

Abstract
Properdin (FP) is an essential positive regulator of the complement alternative pathway (AP) providing stabilization of the C3 and C5 convertases, but its oligomeric nature challenges structural analysis. We describe here a novel FP deficiency (E244K) caused by a single point mutation which results in a very low level of AP activity. Recombinant FP E244K is monomeric, fails to support bacteriolysis, and binds weakly to C3 products. We compare this to a monomeric unit excised from oligomeric FP, which is also dysfunctional in bacteriolysis but binds the AP proconvertase, C3 convertase, C3 products and partially stabilizes the convertase. The crystal structure of such a FP‐convertase complex suggests that the major contact between FP and the AP convertase is mediated by a single FP thrombospondin repeat and a small region in C3b. Small angle X‐ray scattering indicates that FP E244K is trapped in a compact conformation preventing its oligomerization. Our studies demonstrate an essential role of FP oligomerization in vivo while our monomers enable detailed structural insight paving the way for novel modulators of complement.
Keywords: alternative pathway, complement, deficiency, properdin, structure
Subject Categories: Immunology, Structural Biology
Introduction
Complement is of paramount importance for an effective immune defense. The system is activated when pathogens, immune complexes, dying host cells, or damaged tissue (collectively called activators) are recognized (Bajic et al, 2015; Merle et al, 2015). Activation triggers a proteolytic cascade through the classical pathway (CP), the lectin pathway (LP), or the alternative pathway (AP). This leads to assembly of the C3 convertase, which cleaves C3 into C3a and C3b (Fig 1A). Activator‐bound C3b together with zymogen factor B (FB) form the AP proconvertase C3bB, which is subsequently activated by factor D (FD) resulting in the active AP C3 convertase C3bBb. Since each C3b acts as a focal point for convertase assembly, strong amplification occurs through the AP, and the activator is opsonized by multiple C3b molecules. Once a threshold density of C3b is reached on the activator, the C3 convertase changes specificity and becomes a C5 convertase (Fig 1A).
Figure 1. The alternative pathway of complement and a new form of FP deficiency.
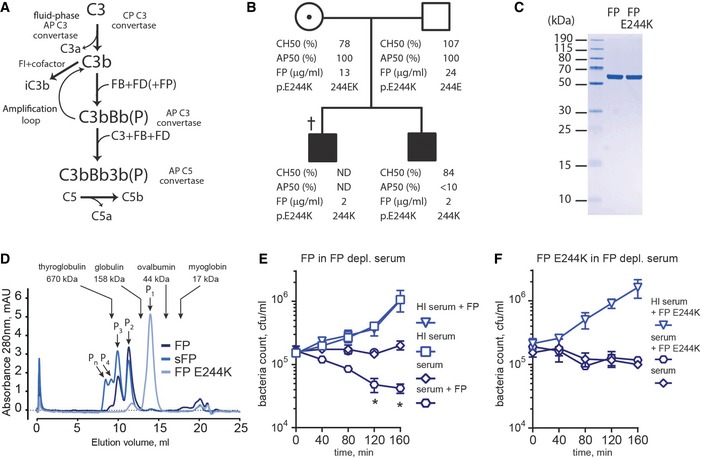
-
AThe AP may be initiated by C3b produced in the CP or by C3(H2O) generated via C3 tick‐over. Upon C3b complex formation with FB and cleavage by FD, the AP C3 convertase C3bBb(P) is formed. The “P” in parenthesis signifies that FP is optional.
-
BPedigree of the FP deficiency family. The father and the heterozygous mother carrying the FP mutation exhibited normal complement activity profiles (AP50/CH50) and FP serum levels. Serum FP levels for both sons were below 10% of normal. AP50 and CH50 were determined for one of the sons and showed normal CP but impaired AP activity. † deceased; ND, not determined.
-
CSDS–PAGE analysis of recombinant purified wt and E244K FP.
-
DSEC analysis (24 ml Superdex 200 increase column) of wt FP, commercial sFP and FP E244K. Oligomeric FP and sFP eluted in multiple peaks between 10.1 and 11.4 ml whereas FP E244K eluted almost entirely in a single peak at 14.0 ml. The elution volumes and molecular weights of standard proteins are marked by arrows.
-
E, FThe contribution of FP (E) or FP E244K (F) during bacteriolysis of N. meningitidis HF85 (serogroup C). FP was present at 10 μg/ml FP in 1:5 FP‐depleted serum. Error bars indicate SDs of bacteria counts in duplicates. *P < 0.05, Student's t‐test. HI, heat inactivated.
Complement is a powerful tool for opsonization of pathogens and dying cells, but it is also potentially a threat to healthy host cells. Hence, the AP is tightly controlled by regulators that exert decay accelerating activity by dissociating Bb irreversibly from C3b, and cofactor activity through assisting factor I (FI) in degrading C3b to iC3b (Fig 1A). FP is the only positive regulator of complement; it stabilizes C3bBb increasing its half‐life fivefold to 10‐fold (Hourcade, 2006). FP deficiency is a rare X‐chromosomal linked disorder with 100–500 known cases characterized by an increased susceptibility to Neisseria infections and sepsis (Skattum et al, 2011), which can be divided into three subtypes: Type I referring to complete lack of FP, type II representing a low FP level in plasma (1–10% of normal concentration), and type III designating normal level of a dysfunctional FP (Fijen et al, 1999). In vivo, FP is organized as dimers, trimers, and tetramers in a ratio of 1:2:1 and present at a plasmatic concentration of 5–25 μg/ml (Blatt et al, 2016a). The FP monomer encompasses seven thrombospondin type I repeats (TSRs) denoted as TSR0–TSR6. A recent EM study of oligomeric FP suggested that vertexes in oligomeric FP are formed by four thrombospondin repeats from two neighboring monomers, but the exact composition of the vertexes could not be determined. It was also suggested that such vertexes stabilize the AP C3 convertase C3bBb by contacting both the C3b C345c and the Bb vWA domain and even approaches the junction between the Bb serine protease and vWA domains (Alcorlo et al, 2013).
Strong binding to a multimeric pattern by a protein containing multiple copies of the recognition domain is known as avidity and plays a prominent role in both innate and adaptive immunity. Avidity is highly relevant for FP since the activator will present multiple copies of C3b, iC3b, the proconvertase C3bB, and the convertase C3bBb to which an oligomeric FP may bind to simultaneously. So far, it is not known whether FP oligomerization is required for function, but both with respect to stimulation of hemolysis (a typical in vitro assay to assess AP activity), the formation of platelet/leukocyte aggregates in vivo, and binding to Chlamydia pneumoniae in vitro, the tetramer exhibits significantly higher activities than both the trimer and the dimer (Pangburn, 1989; Blatt et al, 2016b).
In this study, we show that FP oligomerization is essential for in vivo function. We characterize a patient‐derived monomeric FP, which is dysfunctional due to a single point mutation and compare this monomer to a functional unit excised from oligomeric FP. The resulting protein does not form oligomers and is therefore monomeric although it is formed by TSRs 0–3 from one FP molecule and TSRs 4–6 from a second FP molecule, and in contrast to the patient‐derived monomer, TSR3 and TSR4 are not connected in the excised FP monomer. Whereas both monomers fail to support lysis of Neisseria by complement, biophysical and biochemical studies demonstrate that binding of FP to C3 products and convertase does not require oligomerization although it strongly enhances these functions. The access to the FP monomer allowed us to crystallize the FP‐convertase complex. The resulting structure demonstrates that FP interacts extensively with the very C‐terminal region of C3b through a single thrombospondin repeat (TSR) with possible additional contacts to the catalytic subunit Bb. We propose that the identification of the functional FP monomer can help in the purposeful design of novel complement inhibitors.
Results
FP E244K mutation causes type II properdin deficiency
A patient with a type II FP deficiency was identified in the Complement Diagnostic Laboratory at the HEGP hospital in Paris (with informed consent from the patient's parents for the genetic analyses). The patient was found to carry a novel variant of the FP gene with a single point mutation E244K in thrombospondin repeat 3 (TSR3). The index patient died at the age of 3 years from a fulminant Neisseria meningitidis serogroup Y infection. Family investigation revealed FP deficiency in the patient's brother, in whom the functional activity of the AP was undetectable while CP activity was normal (Fig 1B). Recombinant wild‐type (wt) FP and mutant containing the E244K mutation (FP E244K) were expressed in mammalian cells and purified (Fig 1C). Compared to wt FP the mutant was typically expressed at a fivefold lower level in mammalian cell culture in accordance with the reduced plasma concentration of FP in the two sons compared to their parents (Fig 1B). Size‐exclusion chromatography (SEC) analysis revealed that recombinant wt FP eluted in two high‐molecular weight peaks (10.1 and 11.4 ml) indicating its appearance as dimers, trimers, and a low content of tetramers. The major peak of FP E244K eluted much later (14.0 ml) suggesting that it is monomeric, while a minor peak eluted like the FP dimer (Fig 1D). Clearly the E244K mutation prevents FP oligomerization during secretion from HEK293 cells, and this, together with the decreased FP plasma level in the patient, is likely to underlie its functional defect in the patient. The potency of wt and FP E244K in AP‐mediated lysis of N. meningitidis was evaluated in bacteriolysis assays. Compared to serum alone, addition of oligomeric wt FP resulted in considerable increased bacterial killing in FP‐depleted serum, whereas addition of FP E244K did not stimulate bacteriolysis (Fig 1E and F) in agreement with the phenotype of the patients carrying the mutation.
An excised monomeric unit of FP
Having observed that monomeric FP E244K was non‐functional in vivo and in bacteriolysis, we wanted to investigate whether this was solely due to its lack of oligomerization. As a recent EM study has shown that a FP vertex formed by TSRs from two neighboring FP subunits binds to C3bBb (Alcorlo et al, 2013), we wanted to compare such a vertex and its associated TSRs to FP E244K. We hypothesized that a vertex could be excised from recombinant oligomeric FP by proteolytic cleavage of a TEV linker inserted between TSR3 and TSR4 (Fig 2A). For both FP and FP E244K, this strategy resulted in formation of the monomeric unit that we term FPc which eluted identically in SEC (Fig 2B and D). Strikingly, FPc E244K eluted earlier than its non‐cleaved version indicating that FP E244K is more compact than the monomeric unit excised from oligomeric FP.
Figure 2. Engineering and binding properties of FPc.
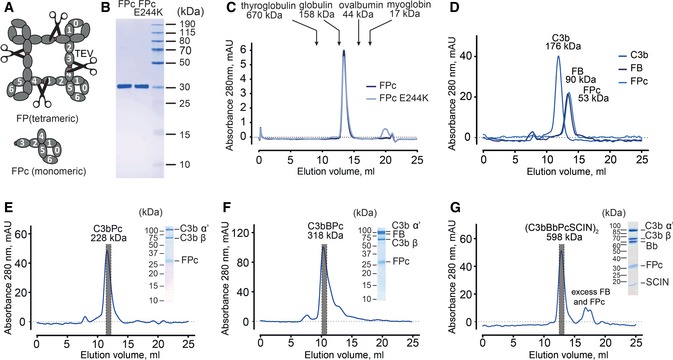
-
AOutline of the strategy for preparing FPc by TEV cleavage between TSR3 and TSR4 of recombinant oligomeric FP.
-
BSDS–PAGE analysis of purified wt and FPc E244K. The two fragments within FPc migrate similarly and appear as one band.
-
CWt and FPc E244K elute identically in SEC.
-
DSEC analysis of C3b (176 kDa), FB (90 kDa), and FPc (53 kDa). All three proteins eluted in single symmetrical peaks at 11.9, 13.3, and 13.4 ml, respectively.
-
E, FSEC analysis of C3bPc (228 kDa) and C3bBPc (318 kDa) complexes and SDS–PAGE analysis of peak fractions.
-
GSEC analysis of SCIN‐stabilized C3 convertase (C3bBbPcSCIN)2 (598 kDa) and SDS–PAGE analysis of the peak fraction.
Bacteriolysis assays demonstrated that like the monomeric FP E244K, FPc did not support bacteriolysis (Fig EV1). We next asked whether the FPc dysfunction was caused by failure to interact with C3b. Reconstituted C3bPc (by convention P is used instead of FP in complexes) and C3bBPc samples were subjected to SEC. This showed that FPc formed stable and well‐defined complexes with C3b and the proconvertase C3bB (Fig 2E and F). The SCIN‐stabilized C3 convertase, where the 10 kDa bacterial protein SCIN induces the formation of a stable dimeric inactive convertase (C3bBbSCIN)2 (Rooijakkers et al, 2005, 2009), was also evaluated for FPc binding by letting FD cleave C3bBPc in the presence of SCIN. SEC analysis revealed that FPc formed a stable complex with the SCIN‐stabilized AP C3 convertase (Fig 2G).
Figure EV1. FPc does not stimulate bacteriolysis of N. meningitidis .
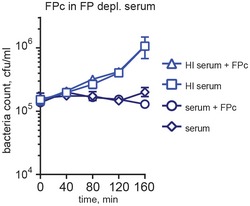
FPc was present at 10 μg/ml in 1:5 FP‐depleted serum during bacteriolysis of N. menigitidis HF85 (serogroup C). Heat‐inactivated serum was used as a negative control. Error bars indicate SDs of bacteria counts in duplicates.
Kinetic analysis of properdin–C3 interactions
Using surface plasmon resonance (SPR), we then compared the binding activity of commercial serum‐derived FP (sFP), recombinant wt FP, FP E244K, and their FPc version toward C3b, iC3b, or methylamine‐treated complement C3 (C3MA), which mimics the tick‐over product C3(H2O). These C3 products were biotinylated through the thioester to allow their immobilization on streptavidin‐coated SPR sensors in an orientation reflecting that of activator‐bound C3b and iC3b. FPc and FPc E244K exhibited similar binding profile and affinities for C3b (Fig 3A). However, when comparing the uncleaved FP variants, the E244K version bound 10–20 times weaker to C3MA, C3b, and iC3b (Fig 3B and C). A kinetic analysis revealed that FPc bound C3b, C3MA, or iC3b with KD values ranging from 4 to 11 μM compared to oligomeric sFP exhibiting apparent KD values of 0.2–1.5 nM (Figs 3D and EV2A, and Table EV1). These KD values were calculated using 1:1 interaction model assuming a conformational change upon ligand–analyte interaction. For all analyzed interactions, this model gave the best fits to the experimental data as compared to a simple 1:1 model.
Figure 3. Binding of FP and FPc (wild type or E244K) to C3 products analyzed by surface plasmon resonance.
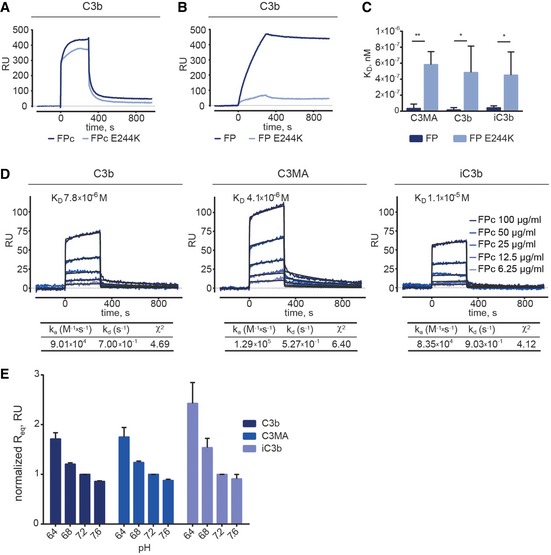
-
A, BSPR analysis of FPc and FPc E244K (20 μg/ml) on immobilized C3b showing that they bind similarly to C3b (A). In contrast, non‐cleaved FP E244K (2.5 μg/ml) bound much weaker to immobilized C3b compared to wild‐type recombinant FP (B).
-
CComparison of KD values observed for FP and FP E244K at 5.0, 2.5, and 1.25 μg/ml upon binding to C3MA, C3b, and iC3b confirms the functional defect of the mutant.
-
DA detailed kinetic SPR analysis using FPc at concentrations ranging from 6.25 to 100 μg/ml. FPc was injected on the C3b/C3MA/iC3b‐coated chip for 300 s followed by 600 s dissociation. The KD values were calculated using a 1:1 interaction model assuming a conformational change upon ligand‐analyte binding.
-
ESPR experiments with wt FPc injected at 100, 50, and 25 μg/ml over immobilized C3b, C3MA, and iC3b at different pH values. The calculated Req values are normalized to Req at pH 7.2.
Figure EV2. sFP and FPc binding to C3 products and homologues.

- SPR analysis with the commercial sFP for comparison with Fig 3D. sFP contains small amounts of non‐natural oligomers (Fig 1D) meaning that it will probably show tighter binding to immobilized C3 products than FP with a natural oligomer distribution. Furthermore, when sFP and recombinant FP are used as analytes, each molecule most likely contacts more than one immobilized C3b. Hence, the assumption of the 1:1 model is not valid, and the derived rate constants and KD values must be considered as apparent.
- sFP and recombinant FPc were attached to the sensor as ligands with amine coupling to a level of 2,680 and 2,320 RU, respectively. All analytes were injected at a concentration of 108 nM. (Top row) sFP and FPc bound to all C3 variants containing the C345c domain except native C3. (Bottom row) sFP and FPc failed to interact with native C3, C4, C5, and CVF.
- Titration of C3b (25–3.125 μg/ml), iC3b (10–1.25 μg/ml), and C3c (12.5–1.56 μg/ml) on immobilized sFP (2,680 RU). Kinetic parameters were calculated by fitting data to a two‐state kinetic model.
- Titration of C3b (25–3.125 μg/ml) on immobilized FPc (2,320 RU) performed as in panel (C).
Furthermore, equilibrium binding analysis showed that at lower pH there is increased binding of FPc to C3b, C3MA and iC3b (Fig 3E). This is in line with previous observations (Fishelson et al, 1987) and suggests that the enhanced AP activation upon serum acidification, as compared to physiological pH, is at least partially driven by a stronger association of FP with C3b.
Surface plasmon resonance analysis was also carried out in the reverse orientation with sFP or FPc covalently attached to the biosensor. This made it possible to investigate the interaction with C3 states devoid of a cleaved thioester, to eliminate the contribution from FP avidity, and to use complement C4, C5, and cobra venom factor (CVF) as controls. CVF is a C3b homologue that forms a stable fluid‐phase C3/C5 convertase with host FB. Both immobilized sFP and FPc bound C3b, iC3b and C3c, but did not interact with native C3, C3d, C4, C5, or CVF (Fig EV2B–D and Table EV1). This confirms prior studies (Lambris et al, 1984; Farries et al, 1988), and suggests that the FP binding site is unavailable in native C3 and C3d, but exposed on the major derivatives C3b, iC3b and C3c.
Assembly and dissociation of the AP C3 convertase were also evaluated using SPR. A dramatic effect of FP oligomerization was demonstrated: As previously observed (Hourcade, 2006), C3bBbsP (the C3bBb in complex with sFP) dissociated 16‐fold slower than C3bBb whereas FPc only decreased the dissociation rate by a factor of 1.4 (Fig EV3A). To reduce the rate of convertase dissociation in the following studies we used the FB mutant D279G‐S699A, which according to SPR binds ~sixfold stronger to C3b which is also the case for FB D279G (Marinozzi et al, 2014). We observed a synergistic effect of FPc and FB upon binding to C3b (Fig 4A–C) in line with previous observations for FP (Farries et al, 1988). At the level of the C3 convertase, FPc decreased the rate of Bb dissociation by 1.7‐ and 1.5‐fold even after its own dissociation from both C3bBb and C3MABb convertases, respectively (Figs 4D and EV3B). The rate of FH‐mediated dissociation of C3bBbPc (Fig 4E) and C3MABbPc (Fig EV3C) was likewise reduced by FPc by 2.8‐ and fourfold as compared to C3bBb and C3MABb. At the time of FH injection, only very little residual FPc, if any, could have remained bound, since FPc dissociated rapidly from the convertase complexes. This suggests that FPc induces a conformational change in the C3 convertase, which is maintained after FPc dissociation. In summary, both the excised monomer and FP E244K bound much weaker than oligomeric FP to immobilized C3b, iC3b and C3MA probably due to lack of avidity. However, FPc bound to all ligands recognized by oligomeric FP and also decreased the rate of both spontaneous and FH‐induced Bb dissociation from the C3bBb and C3MABb convertases.
Figure EV3. SPR analysis of FP‐convertase complexes.
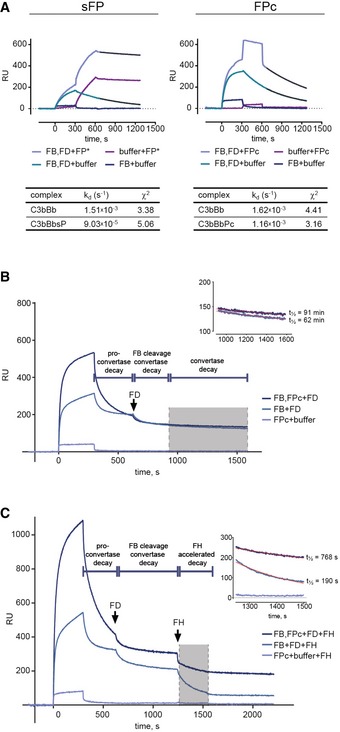
- C3b was immobilized via biotin–streptavidin linkage on the biosensor (1,451 RU). Left: AP C3 convertase was formed by injecting a mixture of inactive FB S699A (10 μg/ml) and FD (2 μg/ml). After 300 s, one channel was injected with sFP (5 μg/ml), while another channel was left to dissociate, KD was calculated by fitting the dissociation phase (t = 650–1,250 s) to a 1:1 model. Right: The C3 convertase was formed by injecting a mixture of FB S699A (20 μg/ml) and FD (4 μg/ml) for 300 s. One channel was injected with FPc (50 μg/ml), and another channel was left to dissociate. C3bBbPc exhibited a two‐state dissociation of which the latter (t = 650–1,250 s) was used for calculating KD. Quality of fit is expressed as χ2.
- SPR experiment similar to Fig 4D but performed with immobilized C3MA. FB D279G/S699A was used to reduce the convertase dissociation rate.
- SPR experiment similar to Fig 4E but performed with immobilized C3MA. FB D279G/S699A was used to reduce the convertase dissociation rate.
Figure 4. SPR analysis of FPc‐convertase interaction and stabilization.
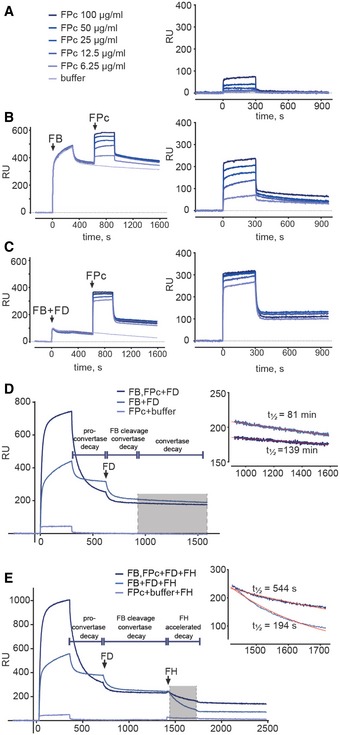
- Binding of FPc (6.25–100 μg/ml) to immobilized C3b.
- Formation of C3bB induced by injecting FB D279G/S699A (20 μg/ml) for 300 s followed by injection of the indicated concentrations of FPc. To the left is shown the raw sensorgram and to the right the buffer (FB injected for 300 s, but no FPc) subtracted sensorgram isolating the FPc signal from binding to decaying C3bB.
- As in panel (B) but with simultaneous injection of FD (2 μg/ml) together with FB for 300 s. After 300 s decay, FPc was injected for 300 s followed by 600 s decay. To the left is shown the raw sensorgram and to the right the buffer (FB and FD injected for 300 s, but no FPc) subtracted sensorgram isolating the FPc signal from binding to decaying C3bBb.
- C3bB was assembled on immobilized C3b in the presence or absence of FPc. FD was injected after 350 s C3bB decay, and the convertase was left to dissociate spontaneously after cleavage. The convertase decay phase (gray shading and zoomed in sensorgram to the right) was fitted to an exponential decay model and the half‐life t½ calculated.
- The effect of FPc stabilization on FH‐mediated decay of C3bBbPc. The convertase was assembled as in panel (D). FH was injected for 350 s to dissociate the convertase. The binding of FH to immobilized C3b was measured in parallel and subtracted from the signal obtained in the channels with the convertase. The FH‐induced decay (gray shading and zoomed in sensorgram to the right) was fitted to an exponential decay model and the half‐life t½ calculated.
FP oligomerization stimulates C3/C5 convertase stabilization
We next asked whether FPc might be partially functional under conditions closer to in vivo than SPR despite its failure to support bacteriolysis. We tested FP and FPc in an assay where rabbit erythrocytes (Er) acted as AP activator in FP‐deficient serum. Both stimulated lysis in a dose‐dependent manner, but FP was more potent since a > fivefold higher FPc concentration was required to obtain the same level of lysis (Fig 5A). This was further corroborated in an assay quantitating C3b deposition in zymosan‐coated microtiter wells, also run at conditions inhibiting CP and LP. Roughly 10‐fold more FPc than FP was required to reach a C3b deposition plateau (Fig 5B). We also investigated the function of oligomeric murine properdin (mFP) and monomeric mouse properdin (mFPc) prepared like the human counterparts (Fig 5C). Er hemolysis and C3 deposition assays were performed in FP‐deficient murine serum. Similarly to the human system, both mFP and mFPc stimulated AP‐mediated lysis of rabbit erythrocytes (Fig 5D), but compared to the human equivalents even more mFPc was needed relative to mFP to obtain similar levels of lysis. Likewise, mFP and mFPc both stimulated C3 deposition on zymosan surfaces (Fig 5E).
Figure 5. FPc support alternative pathway activation on erythrocytes and zymosan but not prolonged convertase stabilization.
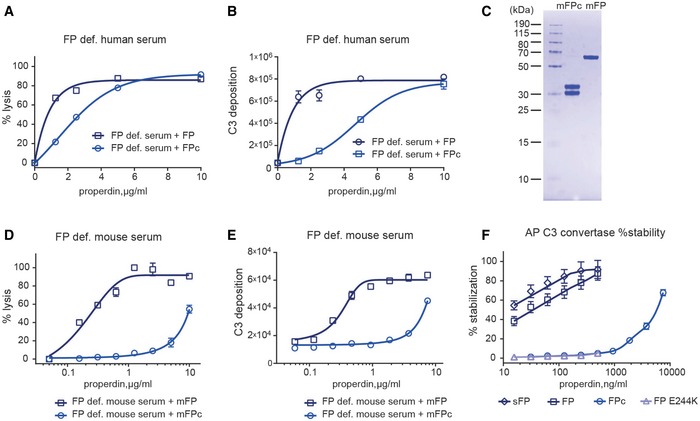
- In a hemolysis assay with FP‐deficient serum using the AP initiating rabbit erythrocytes (Er), the addition of both recombinant human FP and FPc stimulated lysis.
- The addition of FP and FPc to FP‐deficient serum also enhanced AP‐mediated C3b deposition on zymosan‐coated microtiter wells.
- SDS–PAGE analysis of recombinant murine mFP and mFPc.
- mFP and mFPc stimulated AP‐mediated lysis of Er in murine FP‐deficient serum.
- Addition of mFP and mFPc to FP‐deficient murine serum also led to augmentation of C3b deposition in zymosan‐coated microtiter wells.
- C3/C5 convertase stability assay performed with C3b‐covered sheep erythrocytes Es. The efficacy of serum‐derived sFP, FP, FP E244K, and FPc was measured as the % of the residual convertase on the erythrocyte surface compared to a reference without a 30‐min decay period. While recombinant FP and sFP stabilized the C3 convertase in a dose‐dependent manner, FP E244K and wild‐type FPc failed to stabilize the convertase, but with at very high FPc concentration some stabilization is observed.
To more directly investigate the role of FP oligomerization in C3/C5 convertase stability, we coated sheep erythrocytes (Es) with AP convertases and incubated them for 30 min with increasing amounts of either sFP, FP, FP E244K, or FPc. Residual AP convertase on the sheep erythrocytes was subsequently determined by measuring Es lysis induced by lytic pathway reconstitution using rat serum. In contrast to rabbit erythrocytes, Es express large amounts of sialic acid serving as ligand for FH which in turn protect the Es against human complement attack (Goicoechea de Jorge et al, 2007). sFP and recombinant FP exhibited similar stabilizing effect on the C3 convertase. Neither FPc nor FP E244K stabilized the AP convertases at low concentrations (Fig 5F) although at very high concentrations FPc increased convertase life time. In summary, both human and murine FPc could restore FP activity in erythrocyte lysis and C3b deposition assays, but significantly higher concentrations were needed as compared to oligomeric FP. Likewise, convertase stabilization on erythrocytes was dependent on FP oligomerization.
The structure and convertase binding site of FPc
Having shown that FPc was functional in binding the convertase, we prepared crystals of the SCIN‐stabilized C3bBbPc complex, which diffracted X‐rays to a maximum resolution of 6 Å (Fig EV4A and Table 1). The asymmetric unit of the crystals was found to contain two copies of the C3bBbSCIN complex (Fig EV4B). The resulting electron density map revealed for both copies a flat, eye‐shaped electron density, not covered by the C3bBbSCIN complex. This was located on the edge of the C3b C345c domain and extended toward the Bb vWA domain with one major leg oriented in the plane of the eye and protruding from this (Figs 6A and EV4C). A comparison with the EM envelope for FP determined at 23 Å resolution (Alcorlo et al, 2013) revealed a good resemblance in terms of the eye shape and the direction of the major leg (Fig 6B). Due to the resolution of our X‐ray diffraction data, we cannot construct an atomic model of C3bBb‐bound FPc. Nonetheless, we have tentatively docked six identical polyalanine‐model thrombospondin repeats into the density (Figs 6B and EV4C). Two of these TSRs are not fully covered by density and may not necessarily correspond to a repeat, but function as makers for a certain volume of electron density. One of the remaining four repeats is quite well embedded in the density of the major leg, and this repeat must be terminal due to a crystal contact formed in one of the two copies, implying that it is TSR3 or TSR4. A neighboring repeat connected to this terminal repeat forms the vast majority of the interactions with C3b. As multiple lines of evidence points to TSR5 as being deeply involved in C3b interaction (see Discussion), we have made a putative assignment of TSR4 to the major protruding leg, and TSR5 as the connected TSR interacting extensively with C3b (Figs 6B and EV4C). On the convertase side, residues in and around the C‐terminal α2‐helix in the C3b C345c domain are in a position suitable for forming contacts with FPc (Fig 6C). At the N‐terminal end of this helix, negatively charged and highly conserved acidic residues are clustered, which could interact with a positively charged domain such as TSR5. Residues at the other end of the C3b α2‐helix together with a single leucine from Bb form a conserved and predominantly non‐polar patch likewise close to the FPc‐containing electron density (Fig 6C). A comparison with the C3bBbSCIN complex hints at one mechanism for how FP stabilizes the convertase: The C3b C345c domain is rotated significantly relative to Bb in the C3bBbPc complex as compared to the C3bBb complex (Fig 6D).
Figure EV4. Structure determination of the SCIN‐stabilized C3bBbPc complex.
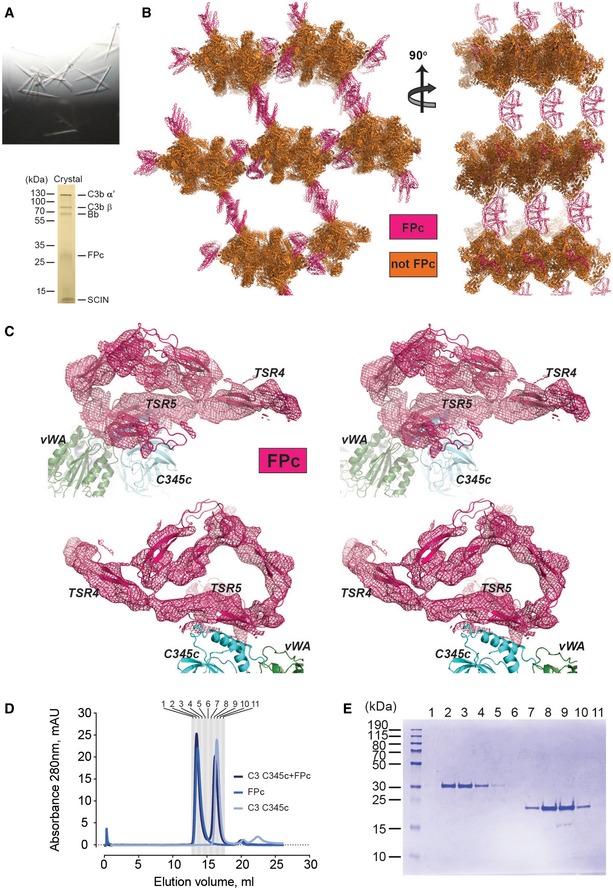
- Crystals of the complex (top), maximum dimensions are 100 × 20 × 5 μm3. An SDS–PAGE analysis of washed crystals with subsequent silver stain of the acrylamide gel (bottom) showed that all components are present.
- Crystal packing lattice for the complex. FPc is essential for lattice contacts, but presumably due to its pronounced non‐globular structure tight crystal packing is not possible resulting in solvent content of approx. 85% explaining the relative poor resolution of the diffraction data.
- Docking of six identical polyalanine TSRs in the real space averaged 2mFo‐DFc electron density. Two of the domains are tentatively assigned as TSR4 and TSR5 based on crystal packing constraints and functional data. The electron density map is calculated without any contribution from the docked TSRs.
- SEC on a 24 ml Superdex 200 Increase column with 0.72 nmol FPc (blue), 1.9 nmol C3 C345c domain (light blue) or combined (bark blue), performed in 10 mM HEPES pH 7.2, 75 mM NaCl.
- Relevant fractions from the combined run were analyzed by SDS–PAGE showing that FPc (25.5 + 23.9 kDa) and C3 C345c (17.4 kDa) did not co‐elute.
Table 1.
Data collection and refinement statistics
| Data collection | |
|---|---|
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 634.9, 122.0, 264.4 |
| α, β, γ (°) | 90.00, 112.9, 90.00 |
| Resolution (Å) | 48.8–6.0 (6.27–6.0) |
| R meas | 0.34 (2.53) |
| I/σI | 5.17 (0.92) |
| Completeness (%) | 99.5 (99.9) |
| Redundancy | 6.2 (6.4) |
| Refinement | |
|---|---|
| Resolution (Å) | 48.8–6.0 |
| No. reflections | 46729 |
| R work/R free | 28.9/32.2 |
| No. atoms | |
| Protein/Ligand | 34,018/422 |
| B‐factors (Å2) | |
| Protein/Ligand | 352.3/528.0 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.911 |
| Clash score | 1.54 |
| Ramachandran plot (%) | |
| Favored/Allowed/Outliers | 95.3/4.2/0.5 |
Data from two crystals were merged. The resolution limit was chosen according to CC(½), which has values of 0.99 and 0.31 for the resolution shells 48.8–6.0 and 6.27–6.0 Å, respectively. Using I/σ(I) = 2 in the outer shell as criteria, the maximum resolution would be around 7 Å. Refinement statistics does not include any contribution from FP, and the model is therefore only 82% complete. R meas is the redundancy independent R‐factor on intensities. Values in parentheses are for highest resolution shell.
Figure 6. Structure of the AP C3 convertase with bound FPc.
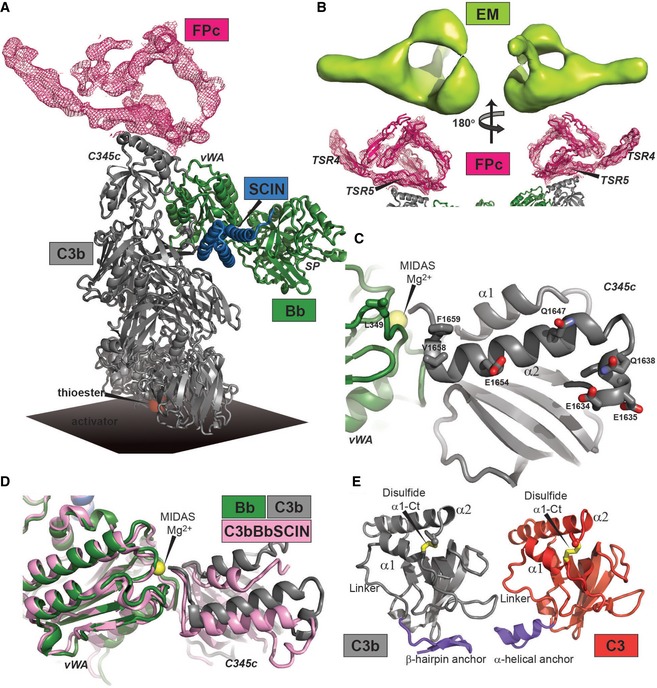
- Crystal structure of the SCIN‐stabilized C3bBbPc complex. The FPc‐containing electron density map is a real space twofold averaged 2mFo‐DFc map contoured at 0.8 σ containing no model information concerning FPc.
- C3b and Bb seen from a FPc perspective. Residues with side chains displayed are facing the FPc‐containing density and are therefore putative interaction partners.
- Comparison of the SCIN‐stabilized C3bBbPc (gray and green, this study) and C3bBb complexes (pink, RCSB entry 2WIN) (Rooijakkers et al, 2009). Binding of FPc causes a significant rotation of the C3b C345c domain relative to Bb.
- Comparison of the anchor region (blue) preceding the C345c domain in the SCIN‐stabilized C3bBbPc complex (left) and in native C3 (right, RCSB entry 2A73) (Janssen et al, 2005). The α‐helical conformation is found only in C3 and in C3b bound to FH or MCP. The β‐hairpin is present in all other known structures of C3b and C3c.
A FP binding site located almost entirely on the C345c domain of C3b explains why we observe comparable interactions between FP/FPc and C3b, iC3b, C3MA, or C3c (Figs 3 and EV2). The fact that native C3 does not bind FP even though it features an exposed C345c domain (Janssen et al, 2005; Fredslund et al, 2006) with a structure similar to that adopted in C3b and C3c (Janssen et al, 2005; Forneris et al, 2016) is possibly related to the so‐called anchor region connecting the C345c domain to the preceding MG8 domain. In native C3, it is α‐helical, whereas in almost all known structures containing C3b or C3c it forms a β‐hairpin (Fig 6E). It is conceivable that the anchor conformation is conformationally coupled with the FP interacting residues at each end of the α2 helix since (i) the anchor regions is connected to the α1 helix through a linker that is flexible in many structures of C3 and C3b; (ii) the α1 helix interacts directly with α2 helix; (iii) the α1 helix is connected through a disulfide bridge to the very C‐terminal end of the α2 helix (Fig 6E). A model stating that the C3 C345c domain has to be presented in a specific structural context to bind FP is also supported by SEC: A recombinant form of the isolated C3 C345c domain did not form a stable complex with FPc (Fig EV4D–E). Overall, our crystallographic data in combination with our SPR data puts forward an unequivocal model for the interaction between FP and C3b/C3bBb stating that the vast majority of the interactions occur between a single TSR (likely to be TSR5) and a small C‐terminal part of the C3b C345c domain possibly with minor contributions from Bb residues and other FP TSRs.
Solution structures of FPc and FP E244K
We next compared the solution conformation of the two types of monomers by small angle X‐ray scattering (SAXS), which suggested that FP E244K is more compact than FPc in agreement with the SEC data (Fig 7A–D). Ab initio modeling revealed that FPc is an elongated particle with a central eye‐like feature close to one end of the particle, whereas FP E244K is more compact but also contains the eye shape (Fig 7E and F). We performed rigid body refinement using homology models of TSR domains. For FPc, TSR0‐3 and TSR4–6 were linked in two separate chains as TSR3 and TSR4 are separated upon TEV cleavage and no other distance restrains were applied. More than 75% of the output models formed an eye‐like structure delimited by 3–4 TSR domains with χ2 = 1.22 ± 0.075. Rigid body refinement of FP E244K was performed with TSR0‐6 linked in a single chain reflecting that TSR3 and TSR4 in this protein are connected. The output models were very homogenous with χ2 = 1.00 ± 0.013, and more than 90% of the models formed an eye‐like structure similar to that of FPc (Fig 7F). In summary, the solution scattering studies provided structural insight into the difference between FPc and FP E244K and confirmed the flat, eye‐shaped feature observed by crystallography in the FPc‐convertase complex and in oligomeric FP by EM (compare Figs 6A and B, and 7E and F). Formation of FP oligomers must involve formation of extensive contacts between thrombospondin repeats from different FP monomers which must require a quite open conformation of the involved monomers. Hence, the observed compact conformation is a likely reason why FP E244K is much less prone to oligomerization during the post‐translational steps taking place prior to secretion.
Figure 7. Solution structures of FPc and FP E244K.

- Experimental SAXS data (dark blue) from FPc and the calculated curve (red) from the rigid body model shown in panel (E).
- Experimental SAXS data (dark blue) from FP E244K and the calculated curve (red) from the rigid body model shown in panel (F).
- The distance distribution functions P(r) calculated from the data indicate Dmax values of 18 and 15 nm for FPc and FP E244K, respectively.
- The Rg‐normalized Kratky plot indicates that FPc has a compact core with flexible legs attached. FP E244K appears more globular and slightly less flexible. The dashed black lines indicate qRg = √(3) and I(q)/I(0)(qRg)2 ≈ 1.104, which is the theoretical maximum value for a globular shape. The FP E244K plot has a maximum much closer to this theoretical maximum compared to FPc.
- Ab initio model (transparent surface) and a representative rigid body model (cartoon) of FPc.
- Similar to panel (E), but based on data from FP E244K.
Discussion
Since its discovery in 1954 (Pillimer et al, 1954), human FP has been subject to extensive research. However, both the structure and crucial aspects of FP function remain unclear, and oligomerization has been a major obstacle for a detailed analysis. Our interest in FP monomers was sparked by the discovery of a novel type II FP deficiency caused by a mutation that upon expression in cell culture resulted in an almost exclusively monomeric protein. The recent EM study of oligomeric FP (Alcorlo et al, 2013) inspired us to engineer another form of monomeric FP which enabled detailed structural information and offered an opportunity to investigate how function depends on oligomerization. Our SPR experiments allowed for the first time a biophysical analysis of the contribution of FP oligomerization to the interaction with its binding partners. This showed that monomeric FPc is able to bind C3b, C3MA, iC3b, C3c, the C3 proconvertase C3bB, and the convertase C3bBb. From these measurements, we conclude that oligomerization lowers the dissociation constant 102–103‐fold for FP in complexes with physiologically relevant C3 products, as compared to the monomeric FPc (Table EV1). Our SPR experiments additionally indicated that FPc stimulates FB binding and assembly of the proconvertase. But after FD cleavage, FPc only modestly prevented spontaneous Bb dissociation from C3bBb, probably because FPc dissociated very rapidly from the convertase. Interestingly, even after its own dissociation, FPc significantly decreased FH‐induced convertase dissociation. Our assays with erythrocyte lysis and C3b deposition on zymosan confirmed the partial functionality of both mouse and human FPc. In bacteriolysis assays, FPc and FP E244K did not stimulate lysis of N. meningitidis suggesting that oligomerization may have additional roles besides increasing the C3/C5 convertase activity during bacteriolysis as compared to lysis of erythrocytes. This role is probably not pattern recognition (see below) since physiological forms of FP does not bind directly to N. meningitidis (Agarwal et al, 2010).
The E244K mutation itself does not alter the fundamental binding properties as evidenced by the comparable binding of FPc and FPc E244K to C3b. Our structural work also showed that FP E244K adopts a compact conformation compared to FPc, but FP E244K still has the central eye shape observed in both free and convertase‐bound FPc. By exploring the functional consequences of the FP E244K mutation, we underscored the importance of FP oligomerization for the positive regulation of the AP. The reduced plasmatic concentration together with the failure of the mutant protein to oligomerize and stabilize the C3 convertase and to induce killing of N. meningitidis explain the clinical phenotype.
FP aggregates with the formation of large oligomers during freeze–thaw cycles, and many prior functional studies have been performed with purified FP containing such large FP oligomers (Ferreira et al, 2010). These cause fluid‐phase AP activation and artifactual binding to biologic surfaces. A minor content of such large FP oligomers was present in the commercial sFP we used, but absent in our recombinant FP. Possibly, such sFP oligomers contributed to an unnaturally strong binding to C3 products in a subset of our SPR experiments—but we merely used sFP as a convenient positive control during analysis of FPc function.
Two groups have reported that FP may bind directly to necrotic and apoptotic human cells, recruit fluid‐phase C3b or C3(H2O), and initiate complement activation—thereby acting as a pattern recognition molecule (Kemper & Hourcade, 2008; Kemper et al, 2008; Xu et al, 2008). FP has also been observed to bind directly to Chlamydophila pneumoniae and strongly stimulate AP C3b deposition (Cortes et al, 2011), and in this case, binding of FP increased with the number of subunits demonstrating the effect of oligomerization and avidity. An important chronic pathological condition is proteinuric renal disease, where FP is believed to acts as C3b‐independent pattern recognition molecule binding to sulfated glycan on tubular epithelial cells and triggering complement activation (Gaarkeuken et al, 2008; Zaferani et al, 2011, 2012). However, doubts have also been raised concerning whether FP actually performs C3b‐independent pattern recognition (Harboe et al, 2012, 2017). Potentially FPc enables detailed structural analysis of FP and its complexes with small molecule ligands mimicking patterns recruiting FP independent of prior C3b deposition. Such structures will greatly facilitate an in depth analysis of the putative pattern recognition function.
An EM study of oligomeric FP bound to C3bBb resulted in a 3D reconstruction with a resolution of 30 Å, suggested that FP vertexes are formed by 4 TSRs from two neighboring monomers interacting in a head‐to‐tail manner. Three suggestions for the composition of the vertexes were presented: TSR0/TSR‐4‐5‐6, TSR0‐1/TSR5‐6 and TSR0‐1‐2/TSR6 (Alcorlo et al, 2013). Our assignment also contains four TSRs in the vertex including TSR5 but has TSR4 outside the vertex, and we cannot decide whether TSR6 is part of the vertex. The EM studies furthermore proposed that a FP vertex forms extensive contacts with both the C3b C345c and the Bb vWA domain and even approaches the junction between the Bb serine protease domain and the vWA domain (Alcorlo et al, 2013). Our crystal structure clearly shows that FPc forms strong contacts to the C3b C345c domain, possibly supplemented with minor interactions to the Bb vWA domain. This is also in agreement with our SPR experiments demonstrating FB independent interaction of FPc and sFP with C3MA, C3b, iC3b, or C3c. Furthermore, a 2D‐average image of the C3bBbP complex presented by the same authors (Alcorlo et al, 2013) appears to be in excellent agreement with our structure further supporting the FP binding site that we observe by crystallography.
We used the SCIN protein to trap the convertase‐FPc complex in a crystallizable state, but we cannot exclude that SCIN partially prevents FPc from exerting its true function with respect to stabilizing the convertase. However, comparison of our structure of SCIN‐stabilized C3bBb bound to FPc with the same structure in its absence (Wu et al, 2009) suggests that FPc induces a rotation of the C3b C345c domain relative to the Bb vWA domain (Fig 6D). If this also occurs in the AP C3 convertase not stabilized by SCIN, it may explain the slower spontaneous and FH‐induced dissociation of Bb we observed by SPR when C3bBb was assembled in the presence of FPc as compared to without FPc (Fig 4D and E). Based on the structure of the C3b‐FH complex, it was proposed that FH destabilization of C3bBb is due to massive steric hindrance and electrostatic repulsion between FH and Bb, but in none of the known C3b‐regulator structures (Wu et al, 2009; Forneris et al, 2016) is there a direct overlap between the FP binding site we observe and the regulator. The simplest explanation for both the slower FH‐induced and spontaneous Bb dissociation from the AP C3 convertase is therefore that Bb binds with higher affinity to a C3bBb assembled and activated in the presence of FPc. However, this will be challenging to verify experimentally due to the irreversible dissociation of Bb from C3b, and structures at higher resolution are required to understand the effects of FP binding to C3bBb in atomic details.
Our crystal structure is also compatible with FP binding to the proconvertase C3bB. A comparison of our C3bBbPc complex with the structures of C3bB and its complex with FD (Forneris et al, 2010) shows that the FP binding mode we observe is fully compatible with FP binding to C3bB and subsequent exosite binding of FD and cleavage of FB. Overlap would not occur between FP and the FB CCP domains or between FP and FD, and FP would be located rather close to the N‐terminus of FB, which offers an explanation for the crosslink formed within the C3bBP complex between FP and the N‐terminal Ba moiety of FB (Farries et al, 1988). This proximity may also underlie the cooperative binding of FB and FP to C3b (DiScipio, 1981) that we and others (Hourcade, 2006) have confirmed by SPR.
We also suggest that the TSR docked on the top of the C3b C345c domain represents TSR5 and that the protruding leg pointing away from Bb encompass TSR4. Deletion of TSR3 does not affect any of the functions ascribed to FP (Higgins et al, 1995) whereas there is ample evidence for the functional importance of TSR4‐5‐6. Recombinant FP lacking TSR6 is unable to form oligomers, and deletion of either TSR5 or TSR6 makes FP unable to bind C3b. FP lacking TSR4 is able to bind C3b but fails to stabilize C3bBb, and proteolytic nicking within TSR5 interferes with C3b binding and convertase stabilization (Higgins et al, 1995). Furthermore, a T414D mutation in TSR6 results in type III FP deficiency characterized by normal plasma level of dysfunctional FP. FP Y414D has an almost normal oligomer distribution, but the Y414D mutation most probably causes a conformational change that prevents it from binding to C3b and the convertases (Fredrikson et al, 1996). Polyclonal anti‐TSR5 antibodies also inhibit binding of FP to immobilized C3b as well as inhibiting FP‐mediated hemolysis of rabbit erythrocytes (Perdikoulis et al, 2001), and a monoclonal antibody binding mouse TSR5‐6 has been shown to block AP‐dependent pathogenesis in a mouse model of abdominal aortic aneurysm (Bertram et al, 2015). Likewise, the anti‐FP antibody CLG561 developed by Alcon as an AP inhibitor also binds TSR5 (Johnson et al, 2016). Finally, a recent study showed that a recombinant TSR4‐5 fragment expressed in bacteria is sufficient for C3b‐binding, but not capable of stabilizing C3bBb (Kouser et al, 2016). These results clearly imply TSR5, and to some extent also TSR6 and TSR4, as principal modules for C3b‐binding and stabilization of C3bBb. In contrast, the epitope of the anti‐FP antibody NM9401 preventing binding to C3b and AP activation is located in TSR1 (Bansal, 2014). A detailed atomic structure of FP is needed to clarify how antibody binding at this repeat can translate into inhibition if our assignment of TSR5 as the primary C3b binding site is correct. Possibly NM9401 alters the overall conformation of FP, and the decreased C3b affinity we observed for FP E244K may also be due the altered conformation detected in our SAXS studies.
The exact role of FP in pathogenesis of complement‐related diseases also remains to be settled. Recent in vivo studies showed that FP inhibition using either anti‐FP‐mAbs or FP gene deletion reduces renal ischemia reperfusion injury in DAF−/− CD59−/− mice and markedly decreases disease severity in the K/B × N model of arthritis (Kimura et al, 2010; Miwa et al, 2013). Similar beneficial effect of FP inhibition is observed in allergen‐induced airway inflammation where FP appeared to contribute to lung injury locally during allergen challenge through AP‐mediated complement activation and C3a production (Wang et al, 2015). On the contrary, in a murine FH‐related C3G model, FP deletion converts a mild C3G to a lethal and rapidly progressing C3G phenotype (Lesher et al, 2013). Thus, anti‐FP therapy may serve as therapeutic target in several complement mediated pathologies but do also require disease specific treatment to avoid adverse effects. Inhibition of FP is currently investigated in a phase 2 clinical trial with the TSR5‐binding anti‐mAb CLG561 for the treatment of age‐related macular degeneration is ongoing (ClinicalTrials.gov Identifier NCT02515942). The two different types of FP monomers presented here offer novel opportunities for obtaining a detailed structure based understanding of FP function, which will greatly facilitate the future development of complement therapeutics targeting the alternative pathway of complement.
Materials and Methods
Protein preparation
Factor P
Human FP and murine FP (mFP) cDNA constructs were designed using the endogenous Kozak sequences and signaling peptides. The constructs were synthesized (Genscript) and subcloned using HindIII and BamHI restriction sites into the pCEP4 mammalian expression vector. To generate the cleavable FPc and mFPc, a TEV site was inserted between TSR3 and TSR4 by site‐directed mutagenesis. For expression, suspension‐adapted HEK293F cells were maintained following manufacturer's instructions at 37°C, 8% CO2, 125 rpm, in serum‐free FreeStyle 293 Expression Medium (Invitrogen). Cells were transiently transfected using final concentrations of 2 mg/l polyethylenimine (PEI – 25 kDa, Polysciences) and 1 mg/l plasmid‐DNA. The conditioned medium was harvested 3–4 days post‐transfection, diluted twofold with water, and adjusted to pH 6.4. Secreted FP or mFP was purified with a human C3b‐Sepharose column equilibrated with 20 mM imidazole, 50 mM NaCl, 5 mM EDTA, pH 6.4. Bound protein was eluted with 15 column volumes (CVs) of the imidazole buffer containing 500 mM NaCl and concentrated to approx. 0.5 mg/ml. To obtain the monomeric forms FPc or mFPc, TEV protease was added in a TEV:protein mass ratio of 1:4 and left to cleave without reducing agent for 3–4 days at 4°C. Complete digestion was confirmed by SDS–PAGE analysis. Mutant FP E244K and FPc E244K were prepared as FP and FPc, but due to the low affinity of FP E244K for the C3b‐Sepharose the flow through was recycled over the column multiple times. Prior to cation exchange chromatography, FP, FPc, mFP, and mFPc were dialyzed against 50 mM phosphate buffer, 2.5 mM EDTA, pH 6.0. The proteins were loaded on a 1 ml MonoS column (GE Healthcare) and eluted with a 20 CV linear gradient from 0 to 500 mM NaCl and subsequently dialyzed into 10 mM HEPES, 75 mM NaCl, pH 7.2. The final yield of FP and FP E244K was 1 mg and 0.2 mg, respectively, per liter HEK293F culture.
Factor B
The FB and FB D279G constructs were previously described (Roumenina et al, 2009; Marinozzi et al, 2014). The FB S699A and the FB D279G, S699A mutants were generated by site‐directed mutagenesis. With the gain‐of‐function mutation D279G, C3b‐binding is enhanced, and with S699A the C3‐cleaving property is lost. Both features were confirmed by SPR and functional hemolytic assays (data not shown). Transient expression was carried out in the Expi293™ expression System (Invitrogen) according to the ExpiFectamin 293 Transfection protocol. The medium was harvested 4 days post‐transfection, diluted twice with water, adjusted to pH 6.0, and added the protease inhibitor benzamidine (BZA) to a final concentration of 5 mM. FB was purified on a 9 ml SP Sepharose FF column (GE Healthcare) equilibrated in 50 mM NaxH3‐xPO4, 2.5 mM EDTA, pH 6.0. Bound FB was eluted with a 10 CV linear gradient from 0 to 500 mM NaCl, and further purified by SEC using a Superdex 200 increase GL 30/100 column (GE Healthcare) equilibrated in 20 mM HEPES, 100 mM NaCl, pH 7.4.
C3b, biotin‐C3b, and biotin‐iC3b
C3 was isolated from outdated human plasma as previously described (Janssen et al, 2005) and stored in 20 mM HEPES, 150 mM NaCl, pH 7.5 at −80°C. For C3b generation, C3 was incubated with 1% (w/w) trypsin for 90 s at 37°C. The reaction was stopped by adding fourfold molar excess of pancreatic trypsin inhibitor (PTI) relative to trypsin. Additionally, PMSF and iodoacetamide were added to 1 and 10 mM final concentration, respectively. The reaction was incubated for 2 h on ice and then diluted fivefold in 20 mM MES pH 6.0. The generated C3b was purified on a 9 ml Source 15S column (GE Healthcare) using a 10 CV ml linear gradient from 0 to 500 mM NaCl. The C3b eluate was concentrated and further purified on a Superdex 200 increase GL 10/300 column (GE Healthcare) equilibrated in 20 mM HEPES, 150 mM NaCl, pH 7.5. Site‐specific biotinylated C3b (biotin‐C3b) was generated by substituting iodoacetamide in the reactions given above with fivefold molar excess of Maleimide‐PEG2‐Biotin reagent, which reacts with the cysteine residue of the C3b thioester. Biotinylation was carried out for 2 h on ice and purification was carried out as described above. Biotin‐iC3b was generated from biotin‐C3b by adding FH and FI in a mass ratio of 1:100 and 1:500, respectively, relative to biotin‐C3b and left at 4°C overnight. The biotin‐iC3b was further purified using a Mono Q 5/50 GL (GE Healthcare) equilibrated in 20 mM HEPES, 150 mM NaCl, pH 7.5, eluting with a 10 CV linear gradient from 150 to 1,000 mM NaCl.
Biotin‐C3MA
Fresh plasma‐derived C3 in 20 mM HEPES, 150 mM NaCl, pH 7.5 was diluted in 1 M Tris, pH 8.0 to 100 mM final concentration. Methylamine pH 8.0 was added to 100 mM final concentration together with fivefold molar excess of the Maleimide‐PEG2‐Biotin reagent and incubated for 4 h at 37°C. The sample was subsequently diluted tenfold in 20 mM MES pH 6.0 and loaded on a 1 ml Source S column. C3 was separated from the newly generated biotin‐C3MA by applying a 10 CV linear gradient from 0 to 500 mM NaCl. Biotin was incorporated on the α‐chain as seen from Western blotting using streptavidin‐HRP (ThermoFisher). One mole biotin was incorporated per mole C3b/C3MA/iC3b as determined by the Pierce™ Biotin Quantitation Kit (ThermoFisher).
C3 C345c domain
The human C3 C345c domain (Ala1492‐Asn1641) cDNA was synthesized (GenScript) with an N‐terminal TEV cleavage site and cloned into the pET‐32a(+) expression vector (Novagen) downstream of thioredoxin A (TrxA) and a His‐tag using BamHI and HindIII restriction sites. Protein was expressed in the E. coli SHuffle T7 strain (New England Biolabs) in LB Broth containing 2% (w/v) glucose and 100 μg/ml ampicillin. Protein expression was induced at OD600nm = 0.6 by adding IPTG to 1 mM final concentration. Expression was carried out for 18–20 h at 18°C. Bacteria were harvested by centrifugation at 6,000 g for 20 min, and subsequently re‐suspended in binding buffer containing 20 mM Tris, 200 mM NaCl, 20 mM imidazole, 1 mM PMSF, pH 8 (10 ml/g pellet), and lysed by sonication. Bacterial debris was removed by centrifugation at 16,000 g for 20 min at 4°C, and the supernatant was loaded on a 5 ml HisTrap FF crude column (GE Healthcare) equilibrated in binding buffer. The column was washed with 10 CV of binding buffer containing 1 M NaCl and the His‐tagged protein was eluted with buffer containing 500 mM imidazole. The eluate containing the TrxA‐His‐TEV‐C345c fusion protein was added TEV protease in a TEV:protein mass ratio of 1:50, and left to cleave overnight at 4°C while being dialyzed against cleavage buffer (20 mM Tris, 200 mM NaCl, 0.5 mM EDTA, pH 8). The sample was again loaded on the HisTrap column, and the untagged C345c protein was collected in the flow through. The flow through was diluted fourfold with 20 mM Tris pH 8 to reach a NaCl concentration of approximately 50 mM before the protein was loaded on a 9 ml Source 15Q anion exchange column (GE Healthcare). The protein was eluted with a 10 CV ml linear gradient from 50 to 400 mM NaCl. Finally, the C345c protein was purified by SEC on a Superdex 75 GL 10/300 column (GE Healthcare) equilibrated in 20 mM HEPES, 100 mM NaCl, pH 7.5.
SCIN
The SCIN protein was expressed from the pETm11 vector (Novagen) including an N‐terminal His‐tag and TEV site. Expression in E. coli BL21 (DE3) was induced at OD600nm = 0.6 by adding IPTG to a final concentration of 1 mM and carried out for 12–16 h at 18°C. Bacteria were harvested by centrifugation at 6,000 g for 20 min. The pellet was re‐suspended in binding buffer containing 50 mM HEPES, 300 mM NaCl, 30 mM imidazole, 1 mM PMSF, 1 mM benzamidine, pH 7.5 before sonication. The supernatant was recovered after centrifugation at 16,000 g for 20 min and loaded on to a 5 ml HisTrap FF crude column (GE Healthcare) equilibrated in binding buffer. After washing with 8 CVs of binding buffer, His‐tagged SCIN was eluted by applying a 10 CV gradient from 30 to 300 mM imidazole. TEV protease was added to the eluted protein in a TEV:protein mass ratio of 1:25, and dialyzed against 20 mM HEPES, 200 mM NaCl, pH 7.5 for 12–16 h at 4°C. The cleaved SCIN was again loaded on the HisTrap column, and the protein‐containing flow through was collected. Finally, SCIN was purified by SEC using a Superdex 75 10/300 column (GE Healthcare) equilibrated in 20 mM HEPES, 150 mM NaCl, pH 7.4.
Size‐exclusion chromatography
Analytical SEC with isolated C3b, FB D279G/S699A, FP, sFP (Complement Technology, catalogue number A139), FP E244K, FPc, and FPc E244K was performed on a Superdex 200 increase GL 30/100 column (GE Healthcare) equilibrated in 10 mM HEPES, 150 mM NaCl, pH 7.2. For the C3bPc complex, C3b was mixed with FPc in a molar ratio of 1:1.1. For the C3bBPc complex, C3b was mixed with FB and FPc in a molar ratio of 1:1.1:1.2. For the SCIN‐stabilized C3‐convertase (C3bBbPcSCIN)2, C3b was mixed with FB, FPc, SCIN, and FD in a molar ratio of 1:1.1:1.2:2:0.02. For C3bPc and C3bBPc SEC experiments, the individual components were mixed in gel filtration buffer approximately 10 min before injection. For C3bBbPcSCIN (FD), the components were mixed and incubated for 2 h at room temperature prior to injection. C3bPc and C3bBPc were purified on a Superdex 200 increase 10/300 GL column (GE Healthcare) equilibrated in 10 mM HEPES, 75 mM NaCl, 5 mM MgCl2, pH 7.2. C3bBbPcSCIN was purified on Superose 6 increase 10/300 GL column (GE Healthcare) equilibrated in the same buffer. Analysis of possible complex formation between the recombinant C3 C345c domain and FPc was performed on a Superdex 200 Increase 10/300 GL column equilibrated in 10 mM HEPES pH 7.2, 75 mM NaCl.
Surface plasmon resonance
Surface plasmon resonance experiments were performed on ProteOn™ XPR36 protein interaction array system (Bio‐Rad) at 25°C. Biotin‐C3b, ‐C3MA, or ‐iC3b were immobilized on NeutrAvidin‐coated ProteOn NCL sensor chips (Bio‐Rad) according to the manufacturer's recommendations. All experiments were performed in running buffer containing 10 mM HEPES, 150 mM NaCl, 5 mM MgCl2, 0.005% Tween 20, pH 7.2 at a flow rate of 30 μl/min. Regeneration of the chip was performed with three 20 s pulses of 4 M NaCl. Reference spots without immobilized protein were used for background subtraction. Binding affinities of FP and FPc to biotin‐C3b, ‐C3MA, and ‐iC3b were determined by global fitting to a two‐state kinetic model assuming 1:1 binding in combination with a conformational change. Data analysis was performed using the manufacturer's software (Proteon Manager™). Prior studies and data obtained in this study suggest that there is a conformational change upon FP binding to the convertase. Therefore, a 1:1 Langmuir binding model (assuming rigid body interactions) for the SPR data fitting was compared to a two‐state model (assuming a conformational change upon interaction), using the ProteOn Manager software. The 1:1 Langmuir binding model gave bad fitting curves with high χ2 values whereas the two‐state model resulted in fitting curves very similar to the experimental data and much lower values of χ2. For these reasons, the two‐state model was used throughout. SPR analysis of the putative monovalent interaction of sFP and FPc with C3, C4, C5, CVF, C3b, iC3b, C3c, and C3d was performed with sFP and FPc covalently bound to a GLC chip surface using a standard amine coupling procedure, as recommend by the manufacturer (Bio‐Rad). A flow rate of 30 μl/min was applied at all times. Human serum‐derived complement proteins (C3, C3b, iC3b, C3c, C3d, CVF, C5, C4, FH, FD, and sFP) were obtained from Complement Technology.
Hemolysis of rabbit erythrocytes
To assess the biological activity of FP and FPc, hemolytic assays were performed using the standard AP target, rabbit erythrocytes (Er). Er suspended in Alsever's solution (Statens Seruminstitut) were washed and re‐suspended in AP assay buffer (5 mM barbital, 145 mM NaCl, 10 mM EGTA, 5 mM MgCl2, pH 7.4, with 0.1% (w/v) gelatin) to obtain a 6% (v/v) suspension. Ten micro liter of the 6% Er‐suspension was transferred to a V‐shaped bottom 96‐well microtiter plate (Nunc). Samples of 20 μl human or murine FP‐deficient serum diluted in assay buffer and supplemented with FP, FPc, mFP, or mFPc were prepared in a separate microtiter plate and then transferred to the assay plate in duplicates. The assay plate was mixed well and incubated for 2 h at 37°C with gentle shaking. Hemolysis was stopped by adding 40 μl ice‐cold 0.9% NaCl, 5 mM EDTA to each well. The plate was centrifuged at 90 g for 10 min, and 60 μl of each supernatant was subsequently transferred to a flat‐bottom microtiter well plate (Nunc). Hemolysis was then determined from the optical density measured at 405 nm on a Victor3 plate reader (PerkinElmer). Results are expressed relative to total hemolysis (obtained with water alone) and to background hemolysis (buffer alone). FP‐deficient serum obtained from a patient suffering from type I FP deficiency (complete deficiency) was used in some experiments. Murine serum was obtained from wild‐type and FP‐deficient mice as described (Stover et al, 2008).
Hemolysis of convertase‐coated sheep erythrocytes
The AP convertase was assembled on sheep erythrocytes as previously described (Roumenina et al, 2009). C3b‐coated sheep erythrocytes, 100 μl with 108 cells/ml, were incubated with 40 ng purified human FD (Sigma) and FB (Complement Technology) in serial dilutions starting from 15 ng to achieve Z values (corresponding to the number of lytic sites per cell) between 1 and 2. Samples were incubated in DGVB2+ at 30°C for 30 min. After incubation, 100 μl sFP, FP, FPc, or FP E244K diluted in GVB‐EDTA buffer was added, and incubation was continued for another 30 min to allow natural decay of the AP C3 convertase. The number of residual convertases was then revealed by adding 300 μl rat serum diluted 1:40 in GVB‐EDTA buffer serving as a source of terminal complement components. The samples were incubated for 45 min at 37°C to allow MAC assembly. Hemolysis obtained during this period was measured by absorbance at 414 nm, and Z values were calculated. A reference sample representing 100% lysis was incubated in GVB‐EDTA only, and the AP C3 convertase sites were developed without the 30 min of spontaneous decay. A negative control was performed without FB. The efficacies of FP, FP E244K, sFP, and FPc are expressed as % of stabilization, calculated as the fraction of residual convertases on the cell surface as compared to the reference sample without decay.
C3 deposition on zymosan
The activity of the AP was analyzed by measuring the deposition of C3b on a zymosan surface (whole yeast cell walls) in a buffer inhibiting both the CP and the LP. A 96‐well MaxiSorp plate (Nunc) was coated with 100 μl of 20 μg/ml zymosan in 50 mM sodium carbonate buffer pH 9.6 overnight at 4°C. Unbound zymosan was removed by washing the wells three times with 300 μl TBS/Tween (10 mM Tris, 145 mM NaCl, 0.05% Tween 20, pH 7.4). Wells were subsequently blocked with 200 μl of 1 mg/ml human serum albumin (HSA) in TBS/Tween for 1 h at room temperature. A 50 μl sample of human or mouse FP‐deficient serum diluted in VBS/EGTA/Mg buffer (5 mM barbital, 145 mM NaCl, 10 mM EGTA, 5 mM MgCl2, pH 7.4) supplemented with either FP/FPc or mFP/mFPc was added to each well and incubated at 37°C for 1 h with gentle shaking. After incubation, the wells were washed three times with 300 μl TBS/Tween. Deposited human C3b was detected by adding of 50 μl of 0.75 μg/ml biotin‐anti‐hC3c (biotinylated version of anti‐C3c antibody from Dako) in TBS/Tween to each well and incubating 12–16 h at 4°C. The following day, wells were washed three times with 300 μl TBS Tween. 50 μl of 1 μg/ml Europium‐labeled streptavidin (Perkin‐Elmer) in TBS/Tween with 25 μM EDTA was then added to each well, and the plates were incubated for 1 h at room temperature. The wells were washed, and 50 μl DELFIA enhancement solution was added to each well. The plates were vortexed vigorously and incubated for 2 min before the fluorescence was measured at 615 nm with a VICTOR5 plate reader (PerkinElmer).
The deposition of mouse C3b on zymosan was detected by using 50 μl of 0.5 μg/ml rat anti‐mouse C3 (Connex GmbH) in TBS/Tween as the primary antibody. After 12‐ to 16‐h incubation, 50 μl of 0.25 μg/ml rabbit biotin‐anti‐rat (Dako) in TBS/Tween was added to each well and the plates were incubated for 1 h at room temperature. After washing the plates, each well was added 50 μl of 1 μg/ml Europium‐labeled streptavidin in TBS/Tween with 25 μM EDTA, and left for incubation for one more hour. The wells were washed, and the fluorescence was measured as described above.
Serum bactericidal assay
Bactericidal assays were performed to investigate the ability of recombinant FP to support lysis of Gram‐negative bacteria. The Neisseria meningitidis strain HF85 (serogroup C, own collection) was cultured on chocolate agar (heated blood agar plate, Statens Serum Institut) at 37°C, 5% CO2, and used to inoculate 5 ml of gonococcus (GC) broth. Bacteria were grown in a 50‐ml falcon tube by slow “end over end” rotation at 12 rpm, 37°C for approximately 2 h until reaching mid‐log phase (A600nm = 0.100, approximately 108 bacteria/ml). 1.25 ml of bacterial culture was then centrifuged at 2,000 g for 10 min and re‐suspended in 1.5 ml pre‐warmed Hanks buffered salt solution (HBSS) containing 1.2 mM CaCl2 and 0.9 mM MgCl2, pH 7.4 (Invitrogen). The bacteria were washed three times before being re‐suspended in HBSS at a concentration of 2.5 × 106 bacteria/ml. For the lysis reaction, normal human serum (NHS) or FP‐depleted serum (Complement Technology) was diluted 1:5 in HBSS buffer and supplemented with 10 μg/ml recombinant human FP, FP E244K, or FPc. Heat‐inactivated sera (56°C for 30 min) were used as negative controls. For 250 μl reactions, 40 μl bacterial suspensions at 2.5 × 106 bacteria/ml were used. Incubation was carried out on a plate mixer at 200 rpm, 37°C. 10 μl of samples was drawn at 0, 40, 80, 120, and 160 min in duplicates. The samples were serially diluted in HBSS and plated onto chocolate agar and incubated for 12–16 h at 37°C, 5% CO2, and the number of colony‐forming units (cfu) were counted.
Crystal structure determination
Crystals of the C3bBbFPcSCIN complex were grown at 4°C in sitting drops made by mixing the SEC purified complex at 6.8 mg/ml in a 1:1 ratio with reservoir solution containing 0.05 M Mg acetate, 0.05 M MES pH 6.5, and 5% PEG 10,000. Crystals were cryo‐protected by soaking in reservoir solution supplemented with 25% glycerol prior to flash cooling in liquid nitrogen. Data were collected at PETRA III P14 using radiation with λ = 0.976 Å at 100 K and processed with XDS (Kabsch, 2010). The structure was determined using the coordinates of the C3bBbSCIN complex (RCSB entry 2WIN) for molecular replacement in Phaser (McCoy et al, 2007). The solvent content of the crystals was 85% with two C3bBbSCIN‐FPc complexes in the asymmetric unit. The solution from Phaser was refined with rigid body refinement in Phenix.refine (Afonine et al, 2012). A few regions were manually rebuilt in Coot (Emsley et al, 2010), and the rebuild structure was then fitted to the electron density map using iMDFF as described (Croll & Andersen, 2016). The iMDFF fitted structure was subsequently refined to the final structure with Phenix.refine. Atom positions were refined with positional refinement, whereas temperature factors were refined with grouped B‐factors and TLS groups. The two copies of the complex in the asymmetric unit were restrained domain‐wise with non‐crystallographic symmetry restraints. The resulting structure displayed superior stereochemistry according to Molprobity (Chen et al, 2010) considering the low resolution of the diffraction data. No model information from the docked TSRs were used at any point, but they served as atomic models underlying the masks used in real space averaging conducted using the RAVE package (Kleywegt et al, 2006). Polyalanine TSRs derived from RCSB entry 1LSL were docked by hand in Coot or Pymol. Figures were prepared with PyMol v1.8 (www.pymol.org).
Small angle X‐ray scattering
Small angle X‐ray scattering measurements were performed in batch mode at the P12 beamline at PETRA III, DESY using a Pilatus 2 M pixel detector (DECTRIS, Switzerland) and λ = 1.240 Å in a temperature‐controlled capillary at 20°C. The sample‐to‐detector distance was 3.0 m, covering a range of momentum transfer 0.028 < q < 4.8 nm−1 q = (4 · π · sin θ) ÷ λ, where 2θ is the scattering angle). Data were collected from samples containing FPc and FP E244K in 10 mM HEPES, 150 mM NaCl, pH 7.2 with twenty exposures of 45 ms. Radial averaging, buffer subtraction, and concentration scaling were performed by the beamline pipeline (Franke et al, 2012). For FP E244K, data collected at 2.7 mg/ml were used for further modeling. Data collected for FPc at 2.7 mg/ml with q < 2 nm−1 were merged with data collected at 9.1 mg/ml with q > 0.15 nm−1 using PRIMUS (Konarev et al, 2003). The pair distribution function was calculated by indirect Fourier transform using GNOM (Svergun, 1992). Ab initio models were modeled using DAMMIF (Franke & Svergun, 2009) and averaged with the DAMAVER suite (Volkov & Svergun, 2003). Rigid body refinements were performed using a momentum transfer range of q < 0.28 Å−1 with CORAL (Petoukhov et al, 2012). For rigid body modeling, the thrombospondin repeats from RCSB entry 1W0R (Sun et al, 2004) were used and extended to an all‐atom model with PULCHRA (Rotkiewicz & Skolnick, 2008). For the FP E244K, TSR0‐6 were connected using LINK statements, whereas FPc was divided into the TSR0‐3 chain and the TSR4‐6 chain, each connected using LINK statements. No other restraints were applied. Calculations of theoretical scattering profiles of atomic structures and their fits to the experimental data (as measured by the χ2‐value) were done using CRYSOL3 (Svergun et al, 1995).
Data availability
Coordinates and structure factors for the crystal structure of FP in complex with the SCIN‐stabilized C3 convertase are available at the RCSB protein data bank in entry 5M6W. The solution structures and scattering curves for FPc and FP E244K are available at the SASBDB as entries SASDB59 and SASDB69, respectively.
Author contributions
DVP, LR, RKJ, TAFG, CM, and TR conducted experiments and analyzed data. LR, ST, UBSS, VF, and GRA supervised experiments and data analysis. CS supplied reagents and CP clinical data. VF‐B and GRA designed research. DVP, LR, and GRA wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
We would like to thank the beamline staff at Petra and ESRF for support during data collection and Lennart Truedsson, Lund University for providing FP‐deficient serum. Gregers R Andersen was supported by the Lundbeck foundation centers LUNA and BRAINSTRUC (R155‐2015‐2666), Danscatt, the Danish Research Council for Independent Research (1323‐00107B and 4181‐00137B) and the Novo Nordisk Foundation (12261). Lubka T. Roumenina was supported by the French National Research Agency ANR‐15‐CE15‐0001. Veronique Fremeaux‐Bacchi was supported by the French National Research Agency ANR PRC‐C3G 2016‐2019R.
The EMBO Journal (2017) 36: 1084–1099
References
- Afonine PV, Grosse‐Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68: 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Ferreira VP, Cortes C, Pangburn MK, Rice PA, Ram S (2010) An evaluation of the role of properdin in alternative pathway activation on Neisseria meningitidis and Neisseria gonorrhoeae . J Immunol 185: 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcorlo M, Tortajada A, Rodriguez de Cordoba S, Llorca O (2013) Structural basis for the stabilization of the complement alternative pathway C3 convertase by properdin. Proc Natl Acad Sci USA 110: 13504–13509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic G, Degn SE, Thiel S, Andersen GR (2015) Complement activation, regulation, and molecular basis for complement‐related diseases. EMBO J 34: 2735–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R (2014) Anti‐properdin antibodies. NovelMed Therapeutics, Inc; United States Patent Application US 20140127204 A1. https://www.google.com/patents/US20140127204 [Google Scholar]
- Bertram P, Akk AM, Zhou HF, Mitchell LM, Pham CT, Hourcade DE (2015) Anti‐mouse properdin TSR 5/6 monoclonal antibodies block complement alternative pathway‐dependent pathogenesis. Monoclon Antib Immunodiagn Immunother 34: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatt AZ, Pathan S, Ferreira VP (2016a) Properdin: a tightly regulated critical inflammatory modulator. Immunol Rev 274: 172–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatt AZ, Saggu G, Kulkarni KV, Cortes C, Thurman JM, Ricklin D, Lambris JD, Valenzuela JG, Ferreira VP (2016b) Properdin‐mediated C5a production enhances stable binding of platelets to granulocytes in human whole blood. J Immunol 196: 4671–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes C, Ferreira VP, Pangburn MK (2011) Native properdin binds to Chlamydia pneumoniae and promotes complement activation. Infect Immun 79: 724–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll TI, Andersen GR (2016) Re‐evaluation of low‐resolution crystal structures via interactive molecular‐dynamics flexible fitting (iMDFF): a case study in complement C4. Acta Crystallogr D Struct Biol 72: 1006–1016 [DOI] [PubMed] [Google Scholar]
- DiScipio RG (1981) The binding of human complement proteins C5, factor B, beta 1H and properdin to complement fragment C3b on zymosan. Biochem J 199: 485–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farries TC, Lachmann PJ, Harrison RA (1988) Analysis of the interactions between properdin, the third component of complement (C3), and its physiological activation products. Biochem J 252: 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira VP, Cortes C, Pangburn MK (2010) Native polymeric forms of properdin selectively bind to targets and promote activation of the alternative pathway of complement. Immunobiology 215: 932–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijen CAP, Van Den Bogaard R, Schipper M, Mannens M, Schlesinger M, Nordin FG, Dankert J, Daha MR, Sjöholm AG, Truedsson L (1999) Properdin deficiency: molecular basis and disease association. Mol Immunol 36: 863–867 [DOI] [PubMed] [Google Scholar]
- Fishelson Z, Horstmann RD, Muller‐Eberhard HJ (1987) Regulation of the alternative pathway of complement by pH. J Immunol 138: 3392–3395 [PubMed] [Google Scholar]
- Forneris F, Ricklin D, Wu J, Tzekou A, Wallace RS, Lambris JD, Gros P (2010) Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science 330: 1816–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forneris F, Wu J, Xue X, Ricklin D, Lin Z, Sfyroera G, Tzekou A, Volokhina E, Granneman JC, Hauhart R, Bertram P, Liszewski MK, Atkinson JP, Lambris JD, Gros P (2016) Regulators of complement activity mediate inhibitory mechanisms through a common C3b‐binding mode. EMBO J 35: 1133–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Svergun DI (2009) DAMMIF, a program for rapid ab‐initio shape determination in small‐angle scattering. J Appl Crystallogr 42: 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Kikhney AG, Svergun DI (2012) Automated acquisition and analysis of small angle X‐ray scattering data. Nucl Instrum Meth A 689: 52–59 [Google Scholar]
- Fredrikson GN, Westberg J, Kuijper EJ, Tijssen CC, Sjoholm AG, Uhlen M, Truedsson L (1996) Molecular characterization of properdin deficiency type III: dysfunction produced by a single point mutation in exon 9 of the structural gene causing a tyrosine to aspartic acid interchange. J Immunol 157: 3666–3671 [PubMed] [Google Scholar]
- Fredslund F, Jenner L, Husted LB, Nyborg J, Andersen GR, Sottrup‐Jensen L (2006) The structure of bovine complement component 3 reveals the basis for thioester function. J Mol Biol 361: 115–127 [DOI] [PubMed] [Google Scholar]
- Gaarkeuken H, Siezenga MA, Zuidwijk K, van Kooten C, Rabelink TJ, Daha MR, Berger SP (2008) Complement activation by tubular cells is mediated by properdin binding. Am J Physiol Renal Physiol 295: F1397–F1403 [DOI] [PubMed] [Google Scholar]
- Goicoechea de Jorge E, Harris CL, Esparza‐Gordillo J, Carreras L, Arranz EA, Garrido CA, Lopez‐Trascasa M, Sanchez‐Corral P, Morgan BP, Rodriguez de Cordoba S (2007) Gain‐of‐function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA 104: 240–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harboe M, Garred P, Lindstad JK, Pharo A, Muller F, Stahl GL, Lambris JD, Mollnes TE (2012) The role of properdin in zymosan‐ and Escherichia coli‐induced complement activation. J Immunol 189: 2606–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harboe M, Johnson C, Nymo S, Ekholt K, Schjalm C, Lindstad JK, Pharo A, Hellerud BC, Nilsson Ekdahl K, Mollnes TE, Nilsson PH (2017) Properdin binding to complement activating surfaces depends on initial C3b deposition. Proc Natl Acad Sci USA 114: E534–E539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JM, Wiedemann H, Timpl R, Reid KB (1995) Characterization of mutant forms of recombinant human properdin lacking single thrombospondin type I repeats. Identification of modules important for function. J Immunol 155: 5777–5785 [PubMed] [Google Scholar]
- Hourcade DE (2006) The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem 281: 2128–2132 [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Huizinga EG, Raaijmakers HC, Roos A, Daha MR, Nilsson‐Ekdahl K, Nilsson B, Gros P (2005) Structures of complement component C3 provide insights into the function and evolution of immunity. Nature 437: 505–511 [DOI] [PubMed] [Google Scholar]
- Johnson L, Splawski I, Baker L, Carrion A, Nguyen A, Twarog M, Wang Y, Jager U, Keating M, Dryja TP, Roguska M (2016) Generation and characterization of CLG561: a fully‐human, anti‐properdin Fab for the treatment of age‐related macular degeneration. Invest Ophthalmol Vis Sci 57: 1116–1116 [Google Scholar]
- Kabsch W (2010) Integration, scaling, space‐group assignment and post‐refinement. Acta Crystallogr D Biol Crystallogr 66: 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper C, Hourcade DE (2008) Properdin: new roles in pattern recognition and target clearance. Mol Immunol 45: 4048–4056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper C, Mitchell LM, Zhang L, Hourcade DE (2008) The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci USA 105: 9023–9028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Zhou L, Miwa T, Song WC (2010) Genetic and therapeutic targeting of properdin in mice prevents complement‐mediated tissue injury. J Clin Investig 120: 3545–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Zou JY, Kjeldgaard M, Jones TA (2006) Around O In International tables for crystallography, Rossmann MG, Arnold E. (eds), Vol. F, 17.1, pp 353–356. Dordrecht: Kluwer Academic Publishers; [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI (2003) PRIMUS: a Windows PC‐based system for small‐angle scattering data analysis. J Appl Crystallogr 36: 1277–1282 [Google Scholar]
- Kouser L, Abdul‐Aziz M, Tsolaki AG, Singhal D, Schwaeble WJ, Urban BC, Khan HA, Sim RB, Kishore U (2016) A recombinant two‐module form of human properdin is an inhibitor of the complement alternative pathway. Mol Immunol 73: 76–87 [DOI] [PubMed] [Google Scholar]
- Lambris JD, Alsenz J, Schulz TF, Dierich MP (1984) Mapping of the properdin‐binding site in the third component of complement. Biochem J 217: 323–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesher AM, Zhou L, Kimura Y, Sato S, Gullipalli D, Herbert AP, Barlow PN, Eberhardt HU, Skerka C, Zipfel PF, Hamano T, Miwa T, Tung KS, Song WC (2013) Combination of factor H mutation and properdin deficiency causes severe C3 glomerulonephritis. J Am Soc Nephrol 24: 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, Cayla M, Tabarin F, Jablonski M, Hue C, Smith RJ, Noris M, Halbwachs‐Mecarelli L, Donadelli R, Fremeaux‐Bacchi V, Roumenina LT (2014) Complement factor B mutations in atypical hemolytic uremic syndrome‐disease‐relevant or benign? J Am Soc Nephrol 25: 2053–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merle NS, Church SE, Fremeaux‐Bacchi V, Roumenina LT (2015) Complement system part I – Molecular mechanisms of activation and regulation. Front Immunol 6: 262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa T, Sato S, Gullipalli D, Nangaku M, Song WC (2013) Blocking properdin, the alternative pathway, and anaphylatoxin receptors ameliorates renal ischemia‐reperfusion injury in decay‐accelerating factor and CD59 double‐knockout mice. J Immunol 190: 3552–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangburn MK (1989) Analysis of the natural polymeric forms of human properdin and their functions in complement activation. J Immunol 142: 202–207 [PubMed] [Google Scholar]
- Perdikoulis MV, Kishore U, Reid KB (2001) Expression and characterisation of the thrombospondin type I repeats of human properdin. Biochim Biophys Acta 1548: 265–277 [DOI] [PubMed] [Google Scholar]
- Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI (2012) New developments in the ATSAS program package for small‐angle scattering data analysis. J Appl Crystallogr 45: 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillimer L, Blum L, Lepow IH, Ross OA, Todd EW, Wardlaw AC (1954) The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomenon. Science 120: 279 [DOI] [PubMed] [Google Scholar]
- Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Wamel WJ, van Kessel KP, van Strijp JA (2005) Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol 6: 920–927 [DOI] [PubMed] [Google Scholar]
- Rooijakkers SH, Wu J, Ruyken M, van Domselaar R, Planken KL, Tzekou A, Ricklin D, Lambris JD, Janssen BJ, van Strijp JA, Gros P (2009) Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat Immunol 10: 721–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotkiewicz P, Skolnick J (2008) Fast procedure for reconstruction of full‐atom protein models from reduced representations. J Comput Chem 29: 1460–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumenina LT, Jablonski M, Hue C, Blouin J, Dimitrov JD, Dragon‐Durey MA, Cayla M, Fridman WH, Macher MA, Ribes D, Moulonguet L, Rostaing L, Satchell SC, Mathieson PW, Sautes‐Fridman C, Loirat C, Regnier CH, Halbwachs‐Mecarelli L, Fremeaux‐Bacchi V (2009) Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood 114: 2837–2845 [DOI] [PubMed] [Google Scholar]
- Skattum L, van Deuren M, van der Poll T, Truedsson L (2011) Complement deficiency states and associated infections. Mol Immunol 48: 1643–1655 [DOI] [PubMed] [Google Scholar]
- Stover CM, Luckett JC, Echtenacher B, Dupont A, Figgitt SE, Brown J, Mannel DN, Schwaeble WJ (2008) Properdin plays a protective role in polymicrobial septic peritonitis. J Immunol 180: 3313–3318 [DOI] [PubMed] [Google Scholar]
- Sun Z, Reid KBM, Perkins SJ (2004) The dimeric and trimeric solution structures of the multidomain complement protein properdin by X‐ray scattering, analytical ultracentrifugation and constrained modelling. J Mol Biol 343: 1327–1343 [DOI] [PubMed] [Google Scholar]
- Svergun DI (1992) Determination of the regularization parameter in indirect‐transform methods using perceptual criteria. J Appl Crystallogr 25: 495–503 [Google Scholar]
- Svergun D, Barberato C, Koch MHJ (1995) CRYSOL – A program to evaluate x‐ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28: 768–773 [Google Scholar]
- Volkov VV, Svergun DI (2003) Uniqueness of ab initio shape determination in small‐angle scattering. J Appl Crystallogr 36: 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Miwa T, Ducka‐Kokalari B, Redai IG, Sato S, Gullipalli D, Zangrilli JG, Haczku A, Song WC (2015) Properdin contributes to allergic airway inflammation through local C3a generation. J Immunol 195: 1171–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Wu YQ, Ricklin D, Janssen BJ, Lambris JD, Gros P (2009) Structure of complement fragment C3b‐factor H and implications for host protection by complement regulators. Nat Immunol 10: 728–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Berger SP, Trouw LA, de Boer HC, Schlagwein N, Mutsaers C, Daha MR, van Kooten C (2008) Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J Immunol 180: 7613–7621 [DOI] [PubMed] [Google Scholar]
- Zaferani A, Vives RR, van der Pol P, Hakvoort JJ, Navis GJ, van Goor H, Daha MR, Lortat‐Jacob H, Seelen MA, van den Born J (2011) Identification of tubular heparan sulfate as a docking platform for the alternative complement component properdin in proteinuric renal disease. J Biol Chem 286: 5359–5367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaferani A, Vives RR, van der Pol P, Navis GJ, Daha MR, van Kooten C, Lortat‐Jacob H, Seelen MA, van den Born J (2012) Factor h and properdin recognize different epitopes on renal tubular epithelial heparan sulfate. J Biol Chem 287: 31471–31481 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Data Availability Statement
Coordinates and structure factors for the crystal structure of FP in complex with the SCIN‐stabilized C3 convertase are available at the RCSB protein data bank in entry 5M6W. The solution structures and scattering curves for FPc and FP E244K are available at the SASBDB as entries SASDB59 and SASDB69, respectively.