– 1661. doi:10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schapira AH. Glucocerebrosidase and Parkinson disease: recent advances. Mol Cell Neurosci 2015; 66(pt A): 37– 42. doi:10.1016/j.mcn.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Srivatsal S, Cholerton B, Leverenz JB, et al. Cognitive profile of LRRK2-related Parkinson’s disease. Mov Disord 2015; 30(5): 728– 733. doi:10.1002/mds.26161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsuang D, Leverenz JB, Lopez OL, et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 2013; 70(2): 223– 228. doi:10.1001/jamaneurol.2013.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mollenhauer B, Rochester L, Chen-Plotkin A, Brooks D. What can biomarkers tell us about cognition in Parkinson’s disease? Mov Disord 2014; 29(5): 622– 633. doi:10.1002/mds.25846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Galasko DR. Dementia with Lewy bodies. Continuum (Minneap Minn) 2007; 13(2 Dementia): 69– 86. doi:10.1212/01.CON.0000267236.07498.38. [Google Scholar]
- 47. Wang HF, Yu JT, Tang SW, et al. Efficacy and safety of cholinesterase inhibitors and memantine in cognitive impairment in Parkinson’s disease, Parkinson’s disease dementia, and dementia with Lewy bodies: systematic review with meta-analysis and trial sequential analysis. J Neurol Neurosurg Psychiatry 2015; 86(2): 135– 143. doi:10.1136/jnnp-2014-307659. [DOI] [PubMed] [Google Scholar]
- 48. Cummings JL, Geldmacher D, Farlow M, et al. High-dose donepezil (23 mg/day) for the treatment of moderate and severe Alzheimer’s disease: drug profile and clinical guidelines. CNS Neurosci Ther 2013; 19(5): 294– 301. doi:10.1111/cns.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA 2014; 311(16): 1670– 1683. doi:10.1001/jama.2014.3654. [DOI] [PubMed] [Google Scholar]
- 50. Gareri P, De Fazio P, Manfredi VG, De Sarro G. Use and safety of antipsychotics in behavioral disorders in elderly people with dementia. J Clin Psychopharmacol 2014; 34(1): 109– 123. doi:10.1097/JCP.0b013e3182a6096e. [DOI] [PubMed] [Google Scholar]
- 51. Braak H, Del Tredici K, Rüb U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24(2): 197– 211. doi:10.1016/S0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 52. Kramer ML, Schulz-Schaeffer WJ. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 2007; 27(6): 1405– 1410. doi:10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nemani VM, Lu W, Berge V, et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010; 65(1): 66– 79. doi:10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gomperts SN. Imaging the role of amyloid in PD dementia and dementia with Lewy bodies. Curr Neurol Neurosci Rep 2014; 14(8): 472 doi:10.1007/s11910-014-0472-6. [DOI] [PubMed] [Google Scholar]
- 55. Brooks DJ, Piccini P. Imaging in Parkinson’s disease: the role of monoamines in behavior. Biol Psychiatry 2006; 59(10): 908– 918. doi:10.1016/j.biopsych.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 56. Marquie M, Locascio JJ, Rentz DM, et al. Striatal and extrastriatal dopamine transporter levels relate to cognition in Lewy body diseases: an (11)C altropane positron emission tomography study. Alzheimers Res Ther 2014; 6(5–8): 52 doi:10.1186/s13195-014-0052-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Petrou M, Kotagal V, Bohnen NI. An update on brain imaging in parkinsonian dementia. Imaging Med 2012; 4(2): 201– 213. doi:10.2217/iim.12.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Delli Pizzi S, Franciotti R, Tartaro A, et al. Structural alteration of the dorsal visual network in DLB patients with visual hallucinations: a cortical thickness MRI study. PLoS One 2014; 9(1): e86624 doi:10.1371/journal.pone.0086624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Boeve BF. Idiopathic REM sleep behaviour disorder in the development of Parkinson’s disease. Lancet Neurol 2013; 12(5): 469– 482. doi:10.1016/S1474-4422(13)70054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gelpi E, Navarro-Otano J, Tolosa E, et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov Disord 2014; 29(8): 1010– 1018. doi:10.1002/mds.25776. [DOI] [PubMed] [Google Scholar]
- 61. Panza F, Solfrizzi V, Imbimbo BP, Logroscino G. Amyloid-directed monoclonal antibodies for the treatment of Alzheimer’s disease: the point of no return? Expert Opin Biol Ther 2014; 14(10): 1465– 1476. doi:10.1517/14712598.2014.935332. [DOI] [PubMed] [Google Scholar]