

Abstract
Heterochromatin is marked by methylation of lysine 9 on histone H3 (H3K9me). A puzzling feature of H3K9me is that this modification localizes not only in promoters but also in internal regions (bodies) of silent transcription units. Despite its prevalence, the biological significance of gene‐body H3K9me remains enigmatic. Here we show that H3K9me‐associated removal of H3K4 monomethylation (H3K4me1) in gene bodies mediates transcriptional silencing. Mutations in an Arabidopsis H3K9 demethylase gene IBM1 induce ectopic H3K9me2 accumulation in gene bodies, with accompanying severe developmental defects. Through suppressor screening of the ibm1‐induced developmental defects, we identified the LDL2 gene, which encodes a homolog of conserved H3K4 demethylases. The ldl2 mutation suppressed the developmental defects, without suppressing the ibm1‐induced ectopic H3K9me2. The ectopic H3K9me2 mark directed removal of gene‐body H3K4me1 and caused transcriptional repression in an LDL2‐dependent manner. Furthermore, mutations of H3K9 methylases increased the level of H3K4me1 in the gene bodies of various transposable elements, and this H3K4me1 increase is a prerequisite for their transcriptional derepression. Our results uncover an unexpected role of gene‐body H3K9me2/H3K4me1 dynamics as a mediator of heterochromatin silencing and epigenome differentiation.
Keywords: gene body, heterochromatin, histone demethylase, histone methylation
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Plant Biology; Transcription
Introduction
In the genome of diverse eukaryotes, constitutively silent sequences, such as transposable elements (TEs) and repeats, are marked by H3K9me (Bender, 2004; Grewal & Elgin, 2007). According to the prevailing paradigm, H3K9me in promoters repress transcription. However, promoters are not the only regions with H3K9me; internal regions (bodies) of constitutively silent transcription units generally have heavy H3K9me as well. Mechanisms to target H3K9me to specific sequences have been extensively studied in multiple model organisms, including Arabidopsis (Saze & Kakutani, 2011; Matzke & Mosher, 2014; Du et al, 2015), but roles of body modifications remain largely unexplored.
Ectopic dimethylation of H3K9 (H3K9me2) in bodies of cellular genes are induced by an Arabidopsis mutation in H3K9 demethylase gene INCREASE IN BONSAI METHYLATION 1 (IBM1; Saze et al, 2008; Miura et al, 2009; Inagaki et al, 2010). In addition to H3K9me2, the ibm1 mutant accumulates DNA methylation in gene bodies, which reflects mutual enhancement of H3K9me2 and DNA methylation (Du et al, 2015). On the other hand, the ibm1 mutation does not affect these chromatin modifications in silent TEs; only transcribed genes are affected. Based on these results, we have proposed that IBM1 protein removes H3K9me2 from bodies of transcribed genes, contributing to differentiation of active and inactive chromatin states (Inagaki et al, 2010; Inagaki & Kakutani, 2012). Remarkably, the ibm1‐induced body H3K9me2 is associated with severe developmental defects, suggesting importance of body H3K9me2 in the gene control, although downstream mechanisms remain to be elucidated.
Here we show through forward genetic approach that the ibm1‐induced developmental defects are suppressed by the second‐site mutations in LYSINE‐SPECIFIC DEMETHYLASE 1‐LIKE 2 (LDL2), which encodes a homolog of H3K4 demethylases conserved among eukaryotes. The LDL2 function is also necessary for ibm1‐induced loss of H3K4me1 and transcriptional repression in genes with body H3K9me2. Furthermore, body H3K9me2 in diverse TEs also directs loss of body H3K4me1. Based on these results, we propose that the body H3K4me1/H3K9me2 dynamics by opposing functions of the histone demethylases induce epigenome differentiation.
Results
The developmental abnormalities of ibm1 depend on LDL2 function
To understand the components functioning downstream of the gene‐body H3K9me2, we mutagenized ibm1 seeds and screened for second‐site mutations that suppress the developmental defects of ibm1. We identified 18 mutants, 15 of which suppressed both developmental phenotypes and ectopic DNA methylation, which concomitantly occurs with the ectopic H3K9me2, in ibm1 (Type I). The Type I mutations are comprised of known factors necessary for methylation of H3K9 and DNA, namely CHROMOMETHYLASE3 (CMT3), KRYPTONITE (KYP)/SUVH4, and HOMOLOGY‐DEPENDENT GENE SILENCING1 (HOG1). Here we characterize the other type of mutants, which suppressed the ibm1‐induced developmental phenotypes with keeping the ectopic DNA methylation (Type II; Fig 1A–C; Appendix Fig S1A and B), because the causal mutations could affect the downstream components.
Figure 1. LDL2 function mediates the developmental phenotypes of ibm1 .

-
A–CThe ldl2 mutations suppress developmental phenotypes, such as small and distorted leaves (A), abnormal inflorescence (B) and low fertility (C), of ibm1. The ibm1 plants are ibm1 LDL2 siblings of the segregating ibm1 ldl2‐11 mutant.
-
DStructure and sites of mutations in the LDL2 gene. SWIRM, Swi3p, Rsc8p, and Moira domain; AOD, amine oxidase domain.
-
E, FLDL2 dsRNA (RNAi) suppresses leaf (E) and fertility (F) phenotypes of ibm1.
To identify the gene responsible for a Type II mutation, we carried out bulked segregant analysis. After backcrossing of the mutant, we examined distortion of SNPs in a sibling population segregating plants with and without the developmental phenotypes (see Materials and Methods). The most distorted region included LDL2 gene. The LDL2 allele in this mutant (hereafter we call ldl2‐11) had a mutation converting Trp531 into a stop codon (Fig 1D). We then sequenced the LDL2 gene in the other Type II mutants and identified another mutant allele of LDL2 (ldl2‐12), which substitutes Gly144 to Glu (Fig 1A–D; Appendix Fig S1A and B).
To test whether the suppression of ibm1 phenotypes is due to the loss of function in LDL2 gene, we knocked down expression of the LDL2 gene in the ibm1 background by a transgene producing double‐strand RNA (dsRNA). In all the transgenic lines (6/6), plants carrying the LDL2 dsRNA transgene showed suppressed ibm1 phenotypes, while sibling segregants lacking the dsRNA transgene showed developmental abnormalities characteristic of ibm1 (Fig 1E and F; Appendix Fig S1C and D). In addition, transgenes with the LDL2 sequence complemented the suppressed phenotypes of ibm1 (Appendix Fig S1E). These results confirmed that LDL2 mediates developmental phenotypes in the ibm1 mutant.
We next investigated H3K9me2 and DNA methylation genome‐wide in the ibm1 and ibm1 ldl2 mutants. The ibm1 ldl2 double mutant induced ectopic H3K9me2 (Fig 2A and B) and DNA methylation at non‐CG sites (Fig 2C) in more than three thousand genes to the level indistinguishable from those of the ibm1 single mutant. These results suggest that LDL2 mediates developmental phenotypes in the downstream of gene‐body heterochromatin marks ectopically accumulated in the ibm1 mutant.
Figure 2. LDL2 functions downstream of ibm1‐induced ectopic H3K9me2 and non‐CG methylation.
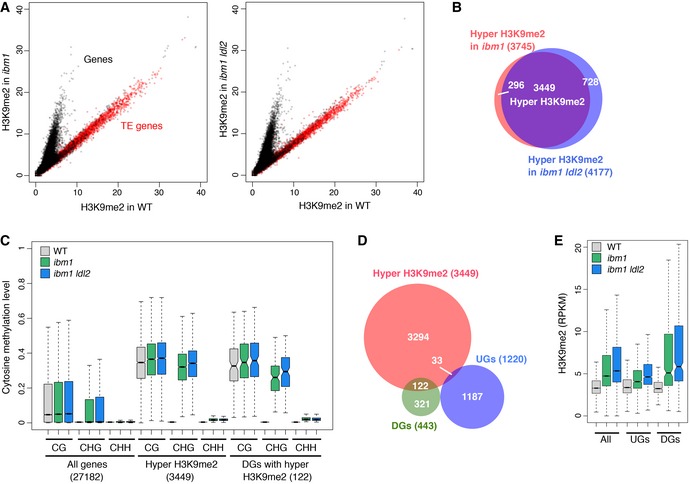
- H3K9me2 levels in ibm1 (left) or ibm1 ldl2 (right) compared to WT. Each dot represents the square root of RPM (read counts per million mapped reads) within each transcription unit (black, protein‐coding genes; red, TE genes).
- Venn diagram showing the overlap between the genes with hyper H3K9me2 in ibm1 and the genes with hyper H3K9me2 in ibm1 ldl2.
- Boxplots showing DNA methylation levels in each context (CG, CHG, and CHH; H can be A, T, or C) in the bodies of all genes, genes with hyper H3K9me2, and DGs with hyper H3K9me2. Numbers in parentheses indicate numbers of genes in each group. Thick horizontal bar corresponds to the median, with the notch representing 95% confidence interval of the median. Upper and lower limits of box correspond to upper and lower quantile, respectively. Whisker indicates data range within 1.5× of the interquantile range and outliers are not shown.
- Venn diagram showing the overlap between the DGs, the UGs, and the genes with hyper H3K9me2. Genes with hyper H3K9me2 are over‐represented (P = 7.6 × 10−25, hypergeometric test) in DGs, while under‐represented (P = 5.9 × 10−25) in UGs.
- Boxplots for H3K9me2 levels (read counts per kilobase of gene per million mapped reads; RPKM) in the bodies of all genes, UGs and DGs. P < 1 × 10−3 for each gene set versus all genes, in both ibm1 and ibm1 ldl2 mutants, two‐sample Kolmogorov–Smirnov test. Boxplot as described in (C).
LDL2 mediates loss of H3K4me1 and down‐regulation of genes accumulating H3K9me2 in ibm1
To understand the molecular basis for the LDL2‐mediated developmental defects, we examined genes up‐ or down‐regulated in ibm1 and restored transcription in ibm1 ldl2 (Appendix Fig S2A and B). We hereafter call these two types of genes as “down‐regulated genes (DGs)” and “up‐regulated genes (UGs)”, respectively (Appendix Fig S2C and D). We analyzed both protein‐coding genes and TEs, but not many TEs were found in this screening; the 443 DGs are comprised of 432 protein‐coding genes, three TE genes, six non‐coding genes, and two pseudogenes, and the 1,220 UG are comprised of 1,155 protein‐coding genes, 47 TE genes, five non‐coding genes, and 13 pseudogenes (protein‐coding genes are significantly over‐represented for both DGs and UGs; by hypergeometric test, P = 5.5 × 10−27 and P = 1.3 × 10−45, respectively). Importantly, a large proportion of DGs (122 out of 443; 119 protein‐coding genes and three non‐coding genes) showed ectopic accumulation of H3K9me2 in their bodies; in contrast, ectopic H3K9me2 accumulation in UG occurred in lower level and lower proportion than in total genes (Fig 2D and E). Most of the ibm1‐induced transcriptional up‐regulation seems indirect effects, while a significant portion of the down‐regulation is likely caused directly by ectopic H3K9me2 and/or non‐CG methylation. Ectopic accumulation of H3K9me2 and non‐CG methylation in DGs was similarly seen in ibm1 and ibm1 ldl2 (Fig 2C and E), again suggesting that the ldl2 mutation affects downstream events, rather than the ectopic accumulation of heterochromatin marks.
The LDL2 gene encodes one of four Arabidopsis homologs of LSD1 class of histone H3K4 demethylases in animals (Shi et al, 2004; Jiang et al, 2007; Rudolph et al, 2007). Among the four LSD1‐like genes in Arabidopsis, mutations in FLD (flowering locus D) are known to cause late‐flowering phenotypes (He et al, 2003), but knowledge about phenotypic effects of mutations in three other members (LDL1, LDL2, and LDL3) has been limited. Increases in H3K4me2 level have been reported for the fld mutant on FLC (flowering locus C) locus (Liu et al, 2007), and an ldl1 ldl2 double mutant on a subset of genes (Greenberg et al, 2013), although effects of any of these Arabidopsis mutations on H3K4me1 have not been reported.
Because these previously reported results and the structure of LDL2 suggest its function as an H3K4 demethylase, we examined tri‐, di‐, and monomethylation of H3K4 genome‐wide using chromatin immunoprecipitation followed by sequencing (ChIP‐seq). H3K4me2 and H3K4me3 were mostly unaffected by the ibm1 or ibm1 ldl2 mutation (Appendix Fig S3). In contrast, many genes showed decreased H3K4me1 levels in ibm1 (Fig 3A, left panel). The H3K4me1 decrease was correlated with the ibm1‐induced H3K9me2 (Fig 3B, left panel), and most of the genes with significant decrease in H3K4me1 accumulate ectopic H3K9me2 (Fig 3C). These results suggest that the ibm1‐induced genic H3K9me2 results in the loss of H3K4me1 in a subset of genes. On the other hand, most genes with decreased H3K4me1 in ibm1 did not show comparable H3K4me1 loss in the ibm1 ldl2 double mutant (Fig 3A and B, right panels; Fig 3C), suggesting that the decrease in genic H3K4me1 in ibm1 depends on the LDL2 function. Taken together, these results suggest that LDL2 induces loss of H3K4me1 as a consequence of the ibm1‐induced genic H3K9me2.
Figure 3. ibm1 mutation induces decrease in H3K4me1 in genes accumulating H3K9me2, and that effect is suppressed by ldl2 mutation.
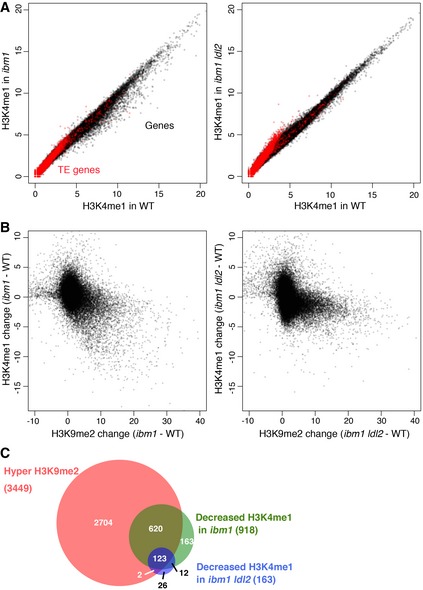
- H3K4me1 levels in ibm1 (left) or ibm1 ldl2 (right) compared to WT, with conditions in Fig 2A.
- Relationships between changes in H3K9me2 and H3K4me1 levels (RPKM) within each gene in ibm1 (left) or ibm1 ldl2 (right) compared to WT.
- Venn diagram showing the overlap between genes with hyper H3K9me2 and genes with decreased H3K4me1 level in ibm1 and ibm1 ldl2. The number of genes with hyper H3K9me2 and decreased H3K4me1 in ibm1 ldl2 (125) is significantly smaller than that in ibm1 (743; P < 2.2 × 10−16, Fisher's exact test, n = 3,449).
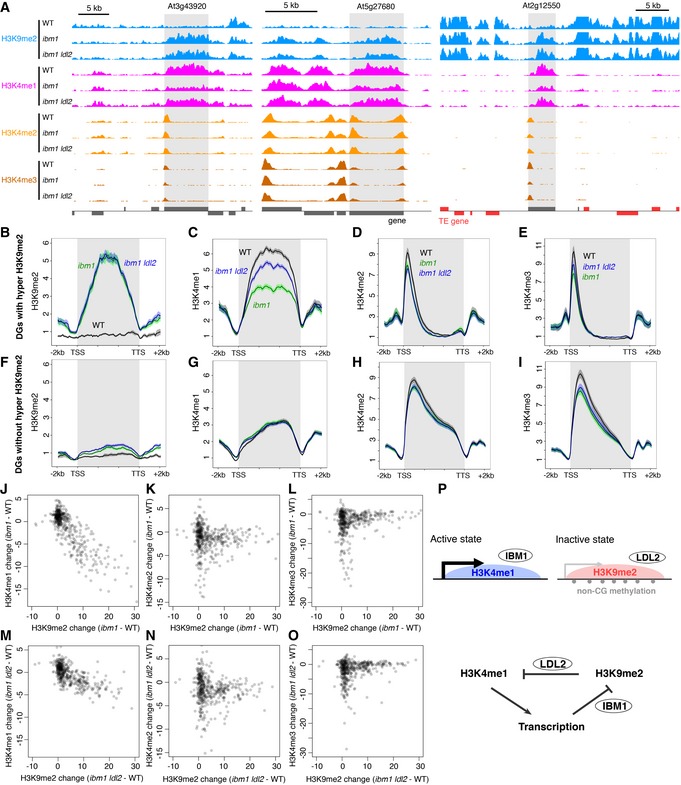
Next we analyzed H3K4 methylations in DGs, expecting that they include genes changing their expression as a response to body H3K9me2. We found that most DGs with gene‐body H3K9me2 showed loss of body H3K4me1 in ibm1 (Fig 4A–C); in contrast, DGs without H3K9me2 accumulation did not show H3K4me1 changes (Fig 4F and G). The decrease in H3K4me1 correlated with H3K9me2 accumulation (Fig 4J). Although the ldl2 mutation did not affect the ibm1‐induced H3K9me2 in DGs (Fig 4A and B), this mutation suppressed most of the ibm1‐induced decrease in H3K4me1 (Fig 4A, C and M). Thus, the LDL2‐dependent loss of H3K4me1 in genes accumulating H3K9me2, which is generally seen many genes (Fig 3), is especially conspicuous in DGs.
Figure 4. H3K9me2 induces LDL2‐dependent loss of H3K4me1.
-
DPatterns of H3K9me2 (range; 0–8), H3K4me1 (0–5), H3K4me2 (0–10), and H3K4me3 (0–15) around representative DGs (shaded). Normalized coverage (per million mapped reads) is shown. Genes (dark gray) and TE genes (red) in forward and reverse orientations are shown above and below the line, respectively.
-
B–IAveraged profiles of H3K9me2 (B, F), H3K4me1 (C, G), H3K4me2 (D, H), and H3K4me3 (E, I) around DGs with (B–E) or without (F–I) hyper H3K9me2. The numbers of genes analyzed are 122 (B–E) and 321 (F–I). The ribbons indicate s.e.m.
-
J–ORelationships between changes in H3K9me2 and H3K4me1 (J, M), H3K4me2 (K, N), and H3K4me3 (L, O) levels in ibm1 (J–L) or ibm1 ldl2 (M–O) compared to WT within each DG represented by each dot. Value represents RPKM.
-
PA model for differentiation of bi‐stable epigenetic states through opposing actions of histone demethylases LDL2 and IBM1. See text for details.
In addition to H3K4me1, some DGs showed decreases in H3K4me2 and H3K4me3 in ibm1 (Fig 4A, D, E, H and I), but effects of ldl2 mutation on H3K4me2/3 were smaller than that on H3K4me1. In addition, changes in H3K4me2/3 were not associated with the accumulation of H3K9me2 (Fig 4K, L, N and O). H3K4me2/3 signals show peaks near transcription start site (Fig 4A, D, E, H and I). That is in contrast to the localization of H3K4me1 in gene body, which largely overlaps with the ibm1‐induced H3K9me2 (Fig 4A–C).
Taken together, these results suggest that LDL2 down‐regulates body H3K4me1 as a consequence of body H3K9me2, and this loss of body H3K4me1 down‐regulates the expression of the DGs (Fig 4P; detailed discussion about the model in the Discussion section).
H3K9me2 directs loss of H3K4me1 in diverse TEs
The results above suggest that ectopic H3K9me2 and non‐CG methylation induce transcriptional gene silencing through decrease in H3K4me1. We wondered whether a similar mechanism silences the transcription of TEs, which are major targets of H3K9me2 and non‐CG methylation in wild type. To see whether H3K9me2 in TEs also controls H3K4me1, we examined H3K4 methylations and RNA in the triple mutant of H3K9 methylase genes KRYPTONITE (KYP)/SUVH4, SUVH5, and SUVH6 (Fig 5 and Appendix Fig S4). These SUVH genes redundantly control H3K9me2 in TEs (Ebbs & Bender, 2006; Stroud et al, 2013). In the triple mutant (suvh456), a large number of TEs showed a drastic increase in the H3K4me1 level, suggesting that H3K9me2 down‐regulates H3K4me1 in TEs (Figs 5A and 6A–D, G and H). In many of the TEs, the increase in H3K4me1 was associated with transcriptional derepression and loss of H3K9me2 (Fig 6K and L). Interestingly, increase in H3K4me1 was also found in TEs losing H3K9me2 without apparent transcriptional activation (Fig 6K and O), suggesting that the suvh456‐induced increase in H3K4me1 does not necessarily depend on the transcription. Instead, the increase in H3K4me1 strongly correlates with reduction in H3K9me2. That was the case for TE populations both with and without the transcriptional derepression (Fig 6L and O). Furthermore, almost all of TEs without H3K4me1 accumulation remain silent (Fig 6K), further suggesting that H3K4me1 functions upstream of transcriptional derepression. Consistent with a previous report (Zhang et al, 2009), transcriptional TE activation in suvh456 also correlated with elevation of H3K4me2 and H3K4me3 levels around the TE promoters (Figs 5B and C, and 6A, B, E and F). However, the correlation of the H3K4me2/3 elevation with loss of H3K9me2 was much weaker than that of H3K4me1 and the elevation was limited to transcriptionally up‐regulated TEs (Fig 6I, J, M, N, P and Q). These features of H3K4me2/3 are in contrast to more tight and transcription‐independent association of H3K4me1 to H3K9me2 in the TE bodies. Taken together, these observations suggest that the H3K9me2 in the TE bodies induces H3K4me1 demethylation, which mediates transcriptional silencing in these TEs, as is the case in the silencing of DGs (Fig 4).
Figure 5. Many TEs gain H3K4me1 in suvh456 .
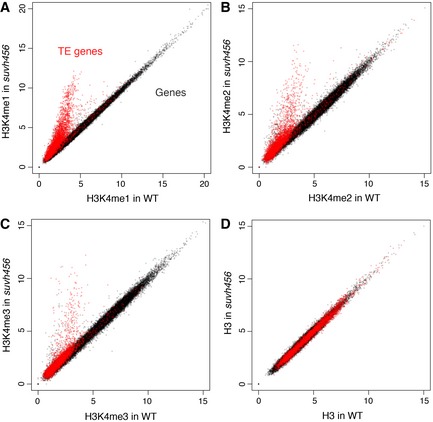
-
A–DComparisons of H3K4me1 (A), H3K4me2 (B), H3K4me3 (C), and H3 (D) in each transcriptional unit between WT and suvh456.
Figure 6. Loss of H3K9me2 induces gain of H3K4me1 in TEs.
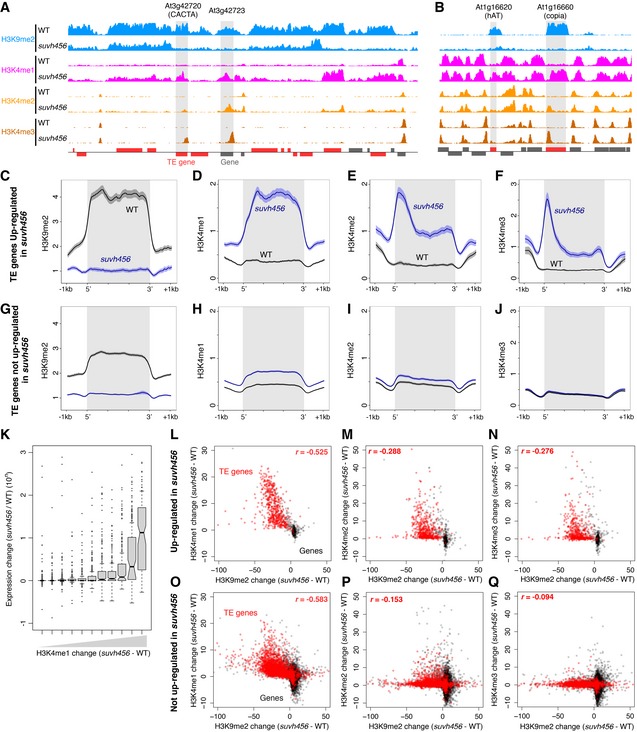
-
A, BHistone H3 methylation patterns in a representative pericentromeric region (A) and in a representative arm region (B). Normalized coverage is shown as in Fig 4A. Shaded region indicates the body of gene or TE gene whose expression is up‐regulated in suvh456 compared to WT.
-
C–JPatterns of H3K9me2 (C, G), H3K4me1 (D, H), H3K4me2 (E, I), and H3K4me3 (F, J) in TE genes whose expressions are up‐regulated (C–F) or not up‐regulated (G–J) in suvh456 compared to WT. The numbers of TE genes analyzed are 549 (C–F) and 3,320 (G–J). The ribbons indicate s.e.m. At2g01022, a TE gene next to the ribosomal DNA cluster in the chromosome 2, was excluded from the analysis to avoid the effect of a large number of reads artificially mapped there.
-
KBoxplots showing relationship between change in H3K4me1 and change in expression level in suvh456 compared to WT in TEs. All TE genes with decreased H3K9me2 in suvh456 are binned into 11 groups according to H3K4me1 change (suvh456‐WT) and boxplot of expression change (suvh456/WT) for each group is shown. Boxplots are shown as in Fig 2C, with outliers indicated by dots.
-
L–QRelationships between suvh456‐induced changes in H3K9me2 and H3K4me1 (L, O), H3K4me2 (M, P), and H3K4me3 (N, Q) levels (RPKM) within TE genes (red) and genes (black). Genes with (L–N) and without (O–Q) transcriptional up‐regulation by suvh456 are shown separately. Spearman's correlation coefficients between changes in H3K9me2 and H3K4me in TE genes are shown.
Discussion
The results of this study suggest that LDL2 down‐regulates H3K4me1 as a consequence of H3K9me2 within gene bodies, and this loss of H3K4me1 mediates reduced expression in many of DGs. We have previously shown that IBM1 removes H3K9me2 from bodies of transcribed genes (Inagaki et al, 2010). In support of this, IBM1 protein is localized in genes with higher expression level, but not in non‐expressed genes or TE genes (Appendix Fig S5). Based on the previous observations and the new results shown above, we propose a model for differentiation of bi‐stable epigenetic states through two opposing activities of histone demethylases on gene bodies (Fig 4P). In gene bodies, transcription induces loss of H3K9me2 thorough action of IBM1, and H3K9me2 induces loss of H3K4me1 through action of LDL2. We posit that these two activities generate positive feedback loop in which H3K4me1 in the gene body ensure transcription (Fig 4P). The positive feedback would lead to differentiation of active and inactive states through the controls of transcription and body modifications.
The connection between H3K4me1 and H3K9me2 was also found in TEs. Mutations in H3K9 methylase genes (suvh456) induced large increase in H3K4me1 level in the bodies of diverse TEs (Fig 5A), and the increased body H3K4me1 seems prerequisite for their transcriptional derepression (Fig 6K). Although this pathway looks comparable to that shown in Fig 4P, LDL2 may not be sufficient for these effects on TEs. The ldl2 mutation induced increased H3K4me1 and transcriptional derepression of TEs (Fig 3A, right panel; Appendix Figs S6 and S7), but these effects were much smaller than those of suvh456. One possible explanation for these observations is that in TEs, LDL2 redundantly function with other H3K4 demethylase(s), such as other LDL(s) or Jumonji‐C domain‐containing protein(s), as the response to H3K9me2.
In regard to the body H3K9me2/H3K4me1 dynamics, important next questions are (i) how H3K9me2 directs the loss of H3K4me1, and (ii) how H3K4me1 within gene bodies ensures transcription. For the question (i), a simple hypothesis would be that H3K9me2 recruits demethylase(s) of H3K4me1. Indeed, it has recently been reported in C. elegans that LSD1 family H3K4 demethylases are recruited to heterochromatic histone modifications (Vandamme et al, 2015). This hypothesis is also consistent with our observations that changes in H3K9me2 and H3K4me1 both occurred in the bodies of transcription units (Figs 4B and C, and 6C and D). Addressing the question (ii) is more challenging and interesting biologically. One possibility is that H3K4me1 facilitates the passage of transcription machinery, which in turn affects transcription initiation and/or RNA processing. A recent report showed that both transcription initiation and elongation in FLC locus are much enhanced by loss of function in another Arabidopsis H3K4 demethylase FLD, and this protein localizes in the body of FLC gene (Wu et al, 2015). An alternative, but not mutually exclusive, possibility would be that gene body might act as a distal controller of transcription initiation, such as intragenic enhancers. Indeed, H3K4me1 is often found in enhancers in the animal genomes (Heintzman et al, 2007; Herz et al, 2012; Whyte et al, 2012), and intragenic transcription controlling elements can be more general in TEs than in genes (Inagaki & Kakutani, 2012). It is tempting to speculate that the novel pathway shown in this work (Fig 4P) links expression and body modifications, and the link could contribute to epigenome differentiation of genes and TEs.
Our genetic and genomic analyses revealed that LDL2 functions downstream of gene‐body H3K9me2 to silence a subset of genes, most likely through decrease in body H3K4me1. Although we identified many genes with ibm1‐induced transcriptional changes, most of the effects can be indirect, and we still do not know which key genes (most likely DGs) trigger the variety of developmental and transcriptional changes. Gene Ontology (GO) analysis showed that genes involved in defense responses to pathogens are highly enriched in UGs (Appendix Fig S8), suggesting that the ibm1 mutant constitutively activates defense responses. Connections between DNA methylation and defense responses to pathogens have been reported (Dowen et al, 2012; Yu et al, 2013), and a recent report shows that immune response loci of Arabidopsis are enriched in genes with natural variations in heterochromatin marks (Kawakatsu et al, 2016). Biological significance of the link between gene‐body heterochromatin and defense responses will be a focus of next study.
Materials and Methods
Plant materials
Arabidopsis thaliana strain Columbia‐0 (Col‐0) was used as “wild type (WT)”. The ibm1‐1 and ibm1‐4 mutants were described previously (Saze et al, 2008). The kyp/suvh4 suvh5 suvh6 triple mutant was described previously (Inagaki et al, 2010); it was constructed from single mutants, kindly provided by Judith Bender. A T‐DNA insertion mutant ldl2 (Jiang et al, 2007) was kindly provided by Yuehui He.
Identification and methylation analyses of mutants suppressing developmental phenotypes of ibm1
Seeds from the first generation (generation segregating from a heterozygote) of ibm1‐4 mutant were mutagenized with ethyl methanesulfonate (EMS), and the M2 plants (the third generation of ibm1‐4) were screened for mutants with suppressed phenotypes (population 1). As another population, we also mutagenized ibm1‐4/ibm1‐4 DDM1/ddm1‐1 seeds and screened M2 plants for the suppressed phenotypes (population 2), because ddm1 mutation generally strengthens the ibm1‐induced developmental defects (Saze et al, 2008).
In population 1, we examined developmental phenotypes and DNA methylation in parallel. DNA methylation was examined using methylation‐sensitive restriction endonuclease McrBC, which cleaves DNA containing methylated cytosine. As target loci, we used two genes, KYP/SUVH4 (At5g13960) and ERECTA‐LIKE 2 (ERL2, At5g07180), which show ibm1‐induced non‐CG methylation (Miura et al, 2009). Compared to wild type, the ibm1 mutant showed less amplification of PCR fragment after McrBC digestion and PCR. We looked for mutants with stronger amplification after McrBC‐PCR. We identified eight mutants that showed reduced DNA methylation (Type I), and two mutants that did not show reduced DNA methylation but showed suppressed developmental phenotypes (Type II). In population 2, we first identified eight mutants that suppressed developmental phenotypes of ibm1. We then analyzed DNA methylation at At2g04860 locus using McrBC digestion and PCR. Seven of eight mutants showed reduced DNA methylation (Type I), while the other one kept the ibm1‐induced DNA methylation (Type II). In the Type I mutants, we identified new mutant alleles of CMT3 (three in population 1 and five in population 2), KYP/SUVH4 (one from population 2), and HOG1 (one from population 1). In order to identify a novel pathway, we concentrated on the Type II mutants in this study. Primers used are listed in Appendix Table S1.
Identification of the LDL2 gene
To identify the gene responsible for a Type II mutation, we carried out bulked segregant analysis. A Type II mutant from population 1 was backcrossed to the parental ibm1‐4 mutant twice and self‐pollinated to obtain progeny segregating the suppressor phenotypes. In this segregating population of 104 plants, we selected 16 plants with clear suppression of the phenotypes and 19 plants with clear phenotypes typical of the ibm1 mutants, and the DNA was sequenced with Illumina GAII sequencer in bulked samples for the plants with and without the phenotypes. SNPs from the reference Col genome were detected for these pools and distortion of the SNP distributions in these pools was assessed by calculating the chi‐squared value. Among SNPs causing amino acid substitution, the fourth most distorted SNP was in the LDL2 gene (Appendix Table S2).
Plasmid construction
RNAi construct for LDL2 was made using pHELLSGATE12 (Helliwell & Waterhouse, 2003). A 466‐bp region around start codon of LDL2 gene was amplified with primers with attB adapters, attB1‐LDL2‐RNAi and attB2‐LDL2‐RNAi, and insert the fragment into pDONR201 vector by using BP Clonase (Thermo Fisher Scientific). Resulting plasmid pDONR201‐LDL2‐RNAi and pHELLSGATE12 were recombined with LR Clonase to produce a hairpin RNA construct pHELLSGATE12‐LDL2‐RNAi. For pLDL2::LDL2‐FLAG, a genomic region spanning the promoter, exon and intron of LDL2 gene until just before its stop codon was amplified using primers, attB1‐LDL2proF and attB2‐LDL2R. The amplified fragment was inserted into pDONR201 to make pDONR201‐LDL2full, and subsequently into pGWB610 (Nakamura et al, 2010) to make pGWB610‐pLDL2::LDL2‐FLAG. pGWB610 containing bar gene identified by Meiji Seika Kaisha, Ltd., was kindly provided by Tsuyoshi Nakagawa. To make pLDL2::LDL2(K436A)‐FLAG, pDONR201‐LDL2full was mutated using a method described previously (Hansson et al, 2008). Briefly, pDONR201‐LDL2full was used as template for PCR with primers, LDL2‐K436A‐F and LDL2‐K436A‐R, to introduce AAA to GCA change. After treating with DpnI (New England Biolabs) to degrade template plasmid, PCR fragment by itself was used to transform E. coli competent cells. Resulting plasmid pDONR201‐LDL2(K436A) was recombined with pGWB610 to make pGWB610‐pLDL2::LDL2(K436A)‐FLAG.
BS‐seq
Whole‐genome bisulfite sequencing (BS‐seq) was conducted as described before (Fu et al, 2013); 100‐bp paired‐end reads were trimmed for the adapter sequences and low‐quality regions using the Trimmomatic program (Bolger et al, 2014). Subsequent trimmed reads were mapped to the Arabidopsis reference genome TAIR10 using Bismark ver. 0.10.1 (Krueger & Andrews, 2011) with ‐n 1 ‐l 20 ‐e 90 parameters. Removal of identical reads (deduplication) and counting of methylated and unmethylated cytosines for each site (methylation extraction) were also performed using Bismark. The “map” function of BEDTools (Quinlan & Hall, 2010) was used to calculate all the methylated and unmethylated cytosines in each transcriptional unit. Methylation level was calculated as the ratio of total methylated cytosines over total cytosines in each region (weighted methylation level; Schultz et al, 2012). Two independent biological replicates were analyzed for each genotype. Control WT data are from our previous report (Ito et al, 2015) and analyzed in parallel.
ChIP‐seq
Chromatin immunoprecipitation (ChIP) was carried out with modifications of Luo & Lam (2014). The material seedlings were grown on Murashige and Skoog (MS) media supplemented with 1% sucrose and solidified with Bacto agar (Difco); 1.5 g of 12‐day‐old whole seedling was frozen with liquid nitrogen, ground into fine powder, cross‐linked, and nuclear‐extracted according to Luo & Lam (2014). Sonication was conducted using Covaris S2 Focused‐ultrasonicator (Covaris) and milliTUBE 1 ml AFA Fiber (Covaris) with following settings: power mode, frequency sweeping; time, 18–20 min; duty cycle, 5%; intensity, 4; cycles per burst, 200; and temperature (water bath), 4–6°C. Sonicated chromatin sample was divided into equal amount by the number of antibodies used. Following antibodies were used: mouse anti‐H3K9me2 (CMA307; Kimura et al, 2008), rabbit anti‐H3K4me1 (ab8895; Abcam), rabbit anti‐H3K4me2 (ab32356; Abcam), rabbit anti‐H3K4me3 (ab8580; Abcam), and rabbit anti‐H3 (ab1791; Abcam). Immunoprecipitation, wash, reverse cross‐linking, and DNA extraction were conducted as described previously (Kimura et al, 2008). Collected DNA was quantified with the Qubit dsDNA High Sensitivity Assay kit (Thermo Fisher Scientific), and 1–2 ng DNA was used to make library for Illumina sequencing. Library was constructed with the KAPA Hyper Prep Kit for Illumina (KAPA Biosystems), and dual size‐selected using Agencourt AMPure XP (Beckman Coulter) to enrich 300–500‐bp fragments. Libraries were pooled and 50‐bp single‐read sequences were obtained with HiSeq4000 sequencer (Illumina) in Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley. Two independent biological replicates were analyzed for each genotype.
Genome‐wide localization pattern of IBM1 protein was analyzed by using FLAG‐His‐HA‐gIBM1 transgenic plants (Saze et al, 2013). Whole seedlings (~1 g) of 12‐day‐old FLAG‐His‐HA‐gIBM1 plants and non‐transgenic control (Col‐0) were first cross‐linked with 1% formaldehyde in HEPES buffer under vacuum for 15 min. After adding glycine to 125 mM, another vacuum infiltration for 5 min was performed. ChIP was performed similar as histone modification ChIP described above with following modifications. Sonication was conducted with following settings: power mode, frequency sweeping; time, 25 min; duty cycle, 10%; intensity, 5; cycles per burst, 200; and temperature (water bath), 4–6°C. Rat anti‐HA antibody clone 3F10 (Sigma‐Aldrich) was used. Instead of high salt wash buffer (500 mM NaCl), medium salt wash buffer (300 mM NaCl) was used for the wash step.
mRNA‐seq
Total RNA was isolated from young tissues including shoot apices and small developing leaves of 12‐day‐old plants grown on MS media, using the RNeasy Plant Mini Kit (Qiagen), and treated with DNase I (Takara). Libraries for mRNA‐seq were constructed using the KAPA Stranded RNA‐seq Library Preparation Kit according to the manufacturer's instruction. First, poly(A) RNA was purified with the Dynabeads mRNA Purification Kit (Thermo Fisher Scientific) and then fragmented by heating purified RNA at 94°C for 7 min in fragment, prime, and elute buffer. Then, double‐strand cDNA was synthesized, and adapter‐ligated libraries were constructed. Libraries were pooled and sequenced as for ChIP‐seq. Three independent biological replicates were analyzed for each genotype.
Data analysis
For mRNA‐seq, quality‐filtered reads were mapped onto cDNA sequences of annotated genes and other transcripts of TAIR10 using Burrows–Wheeler Aligner (BWA) (Li & Durbin, 2009) and default parameters. After alignment, read count in each annotated unit was calculated using a Python script, Sam Gene Counter (http://comailab.genomecenter.ucdavis.edu/index.php/Hybrid_Transcriptome). Deferentially expressed genes (DEGs) are identified in R using the R package edgeR (Robinson et al, 2010), treating biological triplicates as paired samples. Genes with false discovery rate (FDR) < 0.05 in each comparison were identified as DEGs.
For ChIP‐seq, quality‐filtered reads were aligned onto the Arabidopsis reference genome TAIR10, using Bowtie (Langmead et al, 2009) with ‐m 1 –best parameters to report only uniquely mapped reads. Because many TEs contain repeat sequences, many reads derived from TEs cannot be uniquely aligned, and thus, high‐copy TEs cannot be analyzed with this Bowtie setting. Therefore, in Figs 5 and 6 focusing on TEs, we used ‐M 1 –best parameters, which allow non‐unique read to be aligned on a region randomly selected from multiple best hits. The resulting SAM files were converted to sorted BAM files using SAMtools (Li et al, 2009), and then converted to BED files using BEDTools (Quinlan & Hall, 2010). The “slop” function of BEDTools was used to extend the 5′ end of ChIP‐seq reads toward the 3′ direction to fit the average insertion size (250 bp) of the sequenced libraries. Then, the “coverage” function of BEDTools was used to calculate the number of reads that overlapped with each annotation unit. The diffReps program (Shen et al, 2013) was used to identify regions hyper‐accumulating H3K9me2 in ibm1 or ibm1 ldl2 compared to WT incorporating biological replicates. After filtering differentially modified regions with fold enrichment (> 2‐fold increase), the “coverage” function of BEDTools was used to assign identified regions onto annotation units. The same approach was taken to identify genes with decreased H3K4me1 using a fold enrichment threshold of 0.75. For visualization, WIG files were created using Model‐based Analysis for ChIP‐seq (MACS) program (Zhang et al, 2008) from BAM files, and visualized with Integrative Genome Viewer (IGV; Robinson et al, 2011). The ngs.plot program (Shen et al, 2014) was used to make methylation profile around gene bodies. All the downstream analysis including plotting figures and statistic analyses are conducted in R. Figures for ChIP‐seq results were shown for one of biological replicates, as replicates showed very high reproducibility. For IBM1 localization ChIP‐seq, because the total read numbers for the negative controls were small, reads from biological replicates were combined and analyzed as one sample.
The sequence data are deposited to DDBJ (Accession Number: DRA005154).
Author contributions
SI and TK designed the study. AH identified the ldl2‐12 mutant. TI, AT, AF, YT, and TK conducted the bulked segregant analysis and BS‐seq. SI and MT performed all the other experiments. SI analyzed the data. SI and TK wrote the manuscript with incorporating comments from other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
We thank Akiko Terui and Kazuya Takashima for technical assistance; Luca Comai for support with mRNA‐seq analysis; Judith Bender, Yuehui He, Hiroshi Kimura, Tsuyoshi Nakagawa, and Hidetoshi Saze for sharing materials; and Caroline Dean, Yasushi Hiromi, and Eric Richards for comments on the manuscript. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. Computations were partially performed on the NIG supercomputer at NIG, Japan. This study was supported by grants from Mitsubishi Foundation (to T.K.), Japanese Ministry of Education, Culture, Sports, Science and Technology (26221105 and 15H05963, to T.K.), and Systems Functional Genetics Project of the Transdisciplinary Research Integration Center, ROIS, Japan (to A.T., A.F., Y.T., and T.K.).
The EMBO Journal (2017) 36: 970–980
Contributor Information
Soichi Inagaki, Email: soinagak@nig.ac.jp.
Tetsuji Kakutani, Email: tkakutan@nig.ac.jp.
References
- Bender J (2004) DNA methylation and epigenetics. Ann Rev Plant Biol 55: 41–68 [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen RH, Pelizzola M, Schmitz RJ, Lister R, Dowen JM, Nery JR, Dixon JE, Ecker JR (2012) Widespread dynamic DNA methylation in response to biotic stress. Proc Natl Acad Sci USA 109: E2183–E2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Johnson LM, Jacobsen SE, Patel DJ (2015) DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol 16: 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbs ML, Bender J (2006) Locus‐specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell 18: 1166–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Kawabe A, Etcheverry M, Ito T, Toyoda A, Fujiyama A, Colot V, Tarutani Y, Kakutani T (2013) Mobilization of a plant transposon by expression of the transposon‐encoded anti‐silencing factor. EMBO J 32: 2407–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg MV, Deleris A, Hale CJ, Liu A, Feng S, Jacobsen SE (2013) Interplay between active chromatin marks and RNA‐directed DNA methylation in Arabidopsis thaliana . PLoS Genet 9: e1003946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI, Elgin SC (2007) Transcription and RNA interference in the formation of heterochromatin. Nature 447: 399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson MD, Rzeznicka K, Rosenbäck M, Hansson M, Sirijovski N (2008) PCR‐mediated deletion of plasmid DNA. Anal Biochem 375: 373–375 [DOI] [PubMed] [Google Scholar]
- He Y, Michaels SD, Amasino RM (2003) Regulation of flowering time by histone acetylation in Arabidopsis . Science 302: 1751–1754 [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318 [DOI] [PubMed] [Google Scholar]
- Helliwell C, Waterhouse P (2003) Constructs and methods for high‐throughput gene silencing in plants. Methods 30: 289–295 [DOI] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, Voets O, Verrijzer CP, Shilatifard A (2012) Enhancer‐associated H3K4 monomethylation by Trithorax‐related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev 26: 2604–2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki S, Miura‐Kamio A, Nakamura Y, Lu F, Cui X, Cao X, Kimura H, Saze H, Kakutani T (2010) Autocatalytic differentiation of epigenetic modifications within the Arabidopsis genome. EMBO J 29: 3496–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki S, Kakutani T (2012) What triggers differential DNA methylation of genes and TEs: contribution of body methylation? Cold Spring Harb Symp Quant Biol 77: 155–160 [DOI] [PubMed] [Google Scholar]
- Ito T, Tarutani Y, To TK, Kassam M, Duvernois‐Berthet E, Cortijo S, Takashima K, Saze H, Toyoda A, Fujiyama A, Colot V, Kakutani T (2015) Genome‐wide negative feedback drives transgenerational DNA methylation dynamics in Arabidopsis . PLoS Genet 11: e1005154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Yang W, He Y, Amasino RM (2007) Arabidopsis relatives of the human lysine‐specific Demethylase1 repress the expression of FWA and FLOWERING LOCUS C and thus promote the floral transition. Plant Cell 19: 2975–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakatsu T, Huang SS, Jupe F, Sasaki E, Schmitz RJ, Urich MA, Castanon R, Nery JR, Barragan C, He Y, Chen H, Dubin M, Lee CR, Wang C, Bemm F, Becker C, O'Neil R, O'Malley RC, Quarless DX, The 1001 Genomes Consortium et al (2016) Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell 166: 492–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Hayashi‐Takanaka Y, Goto Y, Takizawa N, Nozaki N (2008) The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct 33: 61–73 [DOI] [PubMed] [Google Scholar]
- Krueger F, Andrews SR (2011) Bismark: a flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics 27: 1571–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Quesada V, Crevillén P, Bäurle I, Swiezewski S, Dean C (2007) The Arabidopsis RNA‐binding protein FCA requires a lysine‐specific demethylase 1 homolog to downregulate FLC. Mol Cell 28: 398–407 [DOI] [PubMed] [Google Scholar]
- Luo C, Lam E (2014) Quantitatively profiling genome‐wide patterns of histone modifications in Arabidopsis thaliana using ChIP‐seq. Methods Mol Biol 1112: 177–193 [DOI] [PubMed] [Google Scholar]
- Matzke MA, Mosher RA (2014) RNA‐directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet 15: 394–408 [DOI] [PubMed] [Google Scholar]
- Miura A, Nakamura M, Inagaki S, Kobayashi A, Saze H, Kakutani T (2009) An Arabidopsis jmjC domain protein protects transcribed genes from DNA methylation at CHG sites. EMBO J 28: 1078–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Mano S, Tanaka Y, Ohnishi M, Nakamori C, Araki M, Niwa T, Nishimura M, Kaminaka H, Nakagawa T, Sato Y, Ishiguro S (2010) Gateway binary vectors with the bialaphos resistance gene, bar, as a selection marker for plant transformation. Biosci Biotechnol Biochem 74: 1315–1319 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph T, Yonezawa M, Lein S, Heidrich K, Kubicek S, Schäfer C, Phalke S, Walther M, Schmidt A, Jenuwein T, Reuter G (2007) Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU(VAR)3‐3. Mol Cell 26: 103–115 [DOI] [PubMed] [Google Scholar]
- Saze H, Shiraishi A, Miura A, Kakutani T (2008) Control of genic DNA methylation by a jmjC domain‐containing protein in Arabidopsis thaliana . Science 319: 462–465 [DOI] [PubMed] [Google Scholar]
- Saze H, Kakutani T (2011) Differentiation of epigenetic modifications between transposons and genes. Curr Opin Plant Biol 14: 81–87 [DOI] [PubMed] [Google Scholar]
- Saze H, Kitayama J, Takashima K, Miura S, Harukawa Y, Ito T, Kakutani T (2013) Mechanism for full‐length RNA processing of Arabidopsis genes containing intragenic heterochromatin. Nat Commun 4: 2301 [DOI] [PubMed] [Google Scholar]
- Schultz MD, Schmitz RJ, Ecker JR (2012) “Leveling” the playing field for analyses of single‐base resolution DNA methylomes. Trends Genet 28: 583–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao NY, Liu X, Maze I, Feng J, Nestler EJ (2013) diffReps: detecting differential chromatin modification sites from ChIP‐seq data with biological replicates. PLoS ONE 8: e65598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, Nestler E (2014) ngs.plot: quick mining and visualization of next‐generation sequencing data by integrating genomic databases. BMC Genom 15: 284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953 [DOI] [PubMed] [Google Scholar]
- Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, Patel DJ, Jacobsen SE (2013) Non‐CG methylation patterns shape the epigenetic landscape in Arabidopsis . Nat Struct Mol Biol 21: 64–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandamme J, Sidoli S, Mariani L, Friis C, Christensen J, Helin K, Jensen ON, Salcini AE (2015) H3K23me2 is a new heterochromatic mark in Caenorhabditis elegans . Nucleic Acids Res 43: 9694–9710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA (2012) Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482: 221–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Ietswaart R, Liu F, Yang H, Howard M, Dean C (2015) Quantitative regulation of FLC via coordinated transcriptional initiation and elongation. Proc Natl Acad Sci USA 113: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Lepère G, Jay F, Wang J, Bapaume L, Wang Y, Abraham AL, Penterman J, Fischer RL, Voinnet O, Navarro L (2013) Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc Natl Acad Sci USA 110: 2389–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bernatavichute YV, Cokus S, Pellegrini M, Jacobsen SE (2009) Genome‐wide analysis of mono‐, di‐ and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana . Genome Biol 10: R62 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File