

Abstract
Nuclear FOXO proteins act as tumor suppressors by transcriptionally activating genes involved in apoptosis and cell cycle arrest, and these anticancer functions are inhibited by AKT‐induced phosphorylation and cytoplasmic sequestration of FOXOs. We found that, after AKT‐mediated phosphorylation at serine 319, FOXO1 binds to IQGAP1, a hub for activation of the MAPK pathway, and impedes IQGAP1‐dependent phosphorylation of ERK1/2 (pERK1/2). Conversely, decreased FOXO1 expression increases pERK1/2 in cancer cell lines and correlates with increased pERK1/2 levels in patient specimens and disease progression. Treatment of cancer cells with PI3K inhibitors or taxane causes FOXO1 localization in the nucleus, increased expression of pERK1/2, and drug resistance. These effects are reversed by administering a small FOXO1‐derived phospho‐mimicking peptide inhibitor in vitro and in mice. Our results show a tumor suppressor role of AKT‐phosphorylated FOXO1 in the cytoplasm and suggest that this function of FOXO1 can be harnessed to overcome chemoresistance in cancer.
Keywords: FOXO1, AKT, MAPK, chemoresistance, cancer
Subject Categories: Cancer, Signal Transduction
Introduction
O‐class forkhead factors FOXO1, FOXO3, FOXO4, and FOXO6 (the human orthologs of Caenorhabditis elegans DAF‐16 and Drosophila dFOXO) are a family of proteins that transcriptionally activate genes involved in apoptosis (e.g., Bim and FasL), cell cycle arrest (e.g., p27 KIP1 and p21 CIP1), and oxidative stress detoxification (e.g., MnSOD and Catalase) (Brunet et al, 1999; Medema et al, 2000; Kops et al, 2002; Nemoto & Finkel, 2002; Gilley et al, 2003). These findings imply a tumor suppressor function of FOXOs in the nucleus. This concept is supported by studies in human cancer specimens and mouse genetic models (Dong et al, 2006; Paik et al, 2007).
The PI3K‐AKT pathway is often hyperactivated in human cancers as a result of amplification of the AKT gene, activating mutation in the catalytic subunit of PI3K and loss of the tumor suppressor phosphatase and tension homolog (PTEN) (Vivanco & Sawyers, 2002; Yuan & Cantley, 2008). On activation, AKT phosphorylates FOXO proteins at 3 serine/threonine residues, promoting nuclear exclusion and inactivation of the transactivation‐dependent (genomic) tumor suppressor activities of these proteins in the nucleus (Biggs et al, 1999; Brunet et al, 1999). Unexpectedly, many groups have found that ubiquitination‐dependent degradation of AKT‐phosphorylated FOXO proteins is critical for cell transformation, proliferation, survival, and insulin resistance (Matsuzaki et al, 2003; Plas & Thompson, 2003; Aoki et al, 2004; Huang et al, 2005; Wang et al, 2012). AKT‐phosphorylated FOXO proteins have long been suspected to possess a tumor suppressor function unless they are degraded by E3 ubiquitin ligases (Huang et al, 2005). To date, however, the enigma remains.
The prevalence of PI3K‐AKT hyperactivation in human cancers has prompted the development of small molecule inhibitors of this pathway, and their anticancer efficacy is currently being tested in both preclinical and clinical settings (Engelman et al, 2008). Intriguingly, under certain cellular contexts, inhibition of this pathway triggers “feedback” activation of the MAPK pathway, another pro‐proliferation/survival signaling axis (Chandarlapaty et al, 2011; Serra et al, 2011; Lin et al, 2014). Co‐administration of the MEK1/2 inhibitor AZD6244 has been shown to circumvent tumor resistance to the PI3K/mTOR dual inhibitor NVP‐BEZ235 in animals (Serra et al, 2011). This finding stresses the importance of acquired ERK activation in development of drug resistance to the PI3K and AKT inhibitors in cancer. However, the molecular basis through which PI3K/AKT inhibition promotes ERK activation remains poorly understood.
IQGAP1 is a scaffold protein that integrates signals to regulate cell adhesion, actin cytoskeleton, and cell proliferation, among others (Kim et al, 2011; White et al, 2012). It functions as a hub for c‐Raf, MEK1/2, and ERK1/2 binding and, ultimately, activation of ERK1/2 (Roy et al, 2004, 2005; Ren et al, 2007, 2008). Most importantly, IQGAP1 is required for c‐Raf‐dependent tumorigenesis, as manifested in animal models (Jameson et al, 2013). Through unbiased protein tandem affinity purification and mass spectrometry, we identified IQGAP1 as a binding partner of FOXO1. We provide evidence that AKT‐phosphorylated FOXO1 binds to IQGAP1 and inhibits IQGAP1‐dependent activation of ERK1/2. Conversely, treating cells with PI3K or AKT inhibitors or taxane results in FOXO1 nuclear localization and ERK1/2 activation, which in turn lead to chemotherapy resistance in cancer cells.
Results
Identification of the scaffold protein IQGAP1 as a binding partner of FOXO1
To search for novel functions of FOXO1, we constructed a FOXO1 mammalian expression vector (SFB‐FOXO1) that contains S, Flag, and biotin‐binding‐protein‐(streptavidin)‐binding‐peptide tags. This plasmid and the backbone vector were transfected separately into 293T cells, and cell extracts were prepared for tandem affinity purification and mass spectrometry. A total of 109 proteins were identified with confidence, which include FOXO1 (bait), known FOXO1‐interacting proteins such as USP7/HAUSP, and PLK1 (van der Horst et al, 2006; Yuan et al, 2014), and a large number of new binding partners such as IQGAP1 (Fig 1A and Appendix Table S1). Gene ontology analysis indicates that FOXO1‐associated proteins are involved in many biological processes such as protein biosynthesis, translation elongation, and acetylation (Appendix Fig S1A). Because IQGAP1 is a scaffold protein that is important for activation of the Raf‐MEK‐ERK pathway and tumorigenesis (White et al, 2012), we chose to investigate the molecular basis of the interaction between FOXO1 and IQGAP1 and the biological impact of their interaction on cancer cell growth and therapy resistance.
Figure 1. IQGAP1 interacts with FOXO1 in vitro and in vivo .
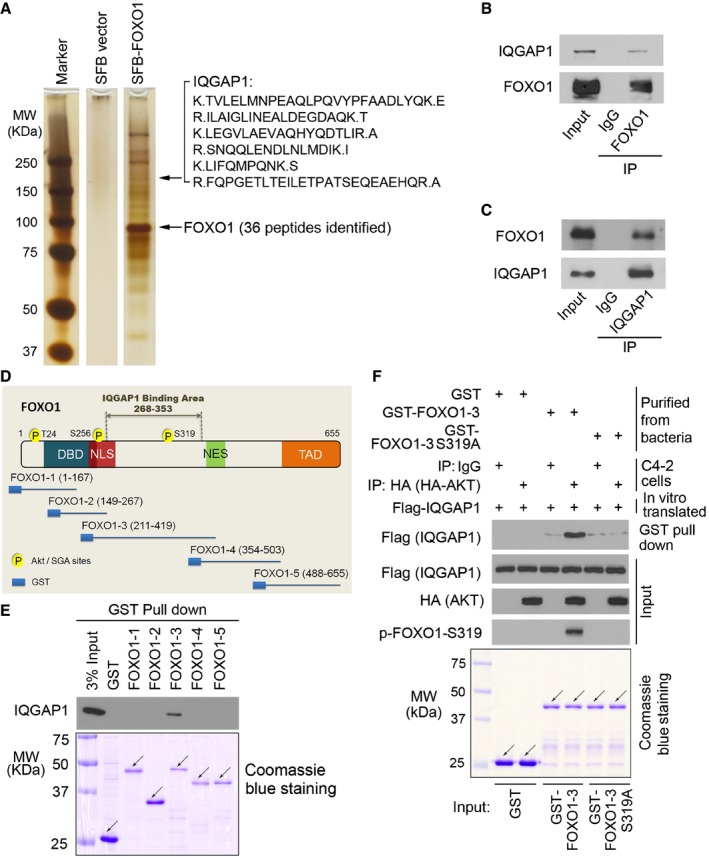
-
ASDS–PAGE and silver staining of proteins purified by tandem affinity purification using lysates of 293T cells transiently transfected with control vector and SFB‐tagged FOXO1 for 24 h. Six IQGAP1 peptides detected by mass spectrometry are shown.
-
B, CWestern blot detection of co‐immunoprecipitated endogenous FOXO1 and IQGAP1 proteins in LNCaP cells.
-
DSchematic diagram depicting a set of GST‐FOXO1 recombinant protein constructs.
-
EWestern blot analysis of IQGAP1 proteins in DU145 whole‐cell lysate pulled down by GST or GST‐FOXO1 recombinant proteins. Arrows indicate the proteins in expected molecular weight.
-
FIn vitro protein binding assay. GST and GST‐FOXO1‐3 (amino acids 211‐419) purified from bacteria were subjected to AKT kinase assay with IgG or HA‐AKT‐CA immunoprecipitated from HA‐AKT‐CA‐transfected C4‐2 cells before incubating with in vitro translated Flag‐IQGAP1 for protein binding assay. Arrows indicate the proteins in expected molecular weight.
Co‐immunoprecipitation (co‐IP) assay confirmed that endogenous FOXO1 and IQGAP1 proteins associated with each other in PTEN‐null LNCaP prostate cancer cells (Fig 1B and C, and Appendix Fig S1B). To define which region in FOXO1 mediates its interaction with IQGAP1, we generated glutathione‐S‐transferase (GST)‐FOXO1 constructs (Fig 1D), purified recombinant proteins from bacteria (Fig 1E, lower panel), and performed GST pull‐down assays. We demonstrated that GST‐FOXO1‐3 (amino acids 211–419), but not GST and other GST‐FOXO1 recombinant proteins, interacted with IQGAP1 (Fig 1E, upper panel), although the binding was relatively weak (see more data below). Nonetheless, these data suggest that the central portion (amino acids 268–353) of FOXO1 is important for its binding to IQGAP1.
Serine‐319 phosphorylation of FOXO1 is important for FOXO1‐IQGAP1 interaction
Given that the interaction between recombinant FOXO1 from bacteria and cellular IQGAP1 was much weaker than the input (Fig 1E), we hypothesized that posttranslational modification such as phosphorylation of FOXO1 is important for FOXO1 binding to IQGAP1. To test this hypothesis, LNCaP cell (PTEN‐negative) lysate was treated with λ protein phosphatase before co‐IP assays. Threonine 24, serine 256, and serine 319 (T24, S256, and S319) residues in FOXO1 are readily phosphorylated by AKT in PTEN‐negative cells (Biggs et al, 1999; Tang et al, 1999). The effectiveness of phosphatase treatment was evident by reduction or depletion of FOXO1 phosphorylation at S256 and S319, respectively (Fig 2A). Phosphatase treatment largely abrogated FOXO1‐IQGAP1 interaction (Fig 2A and Appendix Fig S2A), an indication of the importance of phosphorylation for their interaction. In contrast, ectopic expression of a constitutively active AKT (AKT‐CA), but not the kinase‐dead mutant (AKT‐DN), substantially enhanced FOXO1‐IQGAP1 interaction in DU145 (PTEN‐positive) prostate cancer cells (Fig 2B and Appendix Figs S1B and S2B). Similarly, knockdown of endogenous PTEN in DU145 cells also markedly increased FOXO1‐IQGAP1 interaction (Fig 2C and Appendix Fig S2C). Conversely, inhibition of AKT by the PI3K inhibitor LY294002 decreased the FOXO1‐IQGAP1 interaction to the background level in PTEN‐null LNCaP cells (Fig 2D and Appendix Fig S2D). These data indicate that FOXO1‐IQGAP1 interaction is regulated by the PI3K‐AKT pathway.
Figure 2. AKT phosphorylation of FOXO1 at serine 319 is critical for FOXO1 binding to IQGAP1.
-
AWestern blot analysis of LNCaP whole‐cell lysates (WCL) and co‐IP samples. Cell lysates were treated with or without λ phosphatase before IP.
-
BWestern blot analysis of WCL and co‐IP samples in DU145 cells 24 h after transfection with indicated plasmids.
-
CWestern blot analysis of WCL and co‐IP samples in DU145 cells 48 h after transfection with indicated siRNAs.
-
DWestern blot analysis of WCL and co‐IP samples in LNCaP cells. Cells were treated with 30 μM of LY294002 for 6 h before IP.
-
E, FWestern blot analysis of WCL and co‐IP samples in LNCaP cells 24 h after transfection with indicated plasmids.
To determine whether AKT phosphorylation of FOXO1 is involved in FOXO1‐IQGAP1 interaction, we mutated three AKT phosphorylation sites to alanine individually or together. Interaction of IQGAP1 with S319A and triple mutant (A3), but not T24A and S256A mutants, was decreased to the background level (Fig 2E and Appendix Fig S2E). FOXO1‐A3 and S319A are two transcriptionally active mutants. Next, we sought to rule out the possibility that the inhibition of FOXO1‐IQGAP1 interaction caused by these two mutants was mediated indirectly through their downstream transcription targets. FOXO1‐537 is a transcription‐deficient mutant of FOXO1, in which histidine 215, a key residue for DNA binding, is mutated to arginine and the transactivation domain (amino acids 538–655) is deleted (Liu et al, 2008). Similar to the results shown in Fig 2E, binding of the AKT phosphorylation‐resistant mutant (FOXO1‐537‐A3) to IQGAP1 was much lower compared with the wild‐type counterpart FOXO1‐537 (Fig 2F and Appendix Fig S2F).
To further test the role of AKT phosphorylation of FOXO1 at S319 in mediating FOXO1‐IQGAP1 interaction, we immunoprecipitated HA‐AKT from C4‐2 cells and performed in vitro kinase assays using bacterially purified GST‐FOXO1‐3 (amino acids 211–419) and GST‐FOXO1‐3 S319A as substrates. We then carried out in vitro protein binding assays using AKT‐phosphorylated GST‐FOXO1‐3 and in vitro transcribed and translated Flag‐tagged IQGAP1. GST‐FOXO1‐3 had a basal‐level interaction with IQGAP1 (Fig 1F and Appendix Fig S1C and D), which is consistent with the GST pull‐down result using cellular IQGAP1 proteins (Fig 1E). Importantly, the interaction of IQGAP1 with GST‐FOXO1‐3, but not S319A mutant, was substantially enhanced by AKT‐mediated S319 phosphorylation of FOXO1 (Fig 1F and Appendix Fig S1C and D). Together, these data suggest that S319 phosphorylation of FOXO1 is important for FOXO1‐IQGAP1 interaction and their interaction is unlikely mediated indirectly by its downstream transcription targets.
AKT‐phosphorylated FOXO1 inhibits IQGAP1 binding to c‐Raf, MEK, and ERK proteins
To determine which domain of IQGAP1 is involved in FOXO1 binding, we generated six GST‐IQGAP1 recombinant proteins corresponding to six well‐studied functional domains of IQGAP1 (Fig 3A). GST pull‐down assays showed that the coiled‐coil domain of IQGAP1 specifically interacted with FOXO1 proteins in LNCaP cells (Fig 3B).
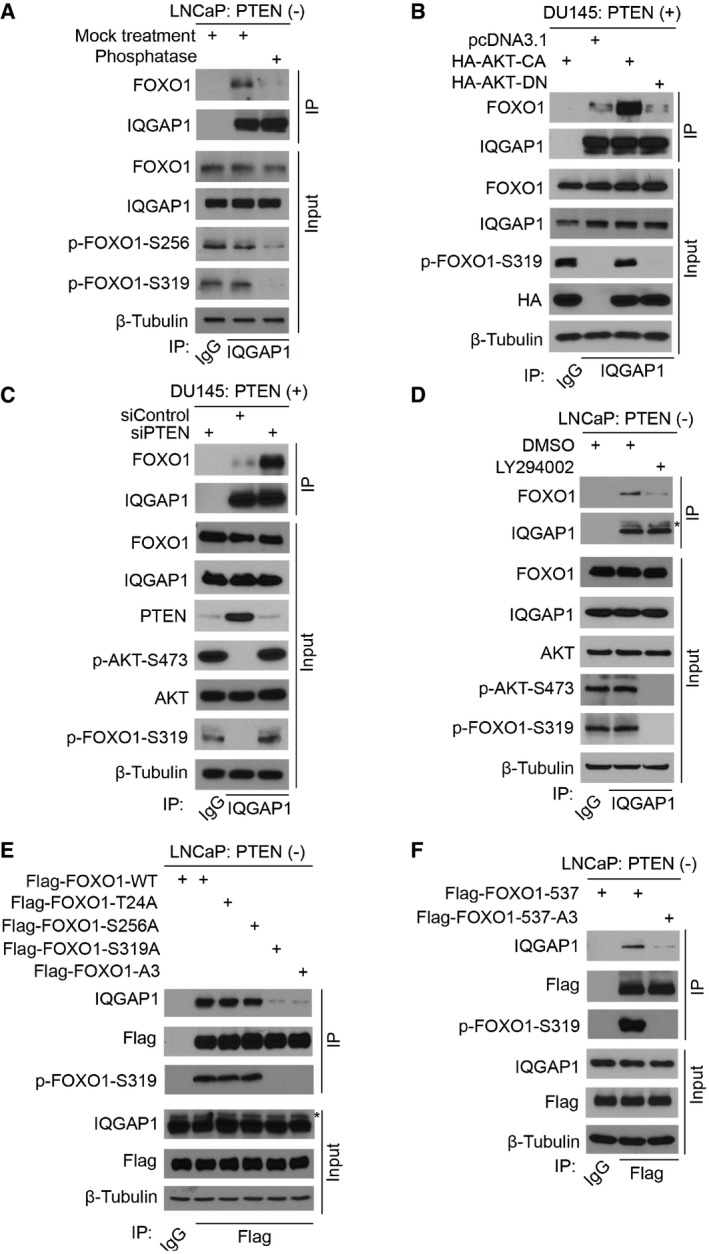
Figure 3. AKT‐phosphorylated FOXO1 binds to IQGAP1 and inhibits IQGAP1 interaction with Raf, MEK, and ERK proteins.
-
ASchematic diagram depicting the domain structure of IQGAP1 and 6 GST‐IQGAP1 constructs. CC, coiled‐coil domain.
-
BLNCaP whole‐cell lysates (WCL) were subjected to GST pull‐down assay by GST or GST‐IQGAP1 recombinant proteins and Western blot analysis of FOXO1 proteins. Arrows indicate the proteins in expected molecular weight.
-
CWestern blot analysis of WCL and co‐IP samples in LNCaP cells 48 h after infection with lentivirus expressing control or FOXO1‐specific shRNA.
-
D–FWestern blot analysis of WCL and co‐IP samples in LNCaP cells 24 h after transfection with indicated plasmids. E.V., empty vector.
Similar to the findings in other cell types (Roy et al, 2004, 2005; Ren et al, 2007; Jameson et al, 2013), IQGAP1 interaction with c‐Raf, MEK2, and ERK2 was readily detected in LNCaP cells (Fig 3C). Importantly, although knockdown of endogenous FOXO1 had no effect on the steady‐state levels of ERK2 and other MAPK proteins, it markedly increased their interaction with IQGAP1 (Fig 3C and Appendix Fig S3A). Moreover, expression of FOXO1‐537 diminished IQGAP1 interaction with c‐Raf, MEK2, and ERK2, whereas almost no inhibitory effect was observed for FOXO1‐537‐A3 (Fig 3D and Appendix Fig S3B). This result is in agreement with the difference in the capacity of FOXO1‐537 and the A3 mutant to bind to IQGAP1 (Fig 2F).
Given that AKT phosphorylation induces cytoplasm localization of FOXO proteins, we examined the impact of FOXO1 cellular localization on IQGAP1 interaction with MAPK proteins. Similar to previous findings (Brunet et al, 2002; Matsuzaki et al, 2003), FOXO1‐NESm and FOXO1‐NLSm, in which the nuclear exportation signal (NES) or the nuclear localization signal (NLS) is mutated, were primarily localized in the nucleus and the cytoplasm, respectively (Fig EV1A and B). Consistent with a previous report (Nakamura et al, 2000), FOXO1‐WT was mainly localized in the cytoplasm in PTEN‐null LNCaP cells (Fig EV1A and B). Co‐IP assays demonstrated that ectopic expression of FOXO1‐WT and cytoplasmic FOXO1‐NLSm largely inhibited IQGAP1 binding to MAPK proteins in these cells (Fig 3E and Appendix Fig S3C). In contrast, the inhibitory effect of the nuclear mutant FOXO1‐NESm on IQGAP1‐MAPK interaction was much less than FOXO1‐NLSm and FOXO1‐WT (Fig 3E and Appendix Fig S3C). Furthermore, we generated a small (30 amino acids) S319 phospho‐mimicking IQGAP1‐binding peptide of FOXO1, termed FOXO1‐IQBP (S319E) or FOXO1‐IQBP (SE) by mutating the serine 319 residue to glutamic acid (E). This peptide and the wild‐type (S319) and S319A counterparts were ubiquitously expressed in both cytoplasm and nucleus (Fig EV1C). Compared with S319, the S319E mutant had higher affinity of binding to IQGAP1, and a similar result was obtained with another phospho‐mimicking mutant S319D in which S319 was mutated to aspartic acid (D) (Fig EV1D and E). Moreover, in comparison with S319, S319E had much greater inhibitory effect on the interaction between endogenous FOXO1 and IQGAP1 (Fig EV1F). In contrast, IQGAP1‐binding affinity of the non‐phosphorylatable mutant S319A and its inhibitory effect on FOXO1‐IQGAP1 interaction was much lower in comparison with S319 (Fig EV1D and F). In line with these findings, expression of the S319E peptide had the greatest inhibitory effect on IQGAP1‐MAPK interaction, whereas the inhibitory effect of S319A was much smaller (Fig 3F and Appendix Fig S3D and E). Together, AKT‐phosphorylated FOXO1 binds to the coiled‐coil domain of IQGAP1 and impedes IQGAP1 interaction with MAPK proteins in cells.
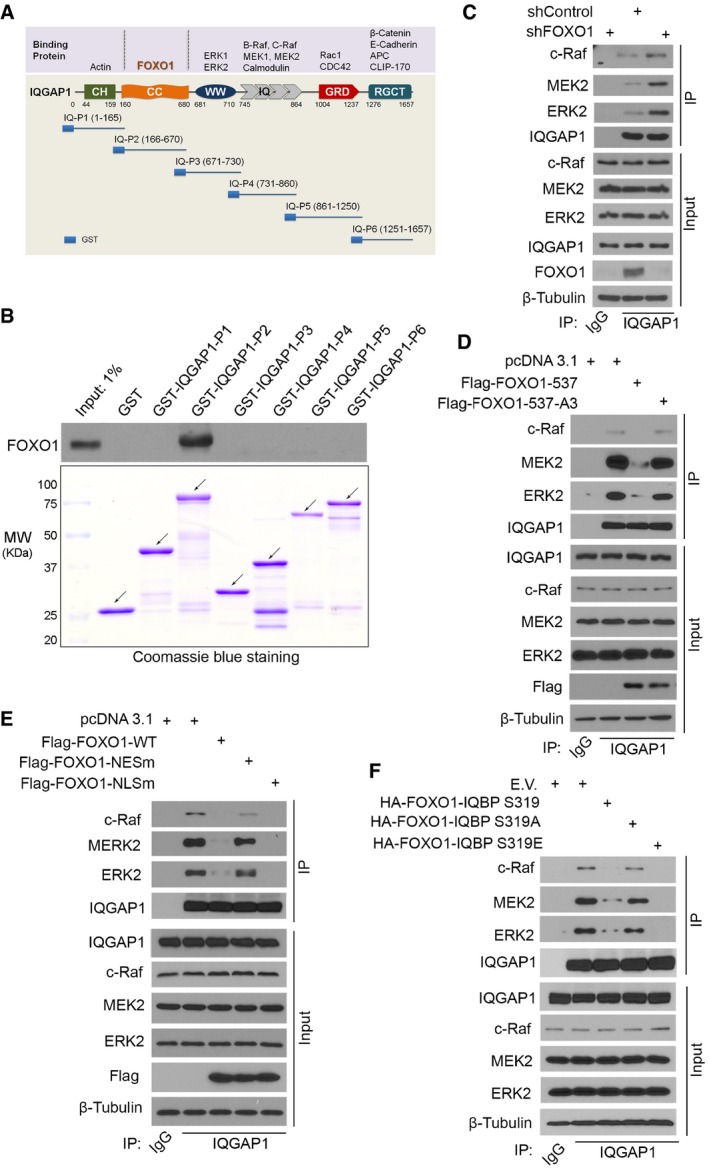
Figure EV1. Assessment of cellular localization of FOXO1 and phospho‐mimicking FOXO1 peptide HA‐FOXO1‐IQBP S319E and its wild‐type (S319) and non‐phosphorylatable (S319A) counterparts in PCa cells.
-
A, BImmunofluorescence cytochemistry using antibodies for anti‐Flag and IQGAP1 in LNCaP cells transfected with control vector (pcDNA3.1) and plasmids for Flag‐FOXO1‐WT, Flag‐FOXO1‐NESm, and Flag‐FOXO1‐NLSm. Cellular localization (nuclear, cytoplasm, or both) was photographed (A) and analyzed in > 100 cells in each experimental condition (B). Scale bars, 20 μm. The experiment was repeated at least once and similar results were obtained.
-
CImmunofluorescence cytochemistry using antibodies for anti‐HA and IQGAP1 in LNCaP cells transfected with empty vector (E.V.) pcDNA3.1 and plasmids for phospho‐mimicking FOXO1 peptide HA‐FOXO1‐IQBP S319E and its wild‐type (S319) and non‐phosphorylatable (S319A) counterparts. > 100 cells in each experimental condition were analyzed. Scale bars, 20 μm. The experiment was repeated at least once and similar results were obtained.
-
DLNCaP cells were transfected with SFB‐tagged empty vector (SFB‐EV) and SFB‐tagged plasmids for phospho‐mimicking FOXO1 peptide SFB‐FOXO1‐IQBP S319E and its wild‐type (S319) and non‐phosphorylatable (S319A) counterparts. 24 h after transfection, cells were harvested and lysed for co‐IP with IgG or anti‐Flag antibodies and immunoprecipitated proteins were analyzed by Western blotting. SFB tags are S, Flag, and biotin‐binding protein (streptavidin) binding peptide tags. These three tags encode an ˜12‐kDa peptide (119 amino acids).
-
ELNCaP cells were transfected with SFB‐tagged plasmids for phospho‐mimicking FOXO1 peptides SFB‐FOXO1‐IQBP S319E and S319D and the non‐phosphorylatable counterpart S319A. 24 h after transfection, cells were harvested and lysed for co‐IP with IgG or anti‐Flag antibodies and immunoprecipitated proteins were analyzed by Western blotting.
-
FLNCaP cells were transfected with SFB‐tagged empty vector (EV) and SFB‐tagged plasmids for SFB‐FOXO1‐IQBP S319E and its wild‐type (S319) and non‐phosphorylatable (S319A) counterparts. 24 h after transfection, cells were harvested and lysed for co‐IP with IgG or anti‐IQGAP1 antibodies and immunoprecipitated proteins were analyzed by Western blotting using indicated antibodies.
-
GLimited proteolysis assay. Limited proteolysis of GST‐IQGAP1 incubated with GST‐FOXO1‐IQBP S319E or GST alone using trypsin. Proteolysis was carried out on ice for the times indicated under each lane. The products of proteolysis were resolved by 13.5% acrylamide SDS–PAGE and visualized by staining with Coomassie blue. The experiments were repeated once, and similar results were obtained.
The observation that c‐Raf, MEK, and ERK all bind to separate sites downstream of the coiled‐coil domain prompted us to test the hypothesis that binding of the short S319E peptide to the coiled‐coil region causes conformation changes in IQGAP1, which in turn impair the binding of MAPK proteins to IQGAP1. Limited proteolysis assay is often used to monitor protein conformation changes (Varne et al, 2002). We incubated recombinant IQGAP1 proteins with GST‐FOXO1‐IQBP S319E or GST alone and performed partial digestion of proteins using trypsin. As shown in Fig EV1G, there were two major proteolytic bands migrated slightly faster than the uncleaved IQGAP1 in the control (GST alone) group, whereas there was only one major band migrated slightly faster than the uncleaved IQGAP1 in the GST‐FOXO1‐IQBP S319E group. These data suggest that binding of the short S319E peptide causes conformation changes in IQGAP1 which therefore provide a plausible explanation for the inhibitory effect of this peptide on the binding of c‐Raf, MEK, and ERK to IQGAP1.
AKT‐phosphorylated FOXO1 inhibits IQGAP1‐augmented phosphorylation of ERK1/2
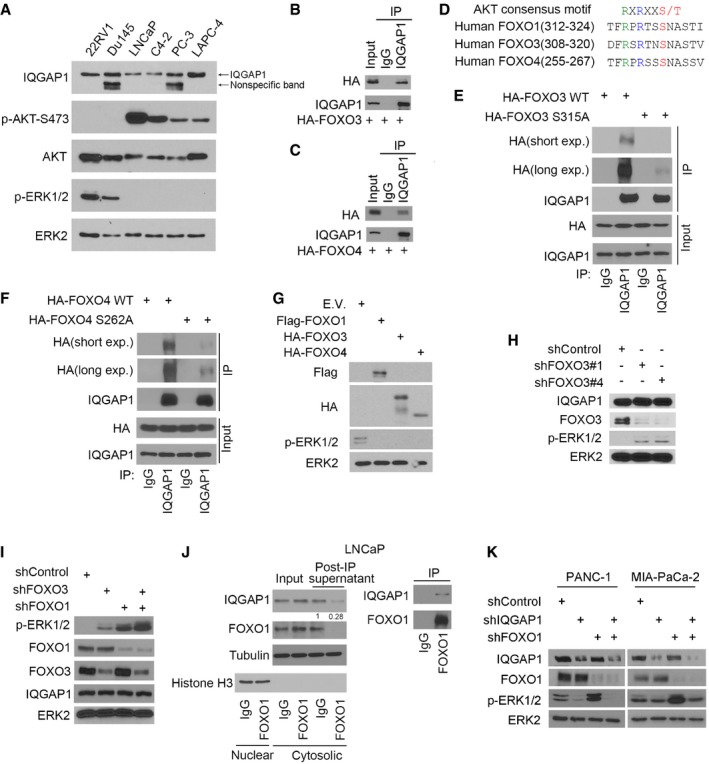
Given that AKT‐phosphorylated FOXO1 dampens IQGAP1‐MAPK protein interaction, we sought to determine whether FOXO1 regulates phosphorylation and activation of ERK1/2. Expression of AKT phosphorylation (pAKT) and ERK1/2 phosphorylation (pERK1/2) was examined in a panel of prostate cancer cell lines. As shown in Fig EV2A, pERK1/2 was high in pAKT‐undetectable cell lines 22Rv1 and DU145, whereas pERK1/2 was hardly detectable in pAKT‐high cell lines LNCaP, C4‐2, PC‐3, and LAPC‐4; thus, there was an inverse relationship between pAKT and pERK1/2 in the cell lines surveyed.
Figure EV2. Assessment of the interaction of IQGAP1 with FOXO3 and FOXO4, their effect on ERK1/2 phosphorylation and effect of FOXO1 on ERK1/2 phosphorylation in non‐prostate cancer cells.
-
AWestern blot analysis of expression of phospho‐AKT (serine 473) and ERK1/2 in prostate cancer cell lines indicated. Arrow indicates non‐specific reaction protein bands.
-
B, CWestern blot analysis of whole‐cell lysate (WCL) and co‐immunoprecipitation (co‐IP) samples of IgG or anti‐IQGAP1 antibody from LNCaP cells 24 h after transfection with HA‐FOXO3 (B) or HA‐FOXO4 (C).
-
DComparison of S319, an AKT phosphorylation site in FOXO1 with the homologous site in FOXO3 (S315) and FOXO4 (S262) by multiple sequence alignments.
-
E, FWestern blot analysis of WCL and co‐IP samples from LNCaP cells 24 h after transfection with indicated plasmids.
-
GWestern blot analysis of WCL from DU145 cells 24 h after transfection with indicated plasmids. E.V., empty vector.
-
H, IWestern blot analysis of WCL from LNCaP cells 48 h after infection with lentivirus expressing indicated shRNAs.
-
JWestern blot analysis of indicated proteins in cytosolic fractionation (input), the supernatant post IP and co‐IP samples from LNCaP cells. Nuclear fractionation was used to indicate the effectiveness of the fractionation.
-
KWestern blot analysis of WCL from PANC‐1 and MIA‐PaCa‐2 cells 48 h after infection with lentivirus expressing indicated shRNAs.
To determine the causal role of FOXO proteins in regulation of pERK1/2, we overexpressed FOXO proteins in DU145, a cell line with high pERK1/2. Similar to FOXO1, FOXO3 and FOXO4 (FOXO6 was not examined because it is primarily expressed in neurons) were also able to interact with IQGAP1 (Fig EV2B and C), and their interaction with IQGAP1 also depends on AKT phosphorylation at S315 in FOXO3 and S262 in FOXO4, which are homologous to S319 in FOXO1 (Fig EV2D–F). Moreover, ectopic expression of these proteins abrogated pERK1/2 in DU145 cells (Fig EV2G).
Next, we examined the effect of knockdown of FOXOs in LNCaP, a cell line with little or no basal level of pERK1/2. Because the expression level of endogenous FOXO4 is extremely low in human prostate cancer cell lines (Modur et al, 2002; Huang et al, 2006), we focused on only FOXO1 and FOXO3. Knocking down endogenous FOXO1 by two independent gene‐specific short hairpin RNAs (shRNAs) markedly increased pERK1/2 in LNCaP cells, and this was completely reversed by restored expression of shRNA‐resistant FOXO1 (Fig 4A and B, and Appendix Fig S4A and B). Knockdown of FOXO3 by two independent shRNAs also increased pERK1/2 (Fig EV2H). Notably, co‐knockdown of FOXO1 and FOXO3 resulted in a much greater induction of pERK1/2 in comparison with each individual knockdown alone (Fig EV2I), an indication of a collaborative rather than redundant role of different FOXO factors in regulating pERK1/2. A plausible explanation for this observation is that IQGAP1 is a highly abundant protein and there are likely enough IQGAP1 molecules in cells for FOXO1 and FOXO3 binding. This notion is further supported by our finding that while cytoplasmic FOXO1 proteins in LNCaP cell lysate were completely pulled down by anti‐FOXO1 antibody used, ~70% of IQGAP1 was pulled down by the same antibody (Fig EV2J). Importantly, FOXO1 knockdown‐induced increase in pERK1/2 was completely reversed by concomitant knockdown of endogenous IQGAP1 (Fig 4C and Appendix Fig S4C). Similar results were obtained in other cancer types such as pancreatic cancer cell lines PANC‐1 and MIA‐PaCa‐2 (Fig EV2K). These data suggest that the effect of FOXO1 on pERK1/2 is mediated through IQGAP1. Conversely, overexpression of IQGAP1 increased pERK1/2 in C4‐2 cells (Fig 4D and Appendix Fig S4D). This effect was largely diminished in cells transfected with FOXO1‐537 but not the non‐phosphorylatable mutant FOXO1‐537 S319A (Fig 4D and Appendix Fig S4D), a finding highlighting the importance of S319 phosphorylation in FOXO1 regulation of pERK1/2. Finally, epidermal growth factor treatment induced pERK1/2 in C4‐2 cells, whereas this effect was largely abolished by ectopic expression of FOXO1‐537 but not the S319A mutant (Fig 4E and Appendix Fig S4E). Together, these findings demonstrate that AKT‐phosphorylated FOXO1 inhibits pERK1/2 in a transactivation‐independent manner and this effect is mediated through IQGAP1.
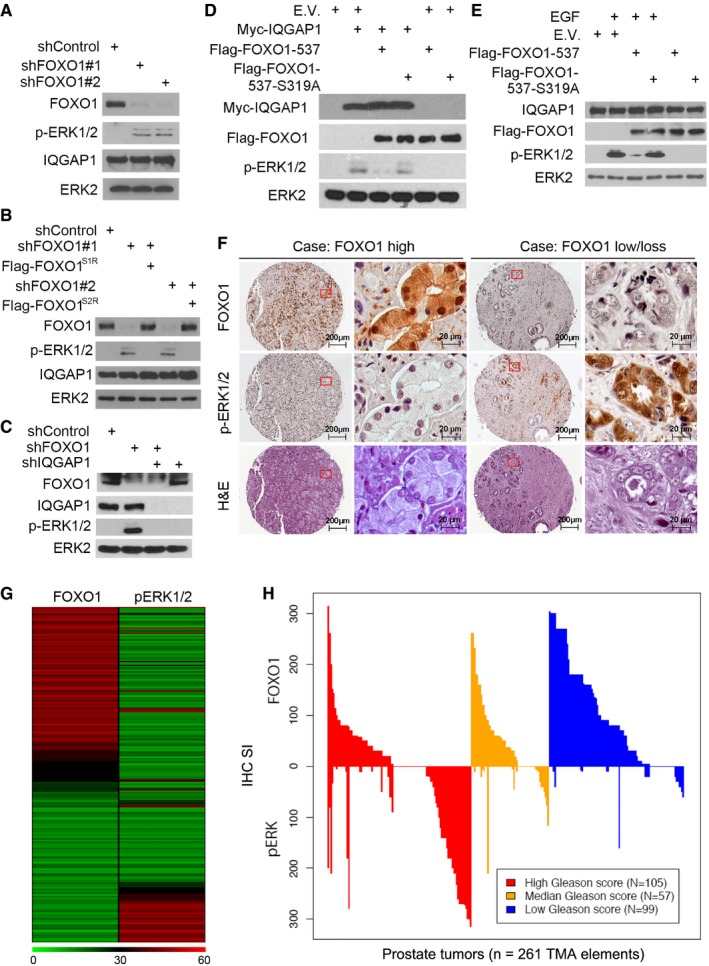
Figure 4. AKT‐phosphorylated FOXO1 inhibits IQGAP1‐dependent pERK1/2.
-
A–CWestern blot analysis of whole‐cell lysates (WCL) from LNCaP cells 48 h after infection with lentivirus expressing indicated shRNA and shRNA‐resistant plasmids.
-
D, EWestern blot analysis of WCL from C4‐2 cells 24 h after transfection with indicated plasmids. Cells were treated with or without 10 ng/ml of epidermal growth factor (EGF) for 10 min before being harvested.
-
FRepresentative images of hematoxylin–eosin staining and immunohistochemical staining of anti‐FOXO1 and anti‐pERK1/2 antibodies on TMA (n = 261) tissue sections.
-
G, HHeat map (G) and waterfall diagram (H) showing the immunohistochemical staining index (IHC SI) of FOXO1 and pERK1/2 in TMA and association with tumor Gleason scores. The scale bar in (G) indicates IHC SI, any of which > 60 were colored in red.
Expression of FOXO1 and pERK1/2 inversely correlates in prostate cancer specimens
FOXO1 is partially deleted or transcriptionally downregulated in ~35% of human prostate cancer cell lines and patient samples (Modur et al, 2002; Dong et al, 2006; Haflidadottir et al, 2013). To explore the clinical relevance of FOXO1‐mediated inhibition of pERK1/2, we sought to determine whether expression of FOXO1 and pERK1/2 correlates in human prostate cancer specimens. We examined the expression of these two proteins immunohistochemically on a tissue microarray (TMA) containing a cohort of prostate cancer samples (n = 261 TMA specimens) obtained from 167 patients. Immunohistochemical staining was evaluated by measuring both percentage of positive cells and staining intensity. Representative images of high and low/no staining of FOXO1 and pERK1/2 and corresponding hematoxylin–eosin staining are shown in Fig 4F. FOXO1 was inversely correlated with pERK1/2 expression in this cohort of patients (Spearman ρ = −0.29, P = 2.6 × 10−6) (Fig 4G). Further analysis indicated that tumors with lower Gleason scores have high FOXO1 expression and FOXO1 expression was negatively associated with Gleason score (Spearman ρ = −0.35, P = 1.0 × 10−8). In contrast, tumors with higher Gleason scores had high pERK1/2 levels, which were positively associated with Gleason score (Spearman ρ = 0.32, P = 1.4 × 10−7) (Fig 4H). These data indicate that loss or reduced expression of FOXO1 correlates with pERK1/2 and prostate cancer progression, at least in a subset of patients.
Nuclear localization of FOXO1 promotes PI3K/AKT inhibition‐induced pERK1/2
As demonstrated in other cancer types such as breast, pancreatic, and nasopharyngeal, among others, inhibition of the PI3K‐AKT pathway often results in an increase in pERK1/2 (Moelling et al, 2002; Robertson et al, 2010; Chandarlapaty et al, 2011; Serra et al, 2011). We demonstrated that inhibition of pAKT by the AKT inhibitor MK2206 increased pERK1/2 in PTEN‐null prostate cancer cell lines LNCaP, C4‐2, and C4‐2B (Fig 5A and Appendix Fig S5A). A similar result was obtained by administering the PI3K/mTOR dual inhibitor NVP‐BEZ235 in LNCaP cells, but its effect was very limited in C4‐2 and almost none in C4‐2B cells. The different effects of NVP‐BEZ235 in these cell lines were not due to the differences in IQGAP1 expression (Fig EV3A), and the precise underlying mechanism warrants further investigation.
Figure 5. AKT‐phosphorylated FOXO1 inhibits PI3K/AKT inhibitor‐induced ERK activation.
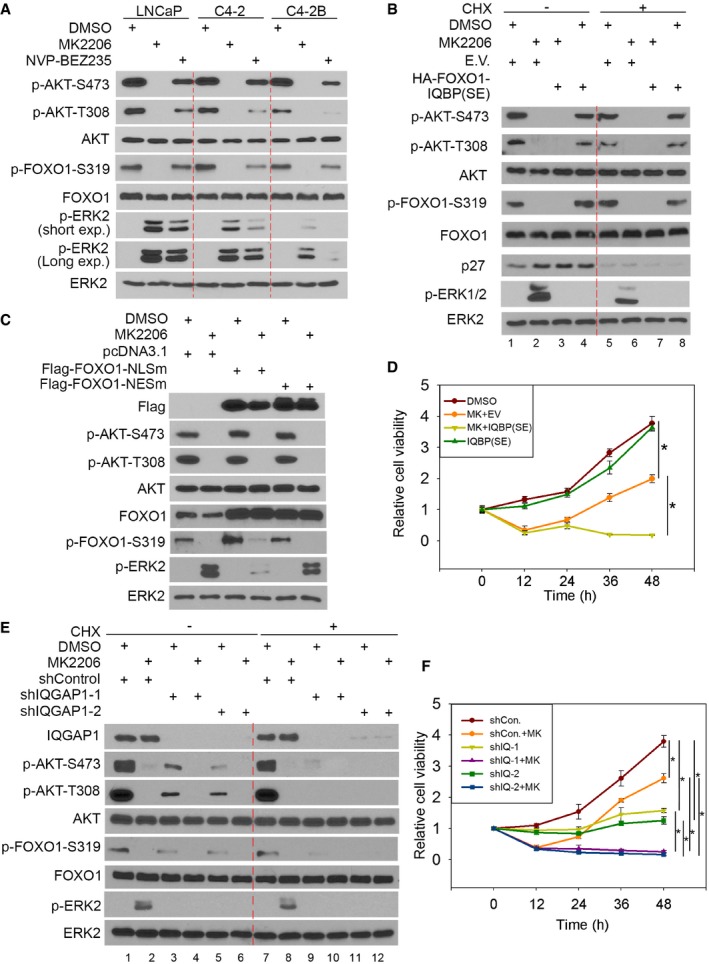
- Western blot analysis of whole‐cell lysates (WCL) of LNCaP, C4‐2, and C4‐2B cells 24 h after treatment with the AKT inhibitor MK2206 (0.5 μM) or the PI3K/mTOR dual inhibitor NVP‐BEZ235 (50 nM).
- Western blot analysis of WCL of LNCaP cells 48 h after transfection with indicated plasmids. Cells were treated with or without MK2206 (0.5 μM) for 24 h prior to harvest. E.V., empty vector.
- Western blot analysis of WCL of LNCaP cells 48 h after transfection with indicated plasmids. Cells were pre‐treated with or without CHX (20 μg/ml) for 30 min and then treated with or without MK2206 (0.5 μM) for 24 h.
- LNCaP cells were transfected with empty vector (E.V.) or a phospho‐mimicking peptide HA‐FOXO1‐IQBP(SE) for 24 h and then were treated with or without MK2206 (0.5 μM) followed by MTS assay at indicated time points (means ± s.d., n = 6). *P = 0.00014608 comparing MK2206 versus DMSO; *P = 4.28734E‐07 comparing MK2206 + EV versus MK2206 + IQBP(SE) (two‐sided Student's t‐test at the 48‐h time point).
- Western blot analysis of WCL of LNCaP cells 72 h after infection with lentivirus expressing indicated shRNAs. Cells were pre‐treated with or without CHX (20 μg/ml) for 30 min prior to being treated with or without MK2206 (0.5 μM) for 24 h.
- LNCaP cells were infected with lentivirus expressing indicated shRNAs for 72 h and then treated with or without MK2206 (0.5 μM) followed by MTS assay at indicated time points (means ± s.d., n = 6). *P = 6.71297E‐07 comparing shControl versus shControl + MK2206; *P = 1.92261E‐12 comparing shIQGAP1‐1 versus shIQGAP1‐1 + MK2206; *P = 6.87256E‐12 comparing shIQGAP1‐2 versus shIQGAP1‐2 + MK2206; *P = 1.64547E‐07 comparing shIQGAP1‐1 versus shControl; *P = 1.68294E‐08 comparing shIQGAP1‐2 versus shControl; *P = 2.91871E‐14 comparing shIQGAP1‐1 + MK2206 versus shControl + MK2206; *P = 8.95133E‐15 comparing shIQGAP1‐2 + MK2206 versus shControl + MK2206 (two‐sided Student's t‐test at the 48‐h time point).
Figure EV3. Assessment of IQGAP1 protein expression in PCa cell lines and effect of PI3K/AKT inhibitors and CHX on cellular localization of FOXO1.
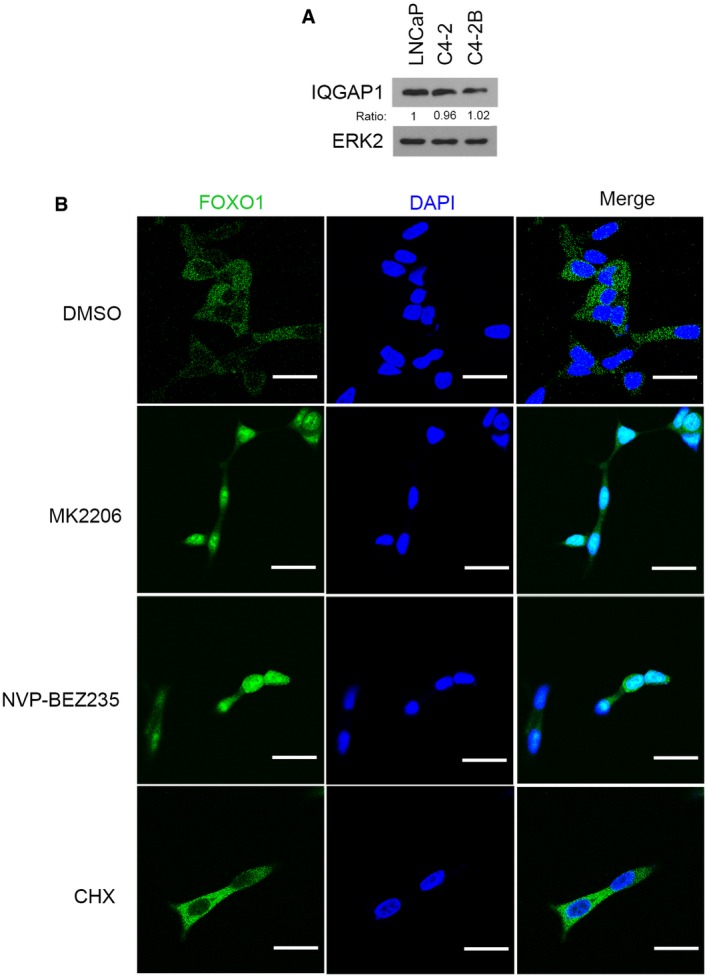
- Western blot analysis of IQGAP1 protein expression in prostate cancer cell lines LNCaP, C4‐2, and C4‐2B. ERK2 was used as a loading control. The density of IQGAP1 was determined by normalizing to ERK2 first and then to the normalized value in LNCaP cells.
- Immunofluorescence cytochemistry using anti‐FOXO1 antibody in LNCaP (PTEN‐negative and FOXO1 in the cytoplasm) cells treated with DMSO, MK2206 (0.5 μM), NVP‐BEZ235 (50 nM), and CHX (20 μg/ml) for 24 h. > 100 cells in each experimental condition were analyzed. Scale bars, 20 μm. The experiment was repeated at least once and similar results were obtained.
In breast cancer cells, treatment with PI3K or AKT inhibitor increases pERK1/2 by inducing nuclear localization of FOXO and FOXO‐dependent transcription of receptor tyrosine kinase genes such as HER3 (Chandarlapaty et al, 2011; Serra et al, 2011). As expected, MK2206 or NVP‐BEZ2235 also induced nuclear localization of FOXO1 in LNCaP cells (Fig EV3B). Surprisingly, treating LNCaP cells with the protein synthesis inhibitor cycloheximide (CHX) resulted in only ~50% reduction in MK2206‐induced pERK1/2 (Fig 5B, lane 2 versus 6 and Appendix Fig S5B). The effectiveness of CHX was evident by the blockage of induction of p27KIP1, a well‐studied FOXO transactivation target (Fig 5B, lane 1, 2 versus 5, 6). No effect of CHX treatment alone on pERK1/2 and FOXO1 nuclear localization was detected (Fig 5B, lane 1 versus 5, Fig EV3B and Appendix Fig S5B). These data indicate the existence of FOXO transcription‐independent pathways responsible for PI3K/AKT inhibition‐induced pERK1/2.
These results prompted us to test the hypothesis that AKT inhibition promotes FOXO1 nuclear localization, which in turn results in dismissal of FOXO1‐mediated inhibition of IQGAP1 in the cytoplasm and subsequent IQGAP1‐dependent hyperactivation of ERK1/2. This hypothesis is supported by our finding that forced expression of FOXO1‐NLSm, a cytoplasmic mutant, but not the nuclear mutant FOXO1‐NESm, largely inhibited MK2206‐induced pERK1/2 in LNCaP cells (Figs 5C and EV1A and B, and Appendix Fig S5C). Thus, loss of inhibition of IQGAP1 by cytosolic FOXO1 represents a crucial mechanism that drives PI3K/AKT inhibitor‐induced ERK1/2 activation.
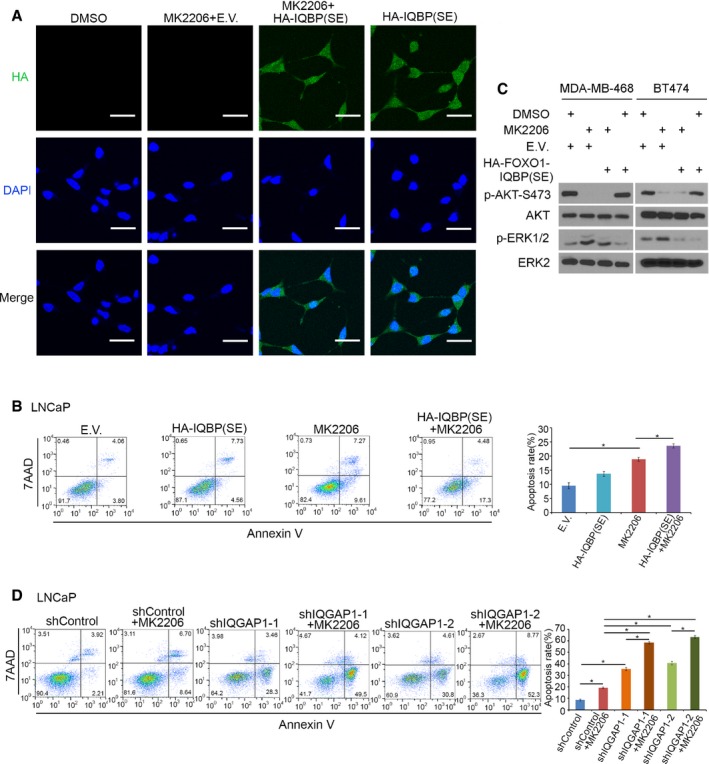
As demonstrated in Fig 3F, expression of FOXO1‐IQBP (S319E or SE), a FOXO1‐derived phospho‐mimicking peptide, impairs the IQGAP1‐MAPK interaction. We next examined the extent to which expression of this peptide affects IQGAP1‐dependent activation of ERK1/2 in PI3K/AKT inhibitor‐treated cells. We found that ectopic expression of FOXO1‐IQBP(SE) completely abolished MK2206‐induced increase in pERK1/2 in LNCaP cells (Fig 5B, lane 2 versus 3, 6 versus 7; Fig EV4A and Appendix Fig S5B). Accordingly, MTS cell viability assays demonstrated that, whereas MK2206 treatment alone only transiently decreased LNCaP cell growth, MK2206 and FOXO1‐IQBP(SE) co‐treatment completely inhibited cell growth (Fig 5D). Annexin V staining showed that more cells underwent apoptosis when treated with both MK2206 and FOXO1‐IQBP(SE) compared with cells treated with each agent alone (Fig EV4B). Expression of FOXO1‐IQBP(SE) also diminished MK2206‐induced increase in pERK1/2 in breast cancer cell lines MDA‐MB‐468 and BT474 (Fig EV4C). These data indicate that a small FOXO1‐derived phospho‐mimicking peptide can enhance AKT inhibitor‐induced cell death by overcoming acquired ERK1/2 activation.
Figure EV4. Assessment of effect of FOXO1 phospho‐mimicking peptide on AKT inhibitor‐induced cell death and pERK1/2.
-
A, BLNCaP cells were transfected with empty vector (E.V.) or the FOXO1 phospho‐mimicking peptide HA‐FOXO1‐IQBP(SE) for 24 h and then treated with or without MK2206 (0.5 μM) for 24 h followed by immunofluorescence cytochemistry using anti‐HA antibody (A) and Annexin V assay and flow cytometry analysis (B). For panel (A), > 100 cells in each experimental condition were analyzed. Scale bars, 20 μm. The experiment was repeated at least once and similar results were obtained. For panel (B), data from two biological replicates were quantified (means ± s.d., n = 2). *P = 0.001687442 comparing MK2206 versus EV; *P = 0.014293987 comparing MK2206 + EV versus MK2206 + IQBP(SE) (two‐sided Student's t‐test at the 48‐h time point).
-
CWestern blot analysis of whole‐cell lysate from indicated cell lines 24 h after transfection with empty vector (E.V.) and the FOXO1 phospho‐mimicking peptide HA‐FOXO1‐IQBP(SE) and then treated with or without MK2206 (0.5 μM) for 24 h.
-
DAnnexin V assay and flow cytometry analysis. 72 h after infection with lentivirus expressing indicated shRNAs, LNCaP cells treated with or without MK2206 (0.5 μM) for 24 h. Quantitative data were obtained from two biological replicates (means ± s.d., n = 2). *P = 0.004378751 comparing shControl versus shControl + MK2206; *P = 0.001300305 comparing shIQGAP1‐1 versus shIQGAP1‐1 + MK2206; *P = 0.000554 comparing shIQGAP1‐2 versus shIQGAP1‐2 + MK2206; *P = 0.000967413 comparing shIQGAP1‐1 versus shControl; *P = 0.000363332 comparing shIQGAP1‐2 versus shControl; *P = 0.000232676 comparing shIQGAP1‐1 + MK2206 versus shControl + MK2206; *P = 0.000180198 comparing shIQGAP1‐2 + MK2206 versus shControl + MK2206 (two‐sided Student's t‐test at the 48‐h time point).
To examine the causal role of IQGAP1 in AKT inhibition‐mediated ERK activation, we knocked down endogenous IQGAP1 using two independent shRNAs before MK2206 treatment. IQGAP1 knockdown decreased pAKT at both serine 308 and 473 in LNCaP cells (Fig 5E, lanes 3, 5, 9, 11 and Appendix Fig S5D), which is consistent with the previous report in other cell types (Chen et al, 2010a). Different from the treatment of MK2066 alone, however, IQGAP1 knockdown did not trigger the elevation of pERK1/2 while decreasing pAKT (Fig 5E and Appendix Fig S5D). This result further indicates the importance of IQGAP1 in AKT inhibition‐caused ERK1/2 activation. Most importantly, depletion of IQGAP1 completely abolished MK2206‐induced pERK1/2 regardless of treatment with CHX (Fig 5E, lane 2 versus 4 and 6, lane 8 versus 10 and 12; Appendix Fig S5D). Accordingly, co‐treatment of LNCaP cells with MK2066 and IQGAP1 shRNAs resulted in greater inhibition of cell growth than MK2066 alone (Fig 5F). As demonstrated by Annexin V staining, MK2206 treatment plus IQGAP1 knockdown induced a much higher percentage of apoptosis than each agent alone (Fig EV4D). Collectively, these data suggest that the FOXO1‐IQGAP1 signaling axis plays an essential role in regulating AKT inhibition‐induced activation of ERK and drug resistance in cells.
A small FOXO1‐derived phospho‐mimicking peptide inhibitor impedes taxane‐induced ERK activation and chemoresistance
Paclitaxel and its semisynthetic analogue docetaxel (DTX) are widely used chemotherapeutic agents for treatment of solid tumors. In both preclinical and clinical settings, paclitaxel treatment is well documented to result in activation of the Ras‐Raf‐MAPK pathway, which confers resistance to paclitaxel (Okano & Rustgi, 2001; Sunters et al, 2006; Mehnert et al, 2011). However, the mechanism underlying paclitaxel‐induced MAPK activation remains poorly understood. Paclitaxel induces nuclear localization of FOXO proteins in various types of human cancer (Sunters et al, 2006; Goto et al, 2008; Gan et al, 2009a). We therefore sought to test the hypothesis that taxane promotes pERK by inducing nuclear localization of FOXO1 and thereby abolishing FOXO1‐mediated inhibition of IQGAP1‐dependent activation of MAPK in the cytoplasm. In agreement with the finding in MCF‐7 breast cancer cell line (Sunters et al, 2006), paclitaxel treatment alone induced inhibition of pAKT, decreased phosphorylation of the 14‐3‐3 binding sites (T24 and S256) responsible for cytoplasmic retention of FOXO1, nuclear localization of FOXO1, and transactivation of p27KIP1 in PTEN‐mutated LNCaP prostate cancer and PIK3α‐mutated BT474 breast cancer cells (Figs 6A and EV5A and B, and Appendix Fig S6A). Importantly, paclitaxel treatment also resulted in an increase in pERK1/2 in these cell lines (Fig 6A and Appendix Fig S6A). Similar results were obtained in LNCaP and PC‐3 cells treated with DTX (Fig EV5C and D). Moreover, knockdown of endogenous IQGAP1 completely abolished paclitaxel‐induced pERK1/2 in both LNCaP and BT474 cell lines without an overt impact on p27KIP1 expression (Fig 6A, lane 2 versus 3, 6 versus 7; Appendix Fig S6A). These data suggest that exclusion of FOXO1 in the cytoplasm and subsequent ablation of FOXO1‐mediated suppression of IQGAP1‐MAPK interaction are responsible for paclitaxel‐induced ERK1/2 activation.
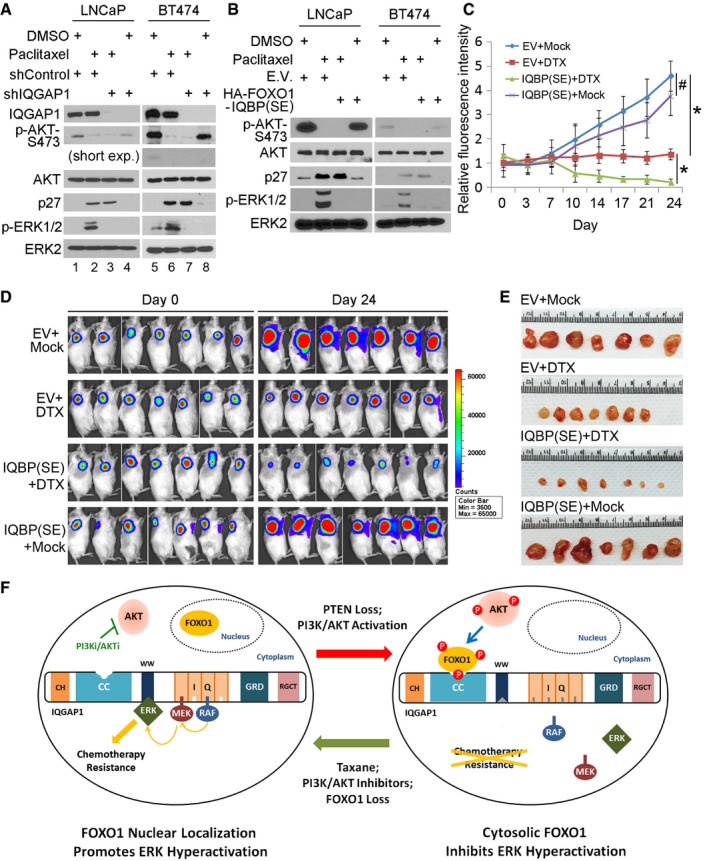
Figure 6. A small FOXO1‐derived peptide inhibitor overcomes taxane‐induced ERK activation and chemoresistance.
-
AWestern blot analysis of whole‐cell lysates (WCL) of LNCaP and BT474 cells 72 h after infection with lentivirus expressing indicated shRNAs. Cells were treated with or without paclitaxel (10 nM) for 24 h before harvest. Short exp., short exposure.
-
BWestern blot analysis of WCL of LNCaP and BT474 cells 72 h after infection with lentivirus expressing indicated plasmids. Cells were treated with or without paclitaxel (10 nM) for 24 h prior to harvest.
-
C–EPC‐3‐Luc cells 72 h after infection with lentivirus expressing an empty vector (EV) or the small FOXO1‐derived peptide FOXO1‐IQBP(SE) were injected subcutaneously into the right flank of NSG mice for 10 days and mice were treated with intravenous DTX (5 mg/kg) or normal saline (mock) twice per week. Luminescent signal intensity in each xenograft at each time point (C), representative luminescent images of xenografts (D), and tumors at the end of treatment (E) are shown. The data are presented as means ± s.d. (n = 7). # P = 0.18457778 comparing EV+MOCK versus IQBP(SE)+MOCK; *P = 0.001033546 comparing EV+MOCK versus EV+DTX, *P = 0.000356333 comparing IQBP(SE)+DTX versus EV+DTX (two‐sided Student's t‐test at the 24‐d time point).
-
FA hypothetical model depicting a rheostat role of AKT‐phosphorylated FOXO1 in the regulation of activation of the MAPK pathway by the PI3K‐PTEN‐AKT signaling axis. P in a small red circle, phosphorylation; PI3Ki/AKTi, PI3K, and AKT inhibitors.
Figure EV5. The FOXO1 small peptide inhibits taxol‐induced ERK activation and chemoresistance.
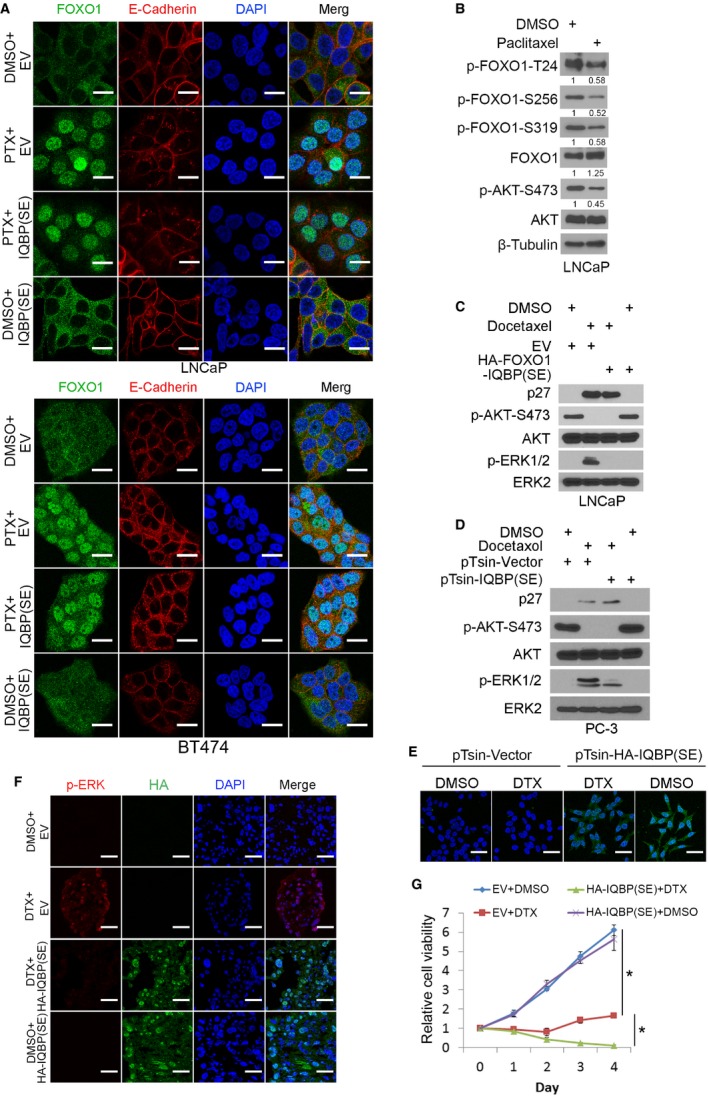
- Immunofluorescence cytochemistry using indicated antibodies in LNCaP and BT474 cells 24 h after transfection with empty vector (EV) or FOXO1‐IQBP(SE) and then treated with or without paclitaxel (PTX) (10 nM) for 24 h. E‐cadherin was used to mark the plasma membrane (cell boundary). > 100 cells were analyzed in each experimental condition. Scale bars, 20 μm. The experiment was repeated at least once and similar results were obtained.
- Western blot analysis of whole‐cell lysate (WCL) from LNCaP cells treated with or without paclitaxel (10 nM) for 24 h before harvest.
- Western blot analysis of WCL from LNCaP cells 24 h after transfection with indicated plasmids and then treated with or without docetaxel (10 nM) for 24 h.
- Western blot analysis of WCL from PC‐3‐Luc cells 72 h after infection with lentivirus expressing empty vector (pTsin) or FOXO1‐IQBP(SE). Cells were then treated with or without docetaxel (10 nM) for 24 h prior to harvest.
- Immunofluorescence cytochemistry using anti‐HA antibody in PC‐3‐Luc cells 72 h after infection with lentivirus expressing indicated empty vector or HA‐FOXO1‐IQBP(SE). Cells were then treated with or without docetaxel (10 nM) for 24 h. > 100 cells were analyzed in each experimental condition. Scale bars, 50 μm. The experiment was repeated at least once and similar results were obtained.
- Immunofluorescence histochemistry using anti‐phospho‐ERK (p‐ERK) and anti‐HA antibodies in frozen sections acquired from indicated mouse xenografts harvested at the end of treatment. > 100 cells were analyzed in each experimental condition. Scale bars, 50 μm. Similar experiments were performed in samples obtained from at least three animals (n = 3).
- MTS assay performed in PC‐3‐Luc cells. 72 h after infection with lentivirus expressing empty vector (EV) or HA‐IQBP(SE), cells were treated with or without docetaxel (10 nM) for 24 h (means ± s.d., n = 6). *P = 1.24674E‐07 comparing EV+DMSO versus EV+DTX; *P = 3.29127E‐07 comparing IQBP(SE)+DTX versus EV+DTX (two‐sided Student's t‐test at the 4‐day time point).
Given that expression of the small FOXO1‐derived peptide FOXO1‐IQBP1(SE) antagonizes AKT inhibition‐induced ERK1/2 activation (Fig 5B), we examined whether it can overcome taxane resistance in vitro and in vivo. Expression of FOXO1‐IQBP1(SE) blocked taxane‐induced ERK1/2 activation in LNCaP, PC‐3, and BT474 cells without affecting pAKT and p27KIP1 expression (Figs 6B and EV5C and D, and Appendix Fig S6B). Similar to the results in PC‐3 cells cultured in vitro (Fig EV5D), DTX treatment increased pERK1/2 in PC‐3 xenografts in mice (Fig EV5F). This result is consistent with the observation that DTX treatment failed to completely block tumor growth in vitro and in vivo (Figs 6C–E and EV5G). In contrast, co‐treatment with DTX and FOXO1‐IQBP(SE) not only blocked pERK1/2 but also inhibited cancer cell growth in culture and in mice (Figs 6C–E and EV5G). Thus, we have identified a small bioactive FOXO1‐derived peptide inhibitor that overcomes chemoresistance in cancer cells by blocking taxane‐induced ERK1/2 activation.
Discussion
Both PI3K‐AKT and MAPK pathways are important for cancer cell proliferation, survival, and resistance to therapies (Kinkade et al, 2008; Chandarlapaty et al, 2011). In this study, we demonstrate in PTEN‐null prostate cancer cells and AKT‐activated other cancer types that phosphorylated FOXO1 binds to the scaffold protein IQGAP1 in the cytoplasm and impedes IQGAP1‐dependent pERK1/2 (Fig 6F, right panel). Our findings pinpoint that whereas AKT‐mediated phosphorylation abolishes the tumor suppressor functions of FOXO1 in the nucleus (Greer & Brunet, 2005; Huang & Tindall, 2007), it also activates a non‐genomic tumor suppressor function of FOXO1 in the cytoplasm by inhibiting the MAPK pathway. Thus, our study identifies a previously uncharacterized tumor suppressor function of AKT‐phosphorylated FOXO1 protein in the cytoplasm.
In several different cancer types, including prostate, breast, pancreatic, and nasopharyngeal, among others, pharmacologic inhibition of the PI3K‐AKT pathway often leads to activation of ERK1/2 (Moelling et al, 2002; Robertson et al, 2010; Chandarlapaty et al, 2011; Serra et al, 2011). Whereas acquired ERK1/2 activation has been recognized as a major cause of resistance to the PI3K/AKT inhibitors (Kinkade et al, 2008; Serra et al, 2011), the molecular mechanism underlying AKT inhibition‐induced ERK activation remains poorly understood. As demonstrated in breast cancer cells, AKT inhibition promotes ERK activation by inducing nuclear localization of FOXO proteins and FOXO‐mediated transcription of receptor tyrosine kinase family members (Chandarlapaty et al, 2011; Serra et al, 2011). We found that blocking the genomic effect of FOXO proteins by treating cells with the protein synthesis inhibitor did decrease AKT inhibition‐induced pERK, but only by 50%. In contrast, ERK activation was completely abolished by treating cells with a small FOXO1‐derived peptide inhibitor or genetic depletion of IQGAP1. Thus, cytoplasmic FOXO1‐mediated suppression of the IQGAP1‐MAPK interaction represents a more proximal mechanism that controls AKT inhibition‐induced activation of ERK (Fig 6F). Given that IQGAP1 is a pleiotropically acting protein with effects on multiple signaling pathways, targeting ERK activation by interfering with the function of IQGAP1 might have adverse side effect. On the contrary, utilization of a small, FOXO1‐derived peptide inhibitor of IQGAP1 could specifically inhibit AKT inhibition‐induced activation of ERK and drug resistance.
Paclitaxel and DTX have emerged as important chemotherapeutic agents for the treatment of cancers such as advanced breast and prostate cancer (Petrylak et al, 2004; Tannock et al, 2004). However, taxane treatment often induces ERK activation (Okano & Rustgi, 2001; Sunters et al, 2006), thereby limiting the efficacy in clinic (Tan et al, 2005; Xu et al, 2009). Despite the recognition of the importance of ERK activation in development of drug resistance, how taxane treatment causes ERK activation remains elusive. Consistent with the previous report that paclitaxel induces nuclear localization of FOXO proteins (Sunters et al, 2006; Goto et al, 2008; Gan et al, 2009a), our study demonstrates that paclitaxel treatment increases pERK1/2 by inducing nuclear localization of FOXO1. Importantly, we show that this effect was completely abolished by co‐treatment of cells with a small FOXO1‐derived peptide inhibitor or IQGAP1 knockdown. Our findings suggest that paclitaxel‐induced nuclear localization of FOXO1 proteins and loss of cytoplasmic FOXO1‐mediated inhibition of the IQGAP1‐MAPK interaction represent a key mechanism responsible for ERK activation and drug resistance in taxane‐treated cancer cells (Fig 6F, left panel). However, our data cannot rule out the possibility that cells can also acquire resistance independent of the FOXO1‐IQGAP1‐MAPK axis.
In summary, we identify FOXO1 as a negative modulator of the IQGAP1‐MAPK signaling axis. We provide evidence that FOXO1 binds to IQGAP1 and their interaction is largely enhanced by S319 phosphorylation on FOXO1 mediated by AKT. We further show that treatment of cancer cells with the PI3K/AKT inhibitors or taxane induces ERK activation by causing nuclear localization of FOXO1 and dissociation of FOXO1 from IQGAP1 in the cytoplasm. Most importantly, we define a bioactive FOXO1‐derived phospho‐mimicking peptide that overcomes PI3K/AKT inhibitor or taxane‐induced ERK activation and chemoresistance. These findings suggest that the cytosolic tumor suppressor function of FOXO1 can be exploited for effective treatment of human cancers.
Materials and Methods
Plasmids, antibodies, and reagents
The mammalian expression vectors Flag‐FOXO1, Flag‐FOXO1‐T24A, S256A, S319A, A3, Flag‐FOXO1‐537 (in which histidine 215, a key residue for DNA binding is mutated to arginine and the transactivation domain (amino acids 538–655) is deleted), FOXO1‐537‐A3, NLSm, hemagglutinin (HA)‐tagged CA‐AKT, HA‐FOXO3, HA‐FOXO4 were described previously (Huang et al, 2005, 2006; Liu et al, 2008; Gan et al, 2009a,b; Zhang et al, 2011). A HA‐tagged small (30 amino acids) FOXO1‐derived IQGAP1‐binding peptide, 304‐NDDFDNWSTFRPRTSSNASTISGRLSPIMT‐333 (S319 in bold), was cloned into the pCMV vector (HA‐FOXO1‐IQBP‐S319) and further mutated to glutamic acid (E) or aspartic acid (D) to generate two phospho‐mimicking mutants HA‐FOXO1‐IQBP‐S319E and HA‐FOXO1‐IQBP‐S319D using site‐specific mutagenesis (Agilent). Lentivirus‐based HA‐FOXO1 IQBP‐S319E or called HA‐FOXO1 IQBP(SE) was cloned into pTsin vector. A Flag‐tagged FOXO1 IQBP(SE) was generated by subcloning phospho‐mimicking FOXO1 IQBP(SE) peptide into SFB vector. Flag‐FOXO1‐537‐S319A, HA‐FOXO3‐S315A, and HA‐FOXO4‐S262A were generated by site‐specific mutagenesis (Agilent). Using KOD‐Plus Mutagenesis Kit (Toyobo), Flag‐FOXO1‐NESm was generated as reported previously (Matsuzaki et al, 2003) by mutating the FOXO1 nuclear export signal motif MENLLDNLNL to AENALDNANA. shFOXO1#1‐resistant Flag‐tagged FOXO1 (FOXO1S1R) and shFOXO1#2‐resistant Flag‐tagged FOXO1 (Flag‐FOXO1S2R) were generated using KOD‐Plus Mutagenesis Kit (Toyobo). Plasmids for HA‐tagged AKT kinase‐dead mutant (K179M) (termed AKT‐DN) and pcDNA3‐Myc‐IQGAP1 were purchased from Addgene. A Flag‐tagged IQGAP1 was generated by subcloning Flag‐IQGAP1 into pcDNA3.1 vector. Bacterial expression vectors for various GST‐tagged FOXO1 recombinant proteins were generated by subcloning the following regions from full‐length FOXO1 (amino acids 1–655) into the pGEX‐4T‐1 vector: FOXO1‐1 (amino acids 1–167), FOXO1‐2 (amino acids 149–267), FOXO1‐3 (amino acids 211–419), FOXO1‐4 (amino acids 354–503), FOXO1‐5 (amino acids 488–655). GST‐FOXO1‐3 (211–419) S319A was generated by KOD‐Plus Mutagenesis Kit (Toyobo). GST‐tagged IQGAP1 recombinant protein constructs were generated by subcloning the full‐length IQGAP1 (amino acids 1–1,657) or the following regions of IQGAP1 into pGEX‐4T‐1 vectors: IQGAP1‐P1 (amino acids 1–185), IQGAP1‐P2 (amino acids 166–670), IQGAP1‐P3 (amino acids 671–730), IQGAP1‐P4 (amino acids 731–860), IQGAP1‐P5 (amino acids 861–1,250), IQGAP1‐P6 (amino acids 1,251–1,657).
Antibodies used were anti‐IQGAP1, anti‐ERK2, anti‐Myc tag, anti‐p27 (Santa Cruz Biotechnology); anti‐FOXO1 (Bethyl); anti‐p‐ERK1/2, anti‐AKT, anti‐p473‐AKT, anti‐p308‐AKT, anti‐p319‐FOXO1, anti‐p256‐FOXO1 (Cell Signaling Technology); anti‐Flag (Sigma‐Aldrich); and anti‐HA (Covance). The chemicals purchased were trypsin (Thermo Fisher Scientific), cycloheximide (CHX), and paclitaxel (Sigma‐Aldrich), MK2206 (Selleckchem), NVP‐BEZ235 (LC‐Laboratories), and docetaxel (Active Biochem).
Cell lines, cell culture, and transfection
The prostate cancer cell lines DU145 and LNCaP and human embryonic kidney cell line 293T were purchased from ATCC. DU145 and LNCaP cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). 293T cells were maintained in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific) supplemented with 10% FBS. The C4‐2 cell line was purchased from UroCorporation and grown in RPMI 1640 supplemented with 10% FBS. Breast cancer cell lines MDA‐MB‐468 and BT474 were acquired from Fergus Couch at Mayo Clinic, and pancreatic cancer cell lines PANC‐1 and MIA‐PaCa‐2 were obtained from Daniel Billadeau at Mayo Clinic. MDA‐MB‐468, PANC‐1, and MIA‐PaCa‐2 cell lines were cultured in DMEM supplemented with 10% FBS. BT474 was cultured in RPMI 1640 medium supplemented with 10% FBS. Cells were cultured at 37°C supplied with 5% CO2. Transfections were performed by electroporation with an Electro Square Porator ECM 830 (BTX) (Chen et al, 2010b) or with Lipofectamine 2000 (Thermo Fisher Scientific). Approximately 75–95% transfection efficiencies were routinely achieved.
Co‐immunoprecipitation (co‐IP) and Western blotting
Immunoprecipitations were performed as described previously (Huang et al, 2006; Wang et al, 2013). Cells were harvested and lysed in cell lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P‐40, 0.5% sodium deoxycholate, and 1% protease inhibitor cocktails, Sigma‐Aldrich). Cell lysates were centrifuged and the supernatant was then incubated with indicated antibodies and protein‐G beads (Thermo Fisher Scientific) at 4°C overnight. The beads were washed more than five times using cell lysis buffer, and the precipitated proteins were used for further analysis. For Western blotting, protein samples were prepared in modified RIPA buffer (1× PBS, 1% NP‐40, 0.1% SDS, and 1% protease inhibitor cocktails). Equal amounts of protein (50–100 μg) from cell lysate were denatured in sample buffer (Thermo Fisher Scientific). Proteins were separated by SDS–polyacrylamide gel electrophoresis and then were transferred to nitrocellulose membranes (Bio‐Rad). After the membranes were immunoblotted with specific primary antibodies and horseradish peroxidase‐conjugated secondary antibodies, they were visualized by SuperSignal West Pico Stable Peroxide Solution (Thermo Fisher Scientific).
GST pull‐down assay using cell lysate
Cells were lysed with cell lysis/protein binding buffer (20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.1% Nonidet P‐40, 1 mM DTT (dithiothreitol), 10% glycerol, 1 mM EDTA, 2.5 mM MgCl2, and 1 μg/ml leupeptin) for 30 min at 4°C. GST fusion proteins and glutathione–sepharose beads (GE Healthcare Life Science) were incubated with cell lysates for 4 h. The beads were then washed four times with binding buffer and resuspended in sample buffer. The bound proteins were subjected to SDS–PAGE.
In vitro transcription and translation of IQGAP1 proteins
Plasmid DNA (Flag‐IQGAP1) was added to the TNT® T7 Quick Master Mix, and then, 1 μl methionine (1 mM) was added, by following the manufacturer's instruction of TNT® Quick Coupled Transcription/Translation Systems (Promega). The in vitro transcribed and translated proteins were subjected to GST pull‐down assay.
In vitro kinase assay
C4‐2 cells were transfected with expression vector for HA‐tagged constitutively active AKT (HA‐AKT‐CA). 24 h after transfection, cells were harvested and lysed in cell lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P‐40, 0.5% sodium deoxycholate, and 1% protease inhibitor cocktails, Sigma‐Aldrich). Cell lysates were centrifuged and the supernatant was incubated with non‐specific IgG or anti‐HA antibody and protein‐G beads (Thermo Fisher Scientific) at 4°C overnight. The beads were washed five times with cell lysis buffer and then washed with 1× kinase buffer. Immunoprecipitated IgG or HA‐AKT were incubated with purified GST or GST‐FOXO1 recombinant proteins (GST‐FOXO1‐3 (211–419) or GST‐FOXO1‐3 S319A) and ATP in kinase buffer by following the manufacturer's instruction of AKT Kinase Assay Kit (Nonradioactive) (Cell Signaling Technology). The supernatant containing phosphorylated protein were subjected to GST pull down.
GST pull‐down assay using in vitro translated protein
In vitro transcribed and translated Flag‐tagged IQGAP1 proteins were incubated with GST or GST‐FOXO1 recombinant proteins undergone AKT kinase assay in protein binding buffer (20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.1% Nonidet P‐40, 1 mM DTT (dithiothreitol), 10% glycerol, 1 mM EDTA, 2.5 mM MgCl2, and 1 μg/ml leupeptin). Glutathione–sepharose beads (GE Healthcare Life Science) were added and further incubated for 4 h. The beads were then washed four times with binding buffer and resuspended in sample buffer. The bound proteins were subjected to SDS–PAGE.
RNA interference
Non‐specific control small interfering RNA (siRNA) and siRNAs for human IQGAP1, FOXO1, and FOXO3 were purchased from Thermo Scientific Dharmacon. siRNA transfection of cells was performed following the manufacturer's instruction. Lentivirus‐based control and gene‐specific shRNAs were purchased from Sigma‐Aldrich. siRNA and shRNA sequence information is provided in Appendix Table S2.
MTS cell viability assay
Cell growth was measured by absorbance using the MTS assay according to manufacturer's instructions (Promega). Cells were plated in 96‐well plates at a density of 1,000 cells per well. At the indicated time points, 20 μl of CellTiter 96R AQueous One Solution reagent (Promega) was added to cells; after incubating for 60 min at 37°C, cell growth was measured in a microplate reader at 490 nm.
Detection of apoptosis using Annexin V assay and flow cytometry
Cells were stained with PE Annexin V and 7‐amino‐actinomycin following the manufacturer's instruction of PE Annexin V Apoptosis Detection Kit I (BD Biosciences). A minimum of 10,000 stained cells were immediately assayed on a flow cytometer. Data were analyzed with FlowJo analysis software.
Immunofluorescence cytochemistry
Immunofluorescence cytochemistry was performed as previously described (Huang et al, 2006). Briefly, cells were rinsed in PBS, fixed in 4% paraformaldehyde for 15 min, and washed in PBS three times. Fixed cells were permeabilized with 0.2% Triton X‐100 for 20 min, washed in PBS, and then blocked in PBS supplemented with 5% goat serum and 10% glycerol. Cells were incubated with indicated primary antibody at 4°C overnight. Cells were washed three times with PBS and incubated with secondary antibody that was conjugated with Alexa Fluor 488 (Thermo Fisher Scientific) for 1 h at room temperature. After the final wash, cells were counterstained with Vectashield (Vector Laboratories) containing DAPI (4′, 6‐diamidino‐2‐phenylindole). Images were captured using Zeiss laser confocal microscope (LSM780).
Prostate cancer tissue specimens, immunohistochemistry (IHC), and staining scoring
Prostate cancer tissue microarrays (TMAs) were purchased from US Biomax, Inc (Cat. # PR2085b and PR803a). TMA specimens were used for antigen retrieval and immunostaining as described previously (Huang et al, 2001; Zhang et al, 2011). Primary antibodies used were anti‐FOXO1 (Bethyl) and anti‐pERK (Cell Signaling Technology). Staining intensity was graded/scored in a blinded fashion: 1 = weak staining at 100× magnification but little or no staining at 40× magnification; 1.5 = weak staining at 40× magnification; 2 = medium staining at 40× magnification; 2.5 = medium plus staining at 40× magnification; 3 = strong staining at 40× magnification; and 3.5 = very strong staining at 40× magnification. A final staining index was obtained by multiplying values of staining percentage and intensity.
Generation, treatment, and imaging of prostate cancer xenografts in mice
Six‐week‐old NOD‐SCID IL‐2‐receptor gamma null (NSG) mice were generated in house and randomly grouped for animal experiments. The animal study was approved by the IACUC at Mayo Clinic. All mice were housed in standard conditions with a 12‐h light/dark cycle and access to food and water ad libitum. PC‐3‐Luc cells (5 × 106) infected with lentivirus expressing empty vector (E.V.) or the FOXO1 peptide HA‐FOXO1‐IQBP(SE) (in 100 μl 1× PBS plus 100 μl Matrigel (BD Biosciences)) were injected s.c. into the right flank of mice. After xenografts reached the size of ~100 mm3 (7 days after implantation), tumor‐positive animals were randomly divided into different treatment groups. Vehicle (0.9% saline/mock treatment) or DTX (10 mg/ml, Sandoz Inc.) at 5 mg/kg was administered by i.v. injection twice a week (first and fourth day of the week). Tumor growth was monitored blindly by living imaging. Generally, luciferin (150 mg/kg) was administrated by i.p. injection 10 min before imaging, and then, mice were anaesthetized with 3% isoflurane and imaged in an IVIS spectrum imaging system (Xenogen). Images were analyzed with Living Image software (Xenogen). Bioluminescent flux (photons/s per sr per cm2) was determined for the xenograft tumors. Upon the completion of treatment, tumor grafts were harvested. Tumor tissues were divided, and a portion was fast‐frozen into OCT for frozen section, a portion was formalin fixed and paraffin embedded and the rest was frozen for protein and RNA extraction.
Statistical analysis
Experiments were carried out with two or more replicates unless otherwise stated. Statistical analyses were performed with Student's t‐test unless otherwise indicated. P‐values < 0.05 are considered statistically significant.
Additional methods
Other methods are described in Appendix Supplementary Methods.
Author contributions
HH, C‐WP, and XJ conceived the study. C‐WP, XJ, YZ, YP, and JY performed the experiments. LW performed bioinformatics and statistics analyses. RJK, JZ, and C‐WP contributed to acquisition and analysis of patient samples. HH, C‐WP, XJ, and YZ wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Georges Mer for suggestions and Jian An for assistance in generating plasmids. This work was supported in part by grants from the National Institutes of Health (CA134514, CA130908, and CA193239 to H.H.), the Department of Defense (W81XWH‐09‐1‐622 and W81XWH‐14‐1‐0486 to H.H.), the Mayo Clinic Center for Individualized Medicine (to H.H.), and the National Natural Science Foundation of China (81101931 to C.‐W.P.).
The EMBO Journal (2017) 36: 995–1010
See also: S Choi & RA Anderson (April 2017)
References
- Aoki M, Jiang H, Vogt PK (2004) Proteasomal degradation of the FoxO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc Natl Acad Sci USA 101: 13613–13617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs WH III, Meisenhelder J, Hunter T, Cavenee WK, Arden KC (1999) Protein kinase B/Akt‐mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA 96: 7421–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868 [DOI] [PubMed] [Google Scholar]
- Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, Dalal SN, DeCaprio JA, Greenberg ME, Yaffe MB (2002) 14‐3‐3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol 156: 817–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik‐Outmezguine V, Grbovic‐Huezo O, Serra V, Majumder PK, Baselga J, Rosen N (2011) AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19: 58–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhu HH, Zhou LF, Wu SS, Wang J, Chen Z (2010a) IQGAP1 is overexpressed in hepatocellular carcinoma and promotes cell proliferation by Akt activation. Exp Mol Med 42: 477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H (2010b) Cyclin‐dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol 12: 1108–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XY, Chen C, Sun X, Guo P, Vessella RL, Wang RX, Chung LW, Zhou W, Dong JT (2006) FOXO1A is a candidate for the 13q14 tumor suppressor gene inhibiting androgen receptor signaling in prostate cancer. Cancer Res 66: 6998–7006 [DOI] [PubMed] [Google Scholar]
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia‐Echeverria C, Weissleder R, Mahmood U, Cantley LC et al (2008) Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 14: 1351–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Chen S, Wang Y, Watahiki A, Bohrer L, Sun Z, Wang Y, Huang H (2009a) Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res 69: 8386–8394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Liu P, Lu H, Chen S, Yang J, McCarthy JB, Knudsen KE, Huang H (2009b) Cyclin D1 promotes anchorage‐independent cell survival by inhibiting FOXO‐mediated anoikis. Cell Death Differ 16: 1408–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coffer PJ, Ham J (2003) FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol 162: 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto T, Takano M, Hirata J, Tsuda H (2008) The involvement of FOXO1 in cytotoxic stress and drug‐resistance induced by paclitaxel in ovarian cancers. Br J Cancer 98: 1068–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Brunet A (2005) FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24: 7410–7425 [DOI] [PubMed] [Google Scholar]
- Haflidadottir BS, Larne O, Martin M, Persson M, Edsjo A, Bjartell A, Ceder Y (2013) Upregulation of miR‐96 enhances cellular proliferation of prostate cancer cells through FOXO1. PLoS ONE 8: e72400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst A, de Vries‐Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM, Burgering BM (2006) FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol 8: 1064–1073 [DOI] [PubMed] [Google Scholar]
- Huang H, Cheville JC, Pan Y, Roche PC, Schmidt LJ, Tindall DJ (2001) PTEN induces chemosensitivity in PTEN‐mutated prostate cancer cells by suppression of Bcl‐2 expression. J Biol Chem 276: 38830–38836 [DOI] [PubMed] [Google Scholar]
- Huang H, Regan KM, Wang F, Wang D, Smith DI, van Deursen JM, Tindall DJ (2005) Skp2 inhibits FOXO1 in tumor suppression through ubiquitin‐mediated degradation. Proc Natl Acad Sci USA 102: 1649–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Regan KM, Lou Z, Chen J, Tindall DJ (2006) CDK2‐dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 314: 294–297 [DOI] [PubMed] [Google Scholar]
- Huang H, Tindall DJ (2007) Dynamic FoxO transcription factors. J Cell Sci 120: 2479–2487 [DOI] [PubMed] [Google Scholar]
- Jameson KL, Mazur PK, Zehnder AM, Zhang J, Zarnegar B, Sage J, Khavari PA (2013) IQGAP1 scaffold‐kinase interaction blockade selectively targets RAS‐MAP kinase‐driven tumors. Nat Med 19: 626–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, White CD, Li Z, Sacks DB (2011) Salmonella enterica serotype Typhimurium usurps the scaffold protein IQGAP1 to manipulate Rac1 and MAPK signalling. Biochem J 440: 309–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkade CW, Castillo‐Martin M, Puzio‐Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon‐Cardo C, Abate‐Shen C (2008) Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone‐refractory prostate cancer in a preclinical mouse model. J Clin Invest 118: 3051–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419: 316–321 [DOI] [PubMed] [Google Scholar]
- Lin A, Piao HL, Zhuang L, dos Sarbassov D, Ma L, Gan B (2014) FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K‐AKT pathway. Cancer Res 74: 1682–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Li S, Gan L, Kao TP, Huang H (2008) A transcription‐independent function of FOXO1 in inhibition of androgen‐independent activation of the androgen receptor in prostate cancer cells. Cancer Res 68: 10290–10299 [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A (2003) Insulin‐induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci USA 100: 11285–11290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM (2000) AFX‐like Forkhead transcription factors mediate cell‐cycle regulation by Ras and PKB through p27kip1. Nature 404: 782–787 [DOI] [PubMed] [Google Scholar]
- Mehnert JM, Tan AR, Moss R, Poplin E, Stein MN, Sovak M, Levinson K, Lin H, Kane M, Gounder M, Lin Y, Shih WJ, White E, Rubin EH, Karantza V (2011) Rationally designed treatment for solid tumors with MAPK pathway activation: a phase I study of paclitaxel and bortezomib using an adaptive dose‐finding approach. Mol Cancer Ther 10: 1509–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modur V, Nagarajan R, Evers BM, Milbrandt J (2002) FOXO proteins regulate tumor necrosis factor‐related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J Biol Chem 277: 47928–47937 [DOI] [PubMed] [Google Scholar]
- Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M (2002) Regulation of Raf‐Akt Cross‐talk. J Biol Chem 277: 31099–31106 [DOI] [PubMed] [Google Scholar]
- Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR (2000) Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol 20: 8969–8982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Finkel T (2002) Redox regulation of forkhead proteins through a p66shc‐dependent signaling pathway. Science 295: 2450–2452 [DOI] [PubMed] [Google Scholar]
- Okano J, Rustgi AK (2001) Paclitaxel induces prolonged activation of the Ras/MEK/ERK pathway independently of activating the programmed cell death machinery. J Biol Chem 276: 19555–19564 [DOI] [PubMed] [Google Scholar]
- Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, Wong WH, Castrillon DH, DePinho RA (2007) FoxOs are lineage‐restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128: 309–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrylak DP, Tangen CM, Hussain MH, Lara PN Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED (2004) Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 351: 1513–1520 [DOI] [PubMed] [Google Scholar]
- Plas DR, Thompson CB (2003) Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem 278: 12361–12366 [DOI] [PubMed] [Google Scholar]
- Ren JG, Li Z, Sacks DB (2007) IQGAP1 modulates activation of B‐Raf. Proc Natl Acad Sci USA 104: 10465–10469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren JG, Li Z, Sacks DB (2008) IQGAP1 integrates Ca2+/calmodulin and B‐Raf signaling. J Biol Chem 283: 22972–22982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson BW, Bonsal L, Chellaiah MA (2010) Regulation of Erk1/2 activation by osteopontin in PC3 human prostate cancer cells. Mol Cancer 9: 260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Li Z, Sacks DB (2004) IQGAP1 binds ERK2 and modulates its activity. J Biol Chem 279: 17329–17337 [DOI] [PubMed] [Google Scholar]
- Roy M, Li Z, Sacks DB (2005) IQGAP1 is a scaffold for mitogen‐activated protein kinase signaling. Mol Cell Biol 25: 7940–7952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, Gili M, Russillo M, Parra JL, Singh S, Arribas J, Rosen N, Baselga J (2011) PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2‐overexpressing breast cancer. Oncogene 30: 2547–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunters A, Madureira PA, Pomeranz KM, Aubert M, Brosens JJ, Cook SJ, Burgering BM, Coombes RC, Lam EW (2006) Paclitaxel‐induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c‐Jun NH2‐terminal kinase and Akt. Cancer Res 66: 212–220 [DOI] [PubMed] [Google Scholar]
- Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves‐Neira W, Bouillet P, Villunger A, Adams JM, White E (2005) Key roles of BIM‐driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 7: 227–238 [DOI] [PubMed] [Google Scholar]
- Tang ED, Nunez G, Barr FG, Guan KL (1999) Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem 274: 16741–16746 [DOI] [PubMed] [Google Scholar]
- Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA (2004) Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 351: 1502–1512 [DOI] [PubMed] [Google Scholar]
- Varne A, Muthukumaraswamy K, Jatiani SS, Mittal R (2002) Conformational analysis of the GTP‐binding protein MxA using limited proteolysis. FEBS Lett 516: 129–132 [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3‐Kinase AKT pathway in human cancer. Nat Rev Cancer 2: 489–501 [DOI] [PubMed] [Google Scholar]
- Wang F, Chan CH, Chen K, Guan X, Lin HK, Tong Q (2012) Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2‐mediated FOXO3 ubiquitination and degradation. Oncogene 31: 1546–1557 [DOI] [PubMed] [Google Scholar]
- Wang L, Zeng X, Chen S, Ding L, Zhong J, Zhao JC, Sarver A, Koller A, Zhi J, Ma Y, Yu J, Chen J, Huang H (2013) BRCA1 is a negative modulator of the PRC2 complex. EMBO J 32: 1584–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CD, Erdemir HH, Sacks DB (2012) IQGAP1 and its binding proteins control diverse biological functions. Cellular Signal 24: 826–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Sato N, Yanai K, Akiyoshi T, Nagai S, Wada J, Koga K, Mibu R, Nakamura M, Katano M (2009) Enhancement of paclitaxel‐induced apoptosis by inhibition of mitogen‐activated protein kinase pathway in colon cancer cells. Anticancer Res 29: 261–270 [PubMed] [Google Scholar]
- Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27: 5497–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C, Wang L, Zhou L, Fu Z (2014) The function of FOXO1 in the late phases of the cell cycle is suppressed by PLK1‐mediated phosphorylation. Cell Cycle 13: 807–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Pan Y, Zheng L, Choe C, Lindgren B, Jensen ED, Westendorf JJ, Cheng L, Huang H (2011) FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res 71: 3257–3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File