

Abstract
In cardiac and skeletal muscle, the troponin complex turns muscle contraction on and off in a calcium-dependent manner. Many small molecules are known to bind to the troponin complex to modulate its calcium binding affinity, and this may be useful in a broad range of conditions in which striated muscle function is compromised, such as congestive heart failure. As a tool for developing drugs specific for the cardiac isoform of troponin, we have designed a chimeric construct (cChimera) consisting of the regulatory N-terminal domain of cardiac troponin C (cNTnC) fused to the switch region of cardiac troponin I (cTnI), mimicking the key binding event that turns on muscle contraction. We demonstrate by solution NMR spectroscopy that cChimera faithfully reproduces the native interface between cTnI and cNTnC.
We determined that small molecules based on diphenylamine can bind to cChimera with a KD as low as 10 μM. Solution NMR structures show that minimal structural perturbations in cChimera are needed to accommodate 3-methyldiphenylamine (3-mDPA), which is probably why it binds with higher affinity than previously studied compounds like bepridil, despite its significantly smaller size. The unsubstituted aromatic ring of 3-mDPA binds to an inner hydrophobic pocket adjacent to the central beta sheet of cNTnC. However, the methyl-substituted ring is able to bind in two different orientations, either inserting into the cNTnC-cTnI interface or “flipping out” to form contacts primarily with helix C of cNTnC. Our work suggests that preservation of the native interaction between cNTnC and cTnI is key to the development of a high affinity cardiac troponin-specific drug.
1. Introduction
Cardiovascular disease is the number one cause of death worldwide[1], and heart failure is the end stage of almost all heart disease. Heart failure occurs when the heart is unable to pump sufficient blood to satisfy the needs of the body. Impaired cardiac muscle contraction results in systolic heart failure, also referred to as heart failure with reduced ejection fraction (HFrEF). By far, the most common cause of HFrEF is ischemic cardiomyopathy. However, in the absence of atherosclerotic coronary disease or valvular abnormalities, a wide range of etiologies encompassing infiltrative, infectious, autoimmune, toxic, hormonal, and genetic causes, can give rise to dilated cardiomyopathy (DCM)[2]. Over time, the volume overload that develops in HFrEF causes the heart muscle to further dilate, thin, and scar, making it even more difficult to generate force.
Positive inotropes, which increase the contractility of heart muscle to enhance cardiac output, should theoretically be useful in the treatment of systolic heart failure. Two inotropes used in acute decompensated heart failure are dobutamine and milrinone, both of which upregulate sympathetic β1-adrenergic stimulated pathways to increases heart rate and stroke volume[3]. However, inotropes acting on these pathways increase the oxygen demand of the heart and further increase mortality by precipitating life-threatening arrhythmias and systemic hypotension. A different class of positive inotropes is the calcium sensitizers, which increase the contractile response of the heart to calcium, in contrast to calcium mobilizers like dobutamine and milrinone. The first calcium sensitizer to reach large-scale clinical trials is levosimendan[4]. It was found to increase cardiac output without increasing oxygen demand[5]. Earlier trials showed improved survival in the treatment of acute decompensated systolic heart failure, but larger phase III trials did not show a mortality benefit [6,7].
Levosimendan and its active metabolite, OR-1896, bind to cardiac troponin (cTn) [8,9], the protein switch that turns contraction on and off in a calcium-dependent manner. The heterotrimeric cTn complex is composed of the calcium-binding subunit troponin C (cTnC), the inhibitory subunit troponin I (cTnI), and the tropomyosin-binding subunit troponin T (cTnT)[10]. Under resting calcium concentrations, the troponin complex maintains tropomyosin in a position that blocks actin–myosin interaction[11]. When the intracellular calcium concentration increases, calcium binding to the N-terminal regulatory domain of cTnC (cNTnC) induces binding of the switch region of cTnI (residues 147–163)[12]. This in turn promotes the dissociation of the cTnI inhibitory region (residues 135–147) from actin, shifting tropomyosin to expose the myosin-binding sites on actin and allowing cardiac contraction to proceed. Since cNTnC is the calcium-dependent switch for contraction and relaxation in cardiac muscle, it is a prime target for developing calcium sensitizing drugs.
While levosimendan has been shown to bind to cNTnC[13,14], it has been shown to impact the activity of other proteins as well, including type 3 phosphodiesterase[15] and ATP-sensitive potassium channels[16,17]. It is possible that off-target binding could potentially give rise to unintended adverse effects, like hypotension. We aim to develop calcium sensitizers that are more specific for cardiac troponin. In this regard, it is interesting to note that tirasemtiv (CK-2017357) is a drug that binds to skeletal troponin, though it shows no activity towards the cardiac isoform[18].
In the current study, we demonstrate that compounds based on the small molecule diphenylamine are able to bind to cardiac troponin with reasonably high affinity (KD 10 μM) given their small size. Our solution NMR structures reveal how one of these compounds, 3-methyldiphenylamine, binds to a deep but narrow hydrophobic cavity in the troponin C-I complex. Optimizing the fit to this tight cavity will be essential to developing high affinity cardiac troponin-modulating compounds.
2. Material and methods
2.1. cChimera design, expression, and purification
Hybrid proteins have been used to simplify systems with complex protein–protein interactions [19,20], including both skeletal and cardiac muscle isoforms of troponin[21,22]. Since dfbp-o and other compounds are known to bind to the interface between NTnC and TnI switch peptide[23], we developed a cardiac troponin C-troponin I chimera, cChimera, in which the switch region of cTnI is fused to the C terminus of cNTnC, building upon the previous design cNTnC-linker-switch peptide 144–173[21]. cChimera begins with residues 1 to 90 of human cNTnC (C35S, C84S), followed by residues 136–163 of cTnI (comprising the inhibitory 136–147 and switch 147–163 regions) and ending with a C-terminal His-tag. The inhibitory region acts as linker between cNTnC and the cTnI switch region, and residues 136 and 137 of the cTnI inhibitory region are coincidentally the same as residues 91 and 92 of cTnC. Unlike the previous chimera, in which the engineered linker region contains a protease cleavage site[21], all of the amino acids residues in our new cNTnC-cTnI chimera are derived from the native cTnC or cTnI sequence, with the exception of the C-terminal Gly and His-tag. Our new 125-amino acid 14.2 kDa cChimera construct has the following advantages over the previous construct[21]: native N-terminus of cTnC, switch region of cTnI extended N-terminally to include the inhibitory region (residues 136–147), which also serves as the linker to cNTnC, C35S and C84S double mutation to avoid issues with cysteine oxidation, and shortened C-terminal tail of cTnI, limiting the number of disordered residues contributing to NMR spectral overlap.
The expression plasmid for the cChimera was produced by DNA 2.0, with a high copy number origin of replication, ampicillin selection, and IPTG-inducible T5 RNA polymerase (which uses native Escherichia coli RNA polymerase) promoter. Isotope-enriched [15N, 13C]-cNTnC-cTnI [136–163] chimera were expressed in E. coli BL21(DE3) as previously described[24]. The expression of cChimera was carried out in 1 L of minimal M9 medium, containing 9 g Na2HPO4 and 2.5 g KH2PO4 at pH 7.3~7.4. To the autoclaved phosphate solution was added a filter-sterilized solution containing 1 mL of 1 M MgSO4, 1 mL of 100 mM CaCl2, 1 mg of biotin, 200 mg of thiamine, 1 g [15N] NH4SO4 (99.9 atom %), 3 g [13C] glucose (99.0 atom %) dissolved in 20 mL ddH2O, and 1 mL of 10% ampicillin. Cells from a 2 L culture were grown to a cell density between A600 0.7 and 1.0 and then induced for six hours with 1 mM IPTG. Cells were harvested by centrifugation at 5,000 rpm (4,420 xg) for 15 minutes and then resuspended in 20 mL buffer containing 50 mM Tris (pH 8.0), 10 mM MgSO4, 10 μg/mL DNase and 1 mM CaCl2. Cells were lysed by adding 20 mg of lysozyme and 200 mg of deoxycholic acid, with mechanical homogenization to break cell clumps. The cell lysate was clarified by centrifugation at 15,000 rpm (27,200 xg) and then syringe-filtered using a 0.8 μm filter. The supernatant was applied to a Ni-NTA column equilibrated with binding buffer (20 mM Tris-HCl, 0.3 M NaCl, 10 mM imidazole, 1 mM CaCl2), washed with the same buffer containing 80 mM imidazole, and then eluted with the same buffer with 250 mM imidazole. Purified fractions were dialyzed against 5 mM ammonium bicarbonate and 10 μM CaCl2 for three days and then lyophilized. Protein identity and purity >95% were confirmed by gel electrophoresis and mass spectrometry. Yield was ~60–100mg purified protein per liter of growth culture.
2.2 Chemicals
Bepridil (N-benzyl-N-[3-(2- methylpropoxy)-2-(pyrrolidin-1-yl)propyl]aniline) and 3-methyldiphenylamine were purchased from Sigma-Aldrich. 3-Chlorodiphenylamine was purchased from Toronto Research Chemicals Inc. All “NCI” compounds in the study were provided by the Developmental Therapeutics Program (DTP) at the National Cancer Institute (NCI). All “Chembridge” compounds were provided by ChemBridge Corp. All “SHG” compounds were synthesized in-house (see Supplementary Materials section), with the identity and purity confirmed by NMR spectroscopy.
2.3. NMR spectroscopy
NMR samples contained 0.1–0.6 mM cChimera in 100 mM KCl, 10 mM imidazole buffer pH ~6.8, 5–10 mM CaCl2, and 0.25 mM 2,2-dimethyl-2-silapentane-5-sulfonate-d6 sodium salt (DSS-d6) as an NMR internal reference in 95/5% H2O/D2O or 100% D2O. Stock solutions of 10 to 50 mM of small compounds were prepared in DMSO-d6 (Cambridge Isotopes Inc.) and added independently in aliquots to the protein sample. By the end of each titration, the addition of DMSO-d6 did not exceed 10% of the sample volume. NMR data were acquired on a Varian Inova 500 MHz or 800 MHz spectrometer at 30 °C. Both spectrometers are equipped with triple resonance probes with pulsed field gradients.
2.3.1. Drug titrations
The binding of 3-methyl diphenylamine (3-mDPA) to cChimera was monitored using 2D-1H,15N-HSQC and 2D-1H,13C-HSQC NMR spectra. We titrated 0.15 mM 15N-cChimera with 0.01, 0.03, 0.06, 0.1, 0.2, 0.4 and 0.7 mM of 3-mDPA while acquiring 2D-1H,15N-HSQC spectrum. We titrated 0.25 mM 13C,15N-cChimera with 0.05, 0.1, 0.25, 0.5 and 0.8 mM of 3-mDPA while acquiring 2D-1H,13C-HSQC spectrum. The solubility of 3-mDPA in NMR buffer is ~0.15 mM, so precipitation became increasingly apparent towards the end of the titration, a phenomenon that complicated the calculation of KD in most of our drug titrations. Dilution by addition of drug stock solution was taken into consideration. Chemical shift changes at each titration point were used by our in-house software, xcrvfit (www.bionmr.ualberta.ca/bds/software/xcrvfit), to calculate dissociation constants, KD.
For drug screening purposes, we used a 3-point titration to estimate KD. 2D-1H,15N-HSQC spectra were used to monitor the titration of 0.2 mM 15N-cChimera with 0, 0.1, and 1 mM drug, yielding 3 points with [drug]/[protein] ratios with 0, 0.5, and 5 equivalents. In the event of drug compound precipitation during the titration, KD would be underestimated because the observed chemical shift changes plateau early due to precipitation instead of protein saturation. Thus, the 3-point titration provides a lower bound estimate of KD. To provide an upper bound estimate for KD for insoluble compounds, the last titration point was removed and replaced by chemical shifts corresponding to cChimera fully saturated with 3-chlorodiphenylamine, the compound with the highest affinity in this study, displaying some of the largest chemical shift changes. This seemed to be a reasonable approximation, given that the observed chemical shift change patterns were similar in all titrations of DPA-based compounds.
2.3.2. NMR experiments for structure calculation
The backbone 1H, 15N, and 13C chemical shift assignments for cChimera (both with and without 3-mDPA) were obtained by analyzing 3D CBCA(CO)NH, 3D HNCACB, 3D HNCACO and 3D HNCO experiments. 3D (H)C(CO)NH-TOCSY (total correlation spectroscopy) and H(C)(CO)NH-TOCSY experiments were used to obtain side-chain 1H and 13C chemical shift assignments. Moreover, 3D HNHB and 3D HN(CO)HB experiments were used in combination with 3D 15N-edited NOESY (75 ms mixing time) to obtain stereospecific assignment for β methylene protons and χ1 dihedral angles. However, this strategy worked reliably only for residues with trans χ1 dihedral angle (Cγ and N at 180°) due to the low inherent sensitivity of the HN(CO)HB experiment, so that we were unable to distinguish between gauche+ and gauche-conformations. Stereospecific assignments of Val, Leu methyl groups were derived from 2D constant time 1H,13C HSQC of [15N, 10% 13C]-cChimera (pro-R methyl groups were in phase with alanine methyl groups)[25]. Aromatic side-chain resonances were assigned using an aromatic 3D 13C-edited NOESY-HSQC.
3D 15N-edited NOESY-HSQC (mixing time 75 ms), and 3D 13C-edited NOESY (mixing time 100 ms) were obtained to provide intramolecular (within the protein cChimera) distance restraints. 2D 13C,15N-double-filtered NOESY (150 ms mixing time) experiments were obtained on 3-mDPA in complex with 13C,15N-labeled cChimera to obtained chemical shift assignments and intramolecular distance restraints for bound 3-mDPA. Intermolecular distance restraints between 3-mDPA and cChimera were derived from a 3D 13C-filtered/edited NOESY-HSQC (mixing times: 75 ms) experiment. Protons from the methyl-containing aromatic ring could not be observed (though the methyl group itself was observable), because of broadening of 3-mDPA resonances. These resonances could still be observed in the 3D 13C-edited NOESY-HSQC, due to the shorter pulse sequence employed. Intermolecular restraints between 3-mDPA and cChimera could be easily identified in this unfiltered experiment (containing both intra- and intermolecular NOEs) by comparing the spectrum derived from cChimera bound to 3-mDPA with that from cChimera alone. This was the most sensitive method for obtaining intermolecular NOEs. VNMRJ v.2.21B (Varian, Inc.) was used for the analysis of one-dimensional NMR spectra, and all 2D and 3D NMR data were processed with NMRPipe[26] and analyzed with NMRViewJ[27], using scripts written in-house.
2.4. Structure calculation
Backbone φ- and ψ-dihedral angle restraints were obtained using TALOS+[28], which relies on backbone chemical shifts (1Hα, 13C′, 13Cα, 13Cβ, 15N, and 1HN). Restraints were used only for those residues for which there was complete agreement from all 10 database hits with a chemical shift-derived backbone order parameter >0.65[29].
NOE-derived distance restraints for the cChimera protein were generated using ARIA 2.3[30,31], using default settings. ARIA automatically generates distance restraints based on peak tables derived from NOESY spectra. Initially, most restraints are ambiguous due to chemical shift degeneracy. With each successive cycle of restrained molecular dynamics with simulated annealing with CNS 1.21[32], generated structures are used to resolve ambiguities, and the proportion of unambiguous distance restraints increases with each cycle (eight in total). The frequency window tolerances for assigning NOEs were 0.02 and 0.03 ppm for direct and indirect proton dimensions, respectively, and 0.2 ppm for nitrogen and carbon dimensions. Fairly narrow tolerance ranges could be employed because the chemical shift assignments were adjusted to exactly match signals in the NOESY spectra. Dihedral angle restraints derived from TALOS+ were incorporated in the calculation. We found that two spectral artifacts created problems for the ARIA program – diagonal peaks needed to be removed (as specified in the ARIA graphical user interface), and sinc wiggles also had to be manually removed from the NOESY peak lists. Moreover, in our experience, mobile regions of the protein would give rise to high intensity peaks that ARIA could erroneously assign to cause structural distortions if chemical shift assignments were not 100% complete. Thus, NOE cross-peaks involving unstructured regions of the protein were also removed from peak lists after manual inspection confirmed that there was no long range NOEs that could contribute to protein folding.
The web-based server, PRODRG[33], was used to generate topology and parameter files for 3-mDPA. Calcium was not included in the ARIA structure calculations, but 3-mDPA was included. Intermolecular NOEs between 3-mDPA and cChimera were not included in the peak lists. Instead, these NOE peaks were manually assigned and used to generate distance restraints that were added to the ARIA structure calculations. All NOEs were calibrated automatically and assigned iteratively by ARIA; the assignments were checked manually for errors. 80 conformers with lowest energy were calculated for each iteration; after the eighth iteration, 40 conformers with the lowest restraint energies were refined in a shell of water, and 8 conformers with the lowest total energies were selected.
2.5. Virtual Screening of DPA-like Compounds
DPA-like compounds (based on diphenylmethylene, diphenylether, and diphenylamine) were extracted from the NCI database. For this the NCI substructure search was used for SMILES strings c2ccc(Nc1ccccc1)cc2, c2ccc(Cc1ccccc1)cc2, and c2ccc(Oc1ccccc1)cc2. A total of 15876 compounds were retrieved. These were prepared for docking using LigPrep with the protocol described previously[34]. This generated 23438 compounds. The compounds were docked into the hydrophobic pocket of the cardiac troponin complex using Glide XP[35]. The compounds were subsequently ordered by predicted docking score and ligand efficiency. Lastly, NCI compounds with every possible single substitution at the 3 (meta) position (based on diphenylmethylene, diphenylether, and diphenylamine) were identified using the NCI substructure search tool. A total of 61 compounds were identified which were again prepared for docking using LigPrep, generating a total of 72 compounds. After Glide XP docking, those 72 compounds were ranked by docking score. Compounds were selected for NMR binding studies if they scored well, were predicted to be soluble, had a molecular weight less than 300 Da, and were available from NCI.
3. Results and discussion
3.1. Structure of cChimera
The NMR solution structure of the cTnC1–90-cTnI136–163 chimera (cChimera) was determined in the absence of any drug (Figure 2a). As shown in Figure 2b, the NMR structure of cChimera superimposes well to the corresponding regions in the X-ray crystal structure of the cardiac troponin complex (1J1E.pdb)[10], with a root-mean-square deviation (RMSD) of 1.2 Å for the backbone heavy atoms of residues 3–85 of cNTnC. The linker region between cNTnC and cTnI switch region is highly mobile by NMR, as indicated by random coil chemical shifts and lack of NOE contacts to the rest of the protein. Val146 is the first structured residue in cTnI immediately following the linker, showing NOEs to Pro52, Glu56 and Met60 of cNTnC (Figure 2c). This is the first time these contacts have been observed in an NMR structure, though they are also present in a single asymmetric unit of the crystal structure, showing that Val146 is actually part of the switch region of cTnI that binds to cNTnC. The conformation and interactions of the rest of the switch region of cTnI are similar in the X-ray and NMR structures, with residues 146–149 in an extended conformation and residues 150–159 forming an alpha helix. Residues C-terminal to Leu159 are flexible according to the X-ray structure (different conformations observed within the asymmetric unit) and as indicated by NMR chemical shifts.
Figure 2.

a) The eight lowest energy solution NMR structures of cChimera (cTnI switch region shown in red). b) The x-ray structure cNTnC-cTnI derived from the cardiac troponin complex (1J1E.pdb, grey) was aligned by secondary structural elements (residues 3–85) to cChimera (cNTnC region: blue, cyan, green, and yellow spectrum, cTnI inhibitory region linker: orange, cTnI switch region: red). c) Residue Val146 of cTnI (red) forms contacts with Pro52, Glu56 and Met60 (green) of cNTnC, shown in sticks.
In many EF-hand proteins, calcium binding triggers a closed-to-open conformational transition[36]. However, in cNTnC, the closed conformation predominates still upon calcium binding, though the open form is still populated to a small degree [37]. Binding of the cTnI 146–158 switch region locks cNTnC into the open conformation, characterized by a rotation of the B–C helical unit away from helices N-A–D. The cTnI switch region binds between the outer C-terminus of helix B and helices A–D (Figure 2a), analogous to wedging a wastebasket between a door and its frame to keep it open. Importantly, this leaves a sizeable cavity between the hinge of the protein (the central β sheet) and the cTnI peptide, where drug binding can occur. The size and configuration of this cavity is virtually identical between cChimera and the X-ray structure.
3.2. Selection of diphenylamine as starting compound
Bepridil is a calcium channel blocker that was previously marketed as a treatment for angina. As an off-target effect, it was previously found to bind to the troponin complex to act as a calcium sensitizer[38]. A crystal structure showed that in the absence of cTnI, bepridil binds to the hydrophobic patch of cNTnC and stabilizes its calcium-bound open state, though its binding site overlaps with where the cTnI switch region normally binds[39]. NMR studies showed that bepridil and cTnI switch region bind to calcium-saturated cNTnC simultaneously but with negative cooperativity[40]. Thus, bepridil binding stabilizes the calcium-bound open state of cNTnC (with a KD of 20 μM), but also destabilizes the complex by displacing the cTnI switch peptide. In the presence of switch peptide, bepridil binds with an affinity of 80 μM[40]. We tested the binding of bepridil to cChimera, but were unable to obtain a binding constant due to signal broadening, consistent with competitive binding of bepridil against the switch peptide. Since the binding affinity of bepridil for cNTnC was the highest of any compound published to date, we decided that it would be a good starting point for drug design.
In the NMR-based structure determination of bepridil bound to cNTnC-cTnI switch peptide, it was evident that most NOE-derived distance restraints localized to the two aromatic rings of bepridil[40]. Therefore, we tested a series of small compounds mimicking the two aromatic rings of bepridil. Of these, benzylaniline is a fragment of bepridil that binds with a KD of 150 μM (Figure 3). Removing the methylene carbon to yield diphenylamine (DPA) improved binding to KD 120 μM, while addition of a methylene carbon (dibenzylamine) completely abolished binding. The effect of adding methylene carbons is at least two-fold, increasing the length of the molecule and increasing the partial positive charge on the central nitrogen atom (increasing pKa), and both of these factors likely contribute to decreased binding to cChimera. While DPA bound to cChimera with slightly lower affinity than what was previously found for bepridil, we noted that the molecular weight of DPA was less than half that of bepridil, making it an excellent starting scaffold for drug development. We initially added single methyl or chloro substituents and found that 3-chlorodiphenylamine (3-ClDPA) bound with the highest affinity with a KD of 10 μM. We decided to pursue the structure of 3-methyldiphenylamine (3-mDPA) by NMR because the methyl group would provide an additional signal for structure determination.
Figure 3.

Chemical structures and cChimera binding affinity (KD) of N-benzyl-N-(3-isobutoxy-2-pyrrolidin-1-yl-propyl)aniline (bepridil), benzylaniline, dibenzylamine, diphenylamine (DPA), 3-methyldiphenylamine (3-mDPA) and 3-chloroldiphenylamine (3-ClDPA). For bepridil, KD* is binding affinity to cTnC-switch cTnI peptide complex (binding to cChimera could not be accurately measured).
3.3 Interaction of cChimera with 3-mDPA
To characterize its interaction with cChimera, 3-mDPA was titrated into 15N-labeled cChimera and monitored by 1H, 15N-HSQC NMR spectra (Figure 4a). Backbone chemical shifts migrated in a linear fashion, indicating a 1:1 stoichiometry. Since 3-mDPA interacts with cChimera on a fast exchange timescale, almost all resonances could be easily followed throughout the titration. Chemical shift changes of the backbone amides of Phe27, Ser37, Ile61, Val64, Asp65, Glu66, Phe77, Val79, and Ala150 were plotted as a function of 3-mDPA-to-cChimera concentrations; a global dissociation constant (KD) of 29 μM was determined (Figure 4b). Chemical shift changes in cChimera upon titration with 3-mDPA are shown in Figure 5. Chemical shift mapping of 1H and 15N signals do not seem to accurately delineate the small molecule binding site in cNTnC. Instead, they are sensitive markers of conformational changes in the protein[41] that are needed to accommodate drug binding. The largest 15N chemical shift changes are seen in Val64 and Met81 in cNTnC, and Ala150 in cTnI, highlighting hotspots of conformational change that occur upon 3-mDPA binding.
Figure 4.

a) Overlay of 2D HSQC spectra of cChimera acquired during titration with 3-mDPA. The first point of each titration is represented with multiple contours and subsequent titration points for the drug are represented by single contours. Residues that experienced large chemical shift changes are labeled. b) Global fit for residues in cChimera that experienced large chemical shift perturbations upon addition of 3-mDPA. KD ~ 30 μM.
Figure 5.
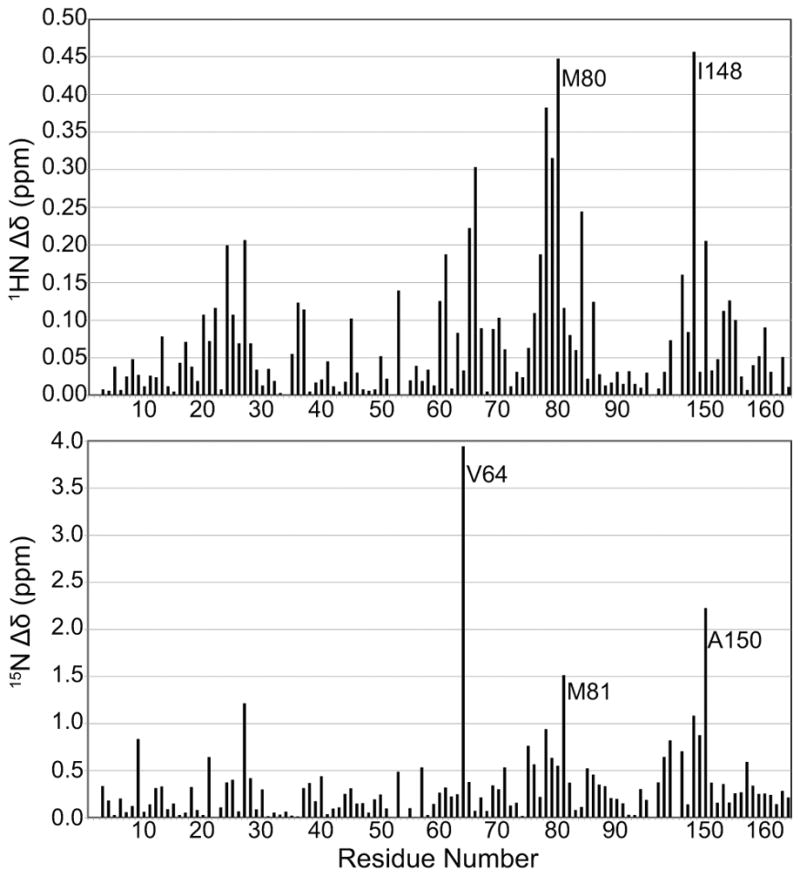
The backbone 1HN and 15N chemical shift differences (Δδ) between cChimera and cChimera-3-mDPA as a function of sequence.
13C chemical shift changes upon 3-mDPA titration were also tracked using 1H,13C-HSQC NMR spectra. The chemical shift changes of the methyl groups of Ile61, Val64, Met80, and Met153 were plotted as a function of 3-mDPA-to-cChimera concentrations; a global dissociation constant (KD) of 25 μM was determined (Figure 6a and 6b). The largest chemical shift changes observed in methyl groups seems to map more consistently to the binding site than the backbone amide chemical shift changes.
Figure 6.

a) Overlay of 2D 13C-HSQC spectra of cChimera acquired during the titration of 3-mDPA. b. Global fit for residues in cChimera that experienced large chemical shift perturbations upon addition of 3-mDPA. KD ~ 25μM.
3.4 Solution structure of the cChimera-3-mDPA complex
Accurate determination of the drug binding site of cChimera requires a complete solution structure determination of the protein-drug complex, complete with a full set of NOE-derived distance restraints and chemical shift-derived dihedral angle restraints (see Table 1). NOEs from the cChimera protein to the methyl substituted ring of 3-mDPA clustered into two groups, with one set of NOEs to a solvent-exposed site in cNTnC (to Met60 and Val64) and another set localizing to the cNTnC-cTnI interface (Val44, Met45, Leu48 of cNTnC and Ile148 and Met153 of cTnI). Both sets could not be simultaneously satisfied within a single structure. Since the two sets of NOEs indicated that 3-mDPA was able to bind in two different orientations, two separate structure calculations were performed for each set. Coincidentally, both NOE distance restraint sets contained 30 out the 43 total 3-mDPA-cChimera intermolecular restraints (17 restraints common to both sets, 13 restraints unique to each set).
Table 1.
Total numbers of structural restraints used in the final round of ARIA calculations.
| cChimera | cChimera+3-mDPApeptide | cChimera+3-mDPAsolvent exposed | |
|---|---|---|---|
| Backbone dihedral angles | 192 | 192 | 192 |
| Sidechain dihedral angles | 6 | 6 | 6 |
| Unambiguous distance restraints | 1730 | 1630 | 1630 |
| Ambiguous distance restraints | 420 | 370 | 370 |
| Intermolecular drug-protein distance restraints | - | 30 | 30 |
Unlike the methyl-substituted ring of 3-mDPA, NOEs to the unsubstituted aryl ring unambiguously position it into a deep pocket, with its para-position (furthest from the central nitrogen) making contacts with the central β-sheet core of cNTnC, centered about Ile36 and Val72 (Figure 7a). This deep binding subsite must be sterically tight, since the presence of a single methyl group on the meta-substituted ring precludes binding in this location. The positioning of the deep binding site corresponds exactly to the binding site of the phenyl ring of bepridil in both the X-ray and NMR structures of the cNTnC-bepridil complex[39,40]. Remarkably, this deep binding pocket is present even in the absence of small molecule binding, as seen in the structure of cChimera alone and in the X-ray structure of the cardiac troponin complex[10]. However, it is occupied by Met80, though its sidechain is too small to perfectly fill the cavity. We postulate that binding of any sizeable molecule to the central cavity of cNTnC requires displacement of the Met80 sidechain, making this the minimal conformational change necessary to support drug binding. Displacement of Met80 is achieved through sidechain χ dihedral angle rotations, with the backbone of Met80 relatively fixed within helix D (Figure 7b). The linear nature of the methionine sidechain makes it the amino acid most able to adapt to a wide range of binding partners, a phenomenon that has been well characterized in calmodulin, a versatile signaling protein homologous to TnC that is able to interact with a plethora of protein targets through its methionine-rich binding surfaces[42]. The importance of Met80 sidechain displacement in cNTnC for drug binding is supported by the fact that NMR signal from the εCH3 group of Met80 moves and broadens more than any other 1H-13C group in the 1H-13C HSQC spectrum of cChimera as 3-mDPA is added (Figure 6a). The conformational change of the Met80 sidechain likely accounts for the large chemical shift changes that occur in its vicinity upon drug binding, including the backbone 15N chemical shift of Met81 (Figure 5).
Figure 7.

a) Deep binding site of 3-mDPA bound to cChimera. The main contact residues Ile36 and Val72 are shown in sticks. b) The sidechain of Met80 is displaced in the presence of 3-mDPA. X-ray troponin complex (Grey), cChimera-3-mDPA (yellow).
In contrast to the unsubstituted aryl ring, NOEs observed for the methyl-substituted ring of 3-mDPA cluster into two distinct groups that can only be satisfied by two distinct modes of 3-mDPA binding (Figure 8a). One set of NOEs localizes to the cNTnC-cTnI interface, with the methyl group of 3-mDPA nestled between residues Val44, Met45, and Leu48 in helix B, and the rest of the ring making contacts with Ile148, Ala150, and Met153 of the cTnI switch peptide. The ring is also spatially close to the sidechains of Met80 and Ser84, but intermolecular NOEs could not be observed due to exchange-induced signal broadening of these residues. The hydrophobic burial of the 3-mDPA methyl group explains its favorable effect on cChimera binding.
Figure 8.

a) cChimera residues with observed NOEs to the methyl-substituted ring of 3-mDPA. cChimera-3-mDPApeptide drug binding site, contacting Val44, Met45, and Leu48 of cNTnC are shown in blue, Ile148 and Met153 of cTnI are shown in blue. cChimera-3-mDPAsolvent exposed drug binding site, contacting Val64 and Met60, are shown in green. b) The sidechain of Ile148 is displaced when 3-mDPA binds at the peptide binding site (cChimera: grey, cChimera-3-mDPApeptide: red). c) The drug binding site of the cTnC-bepridil complex x-ray structure (orange), cChimera-3-mDPApeptide (cChimera: grey, 3-mDPA: blue) and cChimera-3-mDPAsolvent exposed (cChimera: grey, 3-mDPA: green). Note how steric clash of the bepridil isopropyl group pushes helices N and A away from the rest of the protein, increasing the size of the central cNTnC cavity in order to accommodate bepridil.
The sidechain of Ile148 is the most deeply inserted element of cTnI relative to the hydrophobic cavity of cNTnC (Figure 8a). Although the sidechain does not extend deeply enough to fill the cavity, it does need to be displaced to allow insertion of the 3-mDPA methyl-substituted ring. Ile148 is a β-branched amino acid, and displacement of its sidechain by 3-mDPA requires a rotation of the extended backbone around Ile148 along its long axis (Figure 8b). This accounts for the large changes in chemical shift that occur in this region upon titration with 3-mDPA (Figure 5).
In the alternative mode of 3-mDPA binding to cChimera, the methyl-substituted ring is flipped out of the switch peptide interface into a solvent-exposed space adjacent to helix C, resulting in NOEs to Met60 and Val64 of helix C (Figure 8a). Val64 is in an opposite corner of the drug binding site, spatially distant from the interface between cTnI switch peptide and cNTnC helix B. This solvent-exposed binding site corresponds well with the location of the pyrrolidine ring in the cNTnC-bepridil X-ray crystal structure (Figure 8c). It has fewer hydrophobic contacts than the interfacial site, mainly Met60, Val64, and Met80, suggesting that binding here would not be as favorable. However, the intensity of NOEs to Met60 and Val64 suggest that this mode of binding is as favorable as the interfacial mode, likely because it does not require any displacement of the switch peptide. The solvent-exposed subsite has some proximity to charged sidechains from cNTnC: Glu63 from helix B and Arg83 from helix D, which form an ion pair, but there are no groups in 3-mDPA that are available for hydrogen bonding to these.
The eight lowest energy structures of the cChimera-drug complex for both interfacial and solvent-exposed binding modes are shown in figures 9a and 9b, respectively. Both drug-bound models were aligned to cChimera without drug (Figure 9c) and the backbone RMSDs of residues 3–85 were 1.3 Å and 1.2 Å for the interfacial and solvent-exposed binding modes, respectively, similar to the RMSD between the cChimera alone and the X-ray structure of the troponin complex (1J1E.pdb). The binding of 3-mDPA does not impart a large-scale structural perturbation to the backbone of cChimera, contrasting with the considerable opening of cNTnC structure needed to accommodate the larger bepridil molecule, particularly its isopropyl group (see Figure 8c).
Figure 9.

a) The eight lowest energy structures of cChimera-3-mDPApeptide. b) 8 lowest energy structures of cChimera-3-mDPAsolvent exposed. c) The x-ray structure cNTnC-cTnI derived from the cardiac troponin complex (1J1E.pdb, wheat) was aligned by the secondary structural elements (residues 3–85) to the structure of cChimera (grey), cChimera-3-mDPApeptide (blue) and cChimera-3-mDPAsolvent exposed (green). Linker regions are hidden.
3.5 Small molecule binding
We screened many compounds based on DPA for cChimera binding in order to explore its potential as a base structure for drug development (see Table 2). 3-mDPA binds to cChimera through entirely hydrophobic interactions. The choice of compounds to be tested was guided, in part, by the compounds suggested using in silico screening. Based on our structures, it appears that the central nitrogen atom of 3-mDPA does not participate in polar interactions with cChimera. DPA tolerates hydrophobic burial reasonably well because it does not carry appreciable positive charge. The central nitrogen is important for imparting some water solubility to the DPA base structure, but it is replaceable with –O- or –CH2- without a major influence on cChimera binding, even though this changes the central geometry from trigonal planar to tetrahedral.
Table 2.
Binding affinity (KD) of DPA-like compounds for cChimera. KD is the affinity constant calculated from small molecule titration, with the proviso that it provides a lower bound estimate (that is, a KD that is too low) when the compound precipitates out before the final point in the titration. KD* is calculated based on the assumption that if solubility of the compound were not a limiting factor, saturation with the compound would cause chemical shift perturbations similar to 3-ClDPA. “Weak binding” denotes when titration with a compound led to very small chemical shift changes that could not be fit to a saturating curve, indicating a KD > 2 mM.
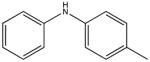
|
4-methyldiphenalamine KD 60 μM KD* 120 μM |
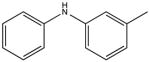
|
3-methyldiphenalamine KD 30 μM KD* 50 μM |
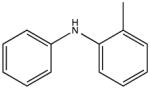
|
2-methyldiphenalamine KD 15 μM KD* 25 μM |
|
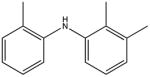
2,2′,3′-trimethyldiphenalamine KD 5 μM KD* 100 μM |
|
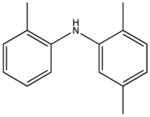
2,2′,5′-trimethyldiphenalamine KD 3 μM KD* 30 μM |
|
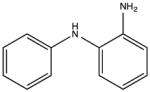
NSC18731 KD 330 μM KD* 320 μM |
|
NSC215211 KD 400 μM KD* 800 μM |
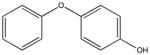
|
NSC25027 KD 80 μM KD* 100 μM |
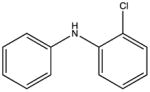
|
2-chlorodiphenylamine KD 10 μM |
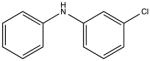
|
3-chlorodiphenylamine KD 10 μM |
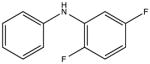
|
NSC50696 KD 40 μM KD* 30 μM |
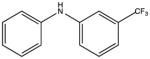
|
NSC50453 KD 20 μM KD* 30 μM |
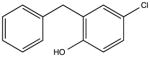
|
NSC59989 KD 20 μM KD* 10 μM |
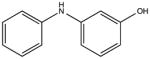
|
NSC56930 KD 300 μM KD* 220 μM |
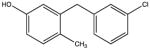
|
NSC11182 KD 10 μM KD* 10 μM |
|
NSC11184 KD 60 μM KD* 70 μM |
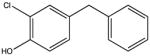
|
NSC17285 Weak binding |
|
NSC11177 KD 60 μM KD* 100 μM |
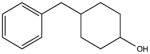
|
NSC71607 KD 180 μM KD* 190 μM |
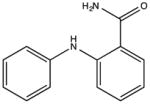
|
NSC75183 KD 80 μM KD* 60 μM |
|
|
NSC167798 Weak binding |
|
NSC89784 Weak binding |
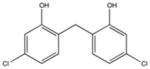
|
NSC3947 KD 180 μM KD* 90 μM |
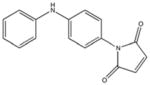
|
NSC39744 KD 140 μM KD* 500 μM |
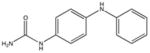
|
NSC24035 KD 600 μM KD* 1340 μM |
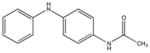
|
NSC40576 KD 1400 μM KD* 2000 μM |
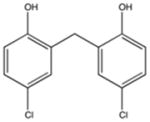
|
NSC38642 KD 270 μM KD* 120 μM |
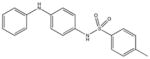
|
NSC41053 Weak binding |
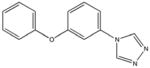
|
ChemBridge 54451398 Weak binding |
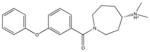
|
ChemBridge 58784194 No binding |
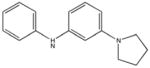
|
SHG70 Weak binding |
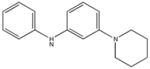
|
SHG72 Weak binding |
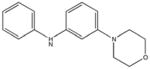
|
SHG74 KD 210 μM KD* 340 μM |
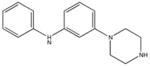
|
SHG78 Weak binding |
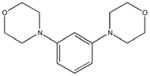
|
SHG82 No binding |
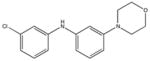
|
SHG83 KD 60 μM KD* 360 μM |
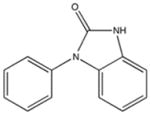
|
SHG102 KD 270 μM KD* 340 μM |
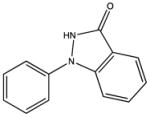
|
SHG115 KD 26 μM KD* 460 μM |
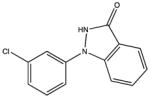
|
SHG116 Weak binding |
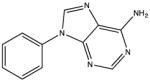
|
SHG118 No binding |
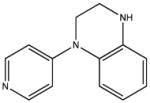
|
SHG123 No binding |
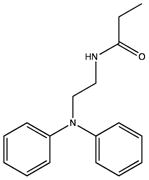
|
SHG_DPA1: KD 280 μM |
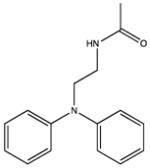
|
SHG_DPA2: KD 130 μM |
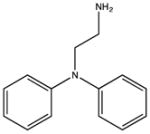
|
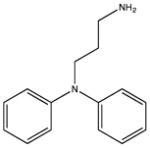
SHG_DPA3: KD 1600 μM |
|
SHG_DPA4: KD 1200 μM |
|
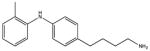
SHG_DPA8 No binding |
|
SHG_DPA9 No binding |
Strategically placed polar groups are important for the design of highly specific and tightly binding drugs. To date, all charged substituents added to the DPA base structure that we have tested have had an unfavorable effect on binding. Furthermore, attachment of charged amino groups to the DPA backbone via flexible linkers also increased the KD. The addition of polar but uncharged substituents to the DPA base structure was better tolerated than the addition of charged groups. It is interesting that –F and –Cl have a favorable effect on binding, with 2-chloro-DPA and 3-chloro-DPA having the tightest affinity of all compounds studied to-date. It is possible that van der Waals interactions between chlorine and protein hydrophobic groups are very favorable. Addition of a hydroxyl substituent to DPA slightly decreases affinity, and addition of an amine is less favorable than a hydroxyl. In the immediate vicinity of an aromatic ring, the amine has a slight positive charge (aniline pKa 4.6), while the hydroxyl has a slight negative charge (phenol pKa 10.0). The preference for –OH over –NH may be related more to the lesser partial charge on OH at the acidic pH (6–7) of the NMR samples, rather than any particular charge preference in the drug binding cavity.
One particularly informative series of compounds is that employing single methyl substitutions to DPA. 4-mDPA, 3-mDPA and 2-mDPA all bind more tightly than unsubstituted DPA itself (KD 60 μM, 30 μM, 15 μM respectively). Much of this is likely due to enhanced hydrophobic interaction to cChimera and decreased water solubility. Nevertheless, the series points toward some important steric effects. The favorable binding of 3-mDPA relative to DPA can be rationalized in terms of the favorable burying of the 3-methyl group in between hydrophobic sidechains of helix B, though this would be partially offset by the loss of interchangeable binding to the deep pocket afforded by the unsubstituted symmetry of DPA. The lowest affinity compound is 4-mDPA, suggesting that a methyl group in the para position is not particularly favorable in any of the three binding subsites highlighted in this study. The best compound of this series is in fact 2-mDPA and not 3-mDPA, perhaps because the 2-methyl-substituted aryl ring is able to bind to any of the three binding subsites. The addition of multiple single atom substituents to the DPA base structure did not appear to increase binding affinity beyond what is observed for a single substituent, though not every possible combination was tested.
Placement of larger substituents on DPA is desirable from a drug development standpoint. Bulky substituents at the para position are not well tolerated, which is not surprising, given that this was the least favorable position for single atom and methyl substituents. Bulky substituents at the meta ring position were better tolerated, but did not improve binding affinity. With the cChimera-3-mDPA structures in hand, it is possible to rationalize why substituents at these positions were not favorable in terms of the three binding sub-sites. Space is very limited in the deep binding pocket, even for single atom substituents. At the switch peptide interface, larger substituents in the meta position cannot fit into the helix B groove like the single methyl group of 3-mDPA, and substituents at the other meta position on the ring would further displace the switch peptide.
4. Conclusion
Our solution NMR structures of cChimera bound to 3-mDPA show that DPA-based compounds bind well to cardiac troponin compared to much larger compounds like bepridil. Binding to the DPA base structure is less perturbing to the troponin C-I complex, requiring only minimal re-positioning of sidechains (Met80 in cTnC, Ile148 in cTnI). Previous cTnC-cTnI-small molecule structures show more extensive opening of cNTnC and more extensive displacement of the cTnI switch peptide, creating a large drug binding cavity. The cavity in the current study is much smaller, explaining the restrictive pattern of binding seen in our collection of DPA-like molecules. This better defined binding site will facilitate in silico screening of potential high affinity compounds.
The flexibility of cNTnC itself and the adaptability of its interface with the cTnI switch region explains why the cNTnC-cTnI complex is able to bind to many structurally unrelated small molecules, albeit with low affinity. Preservation of the preferred native conformation of the troponin complex appears to be critical in designing high affinity cardiac troponin modulators.
Figure 1.

cChimera sequence: residues 1–90 of cNTnC (shown in green, C35S and C84S are shown in blue), followed by residues 136–163 of cTnI (shown in red, overlapping residues are shown in orange), and ending with a C-terminal His-tag (shown in black).
Acknowledgments
This study was supported by grants from Heart and Stroke foundation of Canada (B.D.S., G-14-0005884) and Canadian Institutes of Health Research (CIHR) (B.D.S., 37769). The authors also acknowledge support from the Hwang Professional Corporation and startup funds (to P.M.H.) from the University of Alberta Faculty of Medicine & Dentistry and the Department of Medicine. S.L. is supported by the National Institutes of Health, the National Science Foundation, the Howard Hughes Medical Institute, the National Biomedical Computation Resource, and the NSF Supercomputer Centers. The authors would like to thank Dr. J. Andrew McCammon for helpful discussions.
Footnotes
Disclosures
The authors declare that there no potential conflicts of interest to disclose.
References
- 1.WHO. Global status report on noncommunicable diseases 2014. World Health; 2014. p. 176. [Google Scholar]
- 2.Sisakian H. Cardiomyopathies: Evolution of pathogenesis concepts and potential for new therapies. World J Cardiol. 2014;6:478–94. doi: 10.4330/wjc.v6.i6.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sonnenblick EH, Frishman WH, LeJemtel TH. Dobutamine: a new synthetic cardioactive sympathetic amine. N Engl J Med. 1979;300:17–22. doi: 10.1056/NEJM197901043000105. [DOI] [PubMed] [Google Scholar]
- 4.Cleland JGF, Freemantle N, Coletta AP, Clark AL. Clinical trials update from the American Heart Association: REPAIR-AMI, ASTAMI, JELIS, MEGA, REVIVE-II, SURVIVE, and PROACTIVE. Eur J Heart Fail. 2006;8:105–110. doi: 10.1016/j.ejheart.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Michaels AD, McKeown B, Kostal M, Vakharia KT, Jordan MV, Gerber IL, et al. Effects of Intravenous Levosimendan on Human Coronary Vasomotor Regulation, Left Ventricular Wall Stress, and Myocardial Oxygen Uptake. Circulation. 2005;111:1504–1509. doi: 10.1161/01.CIR.0000159252.82444.22. [DOI] [PubMed] [Google Scholar]
- 6.Mebazaa A, Nieminen MS, Packer M, Cohen-Solal A, Kleber FX, Pocock SJ, et al. Levosimendan vs Dobutamine for Patients With Acute Decompensated Heart Failure. JAMA. 2007;297:1883. doi: 10.1001/jama.297.17.1883. [DOI] [PubMed] [Google Scholar]
- 7.Nieminen MS, Fruhwald S, Heunks LMA, Suominen PK, Gordon AC, Kivikko M, et al. Levosimendan: current data, clinical use and future development. [accessed February 16, 2016];Hear Lung Vessel. 2013 5:227–45. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3868185&tool=pmcentrez&rendertype=abstract. [PMC free article] [PubMed] [Google Scholar]
- 8.Haikala H. Cardiac troponin C as a target protein for a novel calcium sensitizing drug, levosimendan. J Mol Cell Cardiol. 1995;27:1859–66. doi: 10.1016/0022-2828(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 9.Pollesello P, Ovaska M, Kaivola J, Tilgmann C, Lundström K, Kalkkinen N, et al. Binding of a new Ca2+ sensitizer, levosimendan, to recombinant human cardiac troponin C. A molecular modelling, fluorescence probe, and proton nuclear magnetic resonance study. [accessed May 8, 2016];J Biol Chem. 1994 269:28584–90. http://www.ncbi.nlm.nih.gov/pubmed/7961805. [PubMed] [Google Scholar]
- 10.Takeda S, Yamashita A, Maeda K, Maéda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 11.Parry DAD, Squire JM. Structural role of tropomyosin in muscle regulation: Analysis of the X-ray diffraction patterns from relaxed and contracting muscles. J Mol Biol. 1973;75:33–55. doi: 10.1016/0022-2836(73)90527-5. [DOI] [PubMed] [Google Scholar]
- 12.Li MX, Spyracopoulos L, Sykes BD. Binding of cardiac troponin-I147–163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38:8289–98. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 13.Sorsa T, Pollesello P, Permi P, Drakenberg T, Kilpeläinen I. Interaction of levosimendan with cardiac troponin C in the presence of cardiac troponin I peptides. J Mol Cell Cardiol. 2003;35:1055–61. doi: 10.1016/S0022-2828(03)00178-0. [DOI] [PubMed] [Google Scholar]
- 14.Robertson IM, Baryshnikova OK, Li MX, Sykes BD. Defining the binding site of levosimendan and its analogues in a regulatory cardiac troponin C-troponin I complex. Biochemistry. 2008;47:7485–95. doi: 10.1021/bi800438k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szilágyi S, Pollesello P, Levijoki J, Kaheinen P, Haikala H, Édes I, et al. The effects of levosimendan and OR-1896 on isolated hearts, myocyte-sized preparations and phosphodiesterase enzymes of the guinea pig. Eur J Pharmacol. 2004;486:67–74. doi: 10.1016/j.ejphar.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Farmakis D, Alvarez J, Ben Gal T, Brito D, Fedele F, Fonseca C, et al. Levosimendan beyond inotropy and acute heart failure: Evidence of pleiotropic effects on the heart and other organs: An expert panel position paper. Int J Cardiol. 2016;222:303–12. doi: 10.1016/j.ijcard.2016.07.202. [DOI] [PubMed] [Google Scholar]
- 17.Grossini E, Molinari C, Caimmi PP, Uberti F, Vacca G. Levosimendan induces NO production through p38 MAPK, ERK and Akt in porcine coronary endothelial cells: role for mitochondrial K(ATP) channel. Br J Pharmacol. 2009;156:250–61. doi: 10.1111/j.1476-5381.2008.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russell AJ, Hartman JJ, Hinken AC, Muci AR, Kawas R, Driscoll L, et al. Activation of fast skeletal muscle troponin as a potential therapeutic approach for treating neuromuscular diseases. Nat Med. 2012;18:452–5. doi: 10.1038/nm.2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rezvanpour A, Phillips JM, Shaw GS. Design of high-affinity S100-target hybrid proteins. Protein Sci. 2009;18:2528–36. doi: 10.1002/pro.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porumb T, Yau P, Harvey TS, Ikura M. A calmodulin-target peptide hybrid molecule with unique calcium-binding properties. [accessed May 5, 2016];Protein Eng. 1994 7:109–15. doi: 10.1093/protein/7.1.109. http://www.ncbi.nlm.nih.gov/pubmed/8140087. [DOI] [PubMed] [Google Scholar]
- 21.Pineda-Sanabria SE, Julien O, Sykes BD. Versatile cardiac troponin chimera for muscle protein structural biology and drug discovery. ACS Chem Biol. 2014;9:2121–30. doi: 10.1021/cb500249j. [DOI] [PubMed] [Google Scholar]
- 22.Julien O, Mercier P, Allen CN, Fisette O, Ramos CHI, Lagüe P, et al. Is there nascent structure in the intrinsically disordered region of troponin I? Proteins. 2011;79:1240–50. doi: 10.1002/prot.22959. [DOI] [PubMed] [Google Scholar]
- 23.Li MX, Hwang PM. Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene. 2015;571:153–66. doi: 10.1016/j.gene.2015.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gagné SM, Tsuda S, Li MX, Chandra M, Smillie LB, Sykes BD. Quantification of the calcium-induced secondary structural changes in the regulatory domain of troponin-C. Protein Sci. 1994;3:1961–74. doi: 10.1002/pro.5560031108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neri D, Szyperski T, Otting G, Senn H, Wüthrich K. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry. 1989;28:7510–6. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 26.Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 27.Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–14. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 28.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–23. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MVB, Wishart* DS. A Simple Method To Predict Protein Flexibility Using Secondary Chemical Shifts. 2005 doi: 10.1021/ja054842f. [DOI] [PubMed] [Google Scholar]
- 30.Linge JP, Habeck M, Rieping W, Nilges M. ARIA: automated NOE assignment and NMR structure calculation. [accessed May 9, 2016];Bioinformatics. 2003 19:315–6. doi: 10.1093/bioinformatics/19.2.315. http://www.ncbi.nlm.nih.gov/pubmed/12538267. [DOI] [PubMed] [Google Scholar]
- 31.Rieping W, Habeck M, Bardiaux B, Bernard A, Malliavin TE, Nilges M. ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics. 2007;23:381–2. doi: 10.1093/bioinformatics/btl589. [DOI] [PubMed] [Google Scholar]
- 32.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2:2728–33. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 33.Schüttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–63. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 34.Lindert S, Zhu W, Liu YL, Pang R, Oldfield E, McCammon JA. Farnesyl diphosphate synthase inhibitors from in silico screening. Chem Biol Drug Des. 2013;81:742–8. doi: 10.1111/cbdd.12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood Jeremy R, Halgren TA, et al. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. 2006 doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 36.Gifford JL, Walsh MP, Vogel HJ, Berridge MJ, Lipp P, Bootman MD, et al. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 2007;405:199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- 37.Eichmüller C, Skrynnikov NR. A new amide proton R1rho experiment permits accurate characterization of microsecond time-scale conformational exchange. J Biomol NMR. 2005;32:281–93. doi: 10.1007/s10858-005-0658-y. [DOI] [PubMed] [Google Scholar]
- 38.Solaro RJ, Bousquet P, Johnson JD. Stimulation of cardiac myofilament force, ATPase activity and troponin C Ca++ binding by bepridil. [accessed May 10, 2016];J Pharmacol Exp Ther. 1986 238:502–7. http://www.ncbi.nlm.nih.gov/pubmed/2942677. [PubMed] [Google Scholar]
- 39.Li Y, Love ML, Putkey JA, Cohen C. Bepridil opens the regulatory N-terminal lobe of cardiac troponin C. Proc Natl Acad Sci U S A. 2000;97:5140–5. doi: 10.1073/pnas.090098997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, Li MX, Sykes BD. Structure of the regulatory N-domain of human cardiac troponin C in complex with human cardiac troponin I147–163 and bepridil. J Biol Chem. 2002;277:31124–33. doi: 10.1074/jbc.M203896200. [DOI] [PubMed] [Google Scholar]
- 41.Robertson IM, Boyko RF, Sykes BD. Visualizing the principal component of 1H, 15N-HSQC NMR spectral changes that reflect protein structural or functional properties: application to troponin C. J Biomol NMR. 2011;51:115–22. doi: 10.1007/s10858-011-9546-9. [DOI] [PubMed] [Google Scholar]
- 42.Yuan T, Ouyang H, Vogel HJ. Surface Exposure of the Methionine Side Chains of Calmodulin. J Biol Chem. 1999;274:8411–20. doi: 10.1074/jbc.274.13.8411. [DOI] [PubMed] [Google Scholar]