

Summary
Synaptic protein synthesis is essential for modification of the brain by experience and is aberrant in several genetically-defined disorders, notably fragile X, a heritable cause of autism and intellectual disability. Neural activity directs local protein synthesis via activation of metabotropic glutamate receptor 5 (mGlu5), yet how mGlu5 couples to the intracellular signaling pathways that regulate mRNA translation is poorly understood. Here, we provide evidence that β-arrestin2 mediates mGlu5-stimulated protein synthesis in the hippocampus and show that genetic reduction of β-arrestin2 corrects aberrant synaptic plasticity and cognition in the Fmr1−/y mouse model of fragile X. Importantly, reducing β-arrestin2 does not induce psychotomimetic activity associated with full mGlu5 inhibitors, and does not affect Gq signaling. Thus, in addition to identifying a key requirement for mGlu5-stimulated protein synthesis, these data suggest that β-arrestin2-biased negative modulators of mGlu5 offer significant advantages over first-generation inhibitors for the treatment of fragile X and related disorders.
Keywords: metabotropic glutamate receptors, mGlu5, arrestins, β-arrestin2, fragile X, extracellular regulated kinase, ERK, synaptic protein synthesis, biased ligands, long-term depression, autism, intellectual disability
eTOC Blurb
Stoppel et al. find that β-arrestin2 is a critical link between mGlu5 and activity-dependent neuronal protein synthesis. Reducing β-arrestin2 levels corrects many synaptic and cognitive deficits in a mouse model of fragile X.
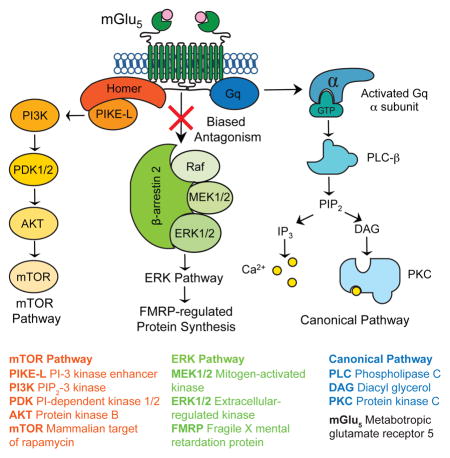
Introduction
Numerous genetic and molecular studies have demonstrated that poorly regulated synaptic protein synthesis downstream of metabotropic glutamate receptor 5 (mGlu5) contributes to the pathophysiology of fragile X (FX), a genetic cause of intellectual disability (ID) and autism spectrum disorder (ASD) (Pop et al., 2014). This work suggests that targeting mGlu5 or its downstream effectors may be a fruitful approach for improving the course of FX and other genetic syndromes with shared pathophysiology (Aguilar-Valles et al., 2015; Auerbach et al., 2011; Barnes et al., 2015; Bozdagi et al., 2010; Tian et al., 2015; Wenger et al., 2016). Indeed, mGlu5-based therapies have been immensely successful at correcting FX in animal models (Bhakar et al., 2012). To date, however, the results of human clinical trials in FX using mGlu5 negative allosteric modulators (NAMs) have been disappointing (Berry-Kravis et al., 2016; Scharf et al., 2015).
Although many factors contribute to the challenge of translating findings from animal models to humans, one factor that is common to all drug trials is the therapeutic window—the range of doses that can treat disease pathophysiology without causing negative side effects. In humans, for example, it has been reported that inhibition of mGlu5 produces dose-limiting psychotomimetic effects (Abou Farha et al., 2014; Pecknold et al., 1982; Porter et al., 2005). The first-generation mGlu5 NAMs were identified based on their ability to inhibit Gq signaling mediated by phosphoinositide hydrolysis and release of Ca2+ from intracellular stores (Cosford et al., 2003; Gasparini et al., 1999; Lindemann et al., 2011). However, available data suggest alternative signaling pathways are central to the regulation of protein synthesis by mGlu5 (Bhakar et al., 2012; Osterweil et al., 2010; Richter et al., 2015). Thus, it is possible that therapeutic effects can be enhanced and separated from side effects by selectively targeting the coupling of mGlu5 to disease-relevant signaling pathway(s).
One pathway that is known to be central to mGlu5-stimulated protein synthesis and FX pathophysiology culminates in activation of ERK1/2 and the phosphorylation of proteins involved in the regulation of cap-dependent mRNA translation (Banko et al., 2006; Osterweil et al., 2013; Osterweil et al., 2010). Activation of this pathway by mGlu5 can occur independently of G-protein signaling, but how this is achieved has remained a mystery. As is the case for many seven-transmembrane domain receptors, G-protein signaling of ligand-bound mGlu5 is terminated by recruitment of β-arrestin to the carboxyl tail of the receptor. In recent years it has become clear that β-arrestin recruitment can also trigger activation of alternative signaling cascades. Of particular relevance is the observation that β-arrestin2 recruitment to the angiotensin II receptor (which, like mGlu5, is also Gq-coupled) stimulates the ERK1/2 pathway and increases mRNA translation rates in both human embryonic kidney 293 and rat vascular smooth muscle cells (Ahn et al., 2009; DeWire et al., 2008). We therefore hypothesized that β-arrestin2 comprises a crucial link between mGlu5 and protein synthesis in neurons.
Results
Heterozygous deletion of β-arrestin2 disrupts mGlu5 stimulated ERK activation and protein synthesis without affecting Gq-signaling
To determine the role of β-arrestin2 in mGlu5-mediated protein synthesis, we stimulated hippocampal slices from male Arrb2+/− and wild-type (WT) littermates with a selective agonist and positive modulator of mGlu5, 3-Cyano-N- (1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB,10 μM, 30 minutes), and measured the incorporation of 35S-methionine/cysteine into new protein as described previously (Henderson et al., 2012; Osterweil et al., 2010). We found that mGlu5 activation caused a parallel increase in protein synthesis (Fig. 1A) and ERK1/2 phosphorylation (Fig. 1B) in WT slices, which were both absent in slices from Arrb2+/− mice. This blunted response to mGlu5 stimulation occurred in the absence of differences in basal protein synthesis rates or ERK phosphorylation levels (Fig. 1A, B). We failed to observe a comparable effect on stimulated protein synthesis in Arrb1+/− mice (Fig. S1), suggesting that β-arrestin2 is the relevant isoform for mGlu5 signaling. From a therapeutic standpoint, it is noteworthy that mGlu5-stimulated protein synthesis is abrogated in mice lacking a single allele of Arrb2; a full knockout is not required to see an effect.
Figure 1. β-arrestin2 is necessary for protein synthesis-dependent mGlu-LTD and ERK1/2 activation.

(A) Schematic illustrates experimental timeline. Protein synthesis was elevated in WT slices stimulated with CDPPB compared with vehicle (two-tailed t test, t = 3.6928, *p = 0.0017, n = 10 animals per group) whereas treatment had no effect in Arrb2+/− slices (two-tailed t test, t = 0.654, p = 0.5214, n = 10 animals per group). Two-way ANOVA, genotype vs. treatment, F = 7.081; *p = 0.012. Mean ± SEM 35S incorporation (%CPM/μg): WT + vehicle = 3.3057 ± 0.2441; WT + CDPPB = 4.4417 ± 0.3196; Arrb2+/− + vehicle = 3.4463 ± 0.3004; Arrb2+/− + CDPPB = 3.3940 ± 0.3397. (B) Representative immunoblots of ERK1/2 phosphorylation and total ERK protein from hippocampal slices ± CDPPB stimulation from WT and Arrb2+/− mice. WT slices stimulated with CDPPB show elevated ERK1/2 phosphorylation compared with vehicle (two-tailed t test, t = 3.1421, *p = 0.0047, n = 12 animals per group), whereas no change was observed in Arrb2+/− mice (two-tailed t test, t = 0.1826, p = 0.8568, n = 12 animals per group). Two-way ANOVA, genotype vs. treatment, F = 6.458, *p = 0.015. Full and uncropped versions of blots underlying the figures are collected in Fig. S4. (C) Quantification of calcium fluorescence over time in WT and Arrb2+/− slices. Data are normalized as ΔF/F as discussed in materials and methods. There is no significant difference in the peak calcium fluorescence measured between WT and Arrb2+/− slices (two-tailed Mann-whitney test, p = 0.7959, Mann-whitney U= 46, n = 10 animals per group). (D) The cumulative probability of peak fluorescence for all cells analyzed is not different between WT and Arrb2+/− slices (Kolmogorov-Smirnov test, p = 0.8334, n = WT, 155 cells, Arrb2−/+, 88 cells). (E, F) DHPG-LTD (25 μM, 5 min) is reduced and unaffected by pretreatment with the protein synthesis inhibitor cycloheximide (CHX, 60 μM) in hippocampal slices from Arrb2+/− animals. Two-way ANOVA, genotype vs. treatment, F = 9.678,*p = 0.003.). Bonferroni multiple comparisons shows a significant effect of genotype under control conditions (*p = 0.005, WT = 13 animals, Arrb2+/− = 12 animals) but not in the presence of CHX (p = 0.125; WT = 9 animals, Arrb2+/− = 11 animals). CHX treatment significantly reduced LTD magnitude in WT slices (*p < 0.001, t = 4.676; control n = 13 animals, CHX n = 9 animals) but not Arrb2+/− slices (p = 0.646, t = 0.463; control n = 12 animals, CHX n = 11 animals). Representative field potential (FP) traces (average of 10 sweeps) were taken at times indicated by numerals. Scales bars equal 0.5 mV, 5 ms. For this and all subsequent figures, data is plotted as mean ± s.e.m. Statistics were performed using each animal as one “n”, with each animal represented by the mean of 1–4 slices, unless otherwise noted. See also Figures S1, S2, and S3.
β-arrestins have also been shown to participate in additional signaling cascades (DeWire et al., 2007), including the Akt-mTOR pathway that has been implicated in the regulation of protein synthesis (Hou and Klann, 2004) and the pathophysiology of FX (Gross et al., 2015; Sharma et al., 2010). However, in agreement with previous studies in the hippocampal slice (Osterweil et al., 2010), we found that mGlu5 activation failed to increase phosphorylation of Akt or ribosomal protein S6, a readout of mTOR activity, in WT mice. These measures of mTOR pathway activity were also unaffected in slices prepared from Arrb1+/− and Arrb2+/− mice (Fig. S2).
To assay the integrity of Gq signaling, we examined calcium mobilization in hippocampal slices from WT and Arrb2+/− animals using the cell-permeable calcium fluorescent dye Fluo4-AM. We found that a brief application of the agonist S-3,5-dihydroxyphenylglycine (DHPG, 25 μM, 1 min) to slices resulted in a rapid increase in Ca2+-mediated fluorescence in area CA1 that was not significantly different between WT and Arrb2+/− slices (Fig. 1C–D). DHPG was employed in these experiments because it activates both of the Gq-coupled metabotropic glutamate receptors, mGlu1 and mGlu5. These DHPG-induced changes in calcium fluorescence were completely blocked by pretreatment with the phospholipase C inhibitor, U73122 (data not shown). These results indicate that a partial reduction in β-arrestin2 does not result in aberrant Gq signaling in response to mGlu5 activation. Moreover, they suggest that modulation of mGlu5 receptor-mediated protein synthesis can be dissociated from G-protein dependent signaling via manipulation of β-arrestin2.
Deficient mGlu5-mediated translation impairs synaptic plasticity in the hippocampus of Arrb2+/− mutants
Activation of mGlu5 results in a form of synaptic long-term depression (LTD) in the hippocampus that requires rapid de novo synaptic protein synthesis (Huber et al., 2000). We therefore investigated the functional relevance of the observed biochemistry by determining if genetic reduction of Arrb2 also alters the expression and/or protein synthesis-dependency of LTD induced with DHPG (25 μM, 5 min) (Huber et al., 2001). Basal synaptic transmission was normal (Fig. S3), but LTD magnitude was significantly reduced in Arrb2+/− slices compared to WT (Fig. 1E–F). Consistent with previous observations, LTD in WT slices was significantly diminished in the presence of the protein synthesis inhibitor cycloheximide (CHX, 60 μM). In contrast, the residual LTD in slices from Arrb2+/− animals was unaffected by CHX (Fig. 1E–F). Therefore, we conclude that the protein synthesis-dependent component of mGlu5-mediated LTD is absent in the Arrb2+/− hippocampus.
In WT mice, the LTD that remains when DHPG is applied in the presence of CHX is expressed via a presynaptic mechanism, revealed by a change in the paired-pulse ratio (Auerbach et al., 2011). This change in paired-pulse ratio after DHPG was similar in Arrb2+/− mice, indicating that this presynaptic, protein synthesis-independent mechanism of LTD is unaffected by reducing signaling through β-arrestin2 (Fig. S3). Another, mechanistically distinct form of hippocampal LTD can be induced by stimulating NMDA receptors. This type of LTD is expressed postsynaptically, but does not require ERK1/2 or immediate translation of mRNA. We found that it is also unaffected by genetic reduction of β-arrestin2 in the hippocampus (Fig. S3). Taken together, these results suggest that the diminished LTD magnitude observed in Arrb2+/− animals is likely a specific consequence of impaired mGlu5-stimulated mRNA translation at the synapse.
Decreasing β-arrestin2 levels reverses synaptic and behavioral deficits in a mouse model of fragile X
Our results indicate that β-arrestin2 couples mGlu5 activation to ERK-dependent protein synthesis and LTD. Aberrantly increased mGlu5-dependent protein synthesis observed in vivo (Qin et al., 2005), brain slices (Dolen et al., 2007; Osterweil et al., 2010) and synaptoneurosomes (Henderson et al., 2012) is believed to be pathogenic in Fmr1-null mice (Bhakar et al., 2012; Dolen et al., 2007). Therefore, we investigated whether a genetic reduction of Arrb2 in Fmr1-null mice could correct FX phenotypes. We crossed Arrb2+/− male mice to Fmr1+/− female mice and found that both the increased protein synthesis (Fig. 2A–B) and exaggerated mGlu-LTD (Fig. 2C–D) characteristic of Fmr1−/y mice were restored to WT levels in Arrb2+/− x Fmr1−/y mice.
Figure 2. Genetic reduction of β-arrestin2 in Fmr1−/y mice corrects exaggerated protein synthesis and mGlu-LTD.

(A) Genetic rescue strategy. (B) Basal protein synthesis is significantly increased in slices from Fmr1−/y mice compared with WT slices (two-tailed t test, t = 3.0689, *p = 0.0078, n = 9 animals each genotype). Basal protein synthesis is comparable in slices from Arrb2+/− x Fmr1−/y mice and WT mice (two-tailed t test, t = 0.4821, p = 0.6363, n = 9 animals each genotype). Basal protein synthesis is significantly increased in slices from Fmr1−/y mice compared with Arrb2+/− x Fmr1−/y slices (two-tailed t test, t = 2.5243, *p = 0.0225, n = 9 animals each genotype). Mean ± SEM 35S incorporation (%CMP/μg): WT = 2.9183 ± 0.1988; Fmr1−/y = 3.7697 ± 0.1934; Arrb2+/− = 3.135 ± 0.0747; Fmr1−/y x Arrb2+/− = 3.0563 ± 0.2060. (C) The magnitude of DHPG-induced LTD in slices from Arrb2+/− x Fmr1−/y mice is significantly different from Fmr1−/y slices and is indistinguishable from WT slices (One-way ANOVA, *p = 0.0001, F = 8.715, with Bonferroni multiple comparisons: Arrb2+/− x Fmr1−/y vs. Fmr1−/y, *p < 0.03, t = 2.971, Arrb2+/− x Fmr1−/y vs. WT, p = 0.999, t = 0.5741, Arrb2+/− vs. Fmr1−/y, *p < 0.0001, t = 5.033, WT n = 15 animals, Fmr1−/y n = 10 animals, Arrb2+/− n = 9 animals, Arrb2+/− x Fmr1−/y n = 16 animals). Representative FP traces (average of 10 sweeps) were taken at times indicated by numerals. Scales bars equal 0.5 mV, 2ms. (D) Summary of LTD data. Bar graphs, percentage decrease from baseline in FP slope.
We next investigated the possibility that restoration of normal protein synthesis and mGlu5-dependent synaptic plasticity could lead to improvements in cognitive and behavioral assays previously shown to be impaired in Fmr1−/y mice. We assayed inhibitory avoidance, a hippocampus-dependent behavior known to be disrupted in Fmr1−/y mice (Dolen et al., 2007; Qin et al., 2002) (Fig. 3A). Memory strength was measured as the latency to enter the dark side of a box that was associated with a foot shock. We discovered that Arrb2+/− as well as Fmr1−/y mice failed to form a strong association between the context and foot shock (between time 0 and 6 hours) indicating impaired memory acquisition. This is consistent with previous results showing that both excessive and deficient hippocampal protein synthesis can manifest similarly at the behavioral level (Auerbach et al., 2011). Remarkably, however, Arrb2+/− x Fmr1−/y mice were indistinguishable from WT and exhibited normal memory acquisition and extinction over the course of 48 hours (Fig. 3B).
Figure 3. Genetic reduction of β-arrestin2 in Fmr1−/y mice corrects behavioral and cognitive deficits.

(A) Experimental design of inhibitory avoidance learning task. (B) Fmr1−/y mice and Arrb2+/− mice show impaired acquisition of inhibitory avoidance learning compared to WT mice (two-way ANOVA, *p < 0.001 for each comparison, wild-type vs. Fmr1−/y (F = 12.760); WT vs. Arrb2+/− (F = 12.525). Arrb2+/− x Fmr1−/y mice show comparable acquisition and extinction of inhibitory avoidance to WT mice (two-way ANOVA, F = 0.145, p = 0.933). There is a statistically significant interaction between genotype and time point across groups (repeated measures two-way ANOVA, F = 12.425, *p = < 0.001). (C) Experimental design of familiar object recognition task. (D) Fmr1−/y mice show impaired novelty detection on experimental test day 2 when presented with a familiar and novel object compared to WT (two-tailed t test, t = 7.1445, *p < 0.00001, n = 10 animals each genotype). In comparison, Arrb2+/− x Fmr1−/y demonstrate a discrimination index that is not significantly different from wild-type mice (two-tailed t test, t = 0.0511, p = 0.9598, n = 10 animals each genotype). (E) Fmr1−/y mice exhibit increased susceptibility to audiogenic seizure activity compared to WT (two-tailed Fisher’s exact test, *p = 0.0001, n = 16, 18 animals) and Arrb2+/− mice (*p = 0.0001, n = 18, 22 animals). Genetic reduction of Arrb2 in Fmr1−/y mice significantly reduces the incidence of seizure activity (*p = 0.0409, n = 18, 17).
We also investigated non-aversive object recognition memory. Mice were first allowed to explore an arena with two identical objects for two sessions. The following day, one of the familiar objects was replaced with a novel object (Fig. 3C). While Fmr1−/y mice explored both the novel and familiar objects to an equal degree, indicating a severe impairment in novelty detection, Arrb2+/− x Fmr1−/y mice as well as Arrb2+/− single mutants showed a strong preference for the novel object similar to WT mice (Fig. 3D).
In an additional series of behavioral experiments, we investigated audiogenic seizures (AGS), as increased susceptibility to AGS is a hallmark phenotype in Fmr1−/y mice. Genetic reduction of Arrb2 in Fmr1-null mice significantly attenuated seizure incidence (Fig. 3E), very similar to what has been observed using mGlu5 and ERK-pathway inhibitors (Dolen et al., 2007; Osterweil et al., 2010; Yan et al., 2005).
Unlike first generation mGlu5 NAMs, β-arrestin2 reduction does not exacerbate MK-801- induced hyperlocomotion
Our data suggest that the mGlu5 signaling relevant to FX pathophysiology passes through β-arrestin2 to activate ERK and protein synthesis. If this conclusion is correct, then modulators that specifically target mGlu5 coupling to β-arrestin2 might avoid side effects that arise from inhibition of Gq and/or mTOR pathway signaling. First-generation mGlu5 NAMs were all identified based on inhibition of Gq signaling, and in humans one adverse side effect reported following treatment with these compounds is derealization and visual hallucinations (Abou Farha et al., 2014; Pecknold et al., 1982; Porter et al., 2005). Similarly in mice, mGlu5 NAMs exacerbate hyperlocomotion in response to treatment with the potent pyschotomimetic MK801 (Homayoun et al., 2004; Pietraszek et al., 2005). Therefore, we examined the effect of genetic reduction of β-arrestin2 and mGlu5 NAM treatment on MK801-induced hyperlocomotion in mice. We confirmed that pretreatment with the selective mGlu5 inhibitor 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine (MTEP) (Cosford et al., 2003) significantly potentiates MK801-induced hyperlocomotion in WT mice. However, we found that baseline locomotor activity was the same in Arrb2+/− and WT mice, as was the synergistic effect of MTEP pretreatment on MK801-induced hyperlocomotion (Fig. 4). The fact that MTEP continues to exacerbate hyper-locomotion in Arrb2+/− mice that lack mGlu5-regulated protein synthesis suggests that the psychotomimetic effects of the NAM are mediated by inhibition of pathways unrelated to FX pathophysiology.
Figure 4. Genetic reduction of β-arrestin2 does not potentiate the psychotomimetic effects of MK801.

WT and Arrb2+/− mice injected intraperitoneally with the NMDAR antagonist MK801 (0.3 mg/kg) show comparable hyperlocomotion 60 minutes post-treatment compared to vehicle (N = 10 mice per group). Data points represent distance travelled in cm over 5 minute bins, averaged as pooled animals per treatment group. Pre-treatment with MTEP (10 mg/kg, i.p.) potentiates hyperlocomotion in both WT and Arrb2+/− mice (N = 9 mice per group). Two-way ANOVA for genotype: F = 0.468, p = 0.499, and for treatment: F = 13.597, *p < 0.001; no significant interaction between genotype and treatment: F = 0.352, p = 0.557. Two-tailed t test, WT + MK801 vs. WT + MTEP + MK801: t = 2.9358, *p = 0.0092. Two-tailed t test, Arrb2+/− + MK801 vs. Arrb2+/− + MTEP + MK801: t = 2.2603, *p = 0.0372.
Discussion
It has been established previously that mGlu5-stimulated protein synthesis and LTD are blocked by inhibitors of MEK and ERK (Banko et al., 2006; Gallagher et al., 2004; Osterweil et al., 2010; Schnabel et al., 1999), but are unaffected by inhibitors of PKC and PLC (Fitzjohn et al., 2001; Rush et al., 2002; Schnabel et al., 1999). It was also known that the ERK pathway is recruited even in the presence of PLC inhibitors (Fitzjohn et al., 2001; Gallagher et al., 2004; Huber et al., 2001). However, it was unknown how mGlu5 can stimulate ERK and protein synthesis independently of Gq/PLC activation. We show here that reducing β-arrestin2 completely blocks mGlu5-stimulated ERK activation, protein synthesis, and protein synthesis-dependent LTD, but has no effect on Gq-dependent mobilization of intracellular Ca2+ via PLC. Thus, β-arrestin2 couples mGlu5 to the ERK signaling pathway and protein synthesis in neurons. This conclusion is in accordance with data on ERK pathway activation and the stimulation of protein synthesis by other Gq-coupled receptors in non-neuronal cells (Ahn et al., 2009; DeWire et al., 2008).
Our findings also are in general agreement with a contemporaneous investigation of β-arrestin involvement in mGlu1 and mGlu5 signaling in the hippocampus (Eng et al., 2016). This study confirmed our finding of impaired mGlu5-dependent synaptic LTD and ERK pathway activation in mice lacking β-arrestin2. One difference is their finding that LTD induced with DHPG (unlike synaptic stimulation) was unaffected in Arrb2−/− mice. However, this discrepancy is likely accounted for by the fact that their slices were not sufficiently rested to observe the protein synthesis-dependent component of agonist-induced LTD (see (Osterweil et al., 2010)). In any case, both studies are in agreement that protein synthesis-independent DHPG-LTD, expressed by a presynaptic modification, is unaffected by reducing β-arrestin2 (see Fig. 1E–F).
The discovery that mGlu5 stimulates protein synthesis via β-arrestin2 has clinical as well as basic biological significance. The core pathophysiology of FX is believed to be excessive synaptic protein synthesis downstream of mGlu5 (Bhakar et al., 2012). In animal models of FX, it has been shown that inhibition of mGlu5 can correct a wide array of mutant phenotypes. This work led directly to human FX clinical trials with mGlu5 inhibitors but, unfortunately, the results of these trials to date have disappointed (Berry-Kravis et al., 2016). With all drug trials, the maximum allowable dosage is determined by the occurrence of adverse side effects. In the case of first generation mGlu5 drugs, a potentially serious dose-limiting psychiatric side effect is derealization and visual hallucinations. To separate the therapeutic effect of mGlu5 inhibition (suppression of protein synthesis) from the unwanted side effects, it is essential to understand the mechanism that specifically couples mGlu5 to the ERK signaling pathway. The correction of multiple FX phenotypes, including excessive basal protein synthesis, in Fmr1−/y mice crossed with β-arrestin2 heterozygous mice indicates that β-arrestin2 is a key component of a pathogenic pathway. Further, the fact that MK-801 induced hyperlocomotion is still augmented in the Arrb2+/− mice by MTEP, a first-generation NAM with high selectivity for mGlu5 (Cosford et al., 2003), indicates that this undesirable effect of inhibiting mGlu5-Gq signaling is likely to be pharmacologically separable from the therapeutic effect of inhibiting mGlu5-β-arrestin2 signaling.
G-protein coupled receptors respond to a wide variety of signals and initiate a large number of distinct cellular signaling pathways. This versatility has made these receptors attractive targets for pharmacological therapies, and over 50% of the current drugs used clinically target these receptors (Insel et al., 2007). The finding that β-arrestin- and G protein-dependent cellular signaling are pharmacologically separable has opened a new vista for the treatment of disease. For some disorders, modulation of only one of these signaling pathways may be therapeutically beneficial, while the other(s) could mediate undesirable and possibly conflicting outcomes (Whalen et al., 2011). Our findings suggest that mGlu5 modulators for the treatment of FX is a case in point. There is little doubt that β-arrestin-biased allosteric modulators of mGlu receptors are feasible (Hathaway et al., 2015; Iacovelli et al., 2014; Sheffler et al., 2011), and their development could lead to the next generation of drugs for the treatment of FX and several other genetically defined causes of ID and ASD (Aguilar-Valles et al., 2015; Auerbach et al., 2011; Barnes et al., 2015; Bozdagi et al., 2010; Tian et al., 2015; Wenger et al., 2016).
Because it is a monogenic disorder, FX has emerged in recent years as a bellwether for the utility of developing medicines for psychiatric diseases by reproducing genetic etiologies in animal models to identify pathophysiology and therapeutic targets. The current study is important because it reveals some of the previously unappreciated limitations of targeting mGlu5 signaling via Gq, and suggests an exciting alternative approach.
Experimental Procedures
Arrb2+/− male and female mutant mice on the C57Bl/6J clonal background were mated to produce the WT and Arrb2+/− offspring used in this study. Fmr1−/+ female mice (Jackson Labs) were crossed with Arrb2+/− male mice to generate double mutant animals. All experimental animals were age-matched male littermates, and were studied with the experimenter blind to genotype and treatment condition. All experimental techniques were approved by The Institutional Animal Care and Use Committee at MIT and all animals were treated in accordance with NIH and MIT guidelines. Hippocampal slice preparation, electrophysiological recordings, metabolic labeling, immunoblotting as well as inhibitory avoidance and audiogenic seizure assays were performed as previously described (Auerbach et al., 2011; Dolen et al., 2007; Osterweil et al., 2010) and are detailed in Supplemental Experimental Procedures. Slices were stimulated with the selective agonist/positive modulator of mGlu5 CDPPB for metabolic labeling and immunoblotting, or the group 1 mGluR agonist (S)-DHPG for electrophysiology and calcium imaging experiments. Calcium mobilization was assessed using the cell-permeable calcium fluorescent dye Fluo4-AM in the presence of TTX and AP-5. The effect of MTEP on MK801-induced hyperlocomotion was assessed using Plexon’sCinePlex® Studio and custom written MATLAB software. Two-way ANOVAs with post-hoc two-tailed t tests or Bonferroni’s test for multiple comparisons were used to determine differences between genotypes and drug treatments unless stated otherwise. All data shown represent mean ± SEM. A full description of the Experimental Procedures can be found in the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
β-arrestin2 is required for stimulated protein synthesis downstream of mGlu5
β-arrestin2 reduction disrupts mGlu5-mediated ERK activation but not Gq-signaling
Decreasing β-arrestin2 in Fmr1-null mice reverses synaptic and behavioral phenotypes
No psychotomimetic effects are associated with β-arrestin2 deletion
Acknowledgments
This work was supported by NIH grants R21NS087225, 2R01HD046943 and R01MH106469 to M.F.B.; L.J.S. and B.D.A. received additional support from T32MH074249; and R.K.S. was supported by a FRAXA postdoctoral fellowship. We thank Arnold Heynen and Robert Komorowski for valuable advice and comments, as well as David Bowen and Amanda Coronado for excellent technical assistance. We are also pleased to acknowledge Suzanne Meagher, Nina Palisano and Erik Sklar for administrative assistance.
Footnotes
Author Contributions:
M.F.B. and R.J.L. conceived the project. R.J.L. provided the Arrb mutant mice and critical input. M.F.B. directed and coordinated the experiments. L.J.S. designed and performed biochemistry and behavioral experiments. B.D.A. and R.K.S. designed and performed electrophysiological recordings. R.K.S. designed and performed fluorescence-based calcium imaging experiments. A.R.P. designed analysis code in MATLAB to analyze hyper-locomotion experiments and assisted with behavioral experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abou Farha K, Bruggeman R, Balje-Volkers C. Metabotropic glutamate receptor 5 negative modulation in phase I clinical trial: potential impact of circadian rhythm on the neuropsychiatric adverse reactions-do hallucinations matter? ISRN Psychiatry. 2014;2014:652750. doi: 10.1155/2014/652750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Valles A, Matta-Camacho E, Khoutorsky A, Gkogkas C, Nader K, Lacaille JC, Sonenberg N. Inhibition of Group I Metabotropic Glutamate Receptors Reverses Autistic-Like Phenotypes Caused by Deficiency of the Translation Repressor eIF4E Binding Protein 2. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:11125–11132. doi: 10.1523/JNEUROSCI.4615-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ. {beta}-Arrestin-2 Mediates Anti-apoptotic Signaling through Regulation of BAD Phosphorylation. J Biol Chem. 2009;284:8855–8865. doi: 10.1074/jbc.M808463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes SA, Wijetunge LS, Jackson AD, Katsanevaki D, Osterweil EK, Komiyama NH, Grant SG, Bear MF, Nagerl UV, Kind PC, et al. Convergence of Hippocampal Pathophysiology in Syngap+/− and Fmr1−/y Mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:15073–15081. doi: 10.1523/JNEUROSCI.1087-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Des Portes V, Hagerman R, Jacquemont S, Charles P, Visootsak J, Brinkman M, Rerat K, Koumaras B, Zhu L, et al. Mavoglurant in fragile X syndrome: Results of two randomized, double-blind, placebo-controlled trials. Sci Transl Med. 2016;8:321ra325. doi: 10.1126/scitranslmed.aab4109. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Dolen G, Bear MF. The pathophysiology of fragile X (and what it teaches us about synapses) Annu Rev Neurosci. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism. 2010;1:15. doi: 10.1186/2040-2392-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, et al. 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J Med Chem. 2003;46:204–206. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Kim J, Whalen EJ, Ahn S, Chen M, Lefkowitz RJ. Beta-arrestin-mediated signaling regulates protein synthesis. J Biol Chem. 2008;283:10611–10620. doi: 10.1074/jbc.M710515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng AG, Kelver DA, Hedrick TP, Swanson GT. Transduction of group I mGluR-mediated synaptic plasticity by beta-arrestin2 signalling. Nature communications. 2016;7:13571. doi: 10.1038/ncomms13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL. A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol. 2001;537:421–430. doi: 10.1111/j.1469-7793.2001.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, Huber KM. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:4859–4864. doi: 10.1523/JNEUROSCI.5407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, et al. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- Gross C, Chang CW, Kelly SM, Bhattacharya A, McBride SM, Danielson SW, Jiang MQ, Chan CB, Ye K, Gibson JR, et al. Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell reports. 2015;11:727–736. doi: 10.1016/j.celrep.2015.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathaway HA, Pshenichkin S, Grajkowska E, Gelb T, Emery AC, Wolfe BB, Wroblewski JT. Pharmacological characterization of mGlu1 receptors in cerebellar granule cells reveals biased agonism. Neuropharmacology. 2015;93:199–208. doi: 10.1016/j.neuropharm.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson C, Wijetunge L, Kinoshita MN, Shumway M, Hammond RS, Postma FR, Brynczka C, Rush R, Thomas A, Paylor R, et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci Transl Med. 2012;4:152ra128. doi: 10.1126/scitranslmed.3004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun H, Stefani MR, Adams BW, Tamagan GD, Moghaddam B. Functional Interaction Between NMDA and mGlu5 Receptors: Effects on Working Memory, Instrumental Learning, Motor Behaviors, and Dopamine Release. Neuropsychopharmacology. 2004;29:1259–1269. doi: 10.1038/sj.npp.1300417. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis--dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Felicioni M, Nistico R, Nicoletti F, De Blasi A. Selective regulation of recombinantly expressed mGlu7 metabotropic glutamate receptors by G protein-coupled receptor kinases and arrestins. Neuropharmacology. 2014;77:303–312. doi: 10.1016/j.neuropharm.2013.10.013. [DOI] [PubMed] [Google Scholar]
- Insel PA, Tang CM, Hahntow I, Michel MC. Impact of GPCRs in clinical medicine: monogenic diseases, genetic variants and drug targets. Biochim Biophys Acta. 2007;1768:994–1005. doi: 10.1016/j.bbamem.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann L, Jaeschke G, Michalon A, Vieira E, Honer M, Spooren W, Porter R, Hartung T, Kolczewski S, Buttelmann B, et al. CTEP: a novel, potent, long-acting, and orally bioavailable metabotropic glutamate receptor 5 inhibitor. J Pharmacol Exp Ther. 2011;339:474–486. doi: 10.1124/jpet.111.185660. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, Bear MF. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron. 2013;77:243–250. doi: 10.1016/j.neuron.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neuroscience. 2010;30:15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecknold JC, McClure DJ, Appeltauer L, Wrzesinski L, Allan T. Treatment of anxiety using fenobam (a nonbenzodiazepine) in a double-blind standard (diazepam) placebo-controlled study. J Clin Psychopharmacol. 1982;2:129–133. [PubMed] [Google Scholar]
- Pietraszek M, Gravius A, Schafer D, Weil T, Trifanova D, Danysz W. mGluR5, but not mGluR1, antagonist modifies MK-801-induced locomotor activity and deficit of prepulse inhibition. Neuropharmacology. 2005;49:73–85. doi: 10.1016/j.neuropharm.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Pop AS, Gomez-Mancilla B, Neri G, Willemsen R, Gasparini F. Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development. Psychopharmacology (Berl) 2014;231:1217–1226. doi: 10.1007/s00213-013-3330-3. [DOI] [PubMed] [Google Scholar]
- Porter RH, Jaeschke G, Spooren W, Ballard TM, Buttelmann B, Kolczewski S, Peters JU, Prinssen E, Wichmann J, Vieira E, et al. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther. 2005;315:711–721. doi: 10.1124/jpet.105.089839. [DOI] [PubMed] [Google Scholar]
- Qin M, Kang J, Burlin TV, Jiang C, Smith CB. Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:5087–5095. doi: 10.1523/JNEUROSCI.0093-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M, Kang J, Smith CB. Increased rates of cerebral glucose metabolism in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:15758–15763. doi: 10.1073/pnas.242377399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Bassell GJ, Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci. 2015;16:595–605. doi: 10.1038/nrn4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Wu J, Rowan MJ, Anwyl R. Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:6121–6128. doi: 10.1523/JNEUROSCI.22-14-06121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf SH, Jaeschke G, Wettstein JG, Lindemann L. Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr Opin Pharmacol. 2015;20:124–134. doi: 10.1016/j.coph.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick IC, Collingridge GL. An investigation into signal transduction mechanisms involved in DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology. 1999;38:1585–1596. doi: 10.1016/s0028-3908(99)00062-3. [DOI] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffler DJ, Gregory KJ, Rook JM, Conn PJ. Allosteric modulation of metabotropic glutamate receptors. Adv Pharmacol. 2011;62:37–77. doi: 10.1016/B978-0-12-385952-5.00010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Stoppel LJ, Heynen AJ, Lindemann L, Jaeschke G, Mills AA, Bear MF. Contribution of mGluR5 to pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat Neurosci. 2015;18:182–184. doi: 10.1038/nn.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger TL, Kao C, McDonald-McGinn DM, Zackai EH, Bailey A, Schultz RT, Morrow BE, Emanuel BS, Hakonarson H. The Role of mGluR Copy Number Variation in Genetic and Environmental Forms of Syndromic Autism Spectrum Disorder. Sci Rep. 2016;6:19372. doi: 10.1038/srep19372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol Med. 2011;17:126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.