

Localization of a regulator of RhoGTPases (ARHGAP18) is important for microtubule stability and endothelial cell function. The localization is demonstrated by advanced imaging and biochemical techniques.
Abstract
RhoGTPases are important regulators of the cell cytoskeleton, controlling cell shape, migration and proliferation. Previously we showed that ARHGAP18 in endothelial cells is important in cell junctions. Here we show, using structured illumination microscopy (SIM), ground-state depletion (GSD), and total internal reflection fluorescence microscopy (TIRF) that a proportion of ARHGAP18 localizes to microtubules in endothelial cells, as well as in nonendothelial cells, an association confirmed biochemically. In endothelial cells, some ARHGAP18 puncta also colocalized to Weibel–Palade bodies on the microtubules. Depletion of ARHGAP18 by small interfering RNA or analysis of endothelial cells isolated from ARHGAP18-knockout mice showed microtubule destabilization, as evidenced by altered morphology and decreased acetylated α-tubulin and glu-tubulin. The destabilization was rescued by inhibition of ROCK and histone deacetylase 6 but not by a GAP-mutant form of ARHGAP18. Depletion of ARHGAP18 resulted in a failure to secrete endothelin-1 and a reduction in neutrophil transmigration, both known to be microtubule dependent. Thrombin, a critical regulator of the Rho-mediated barrier function of endothelial cells through microtubule destabilization, enhanced the plasma membrane–bound fraction of ARHGAP18. Thus, in endothelial cells, ARHGAP18 may act as a significant regulator of vascular homeostasis.
INTRODUCTION
Microtubules (MTs) are a crucial component of the cell cytoskeleton. They provide structural support and are dynamically regulated in response to stimuli. The MT and the actin cytoskeleton cross-talk to regulate cell shape associated with proliferation, migration, cell division, and differentiation (Akhshi et al., 2014). Microtubule-dependent shape changes occur rapidly, whereas the actin-mediated shape changes are slower to develop (Alieva et al., 2013). MTs also regulate the tensile force between cells necessary for the establishment of stable cell–cell junctions (Alieva et al., 2013). In endothelial cells (ECs), MTs control the barrier integrity and the response to inducers such as thrombin (Birukova et al., 2004a). MTs also play a significant role in the transport of cargo. Weibel–Palade bodies (WPBs) are the principal secretory granules of ECs, and a significant proportion localize to MTs, especially at the plus ends (Manneville et al., 2003). On physiological stimuli such as histamine, thrombin, and raised intracellular calcium levels, WPBs translocate along MTs to the plasma membrane, where a dynamic process of membrane fusion and release of cargo proteins, such as endothelin-1 (ET-1), occurs (Sinha and Wagner, 1987; Ozaka et al., 1997; Valentijn et al., 2010; reviewed in Valentijn et al., 2011). The movement and secretion of WPBs are also regulated by the interplay between the actin and MT cytoskeleton (Manneville et al., 2003). Furthermore, MTs are important for mediating leucocyte transendothelial cell migration such that disruption of MT stability inhibits leucocyte migration across ECs (Mamdouh et al., 2008).
One of the principal control points of both the actin and MT cytoskeletons is the RhoGTPase cycle (Akhshi et al., 2014). RhoGTPases are members of the Ras superfamily, acting as molecular switches to activate multiple downstream signaling targets, such as mDia and ROCK (Pertz, 2010). In particular, ROCK has been widely implicated in disease (reviewed in Schofield and Bernard, 2013), including cardiovascular disease (Noma et al., 2008). The activation state of RhoGTPases is tightly regulated by two broad families of proteins—Rho guanine nucleotide exchange factors (RhoGEFs), which promote the conversion of Rho to the active GTP-bound state, and Rho GTPase-activating proteins (RhoGAPs), which accelerate GTP hydrolysis and convert Rho to the inactive GDP-bound state (reviewed in Brandão et al., 2006; Cook et al., 2014). RhoGEFH1 has been localized to MTs (Ren et al., 1998; Krendel et al., 2002). This localization is important to inhibit the activity of the GEF, thus limiting Rho activity (Birkenfeld et al., 2008; Chang et al., 2008). ROCK, associated with active Rho, triggers MT destabilization through increased activity of histone deacetylase 6 (HDAC6) to deacetylate tubulin (Schofield et al., 2012, 2013; Aslan et al., 2013). Conversely, inhibition of ROCK increases MT acetylation and MT stabilization (Birukova et al., 2004a; Takesono et al., 2010). Thus Rho activity is both a downstream consequence of MT disassembly and drives MT disassembly through HDAC6 activation (Saito et al., 2011). Although much work has been done to illuminate localization and activation of the GEFs that regulate the cross-talk between the MT and the actin cytoskeletons, the RhoGAPs that attenuate this response have not been identified.
We previously identified ARHGAP18 as a ubiquitously expressed RhoGAP protein that is highly expressed in ECs (Coleman et al., 2010, 2013; Chang et al., 2014; Powter et al., 2014). ARHGAP18 has both RhoGAP-dependent and -independent roles in human and mouse cells, as well as in Drosophila (Neisch et al., 2013). Overexpression of ARHGAP18 (or SENEX) in primary ECs induces senescence in a GAP-independent manner (Coleman et al., 2010, 2013; Powter et al., 2014). In contrast, its GAP activity directed to RhoC in ECs is important in the reorganization of cell–cell interactions and actin after inflammatory stimuli. Further, ARHGAP18 is involved in the angiogenic process, limiting tip formation and junctional stability in angiogenic sprouts (Chang et al., 2014). In contrast, in non-ECs and in particular in transformed cell lines, ARHGAP18 acts on RhoA to regulate the actin cytoskeleton to control spreading and migration (Maeda et al., 2011). Thus cell type–specific targets for ARHGAP18, together with GAP-independent roles, add to its complexity of action. We identify here that ARHGAP18 localizes to MTs, and depletion results in the loss of MT stability in ECs. In support of this, we show that loss of ARHGAP18 in ECs results in perturbation of two functions known to be controlled by MTs, namely the delivery and secretion of inflammatory cargo associated with WPB and leucocyte transendothelial cell migration.
RESULTS
ARHGAP18 is localized to four distinct areas in ECs
Wide-field imaging was initially used to investigate the subcellular localization of ARHGAP18 (Supplemental Figure S1) in ECs, using human umbilical vein endothelial cells (HUVECs) as the model system. Imaging at high magnification showed four patterns of localization: 1) to membrane ruffles (Figure 1A, light blue arrows), 2) to the perinuclear region (Supplemental Figure S1, B and D), 3) to elongated processes and protrusions characteristic of migrating cells (Supplemental Figure S1, C and D, arrows), and 4) to filamentous structures resembling MTs (Supplemental Figure S1E). ARHGAP18 and tubulin showed a largely coincident staining pattern, suggesting that ARHGAP18 may colocalize along MT filaments (Supplemental Figure S1, E, F, and I).
FIGURE 1:

ARHGAP18 localizes to the cytosol, membrane ruffles, and microtubules in cultured ECs. (A–F) Wide-field imaging of ARHGAP18 shows broad expression in nonconfluent ECs. (A) Spread EC with ARHGAP18 expression at the peripheral membrane ruffles (light blue arrows) and perinuclear region; n, nucleus. Magnification, 630× (A), 1000× (B–F). TIRF imaging of ARHGAP18 (green; B) and α-tubulin–Alexa 647 (magenta; C) to identify MTs; wide-field view is shown at the top right. Scale bar, 10 μm (B). (D) Merge of B and C. ARHGAP18–Alexa 488 immunopositivity is evident to the left side of an EC (light blue arrows), whereas the fringe of the right side of the same cell shows even, diffuse staining (yellow arrows). Reduced ARHGAP18 immunopositivity is seen in some areas adjacent to the cell periphery (red arrowheads in D; n, nucleus). (E, F) Enlarged blue and yellow boxes from G, showing ARHGAP18 puncta along microtubules (white arrows); yellow arrowheads indicate puncta that appear to be cytosolic. Scale bar, 10 μm (D), 2.5 μm (F). (G–K) Localization of ARHGAP18 to MTs by SIM imaging. (G) ARHGAP18–Alexa 488 and (H) α-tubulin–Alexa 647 staining in SIM. Wide-field view of each image is shown at the top right. (I) Merge of SIM images. Small yellow and blue boxes in I are enlarged in J and K, respectively, to show representative contents of that region. White arrows indicate puncta localized to MTs; yellow arrows indicate cytosolic ARHGAP18 puncta not bound to MTs. Scale bar, 5 μm (I), 1.25 μm (J).
To investigate further the localization of ARHGAP18, we used total internal reflection fluorescence microscopy (TIRF), which allows imaging within a defined optical section of up to 250 nm from the plasma membrane. This showed a broad distribution within the evanescent field (Figure 1B) and confirmed localization to the plasma membrane and membrane ruffles (Figure 1D, light blue arrows, and Supplemental Figure S1, G and I). TIRF imaging (Figure 1D, nucleus [n]), also substantially reduced perinuclear labeling (Figure 1A), which is outside the evanescent field of TIRF excitation. In peripheral areas devoid of membrane ruffles (Figure 1D, yellow arrows), images showed a diffuse pattern of staining, whereas some areas adjacent to membrane ruffles (Figure 1, D and E; red arrows) showed a lack of immunopositivity. Cytosolic SENEX puncta were also present (Figure 1, E and F, yellow arrows). Thus the localization of ARHGAP18 in the ECs did not appear random.
ARHGAP18 localizes to MTs in ECs
We next explored whether ARHGAP18 was indeed localized to EC MTs. Imaging at a range of penetration depths <250 nm showed that both ARHGAP18 and the MTs were barely visible (Supplemental Figure S2, H, K, and N), and hence, although EC MTs lie close to the plasma membrane, they are spatially separated. In contrast, ARHGAP18 staining persists at lower penetration depths closer to the plasma membrane (Supplemental Figure S2, A–O). Therefore TIRF imaging was performed at a penetration depth of 250 nm, allowing both MT and ARHGAP18 visualization (Supplemental Figure S2, D–F). When merged and zoomed images of ARHGAP18 and tubulin staining were compared, ARHGAP18 puncta were localized in individual puncta along MT filaments (Figure 1, E and F, white arrows), although non–MT-associated puncta are also present (yellow arrows).
Superresolution microscopy techniques confirm the association of ARHGAP18 with MTs
Superresolution microscopy (SRM) is performed at very high magnification and thus allows high resolution of protein associations. We therefore used two SRM techniques—structured illumination imaging (SIM; approximately doubles the achievable spatial resolution of confocal or wide-field microscopy to ∼100 nm; Brunstein et al., 2013) and ground-state depletion (GSD; Hell and Kroug, 1995; Fölling et al., 2008; Lippincott-Schwartz and Manley, 2009; a technology allowing imaging at resolutions ∼20 nm/pixel, that is, 10 times that of conventional microscopy technologies) to investigate the possibility of ARHGAP18 localization with MTs.
Wide-field imaging of an EC showed poor resolution of individual puncta (Figure 1G, top right) and microtubules (Figure 1H, top right). However, SIM imaging of the same cells suggested that some ARHGAP18 was localized along the MT filaments (Figure 1, I–K), with puncta along the filaments (white arrows) and other ARHGAP18 seen as cytosolic puncta as with TIRF imaging (yellow arrows). However, with this imaging, the individual ARHGAP18 puncta appeared blurred, suggesting that the puncta size may still be below the resolving power of the technique. Therefore, to obtain additional information about the shapes and size of individual puncta, we performed imaging at 1600× magnification using GSD SRM and assigned pixels a size of 20 nm. After first optimizing the reconstruction of images for both the ARHGAP18 and α-tubulin labels in two-dimensional (2D) GSD (and antibody controls shown in Supplemental Figure S3, A–G), we imaged two example separate fields of view with GSD—the periphery of an EC (Figure 2, A–E) and a complex MT network in the fringe of an EC (Supplemental Figure S4, A–E). The 2D GSD imaging resolved individual puncta dotted along the microtubules (Figure 2, D and E, white arrows, and Supplemental Figure S4E), although there is also a cytosolic fraction of ARHGAP18 (Figure 2, D and E, yellow arrows). In addition, the resolution gained by 2D GSD imaging revealed that ARHGAP18 associated with MT filaments had an oval or a flattened, elongated morphology that corresponded to the orientation of the MT filament. Most puncta were ∼80–160 nm in size. Different regions of ECs were imaged, such as MT-organizing centers and processes that consistently confirmed the association of ARHGAP18 with MTs (Supplemental Figure S4, F and G). Line scan fluorescence analysis of the ARHGAP18 channel along MT “tracks” further confirmed the regularly spaced distribution of ARHGAP18 puncta along MTs in the periphery of ECs, with each peak correlating to a single punctum (Figure 2, F–I). In confluent cells imaged with a different combination of antibodies to the same targets, ARHGAP18 was also localized to the MT (Figure 2, J–M). Thus ARHGAP18 puncta are regularly spaced along microtubules, with no apparent preferential distribution to minus or plus ends.
FIGURE 2:

Two-dimensional SRM revealed that ARHGAP18 puncta are regularly localized along MTs. GSD imaging of the periphery of an EC using the markers (A) α-tubulin–Alexa 647 and (B) ARHGAP18–Alexa 488; wide-field view is shown at the top right. (C) Merge of A and B. By removing out-of-focus light and reconstructing at high magnification, a significant improvement in the resolution of the specific labeling for each marker is observed. (D, E) Blue and yellow boxes indicate areas that are magnified and shown in D and E, respectively. Examples of ARHGAP18 puncta aligned in the orientation of the MTs are indicated by white arrows, and examples of ARHGAP18 puncta that are cytosolic are indicated by yellow arrowheads. Scale bar, 2.5 μm (C), 1.25 μm (E). (F–I) ARHGAP18 tracks along MTs as imaged by ImageJ software (same image as that C). Two single MTs (tracks indicated by yellow line) on which ARHGAP18 puncta are localized were analyzed in ImageJ, and line scans of fluorescence intensity along the tracks in the ARHGAP18 channel were generated (G and I, respectively). Data were acquired starting from the end of the MT closest to the minus end (–) and proceeding along the MT to the plus end (+). (J–M) GSD imaging of confluent ECs revealed a similar regular staining pattern at the cell–cell boundary. The boundary between two adjacent cells was imaged (wide-field ARHGAP18-488 image in J; white box is the area imaged in K–M). GSD of ARHGAP18 primary and Alexa 488 secondary antibodies and tubulin–Cy3 direct conjugate antibody are shown in K and L, respectively, with the wide-field images shown at the top right. Merged image is shown in M. The approximate cell boundary between the two cells is indicated by the dashed gray line. The contents of top yellow and blue boxes are shown zoomed in respective larger boxes to demonstrate the regular presence of SENEX puncta along the microtubules as seen in spread ECs. Scale bar, 2.5 μm (M).
Three-dimensional (3D) GSD imaging (Figure 3, A–E), followed by 3D reconstruction in Imaris software, confirmed that ARHGAP18 puncta are associated with MTs. The colocalization of ARHGAP18 with α-tubulin was confirmed biochemically by coimmunoprecipitation (coIP; Figure 3, F and G). Consistent with the reported interaction between α-tubulin and actin (Lee and Gotlieb, 2003; Nagae et al., 2013), the α-tubulin IP yielded a strong actin band.
FIGURE 3:
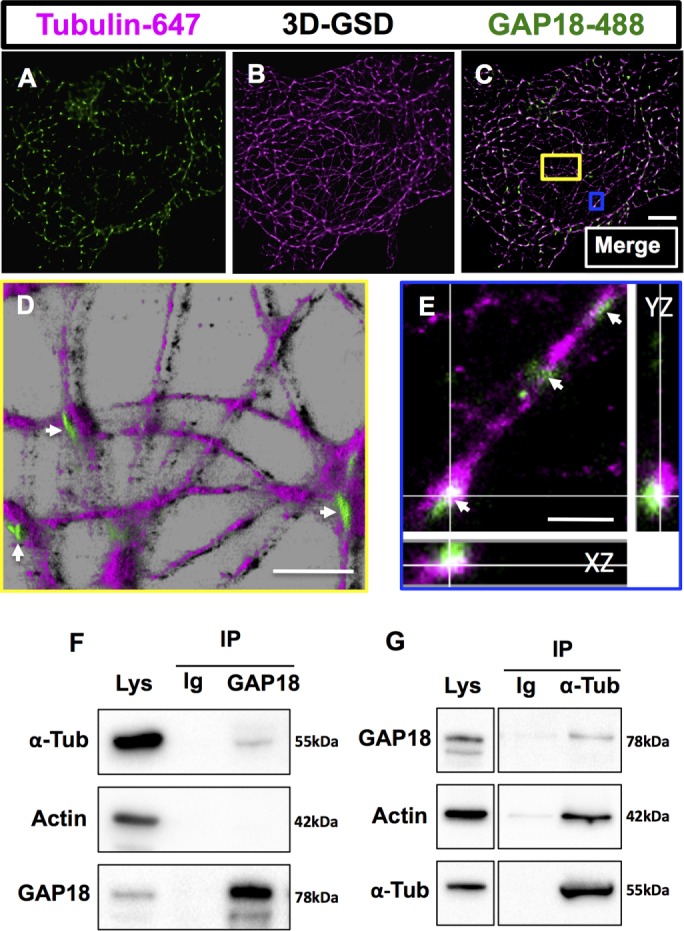
ARHGAP18 puncta are localized along MTs as confirmed by 3D GSD. SRM and coIP. The 3D GSD of an EC reveals that ARHGAP18 puncta wrap around microtubules (z-slices = 16; field of view is 40 μm × 40 μm). Single-channel 3D GSD of (A) ARHGAP18–Alexa 488 direct conjugate and (B) tubulin–Alexa 647. (C) Merge of A and B. (D, E) Zoomed and reconstructed 3D segments (in Imaris software) of the areas indicated by the colored boxes in C. (D) ARHGAP18 puncta are seen on top of the MTs (white arrowheads) in a reconstruction in Imaris (blend mode). (E) ARHGAP18 puncta can be seen localized around the MT filaments (white arrowheads). XZ and YZ are z-projections in each axis at the cross-hair point indicated. Scale bar, 5 μm (C), 1.25 μm (D), 500 nm (E). Note that due to photobleaching and the extended imaging times required for 3D GSD microscopy, cytosolic ARHGAP18 puncta do not resolve well in 3D GSD mode, as not enough positive thresholded events are collected. (F, G) ARHGAP18, actin, and α-tubulin were detected with specific antibodies in EC whole-cell lysate (Lys). (F) The coIP of ARHGAP18 and tubulin, IP of ARHGAP18, and probing for α-tubulin. (G) The coIP of ARHGAP18 and tubulin, IP of α-tubulin, and probing for ARHGAP18. IgG (Ig) antibody used as a control in pull downs showed no bands.
Depletion of ARHGAP18 in ECs causes disruption of MT stability
Because RhoGTPases have previously been linked with control of MT stability (Cook et al., 1998), we investigated whether depletion of ARHGAP18 resulted in changes to MTs. ARHGAP18 was depleted using a mixture of two small interfering RNAs (siRNAs), reducing the endogenous expression of the protein by ∼80–90% (Chang et al., 2014; Supplemental Figure S5). Double immunostaining for α-tubulin and acetylated α-tubulin showed morphologically a pattern consistent with destabilized MTs, with a decrease in the area of acetylated-tubulin detected (Figure 4A). This was reflected in a significant decrease in the extent of coverage of acetylated tubulin as a percentage of total cellular tubulin (Figure 4, B and C) and coverage of tubulin network per total cell area (Figure 4D), as determined by ImageJ analysis of thresholded fluorescence intensity for acetylated α-tubulin (Ac-Tub) and α-tubulin per cell. ECs cultured from the lungs of ARHGAP18-knockout mice (Chang et al., 2014) and identified by von Willebrand factor (vWF)–positive staining showed a similar altered pattern of MT morphology (Figure 4E) and a decrease in Ac-Tub coverage (Figure 4, F–H).
FIGURE 4:

Depletion of ARHGAP18 in ECs decreases Ac-Tub (A–D) ECs were transfected with control siRNA (siC) or siRNA for ARHGAP18 (siGAP18). After 48 h, cells were fixed and immunostained. (A) Representative images stained for Ac-Tub (Alexa 488; green) and α-tubulin (Alexa 647; magenta; 630× magnification). (B) Quantitation of the Ac-Tub levels expressed as percentage coverage of α-tubulin in the same cell; data shown are averages of 30 cells from three separate EC lines. The method for assessment of tubulin coverage was based on that used in Birukova et al. (2004a). Coverage is determined by setting a minimum threshold of fluorescence intensity (to exclude background) and analyzing the percentage of thresholded acetylated α-tubulin area/thresholded α-tubulin area for each cell. This gives a normalized measure of the level of tubulin acetylation in each cell (expressed as a percentage); therefore differences in larger vs. smaller cells are accounted for, and differences between SiControl and SiARHGAP18 can be directly compared. (C) Normalization of averaged Ac-Tub data with siC set to 100%. (D) Quantitation of tubulin area as a percentage of total cell area (as assessed by the extent of the MT network) in the same cells. (E) Mouse lung ECs explanted from WT or ARHGAP18 KO mice, with ECs identified by punctate vWF immunostaining (Alexa 555; red). (F–H) Ac-Tub (green) and α-tubulin (magenta) were detected and quantified for coverage per the method described. Data are an averages of 35 (WT) and 29 cells (KO) from two separate extractions. *p < 0.05, **p < 0.01. Scale bar, 25 μm (A), 10 μm (E).
Tubulin posttranslational modifications are surrogate measures of microtubule stability. Both tubulin acetylation (Maruta et al., 1986) and detyrosination (Kumar and Flavin, 1981) are associated with stable microtubules (reviewed in Janke and Bulinski, 2011). Consistent with the immunofluorescence analysis indicating that ARHGAP18 depletion is associated with a loss of MT stability (Figure 4), depletion of ARHGAP18 resulted in a decrease in the level of Ac-Tub (ac) and detyrosinated (glu) tubulin protein (Figure 5A) in whole-cell lysates. Conversely, overexpression of ARHGAP18 in HUVECs resulted in an increase in Ac-Tub, consistent with MT stabilization (Figure 5B).
FIGURE 5:

Depletion of ARHGAP18 from ECs results in decreased MT stability. (A) ECs were transfected with control siRNA (siC; ■) or siRNA for ARGAP18 (siGAP18; □) and analyzed after 48 h for Ac-Tub or glu-tubulin expression. Western blot is representative of a single HUVEC line. Densitometry analysis of protein expression normalized to untreated control. Results are ± SEM; three independent HUVEC lines in duplicate; *p < 0.05. (B) Analysis of ARHGAP18 in ARHGAP18-overexpressing cells. Overexpression was achieved by infecting HUVECs with ARHGAP18-containing adenovirus. After 48 h, the lysates were analyzed for Ac-Tub expression. Western blot is representative of a single HUVEC line. Densitometry analysis of protein expression normalized to untreated control. Results are ± SEM; three independent HUVEC lines in duplicate; *p < 0.05. (C) ECs were transfected with control siRNA (siC) or siRNA for ARGAP18 (siGAP18), lysed after 48 h, and soluble and polymerized tubulin fractions were separated as given in Materials and Methods. Fractions were then analyzed by Western blotting and probed for ARHGAP18 and α-tubulin. (i) Representative Western blot and comparative densitometry levels of ARHGAP18 in soluble and polymerized fractions from control or ARHGAP18-depleted cells. Densitometry readings from three independent experiments. The levels of ARHGAP18 in the siC cells in both the soluble and polymerized fractions are normalized to 1.0. Representative of three independent experiments. P, polymerized tubulin fraction; S, soluble tubulin fraction. (ii) Percentage of polymerized tubulin. Mean ± SEM, *p < 0.05.
To measure directly the shift in microtubule stability upon ARHGAP18 suppression, we separated soluble and polymerized tubulin fractions in control and ARHGAP18-depleted HUVECs. Consistent with observations on ARHGAP18 localization in HUVECS, a large proportion of ARHGAP18 expression was localized to the soluble fraction, whereas a small amount of ARHGAP18 was visible in the polymerized fraction (Figure 5C). This approach further confirmed that depletion of ARHGAP18 reduced the proportion of tubulin present in its polymerized form (Figure 5C, i and ii), consistent with a reduction in microtubule stability. The effect of ARHGAP18 depletion in reducing microtubule stability was further confirmed in HUVECs pretreated with the microtubule-stabilizing agent paclitaxel to improve the detection of polymerized tubulin (Supplemental Figure S6). ARHGAP18 depletion resulted in loss of the protein from both the soluble and polymerized fractions (Figure 5C and Supplemental Figure S6). Taken together, the data indicate that ARHGAP18 expression is required for the maintenance of stable MTs.
ARHGAP18 depletion affects the function of ECs
WPB structures reside on the MTs and use MTs for exocytosis of their cargo proteins (Sinha and Wagner, 1987; Ozaka et al., 1997; Kayashima et al., 1999). An EC on the periphery of the cultures showing a proliferative elongated morphology has been imaged, as the morphology allows ease of imaging (Figure 6, A–C). With confocal microscopy, some but not all ARHGAP18 puncta also colocalize with WPBs on the MTs.
FIGURE 6:

ARHGAP18 colocalizes with vWF+ WPBs and contributes to regulation of cargo release. (A) EC stained with ARHGAP18 (green) and tubulin antibodies (red) and imaged with confocal microscopy (630× magnification). White box indicates region zoomed in B. (B) Extended process of an EC; tubulin staining (Alexa 647; red), ARHGAP18 (Alexa 488; green), and ARHGAP18 puncta shown along MTs in Merge. Images are rendered in Imaris software (shadow projection mode). vWF immunopositivity is shown in cyan (Alexa 594). All Merge, ARHGAP18, tubulin, and vWF channels merged together. Yellow box indicates region of interest zoomed in C to show colocalization of ARHGAP18 (green) with vWF (cyan) in a WPB attached to the MT filament. Smaller ARHGAP18 puncta (as observed in GSD imaging) are seen to the right of the WPB (white arrowhead). XZ- and YZ-projections at the cross-hair point indicate colocalization of immunopositivity and the WPB localization to the MT. Scale bar, 30 μm (A), 10 μm (B), 1.5 μm (C). (D) ECs were transfected with control siRNA (siC; ■) or siRNA for ARHGAP18 (siGAP18; □) and were stimulated after 48 h with PMA (+) for 30 min. The supernatant was collected and analyzed for expression of soluble endothelin-1 by ELISA. Results are mean ± SEM of four individual HUVEC lines, *p < 0.05, **p < 0.01. (E) ECs were transfected with control siRNA (siControl; □) or siRNA for ARHGAP18 (siGAP18; ■) and were stimulated after 72 h with TNFα for 4 h. Neutrophils were allowed to transmigrate through ECs for 1 h and then collected; the number of neutrophils was determined and is expressed as a percentage. Results are mean ± SEM of triplicate determinations of each group from one experiment representative of three experiments performed; **p < 0.01.
The vasoconstriction agent endothelin-1 (ET-1) depends on MTs for its release (Malek et al., 1999), and, consistent with this, knockdown of ARHGAP18 resulted in a decrease in the amount of ET-1 released from ECs after stimulation with phorbol 12-myristate 13-acetate (PMA; Figure 6D). MTs have also been implicated in the process of leucocyte transendothelial cell migration (Mamdouh et al., 2008). Indeed, depletion of ARHGAP18 resulted in inhibition of neutrophil transmigration (74.2 ± 13.1% decrease in three experiments) through tumor necrosis factor α (TNFα)–activated EC monolayers (Figure 6E). Together these results are consistent with ARHGAP18 being involved in MT stability and MT-dependent functions in ECs.
ARHGAP18 stabilizes MTs through ROCK and HDAC6
ROCK, a downstream effector of Rho, regulates MT stability (Birukova et al., 2004c; Martins and Kolega, 2012). To determine whether the Rho-ROCK pathway is involved in ARHGAP18-mediated MT stability, we assessed whether the ROCK inhibitor Y-27632 was able to reverse the destabilized MT phenotype in cells depleted of ARHGAP18. The decrease in the level of Ac-Tub upon ARHGAP18 depletion was reversed by Y-27632 pretreatment, indicating ROCK involvement (Figure 7A). The activity of the deacetylase HDAC6 is an important negative regulator of acetylated levels of tubulin (Schofield et al., 2012). Inhibition of HDAC6 with the specific inhibitor tubacin also reversed the loss of Ac-Tub as a result of ARHGAP18 knockdown (Figure 7B).
FIGURE 7:

Changes in Ac-Tub by ARHGAP18 depletion are reversed by ROCK or HDAC6 inhibitor treatment. (A–D) ECs were transfected with control siRNA (siC) or siRNA against ARHGAP18 (siGAP18) for 48 h. (A) Cells were left untreated (–) or treated with ROCK inhibitor (Y-27632; +) for 1 h and then lysed. Protein expression of acetylated tubulin (Ac. tub) or β-actin was analyzed. Right, densitometry analysis of protein expression normalized to β-actin and untreated control. Results are ± SEM; five to seven independent HUVEC lines; *p < 0.05, **p < 0.01. (B) ECs were treated with HDAC6 tubulin deacetylase inhibitor (tubacin) for 30 min and then lysed. Protein expression of Ac-Tub or β-actin was analyzed. Densitometry analysis of protein expression from SiC (■) or SiGAP18 (□) cells normalized to β-actin and untreated control. Results are ± SEM; four independent HUVEC lines; *p < 0.05. (C) ECs were treated with thrombin for 10 min and then lysed. Protein expression of Ac. tub or β-actin was analyzed. Right, densitometry analysis of protein expression normalized to β-actin and untreated control. Results are ± SEM; five independent HUVEC isolates; **p < 0.01. (D) ECs were treated with thrombin for 10 min, lysed, separated into membrane and cytosolic fractions, and analyzed for ARHGAP18 expression. VE-cadherin was used as membrane control and GAPDH as cytosolic control. Representative Western blot shown with densitometry analysis of protein expression normalized to VE-cadherin or GAPDH. Results are ± SEM; three independent HUVEC lines; *p < 0.05, **p < 0.01, ****p < 0.0001.
In ECs, thrombin destabilizes MTs (Birukova et al., 2004a). Depletion of ARHGAP18 showed a significantly reduced level of Ac-Tub, and this amount was not significantly changed with thrombin treatment (Figure 7C), suggesting no additive effects of these pathways. Thrombin activation is known to induce the translocation of RhoGTPase to the plasma membrane, resulting in localized activation of Rho (Birukova et al., 2004b). We also previously showed by confocal imaging that ARHGAP18 translocates to the membrane after thrombin stimulation, consistent with the need for negative regulation of this event (Chang et al., 2014). Fractionation studies showed increased localization of ARHGAP18 to the plasma membrane fraction and little change in the cytoplasmic fraction after thrombin treatment (Figure 7D). Depletion of ARHGAP18 occurred to a similar extent in both the membrane and cytosolic fractions, and no change in localization was seen after thrombin stimulation. The integrity of the fractionations was confirmed by use of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a cytosolic and VE-cadherin as a membrane marker.
ARHGAP18 localization to MTs requires a functional RhoGAP domain
Localization of ARHGAP18 to MTs was not restricted to ECs, as ARHGAP18 was also localized to MTs in the human epithelial HeLa cell line (Supplemental Figure S7, A–E). These cells were also used to address whether localization of ARHGAP18 on MTs depends on GAP activity. Because green fluorescent protein (GFP) fluorescence is not strong and photobleaches quickly in superresolution microscopy, we used an anti-GFP camelid antibody conjugated to the ATTO488 dye to boost the fluorescence signal of GFP. GFP control showed a scattered, random localization with no immunopositivity along MTs (Figure 8, A and D). Transfection with wild-type (WT) ARHGAP18-GFP showed some localization to MTs and some to the cytosol, identical to that seen for endogenous ARHGAP18 (Figure 8, B and E). In contrast, full-length ARHGAP18 with a single amino acid mutation (R365A; Coleman et al., 2010) that abrogates GAP activity lacked binding to MTs, and the puncta appear scattered in the cytosol and oriented at different angles to the direction of adjacent MTs (Figure 8, C and F). HeLa cells have a much denser MT network than ECs. This made it necessary to image on the fringe of cells to obtain adequate separation of individual filaments. Our findings show a clear phenotype between the full-length (MT bound and cytosolic; broadly mimicking endogenous ARHGAP18 localization) and R365A (wholly cytosolic) constructs.
FIGURE 8:
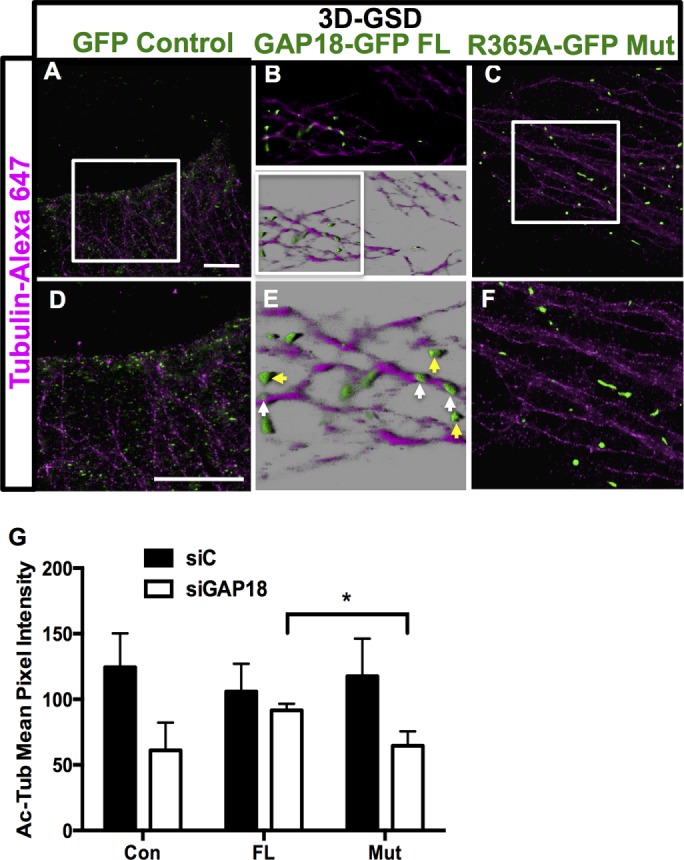
A functional RhoGAP domain in ARHGAP18 is required for MT localization and stability. GFP-ARHGAP18 or GFP-control plasmid was transfected into HeLa cells. (A) The 3D GSD of control plasmid; zoom of the white box is shown in D. (B) The 3D GSD imaging of full-length, wild-type ARHGAP18-GFP puncta in the fringe of a HeLa cell. Maximum intensity projection (top) and 3D reconstruction in Imaris software blend mode (bottom). (E) Zoom of white box in B. Individual ARHGAP18 puncta elongated along MTs are indicated by white arrows; cytosolic puncta are shown in yellow. (C) Lack of localization of ARHGAP18 R365A mutant constructs to MTs is observed. (F) Zoom of white box in C. Scale bar, 5 μm (A, D). (G) Rescue of the disrupted MT phenotype by functional ARHGAP18. ECs were transfected with siRNAs for control (■) or siARHGAP18 (□). At 24 h later, they were transfected with either GFP-control plasmids (Con), wild-type full-length GFP-ARHGAP18 (FL), or GFP-mutant ARHGAP18 (Mut). Eighteen hours later, cells were fixed and stained for Ac-Tub. Cells that were GFP positive, indicating transfection with the plasmids, were analyzed for the extent of Ac-Tub area and pixel intensity. Results from 60 GFP-positive cells from three separate experiments; *p < 0.05.
Consistent with the GAP region being essential for localization to MTs, we show, using a rescue experiment, that the GAP activity is necessary for MT stability. ARHGAP18 was depleted in ECs using an siRNA directed to the 3′ untranslated region (UTR) of ARHGAP18, thus targeting the endogenous ARHGAP18 and not the transfected ARHGAP18. Similar levels of depletion were obtained with this siRNA as for our previous siRNAs (unpublished data). The cells were then transfected with control, full-length WT, or R365A mutant ARHGAP18 constructs, which were GFP tagged. Cells were assessed 24 h later by confocal imaging for both the morphology and level of Ac-Tub. Green cells only were analyzed, as these contained the GFP constructs. We assessed the area and pixel intensity of the Ac-Tub in these cells as for Figure 4. The results in Figure 8G show that expression of the full-length WT constructs in the ARHGAP18-depleted cells reversed the Ac-Tub phenotype induced by ARHGAP18 depletion. In contrast, expression of the mutant ARHGAP18 construct failed to rescue the Ac-Tub phenotype in the ARHGAP18-depleted cells. Thus the GAP activity of ARHGAP18 is required for maintaining MT stability in ECs, as assessed by the level and morphology of Ac-Tub.
DISCUSSION
ARHGAP18 in ECs is important in the response to stimuli, controlling junctional stabilization and degree of angiogenesis (Chang et al., 2014) and is able to induce EC senescence when overexpressed (Coleman et al., 2013; Powter et al., 2014). These responses are all characterized by an alteration in cell shape mediated by cytoskeletal changes. We describe here the potential molecular basis for the cytoskeletal changes, with ARHGAP18 localizing to MTs and its expression being essential for MT stability.
Our results show ARHGAP18 is abundant within ECs in a number of different areas of the cell, including membrane ruffles, lamellipodia, and the cell membrane. The staining pattern is similar to that reported for other ARHGAPs in a variety of non-ECs (Foletta et al., 2002; Vogt et al., 2007; Barcellos et al., 2013; Lazarini et al., 2013). SRM techniques also showed that ARHGAP18 puncta were MT associated, and this was confirmed by biochemical analysis. To our knowledge, this is the first report of the use of SRM to localize RhoGAPs or GEFs, allowing individual puncta smaller than the diffraction limit to be visualized.
MTs are important in EC function, particularly inflammation and angiogenesis. They are altered by oxidative stress and inflammatory stimuli such as TNFα and thrombin and are implicated in the loss of barrier function in the lung (Petrache et al., 2003; Birukova et al., 2004b; Kratzer et al., 2012; Alieva et al., 2013). MTs are critical in inflammatory responses because they transport WPBs and their cargo for release after inflammatory stimulation (Sinha and Wagner, 1987), an intact MT network is needed for the migration of inflammatory cells through the EC barrier (Mamdouh et al., 2008), and they associate with and coordinate the subcellular sorting of VEGFR2 (Czeisler and Mikawa, 2013). Our data show that ARHGAP18 is important for MT stability in ECs. In other cell types, ARHGAP4 was localized to MTs in rat NRK epithelial cells, 3T3 fibroblasts (Vogt et al., 2007), and the PC12 adrenal cell line (Foletta et al., 2002), in addition to ARHGAP21 coimmunoprecipitating with α-tubulin in the DU145 human prostate cancer cell line (Barcellos et al., 2013).
ECs depleted of ARHGAP18 by siRNA treatment and ECs from ARHGAP18-knockout (KO) mice showed a destabilized MT phenotype, as assessed by a reduction in the proportion of tubulin in its polymerized form and in the levels of acetylated and detyrosinated tubulin. Furthermore, we show that the GAP activity is essential for localization of ARHGAP18 to MTs and for MT stability, as the mutant form of ARHGAP18 (R365A), which loses GAP activity, failed to localize to MTs and also failed to rescue the stability of ECs after ARHGAP18 knockdown (Figure 8). However, there are no known MT-binding consensus domains within ARHGAP18. At this stage, it is not known whether ARHGAP18 binds directly via the GAP domain through a nonclassical consensus sequence, whether the binding requires the GAP activity but is mediated through another, uncharacterized protein domain, or whether the binding is indirect through an associated protein. Further detailed structure–function analysis of ARHGAP18 is thus required.
High-resolution imaging techniques revealed ARHGAP18 in multiple compartments in ECs in addition to the MTs. We have not delineated the activity and function of ARHGAP18 in these different compartments. However, the importance of ARHGAP18 and its association with MTs in stimulated EC function was demonstrated in two ways. First, depletion of ARHGAP18 resulted in a reduction in neutrophil transendothelial cell migration after stimulation with TNF, consistent with previous work showing that leukocyte diapedesis requires recycling of membrane at the lateral cell borders and depends on MT function (Mamdouh et al., 2008). Second, we showed that ARHGAP18 is required for the release of WPB contents, which are known to associate with MTs and require intact MTs for their regulated secretion (Sinha and Wagner, 1987). Cargoes of WPBs such as ET-1 are rapidly released upon stimulation, and depletion of ARHGAP18 impairs ET-1 release after stimulation. In this regard, it is interesting that a fraction of ARHGAP18 colocalizes with WPBs, as identified by imaging. Confirmation by biochemical means that ARHGAP18 can be a cargo protein of WPBs and the possibility that a specific pool of ARHGAP18 is localized to WPBs—for example, mature WPBs (Hannah et al., 2003; Bierings et al., 2012) or a pool that is involved in their differential direction of movement (Vinogradova et al., 2000)—remains to be determined.
Localization of GEF-H1 to MTs occurs through the binding to the dynein light chain protein Tctex-1 and this interaction, together with polymerized MTs, renders GEF-H1 inactive (Meiri et al., 2014). Activation by inflammatory stimuli such as lipopolysaccharide and oxidative stress results in changes to the stability of MTs, release of the RhoGEF from the MT, and activation of the GEF activity to subsequently activate Rho at the specific location (Meiri et al., 2012). Of interest is the observation that GEF-H1 can be activated through release from the MT, independent of MT disassembly (Meiri et al., 2014). Further, GEF-H1 activity promotes MT instability (Yoshimura and Miki, 2011). ARHGAP18 may act in a similar manner. Our previous studies showed that loss of ARHGAP18 resulted in failure to form stable cell–cell junctions (Chang et al., 2014), a process known to be regulated through RhoGTPases and through the MT–actin cytoskeleton. Indeed, ARHGAP18 is translocated to the membrane fraction after thrombin stimulation, as shown here and as we showed previously, with ARHGAP18 transiently in the membrane within 10 min of stimulation (Chang et al., 2014). These results are consistent with its localization to areas where suppression of Rho activity would be required. MTs may act to convey ARHGAP18 to areas of the plasma membrane, where it functions to limit localized Rho activation. These sites are also sites of MT stabilization, and indeed Rho activity regulates MT stabilization (Wojnacki et al., 2014). Further, ROCK-Rho-HDAC activity is also crucial for MT stability and dynamics (Saito et al., 2011; Schofield et al., 2012, 2013). Thus, taken together with our data showing translocation of ARHGAP18 to the membrane after thrombin stimulation, the loss of ARHGAP18 (and rescue by ROCK and HDAC inhibition) affects MT stability, and lack of stable junctions, suggests that ARHGAP18 is critical to maintaining MT integrity and limiting localized Rho activation.
Porazinski et al. (2015) showed a role for ARHGAP18 in the determination of vertebrate body shape through actomyosin. In ECs, ARHGAP18 has important roles in processes that also involve changes in cell shape, such as angiogenesis and senescence (Coleman et al., 2010; Chang et al., 2014). In addition, it undergoes spatial movement to regions of Rho signaling, such as the plasma membrane, in response to inflammatory and angiogenic signals. Our evidence showing that it localizes to MTs gives insight into its mechanism of action in regulating cell shape because the MT and actin cytoskeletons are critical in this process. In ECs, ARHGAP18 also contributes to the regulation of exocytosis of WPBs and their cargo and to transmigration of neutrophils. Thus ARHGAP18 is likely to be a control site for inflammation. Consistent with this, the senescence induced by ARHGAP18 overexpression is predominantly an anti-inflammatory phenotype (Coleman et al., 2010), and depletion of ARHGAP18 results in impaired junctional stability of vessels and promotion of angiogenesis and permeability (Chang et al., 2014).
MATERIALS AND METHODS
HUVEC cell culture, transfection, and preparation for confocal and superresolution imaging
Unless otherwise indicated, all chemical reagents were obtained from Sigma-Aldrich. Umbilical cords were obtained from Royal Prince Alfred Hospital and used under its human ethics approval (X11-0211 and HREC/11/RPAH/311) and in accordance with the Declaration of Helsinki. HUVECs (passages 2 and 3) were grown as detailed previously (Coleman et al., 2010) before passaging onto coverslips or Lab-Tek 8 Chamber Slides (Thermo Fischer Scientific) for immunostaining (Lovelace et al., 2015). The cells were seeded at 3 × 104 (chamber slides), 4 × 104 (for 13 mm), or 8 × 104 cells (18 mm) for fibronectin-coated, round, no. 1.5 thickness high-precision coverslips (Marienfeld, Germany) and placed individually in 24- or 12-well culture plates. The cells were allowed to attach overnight before fixation with methanol:acetone (1:1) or methanol only (for tubulin-Cy3 staining) for 20 min on ice.
Culture of mouse primary lung endothelial cells
ECs were isolated from lungs of WT and ARHGAP18-deficient mice using a published method (Fehrenbach et al., 2009), Briefly, after being killed with CO2, mice were perfused with cold phosphate-buffered saline (PBS) through the left ventricle to flush out red blood cells. A 1- ml amount of collagenase A (1.0 mg/ml) was instilled through the trachea to inflate the lungs, and the trachea was then tied off. The lungs were incubated with 5 ml of collagenase A for 1 h at 37°C. PBS was added to collect the digested cells, which were then filtered through a 70-μm strainer (BD Biosciences). The suspension was spun at 700 × g for 10 min at 4°C and washed once. Cells were resuspended in RPMI growth medium (Life Technologies) supplemented with 10% Hyclone fetal bovine serum (FBS; Thermo Scientific), glutamine, 200 U/ml gentamicin, 100 U/ml penicillin, and 100 μg/ml streptomycin and cultured at 37°C and 5% CO2.
siRNA knockdown of ARHGAP18 and adenoviral overexpression in HUVECs
HUVECs were transfected with stealth siRNA control (low GC content; 5 nM) or two stealth siRNAs (HSS132562, HSS190252; 5 nM; Life Technologies) using Lipofectamine RNAiMAX (Life Technologies) as previously described (Chang et al., 2014). For overexpression of ARHGAP18 in HUVECs, an adenovirus encoding ARHGAP18 with a separate GFP reporter was used. Cells were routinely used at 48–72 h after siRNA knockdown and adenoviral overexpression (Coleman et al., 2010, 2013; Powter et al., 2014).
Generation of ARHGAP18 plasmid constructs
pcDNA3GFP-ARHGAP18, and pcDNA3GFP-ARHGAP18(R365A) were described previously (Coleman et al., 2010).
Transfection of HeLa cells with ARHGAP18 constructs
HeLa cells were cultured in DMEM (Life Technologies) supplemented with 10% FBS. Cells were transfected with plasmids encoding full-length or truncated ARHGAP18 using X-tremeGENE HP transfection reagent (Roche Applied Science, Germany) according to the manufacturer’s instructions. Transfection was performed in DMEM. After overnight incubation, the cells were fixed with formaldehyde or methanol/acetone 1:1 (15 min on ice) and further immunostained.
ARHGAP18 protein coIP assay and Western blotting for α-tubulin
HUVECs were lysed with ice-cold IP lysis buffer (0.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate [wt/vol], 1 mM EDTA, 150 mM NaCl, 10 mM NaH2PO4, pH 7.2) containing protease inhibitor cocktail (Sigma-Aldrich) and phosphatase inhibitor (PhosSTOP; Roche). The lysates were precleared with protein G–Sepharose (Life Technologies) and then incubated with mouse monoclonal anti-ARHGAP18 antibody (clone 2A3, in-house), anti–α-tubulin or control immunoglobulin G (IgG; Cell Signaling Technology) for 16 h at 4°C. The bound complexes were captured with protein G–Sepharose for 2 h at 4°C and then washed five times with IP buffer and eluted with LDS sample buffer and reducing agent (Life Technologies) at 70°C for 10 min.
HUVEC lysates for Western blotting were prepared in ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, with 1% NP-40, 150 mM NaCl, 2 mM EDTA, 100 mM NaF, 10 mM sodium pyrophosphate, protease inhibitor cocktail [Sigma-Aldrich], and phosphatase inhibitor [Roche]). The sample buffer contained lithium dodecyl sulfate. HUVEC lysates for Western blotting were prepared in ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, with 1% NP-40, 150 mM NaCl, 2 mM EDTA, 100 mM NaF, 10 mM sodium pyrophosphate, protease inhibitor cocktail [Sigma-Aldrich], and phosphatase inhibitor [Roche]). Total protein was measured using a BCA Kit (Thermo Scientific) and diluted in 4× Laemmli sample buffer (250 mM Tris-HCl, pH 6.8, 40% glycerol, 5% SDS, 0.005% bromophenol blue, and 0.1% 2-β-mercaptoethanol). After heating at 90°C for 10 min, equal amounts of protein were loaded onto 4–15% Mini-PROTEAN TGX Precast Gel (Bio-Rad) or 4–12% NuPAGE gels (Life Technologies), separated by SDS–PAGE in 1× SDS–PAGE buffer (25 mM Tris base, 192 mM glycine, and 0.1% SDS), transferred to a polyvinylidene difluoride, membrane, and blocked with 5% skim milk powder and 0.1% Tween-20 in PBS. The membranes were incubated with primary antibodies overnight, and after washing, membranes were incubated with corresponding secondary goat anti-rabbit or anti-mouse antibody conjugated to horseradish peroxidase (HRP; Cell Signaling Technology) and reactive bands detected by chemiluminescence (ECL Western Blotting Detection Reagents; Thermo Fisher Scientific) on the ChemiDoc MP gel imaging system (Bio-Rad). Primary antibodies used for Western blotting include custom polyclonal rabbit anti-ARHGAP18 or mouse monoclonal clone 2A3 (in-house), rabbit anti–α-tubulin (Cell Signaling Technology), mouse anti–α-tubulin, mouse anti–acetylated tubulin (Sigma-Aldrich), mouse anti–tyrosinated tubulin and HRP-conjugated anti–β-actin (Abcam).
Tubulin polymerization assay in HUVECs
Soluble and polymerized tubulins were separated as described (Kavallaris et al., 2001). Briefly, 48 h posttransfection, HUVEC cells treated were washed twice with warm (37°C) PBS and lysed in 100 μl of hypotonic buffer (1 mM MgCl2, 2 mM ethylene glycol tetraacetic acid [EGTA], 0.5% NP-40, 1% protease inhibitor cocktail, 1 μg/ml paclitaxel, and 20 mM Tric-Cl, pH 6.8) for 10 min at 37°C in the dark. Cells were then scraped from the wells and transferred to Eppendorf tubes. Each well was then rinsed with a further 100 μl of hypotonic buffer to ensure that all cellular components were collected. The lysates were centrifuged at 14,300 × g for 10 min at room temperature. Supernatant containing the soluble tubulin was transferred to a separate Eppendorf tube, and both tubes were subsequently kept on ice. The polymerized tubulin fraction was resuspended in 200 μl of hypotonic buffer by gentle agitation. Both the soluble and polymerized tubulin fractions were mixed with 1× sample buffer (10% glycerol, 5% 2-mercaptoethanol, 2% SDS, 0.05% bromophenol blue, and 0.0625 M Tris HCl, pH 6.8) as per Minotti et al. (1991) and gently sonicated on ice using an ultrasonic homogenizer (Misonix) until the polymerized fraction was completely resuspended. The fractions were then boiled at 100°C for 10 min, and equal volumes of each fraction were then processed by Western blotting. Western blot membranes were probed for α-tubulin (DM1A clone, Sigma-Aldrich; primary antibody, 1:10,000 in 0.1% skim milk/Tris-buffered saline/Tween 20 [TBST] overnight; secondary antibody, 1:10,000 goat anti-mouse HRP-conjugated antibody in 0.1% skim milk/TBST for 1 h at room temperature) and ARHGAP18 (primary antibody, 1:10,000 or 1:1000 in 5% BSA/TBST; secondary antibody, 1:10,000 or 1:1,000 anti-mouse HRP-conjugated antibody in 5% BSA/TBST for 1 h at room temperature) and visualized using Pierce ECL Prime and x-ray development. Western blot densitometry was performed using ImageJ. For paclitaxel-pretreated cells, HUVECs were treated with paclitaxel (2.48 μM) in complete medium for 4 h at 37°C and 5% CO2 before harvesting in hypotonic buffer not containing any additional paclitaxel (1 mM MgCl2, 2 mM EGTA, 0.5% NP-40, 1% protease inhibitor cocktail, and 20 mM Tris-Cl, pH 6.8). Samples were then processed as described.
EC fractionation protocol
Cell fractionation was performed using a FractionPREP Cell Fractionation Kit (BioVision) according to the manufacturer’s protocol. In brief, ECs were lysed in cytosol extraction buffer. Cells were centrifuged and the supernatant collected as the cytosolic fraction. The pellet was then resuspended in membrane extraction buffer and the supernatant collected as the membrane/particulate fraction. The pellet was resuspended in 0.2% SDS with 10 mM dithiothreitol as the cytoskeletal fraction. Total protein was measured using the BCA Kit (Thermo Scientific) and diluted in 4× Laemmli sample buffer, and equal amounts of protein were subject to Western blotting with SDS–PAGE running buffer.
Cell treatments
Thrombin (1 U/ml; Sigma-Aldrich) was added to ECs for 10 min before cells were fixed for imaging or lysed for protein extraction and Western blotting. For phenotype-rescue experiments, preincubations with tubacin (0.5 μM, 15 min; Sigma-Aldrich) and ROCK inhibitor (Y-27632; 1 U/ml, 1 h) were performed. Nocodazole (Sigma-Aldrich) was added to ECs for 30 min before harvest.
Immunocytochemical staining and mounting
Antibodies used for immunostaining included α-tubulin (mouse monoclonal IgG1; 1:200), β-tubulin–Cy3 direct conjugate (1:25; both from Sigma-Aldrich), acetylated α-tubulin (mouse monoclonal IgG2a; 1:200; Abcam or Sigma-Aldrich), goat anti-vWF antibody (1:200; Santa Cruz Biotechnology), and custom-produced mouse anti-human ARHGAP18 antibodies (1:200; clone 2A3; Chang et al., 2014; immunogen for both is the full-length human ARHGAP18 protein). The 2A3 antibody was directly conjugated to Alexa 488 using an antibody labeling kit (Life Technologies). Blocking peptide for human ARHGAP18 was obtained from Santa Cruz Biotechnology. Alexa Fluor–conjugated secondary antibodies were obtained from Life Technologies and used at 1:200 dilution. An anti-GFP camelid antibody directly conjugated to the ATTO488 dye (GFP booster, 1:200; Chromotek, Martinsried, Germany) was used to amplify the signal from the GFP fluorophore.
Methanol/acetone-fixed coverslips were permeabilized for 10 min with 0.1% Triton X-100/PBS before blocking (3% BSA, 30 min). Formaldehyde-fixed coverslips were permeabilized for 10 min with 0.5% Triton X-100/PBS before blocking. Primary antibodies were incubated overnight at 4°C, washed, and then incubated with secondary antibodies for 2 h at room temperature. Once ready for imaging, coverslips were mounted in Vectashield fluorescent mounting media (Vector Labs) before imaging. Coverslips for GSD/TIRF imaging were mounted inverted over a depression slide (Proscitech, Australia), which contained 70 μl of 100 mM β-ethylamine (MEA; Sigma-Aldrich) dissolved 0.1× PBS, pH 7.4, as a mounting medium, or, for tubulin–Cy3 imaging, β-mercaptoethanol was used as recommended (Dempsey et al., 2011). The edges of the coverslip were sealed with a binary silicone sealing agent (Twinsil, Picodent, Germany) and then allowed to dry before imaging.
Wide-field and confocal microscopy
Wide-field images were acquired using AxioVision software with an Axiovert deconvolution microscope (Carl Zeiss, Germany) at the Bosch Institute Advanced Microscopy Facility (University of Sydney) or LAS AF software with a DM6000 upright microscope (Leica Microsystems, Germany). Confocal images and Z-stacks were acquired with LAS AF software on an SP5 confocal microscope (Leica) equipped with HyD hybrid detectors. A pinhole size of 1 Airy unit was used for all images, with a 0.13-μm Z-stack slice thickness (Nyquist sampling optimized).
TIRF imaging of cultured HUVECs
Coverslips were sealed with Twinsil. LAS AF software was used for image acquisition, and consistent acquisition settings in both wide-field and TIRF imaging modalities were used between different fields of view and different penetration depths. Samples were imaged at 1000× total magnification on the GSD microscope, with the 488-nm laser used to excite Alexa 488 and the 642-nm laser used for Alexa 647.
SIM SRM
SIM images were captured using the Nikon Ti-E PFS inverted microscope (Nikon) equipped with 488- and 642-nm lasers (for ARHGAP18–Alexa 488 and tubulin–Alexa 647, respectively), and Nikon NIS Elements software at 1000× total magnification. Wide-field images were captured along with superresolution images for comparison to the staining pattern observed.
Two- and three-dimensional GSD SRM
GSD imaging was performed on a Leica inverted DM16000 microscope equipped with 1.5 numerical aperture/100× oil objective and 405-, 488- (for ARHGAP18–Alexa 488 excitation), 532- (used for tubulin–Cy3), and 642-nm (for tubulin–Alexa 647 excitation) laser lines. A DU897 iXon electron-multiplying charge-coupled device camera (Andor Technology, Northern Ireland) was used for high-sensitivity image acquisition at ∼90 Hz and 512 × 512 pixel resolution at 1600× magnification. The 3D reconstruction of slices acquired on the 3D GSD microscope was performed using Imaris software, version 7.6.3 (Bitplane).
Generation of MT line scans
Images of ARHGAP18 staining were converted to grayscale 8-bit images in Adobe Photoshop CS 3 software before importing into ImageJ software 1.48 (National Institutes of Health, Bethesda, MD). Segmented lines were then drawn through the center of the ARHGAP18 puncta, mimicking the exact path taken by the microtubule filament. Line scan data were generated using the plot profile tool, data exported, and graphed in GraphPad Prism 6 software (GraphPad, San Diego, CA).
Soluble endothelin-1 enzyme-linked immunosorbent assay
Culture medium was assayed for the presence of soluble endothelin-1 after a 30-min stimulation with PMA (20 ng/ml) using an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems) as per the manufacturer’s recommendations.
Neutrophil transmigration
HUVECs were transfected with stealth siRNA and then seeded into 8.0-μm Transwell filters (Corning, Corning, NY). Neutrophils were prepared from fresh blood donated by healthy donors. Blood was dextran sedimented, and cells were separated by Histopaque (Sigma-Aldrich) gradient centrifugation. Neutrophils were purified from the cell pellet with hypotonic lysis of the remaining red blood cells. HUVECs were treated with 5 ng/ml TNFα for 4 h; neutrophils were added, and after 60 min, the number of transmigrated neutrophils was determined by MTS assay.
Rescue experiment
HUVECs were transfected with 5 nM of either stealth siRNA control (low GC content) or siRNA targeting the 3′ UTR of ARHGAP18 designed by BLOCK-iT RNAi Designer (Life Technologies) using Lipofectamine RNAiMAX (Life Technologies) as previously described (Chang et al., 2014). After 24 h the HUVECs were transfected with plasmids encoding WT, R365A mutant ARHGAP18, or control plasmid (pcDNA3) using X-tremeGENE HP transfection reagent (Roche Applied Science) according to the manufacturer’s instructions. Transfection was performed in EBM2 medium. After overnight incubation, the cells were fixed with methanol/acetone 1:1 (15 min on ice) and immunostained.
Statistics
Statistical analysis was performed using GraphPad Prism version 6.0 for Macintosh (GraphPad, San Diego, CA). For direct comparison between two groups, statistical significance was calculated using a two-tailed t test unless otherwise stated. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are presented as mean ± SEM.
Supplementary Material
Acknowledgments
We thank Elena Zaporoshenko and Lutfun Khan for preparation of the HUVECs; Kristina Jahn and Adrian Smith for assistance with confocal and wide-field imaging experiments; and Tristen Tan (Coherent Scientific Australia) and Louise Cole (Core Facilities Manager, Bosch Institute Advanced Microscopy Facility, University of Sydney) for assistance with the SIM experiments. For the GSD superresolution experiments (including validation of z-drift), we acknowledge the Australian Microscopy and Microanalysis Research Facility, Australian Centre for Microscopy and Microanalysis, University of Sydney, and the scientific and technical assistance of Paul Xu, Minh Huynh, Pamela A. Young, Yingying Su, Jeffrey Henriquez, Eleanor Kable, and Filip Braet of the Facility. We also acknowledge the technical advice of Eric Chung, Amgad Habib, Tamara Straube, and Marko Lampe of Leica Microsystems for the GSD experiments. This work was funded by National Health and Medical Research Council Program Grant 571408 and National Heart Foundation G 10S 5140 grant. A.P.M. is supported by a National Health and Medical Research Council CJ Martin Postdoctoral Research Fellowship. J.R.G. holds the Wenkart Chair of the Endothelium at the Centenary Institute.
Abbreviations used:
- Ac or Ac-Tub
acetylated tubulin
- ARHGAP18
RhoGTPase-activating protein 18
- ECs
endothelial cells
- ET-1
endothelin-1
- FBS
fetal bovine serum
- Glu
detyrosinated tubulin
- GSD
ground-state depletion
- HDAC
histone deacetylase
- HUVECs
human umbilical vein endothelial cells
- MTOC
microtubule-organising center
- MTs
microtubules
- siC
siRNA control
- SiGAP18
siRNA ARHGAP18
- SIM
structured illumination microscopy
- TIRF
total internal reflection fluorescence
- vWF
von Willebrand factor
- WPB
Weibel-Palade body.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-05-0285) on March 1, 2017.
REFERENCES
- Akhshi TK, Wernike D, Piekny A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton. 2014;71:1–23. doi: 10.1002/cm.21150. [DOI] [PubMed] [Google Scholar]
- Alieva IB, Zemskov EA, Smurova KM, Kaverina IN, Verin AD. The leading role of microtubules in endothelial barrier dysfunction: disassembly of peripheral microtubules leaves behind the cytoskeletal reorganization. J Cell Biochem. 2013;114:2258–2272. doi: 10.1002/jcb.24575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslan JE, Phillips KG, Healy LD, Itakura A, Pang J, McCarty OJ. Histone deacetylase 6-mediated deacetylation of alpha-tubulin coordinates cytoskeletal and signaling events during platelet activation. Am J Physiol Cell Physiol. 2013;305:C1230–C1239. doi: 10.1152/ajpcell.00053.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos KSA, Bigarella CL, Wagner MV, Vieira KP, Lazarini M, Langford PR, Machado-Neto JA, Call SG, Staley DM, Chung JY, et al. ARHGAP21 protein, a new partner of α-tubulin involved in cell-cell adhesion formation and essential for epithelial-mesenchymal transition. J Biol Chem. 2013;288:2179–2189. doi: 10.1074/jbc.M112.432716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierings R, Hellen N, Kiskin N, Knipe L, Fonseca A-V, Patel B, Meli A, Rose M, Hannah MJ, Carter T. The interplay between the Rab27A effectors Slp4-a and MyRIP controls hormone-evoked Weibel-Palade body exocytosis. Blood. 2012;120:2757–2767. doi: 10.1182/blood-2012-05-429936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenfeld J, Nalbant P, Yoon S-H, Bokoch GM. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: is altered GEF-H1 activity a crucial determinant of disease pathogenesis. Trends Cell Biol. 2008;18:210–219. doi: 10.1016/j.tcb.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Birukov KG, Smurova K, Adyshev D, Kaibuchi K, Alieva I, Garcia JGN, Verin AD. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J. 2004a;18:1879–1890. doi: 10.1096/fj.04-2328com. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JGN, Verin AD. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004b;67:64–77. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Smurova K, Birukov KG, Usatyuk P, Liu F, Kaibuchi K, Ricks-Cord A, Natarajan V, Alieva I, Garcia JG, Verin AD. Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: role of Rho-dependent mechanisms. J Cell Physiol. 2004c;201:55–70. doi: 10.1002/jcp.20055. [DOI] [PubMed] [Google Scholar]
- Brandão MM, Silva-Brandão KL, Costa FF, Saad STO. Phylogenetic analysis of RhoGAP domain-containing proteins. Genomics Proteomics Bioinformatics. 2006;4:182–188. doi: 10.1016/S1672-0229(06)60031-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunstein M, Wicker K, Hérault K, Heintzmann R, Oheim M. Full-field dual-color 100-nm super-resolution imaging reveals organization and dynamics of mitochondrial and ER networks. Opt Express. 2013;21:26162–26173. doi: 10.1364/OE.21.026162. [DOI] [PubMed] [Google Scholar]
- Chang GH, Lay AJ, Ting KK, Zhao Y, Coleman PR, Powter EE, Formaz-Preston A, Jolly CJ, Bower NI, Hogan BM, et al. ARHGAP18: an endogenous inhibitor of angiogenesis, limiting tip formation and stabilizing junctions. Small GTPases. 2014;5:1–15. doi: 10.4161/21541248.2014.975002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y-C, Nalbant P, Birkenfeld J, Chang Z-F, Bokoch GM. GEF-H1 couples nocodazole-induced microtubule disassembly to cell contractility via RhoA. Mol Biol Cell. 2008;19:2147–2153. doi: 10.1091/mbc.E07-12-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman PR, Chang G, Hutas G, Grimshaw M, vadas MA, Gamble JR. Age‐associated stresses induce an anti‐inflammatory senescent phenotype in endothelial cells. Aging. 2013;5:1–12. doi: 10.18632/aging.100622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman PR, Hahn CN, Grimshaw M, Lu Y, Li X, Brautigan PJ, Beck K, Stocker R, vadas MA, Gamble JR. Stress-induced premature senescence mediated by a novel gene, SENEX, results in an anti-inflammatory phenotype in endothelial cells. Blood. 2010;116:4016–4024. doi: 10.1182/blood-2009-11-252700. [DOI] [PubMed] [Google Scholar]
- Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene. 2014;33:4021–4035. doi: 10.1038/onc.2013.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook TA, Nagasaki T, Gundersen GG. Rho guanosine triphosphatase mediates the selective stabilization of microtubules induced by lysophosphatidic acid. J Cell Biol. 1998;141:175–185. doi: 10.1083/jcb.141.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeisler C, Mikawa T. Microtubules coordinate VEGFR2 signaling and sorting. PLoS One. 2013;8:e75833. doi: 10.1371/journal.pone.0075833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrenbach ML, Cao G, Williams JT, Finklestein JM, Delisser HM. Isolation of murine lung endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;296:L1096–L1103. doi: 10.1152/ajplung.90613.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foletta VC, Brown FD, Young WS. Cloning of rat ARHGAP4/C1, a RhoGAP family member expressed in the nervous system that colocalizes with the Golgi complex and microtubules. Brain Res Mol Brain Res. 2002;107:65–79. doi: 10.1016/s0169-328x(02)00448-5. [DOI] [PubMed] [Google Scholar]
- Fölling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, Hell SW. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5:943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- Hannah MJ, Hume AN, Arribas M, Williams R, Hewlett LJ, Seabra MC, Cutler DF. Weibel-Palade bodies recruit Rab27 by a content-driven, maturation-dependent mechanism that is independent of cell type. J Cell Sci. 2003;116:3939–3948. doi: 10.1242/jcs.00711. [DOI] [PubMed] [Google Scholar]
- Hell SW, Kroug M. Ground-state-depletion fluorscence microscopy: a concept for breaking the diffraction resolution limit. Appl Phys B. 1995;60:495–497. [Google Scholar]
- Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol. 2011;12:773–786. doi: 10.1038/nrm3227. [DOI] [PubMed] [Google Scholar]
- Kavallaris M, Tait AS, Walsh BJ, He L, Horwitz SB, Norris MD, Haber M. Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Res. 2001;61:5803–5809. [PubMed] [Google Scholar]
- Kayashima K, Doi Y, Kudo H, Kiyonaga H, Fujimoto S. Effects of endothelin-1 on vasoactivity and its synthesis, storage, and acting sites in the rat superior mesenteric vasculature: an ultrastructural and immunocytochemical study. Med Electron Microsc. 1999;32:36–42. doi: 10.1007/s007950050006. [DOI] [PubMed] [Google Scholar]
- Kratzer E, Tian Y, Sarich N, Wu T, Meliton A, Leff A, Birukova AA. Oxidative stress contributes to lung injury and barrier dysfunction via microtubule destabilization. Am J Respir Cell Mol Biol. 2012;47:688–697. doi: 10.1165/rcmb.2012-0161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krendel M, Zenke FT, Bokoch GM. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol. 2002;4:294–301. doi: 10.1038/ncb773. [DOI] [PubMed] [Google Scholar]
- Kumar N, Flavin M. Preferential action of a brain detyrosinolating carboxypeptidase on polymerized tubulin. J Biol Chem. 1981;256:7678–7686. [PubMed] [Google Scholar]
- Lazarini M, Traina F, Machado-Neto JA, Barcellos KSA, Moreira YB, Brandão MM, Verjovski-Almeida S, Ridley AJ, Saad STO. ARHGAP21 is a RhoGAP for RhoA and RhoC with a role in proliferation and migration of prostate adenocarcinoma cells. Biochim Biophys Acta. 2013;1832:365–374. doi: 10.1016/j.bbadis.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Lee T-YJ, Gotlieb AI. Microfilaments and microtubules maintain endothelial integrity. Microsc Res Tech. 2003;60:115–127. doi: 10.1002/jemt.10250. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Manley S. Putting super-resolution fluorescence microscopy to work. Nat Methods. 2009;6:21–23. doi: 10.1038/nmeth.f.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovelace MD, Gu BJ, Eamegdool SS, Weible MW, 2nd, Wiley JS, Allen DG, Chan-Ling T. P2X7 receptors mediate innate phagocytosis by human neural precursor cells and neuroblasts. Stem Cells. 2015;33:526–541. doi: 10.1002/stem.1864. [DOI] [PubMed] [Google Scholar]
- Maeda M, Hasegawa H, Hyodo T, Ito S, Asano E, Yuang H, Funasaka K, Shimokata K, Hasegawa Y, Hamaguchi M, Senga T. ARHGAP18, a GTPase-activating protein for RhoA, controls cell shape, spreading, and motility. Mol Biol Cell. 2011;22:3840–3852. doi: 10.1091/mbc.E11-04-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek AM, Zhang J, Jiang J, Alper SL, Izumo S. Endothelin-1 gene suppression by shear stress: pharmacological evaluation of the role of tyrosine kinase, intracellular calcium, cytoskeleton, and mechanosensitive channels. J Mol Cell Cardiol. 1999;31:387–399. doi: 10.1006/jmcc.1998.0873. [DOI] [PubMed] [Google Scholar]
- Mamdouh Z, Kreitzer GE, Muller WA. Leukocyte transmigration requires kinesin-mediated microtubule-dependent membrane trafficking from the lateral border recycling compartment. J Exp Med. 2008;205:951–966. doi: 10.1084/jem.20072328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manneville J-B, Etienne-Manneville S, Skehel P, Carter T, Ogden D, Ferenczi M. Interaction of the actin cytoskeleton with microtubules regulates secretory organelle movement near the plasma membrane in human endothelial cells. J Cell Sci. 2003;116:3927–3938. doi: 10.1242/jcs.00672. [DOI] [PubMed] [Google Scholar]
- Martins GG, Kolega J. A role for microtubules in endothelial cell protrusion in three-dimensional matrices. Biol Cell. 2012;104:271–286. doi: 10.1111/boc.201100088. [DOI] [PubMed] [Google Scholar]
- Maruta H, Greer K, Rosenbaum JL. The acetylation of alpha-tubulin and its relationship to the assembly and disassembly of microtubules. J Cell Biol. 1986;103:571–579. doi: 10.1083/jcb.103.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiri D, Marshall CB, Greeve MA, Kim B, Balan M, Suarez F, Bakal C, Wu C, Larose J, Fine N, Ikura M, Rottapel R. Mechanistic insight into the microtubule and actin cytoskeleton coupling through dynein-dependent RhoGEF inhibition. Mol Cell. 2012;45:642–655. doi: 10.1016/j.molcel.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Meiri D, Marshall CB, Mokady D, Larose J, Mullin M, Gingras A-C, Ikura M, Rottapel R. Mechanistic insight into GPCR-mediated activation of the microtubule-associated RhoA exchange factor GEF-H1. Nat Commun. 2014;5:4857. doi: 10.1038/ncomms5857. [DOI] [PubMed] [Google Scholar]
- Minotti AM, Barlow SB, Cabral F. Resistance to antimitotic drugs in Chinese hamster ovary cells correlates with changes in the level of polymerized tubulin. J Biol Chem. 1991;266:3987–3994. [PubMed] [Google Scholar]
- Nagae S, Meng W, Takeichi M. Non-centrosomal microtubules regulate F-actin organization through the suppression of GEF-H1 activity. Genes Cells. 2013;18:387–396. doi: 10.1111/gtc.12044. [DOI] [PubMed] [Google Scholar]
- Neisch AL, Formstecher E, Fehon RG. Conundrum, an ARHGAP18 orthologue, regulates RhoA and proliferation through interactions with Moesin. Mol Biol Cell. 2013;24:1420–1433. doi: 10.1091/mbc.E12-11-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Rikitake Y, Oyama N, Yan G, Alcaide P, Liu PY, Wang H, Ahl D, Sawada N, Okamoto R, et al. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest. 2008;118:1632–1644. doi: 10.1172/JCI29226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaka T, Doi Y, Kayashima K, Fujimoto S. Weibel-Palade bodies as a storage site of calcitonin gene-related peptide and endothelin-1 in blood vessels of the rat carotid body. Anat Rec. 1997;247:388–394. doi: 10.1002/(SICI)1097-0185(199703)247:3<388::AID-AR10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Pertz O. Spatio-temporal Rho GTPase signaling—where are we now. J Cell Sci. 2010;123:1841–1850. doi: 10.1242/jcs.064345. [DOI] [PubMed] [Google Scholar]
- Petrache I, Birukova A, Ramirez SI, Garcia JGN, Verin AD. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol. 2003;28:574–581. doi: 10.1165/rcmb.2002-0075OC. [DOI] [PubMed] [Google Scholar]
- Porazinski S, Wang H, Asaoka Y, Behrndt M, Miyamoto T, Morita H, Hata S, Sasaki T, Krens SF, Osada Y, et al. YAP is essential for tissue tension to ensure vertebrate 3D body shape. Nature 521, 217–21. 2015 doi: 10.1038/nature14215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powter EE, Coleman PR, Tran MH, Lay AJ, Bertolino P, Parton RG, Vadas MA, Gamble JR. Caveolae control the anti-inflammatory phenotype of senescent endothelial cells. Aging Cell. 2014;14:102–111. doi: 10.1111/acel.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Li R, Zheng Y, Busch H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J Biol Chem. 1998;273:34954–34960. doi: 10.1074/jbc.273.52.34954. [DOI] [PubMed] [Google Scholar]
- Saito S, Lasky JA, Guo W, Nguyen H, Mai A, Danchuk S, Sullivan DE, Shan B. Pharmacological inhibition of HDAC6 attenuates endothelial barrier dysfunction induced by thrombin. Biochem Biophys Res Commun. 2011;408:630–634. doi: 10.1016/j.bbrc.2011.04.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield AV, Bernard O. Rho-associated coiled-coil kinase (ROCK) signaling and disease. Crit Rev Biochem Mol Biol. 2013;48:301–316. doi: 10.3109/10409238.2013.786671. [DOI] [PubMed] [Google Scholar]
- Schofield AV, Gamell C, Suryadinata R, Sarcevic B, Bernard O. Tubulin polymerization promoting protein 1 (Tppp1) phosphorylation by Rho-associated coiled-coil kinase (rock) and cyclin-dependent kinase 1 (Cdk1) inhibits microtubule dynamics to increase cell proliferation. J Biol Chem. 2013;288:7907–7917. doi: 10.1074/jbc.M112.441048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield AV, Steel R, Bernard O. Rho-associated coiled-coil kinase (ROCK) protein controls microtubule dynamics in a novel signaling pathway that regulates cell migration. J Biol Chem. 2012;287:43620–43629. doi: 10.1074/jbc.M112.394965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Wagner DD. Intact microtubules are necessary for complete processing, storage and regulated secretion of von Willebrand factor by endothelial cells. Eur J Cell Biol. 1987;43:377–383. [PubMed] [Google Scholar]
- Takesono A, Heasman SJ, Wojciak-Stothard B, Garg R, Ridley AJ. Microtubules regulate migratory polarity through Rho/ROCK signaling in T cells. PLoS One. 2010;5:e8774. doi: 10.1371/journal.pone.0008774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood. 2011;117:5033–5043. doi: 10.1182/blood-2010-09-267492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentijn KM, van Driel LF, Mourik MJ, Hendriks G-J, Arends TJ, Koster AJ, Valentijn JA. Multigranular exocytosis of Weibel-Palade bodies in vascular endothelial cells. Blood. 2010;116:1807–1816. doi: 10.1182/blood-2010-03-274209. [DOI] [PubMed] [Google Scholar]
- Vinogradova TM, Roudnik VE, Bystrevskaya VB, Smirnov VN. Centrosome-directed translocation of Weibel-Palade bodies is rapidly induced by thrombin, calyculin A, or cytochalasin B in human aortic endothelial cells. Cell Motil Cytoskeleton. 2000;47:141–153. doi: 10.1002/1097-0169(200010)47:2<141::AID-CM5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Vogt DL, Gray CD, Young WS 3rd, Orellana SA, Malouf AT. ARHGAP4 is a novel RhoGAP that mediates inhibition of cell motility and axon outgrowth. Mol Cell Neurosci. 2007;36:332–342. doi: 10.1016/j.mcn.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojnacki J, Quassollo G, Marzolo MP, Caceres A. Rho GTPases at the crossroad of signaling networks in mammals: impact of Rho-GTPases on microtubule organization and dynamics. Small GTPases. 2014;5:e28430. doi: 10.4161/sgtp.28430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura Y, Miki H. Dynamic regulation of GEF-H1 localization at microtubules by Par1b/MARK2. Biochem Biophys Res Commun. 2011;408:322–328. doi: 10.1016/j.bbrc.2011.04.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.