

Abstract
Parietal cell atrophy is considered to cause metaplasia in the stomach. We developed mice that express the diphtheria toxin receptor specifically in parietal cells to induce their death, and found this to increase proliferation in the normal stem cell zone and neck but not to cause metaplastic reprogramming of chief cells. Furthermore, the metaplasia-inducing agents tamoxifen or DMP-777 still induced metaplasia even following previous destruction of parietal cells by diphtheria toxin. Atrophy of parietal cells alone is therefore not sufficient to induce metaplasia—completion of metaplastic reprogramming of chief cells requires mechanisms beyond parietal cell injury or death.
Keywords: SPEM, organoids, CD44 variant 9, Atp4b-Cre
Gastric metaplasia consistently occurs following parietal cell atrophy: in autoimmune gastritis patients1, Helicobacter pylori induced atrophic gastritis2, and animal models of acute injury3, 4. Thus, it has been proposed that parietal cell death causes metaplasia, perhaps because parietal cells elaborate gastric-differentiation-promoting factors whose loss elicits aberrant (metaplastic) differentiation of remaining cells. Alternatively, parietal cell death could cause a metaplasia-promoting immune response, or injured parietal cells might release metaplasia-promoting factors before dying.
Here, to test the role of parietal cells in metaplasia, we developed a method to precisely kill parietal cells in adults. We bred parietal cell-specific, Cre-inducible simian Diphtheria Toxin Receptor (Atp4b-Cre;LSL-DTR) mice [“DTR mice” (Supp. Fig. 1)], in which parietal cells alone respond to apoptosis-inducing diphtheria toxin. As a positive control for parietal cell atrophy and Spasmolytic Polypeptide-Expressing Metaplasia (SPEM), the metaplasia seen in direct temporal and spatial correlation with human and mouse parietal cell atrophy2, we used a previously described system3, 5, 6 involving three daily injections of high-dose (5 mg/20 g body mass) tamoxifen (“TAM”). Consistent with previous results, TAM caused >90% parietal cell atrophy and increased proliferation throughout the gastric unit. The pathognomonic pattern for SPEM was identified in >75% of units: GIF+ chief cells at the unit base co-expressing the epitope for the lectin GSII. Many SPEM cells were proliferative (yellow arrowheads, Fig. 1A,B). Three daily injections with 225 ng DT also killed >90% parietal cells and increased proliferation from the isthmus through the neck (Fig. 1A–C). Both atrophy and proliferation were maintained up to 14 days, whereas complete recovery occurred at that timepoint if injections were ceased at D3 (Fig. 1C).
Figure 1.
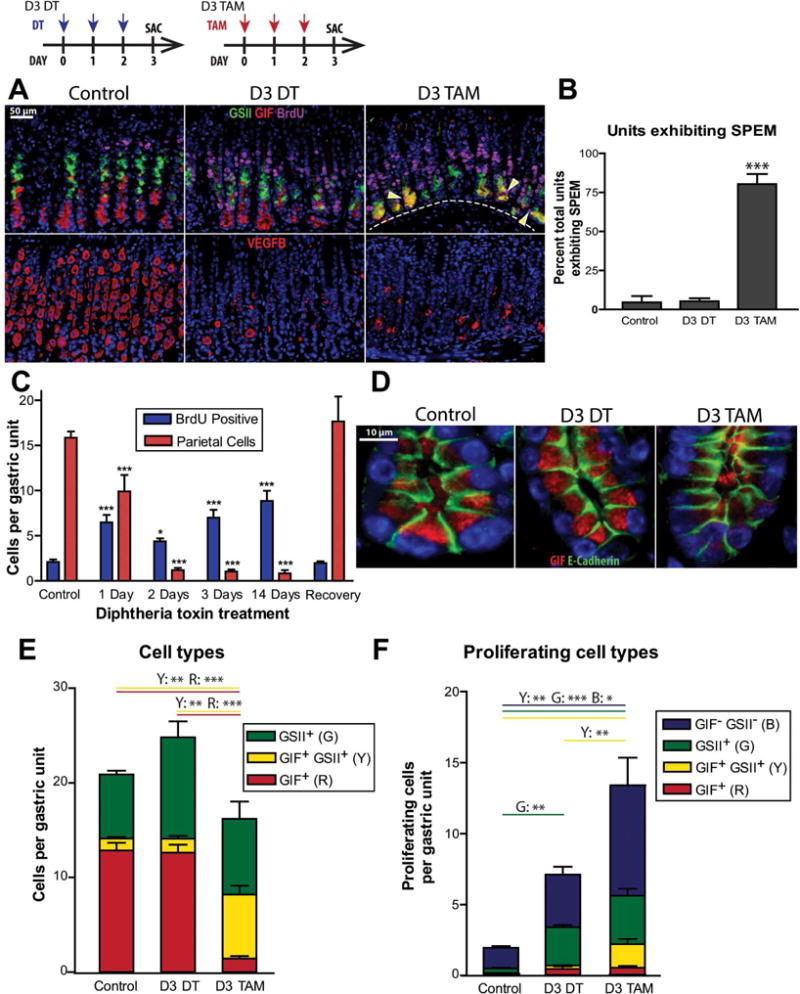
A) Stomachs following three days of vehicle, DT, or TAM (top: green: GSII, red: anti-GIF, magenta: anti-BrdU); arrowheads = proliferating SPEM cells. Bottom: red anti-VEGFB (parietal cells). B,C) Quantification. D) Unit bases with anti-E-Cadherin (green) and anti-GIF (red). E) Quantification of chief cells (GIF+, red), SPEM cells (GIF+/GSII+, yellow), and neck cells (GSII+, green) per unit. F) Quantification of proliferating cell types per unit with isthmal (double negative) cells in blue. “*” (p<0.05), “**” (p<0.01), “***” (p<0.001) in ANOVA; (B,C) with Dunnett; (E,F) with Tukey; n≥3 mice/group.
To confirm that DT directly targeted parietal cells, we grew gastroids from DTR × mTmG reporter mice in which DTR-expressing parietal cells also express membrane-associated eGFP (Supp. Fig. 1). Control gastroids showed negligible death (Supp. Fig. 2), whereas DT caused specific extrusion of eGFP+ cells without change in gastroid size or number. Thus, DT specifically kills parietal cells.
In contrast to TAM, DT never caused substantial SPEM at any timepoint (n>40 total mice examined). Proliferation occurred in the isthmus and neck but not in the base (Fig. 1A, B). SPEM is thought to arise in part from reentry of chief cells into the cell cycle7, 8. We observed that chief cells following TAM had the expected simple columnar morphology with scant GIF observed in SPEM cells, while chief cells following DT maintained largely normal morphology with apical GIF granules still apparent, even through day 14 (Fig. 1D, Supp. Fig. 3A). We quantified neck cells (GSII+), chief cells (GIF+), and GSII+/GIF+ cells, and their proliferative activity (Fig. 1E,F). DT did not significantly change GIF+ or GSII+/GIF+ cell census vs. control; however, TAM caused loss of chief cells and increased costaining cells. DT and TAM both increased proliferation in unlabeled isthmal cells and neck cells, while TAM additionally increased proliferating, GSII+/GIF+ SPEM cells.
We next analyzed additional markers of SPEM. The mouse ortholog of CD44 variant 9 (CD44v)9, neck cell marker TFF2, and secreted SPEM marker Clusterin10 were all increased only in the proliferative neck of DT treated mice, whereas TAM increased expression in both the neck and base (Supp. Fig. 3B,C,D). Thus, by all markers, parietal cell apoptosis alone was insufficient to cause metaplasia.
We next performed quantitative analyses of normal and metaplastic differentiation markers. GIF and the critical chief cell differentiation factor MIST1 (BHLHA15) decreased across the gastric corpus in DT mice; however, both were substantially lower in TAM mice (Supp. Fig. 4). SPEM markers Clusterin and HE4 (Wfdc2)11 were also significantly increased only following TAM (Supp. Fig. 4). TAM alone caused significantly increased expression of 6 other genes involved in metaplasia and immune response (Cd14, Ceacam10, Cftr, Ctss, Dmbt1, Vil1), with both treatments increasing proliferation-related transcripts (Ccnb2, Chek2) (Supp. Fig. 4).
These results argued against the model wherein parietal cells constitutively elaborate differentiation-promoting factors, as chief cells were maintained after parietal cell death. However, it was still possible parietal cell atrophy causes metaplasia: perhaps parietal cells dying via H pylori infection or TAM – but not DT – release metaplasia-inducing signals when injured. If true, metaplasia should not occur in mice with parietal cells already killed. Thus, we injected DTR mice with DT to kill parietal cells first and then co-injected DT and TAM for three days (DT+TAM). Five days of DT injection caused increased isthmal/neck proliferation without SPEM; however, TAM following DT caused proliferative SPEM similar to TAM alone (Fig. 2A–C). Similar results were obtained with another atrophy/SPEM-inducing agent, DMP-7774. DMP-777 treatment caused SPEM equally effectively even with parietal cells already killed (Fig. 2D–F; Supp. Fig. 5). Therefore, SPEM can occur without substances released from injured parietal cells.
Figure 2.
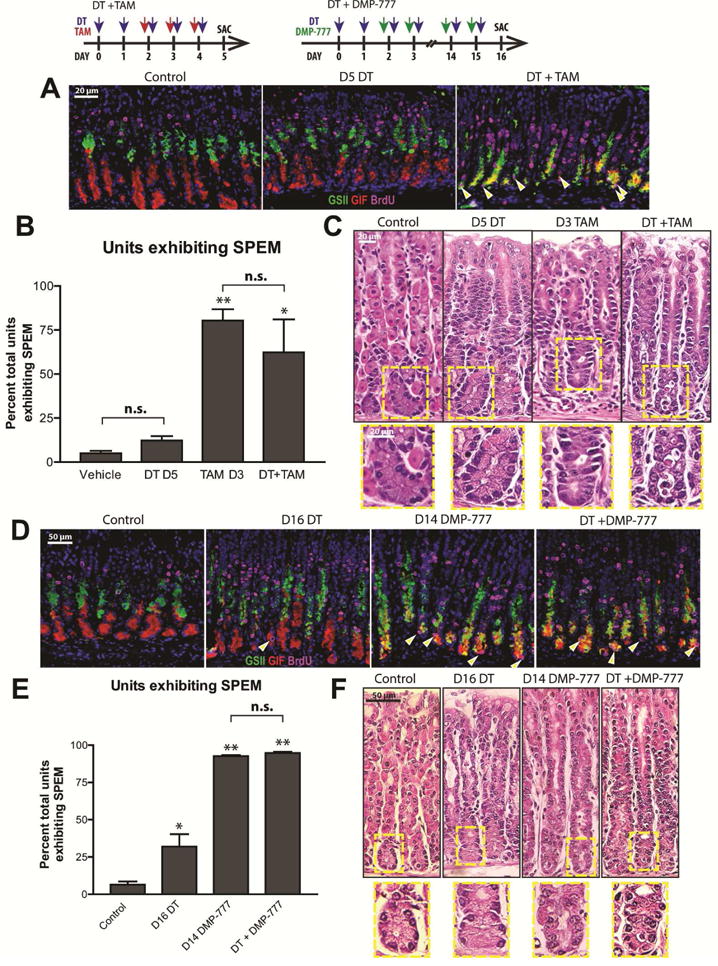
A Stomachs following five days control, DT, or DT then TAM (green: GSII, red: anti-GIF, magenta: anti-BrdU; arrowheads = proliferating SPEM cells. B) Quantification. C) H&E. D) Stomachs following 16 days control, DT, or DT then DMP-777, stained as panel A. E) Immunofluorescence data quantified. F) H&E. “*” (p<0.05), “**” (p<0.01), “***” (p<0.001) vs. Control, ANOVA with Dunnett; n≥3 mice/group.
Overall, our results show parietal cell atrophy alone is insufficient to induce metaplasia, and signals from injured/dying parietal cells are not necessary for metaplasia induction. Additionally, DTR mice increased proliferation only in the isthmal progenitor zone and neck, whereas TAM/DMP777 treatment showed these plus proliferative basal metaplastic cells. The number of metaplastic (GIF+/GSII+) cells arising in the base was approximately equivalent to the decrease in differentiated GIF+ only chief cells (Fig. 1E,F). Thus, parietal cell atrophy alone can cause isthmal stem cell and mucous neck cell proliferation; however, the rapid emergence of basal metaplastic cells likely involves an additional basal cellular source. Our results, therefore, favor a model (supported by Ito and colleagues6) identifying two distinct zones of proliferation that can expand during injury: 1) the isthmus/neck12, 13; and 2) a more mature cell of the chief cell lineage that reprograms to co-label with neck cell markers and reenter the cell cycle6–8. The reentry of differentiated secretory cells to serve as progenitors resonates with emerging work on pancreatic acinar cell plasticity and quiescent intestinal stem cells7.
Earlier work showed that, with constitutive absence of parietal cells throughout development, mature chief cells never emerge, and gastric units show abundant isthmal, pre-neck, neck, and neck/chief co-labeled cells14. It is unclear whether that signifies SPEM or simply a failure of parietal cell-less units to ever adopt the adult pattern with distinct neck and chief cell zones (juvenile gastric units are SPEM-like15). The usefulness of those mice as a model for adult-onset atrophy/metaplasia may thus be limited.
Future progress in understanding the process of chief cell reprogramming will require a system allowing perpetual induced parietal cell atrophy to determine if long-term parietal cell absence is sufficient to induce SPEM. In any case, our current results suggest that parietal cell loss alone is insufficient to directly induce SPEM and that metaplasia induction may require additional unidentified factors (e.g. cytokines or specific immune cell activation) that recruit chief cells back into the cell cycle.
Supplementary Material
Acknowledgments
J.C.M. is funded by the NIH National Institute of Diabetes and Digestive and Kidney Diseases (R01s DK094989 and DK105129) and the Siteman Cancer Center Investment Program, J.B. is supported by the NIGMS Cell and Molecular Biology Training Grant (GM007067); Histology performed by Digestive Disease Research Core Centers (Grant #P30DK052574, AITAC Core); J.R.G. is supported by grants from a Department of Veterans Affairs Merit Review Award (I01BX000930) and NIH R01 DK071590.
Abbreviations
- DT
diphtheria toxin
- DTR
Diphtheria toxin receptor
- SPEM
Spasmolytic polypeptide-expressing metaplasia
- TAM
Tamoxifen
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts to disclose.
Author contributions: JB (study concept and design; data acquisition, analysis, and interpretation for all mouse work; drafting of the manuscript), LHO (data acquisition and analysis), DL (data collection and analysis), JRG (data interpretation, supplied reagent, manuscript editing), JCM (study concept and design, analysis and interpretation of data, statistical analysis, funding, supervision, editing)
References
- 1.Adams JF, et al. Lancet. 1964;1:401–403. doi: 10.1016/s0140-6736(64)92785-0. [DOI] [PubMed] [Google Scholar]
- 2.Lennerz JK, et al. Am J Pathol. 2010;177:1514–1533. doi: 10.2353/ajpath.2010.100328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huh WJ, Khurana SS, et al. Gastroenterology. 2012;142:21–24.e27. doi: 10.1053/j.gastro.2011.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nomura S, Yamaguchi H, et al. Am J Physiol Gastrointest Liver Physiol. 2005;288:G362–375. doi: 10.1152/ajpgi.00160.2004. [DOI] [PubMed] [Google Scholar]
- 5.Saenz JB, et al. Methods Mol Biol. 2016;1422:329–339. doi: 10.1007/978-1-4939-3603-8_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuo J, et al. Gastroenterology. 2016 [Google Scholar]
- 7.Mills JC, Sansom OJ. Sci Signal. 2015;8:re8. doi: 10.1126/scisignal.aaa7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nam KT, et al. Gastroenterology. 2010;139:2028–2037 e2029. doi: 10.1053/j.gastro.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wada T, et al. Cancer Sci. 2013;104:1323–1329. doi: 10.1111/cas.12236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weis VG, et al. Gut. 2013;62:1270–1279. doi: 10.1136/gutjnl-2012-302401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nozaki K, et al. Gastroenterology. 2008;134:511–522. doi: 10.1053/j.gastro.2007.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayakawa Y, Ariyama H, et al. Cancer Cell. 2015;28:800–814. doi: 10.1016/j.ccell.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khurana SS, et al. The Journal of biological chemistry. 2013;288:16085–16097. doi: 10.1074/jbc.M112.445551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Q, et al. Journal of Biological Chemistry. 1996;271:3671–3676. [PubMed] [Google Scholar]
- 15.Keeley TM, Samuelson LC. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1241–1251. doi: 10.1152/ajpgi.00239.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.