

The kinetics of sterol transport between the plasma membrane and the endocytic recycling compartment is measured using fluorescence microscopy. STARD4, a small, soluble sterol transport protein, is responsible for 25% of the total transport and 33% of nonvesicular transport. Elevated cholesterol dramatically increases sterol transport rate constants.
Abstract
Cholesterol is an essential constituent of membranes in mammalian cells. The plasma membrane and the endocytic recycling compartment (ERC) are both highly enriched in cholesterol. The abundance and distribution of cholesterol among organelles are tightly controlled by a combination of mechanisms involving vesicular and nonvesicular sterol transport processes. Using the fluorescent cholesterol analogue dehydroergosterol, we examined sterol transport between the plasma membrane and the ERC using fluorescence recovery after photobleaching and a novel sterol efflux assay. We found that sterol transport between these organelles in a U2OS cell line has a t1/2 =12–15 min. Approximately 70% of sterol transport is ATP independent and therefore is nonvesicular. Increasing cellular cholesterol levels dramatically increases bidirectional transport rate constants, but decreases in cholesterol levels have only a modest effect. A soluble sterol transport protein, STARD4, accounts for ∼25% of total sterol transport and ∼33% of nonvesicular sterol transport between the plasma membrane and ERC. This study shows that nonvesicular sterol transport mechanisms and STARD4 in particular account for a large fraction of sterol transport between the plasma membrane and the ERC.
INTRODUCTION
Cholesterol is an essential constituent of biological membranes. Structurally, cholesterol has a single hydroxyl as the sole polar component, four rings, and a short alkyl chain, whereas most other lipids, such as glycerophospholipids and sphingolipids, are composed of large polar head groups and two long hydrocarbon chains (van Meer, 2005; Iaea and Maxfield, 2015). The unique chemical structure of cholesterol allows it to be absorbed into and extracted from membranes easily (Phillips et al., 1987). Cholesterol substantially affects the biophysical properties of membranes because it is able to pack against the acyl chains of phospholipids, reduce membrane permeability, and facilitate the formation of liquid-ordered domains in membranes (Huang and Feigenson, 1999; Pani et al., 2008; Galan et al., 2010). The abundance and distribution of cholesterol within membranes are tightly controlled by complex homeostatic mechanisms (Mesmin and Maxfield, 2009; Iaea et al., 2014; Iaea and Maxfield, 2015).
Understanding the intracellular dynamics of cholesterol is critical because the proper distribution among the plasma membrane and cellular organelle membranes is important for cellular function (van Meer and Sprong, 2004; Maxfield and Menon, 2006; Simons and Gerl, 2010). Cholesterol distribution among cellular compartments and within the membrane is heterogeneous (Iaea and Maxfield, 2015). The plasma membrane is the largest cholesterol pool in the cell and contains ∼60% of total cellular cholesterol in cells such as fibroblasts (Liscum and Munn, 1999). Cholesterol can flip rapidly between two membrane leaflets in a bilayer, and it has been shown that the half-time is <1 s in erythrocytes (Lange et al., 1981; Steck et al., 2002). Cholesterol is also highly enriched in the endocytic recycling compartment (ERC), which can contain ∼30–35% of cellular cholesterol (Hao et al., 2002). In contrast, the endoplasmic reticulum (ER), which is the central organelle in maintaining cholesterol homeostasis, has relatively low cholesterol content (Maxfield and Menon, 2006; Mesmin and Maxfield, 2009; Iaea and Maxfield, 2015). This creates a paradigm in which cholesterol levels sensed in the ER reflect the cholesterol distribution in other organelles, such as the plasma membrane and endosomes, and so there must be a mechanism for rapid redistribution of cholesterol among these organelles (Iaea et al., 2014; Iaea and Maxfield, 2015).
Several lines of evidence indicate that nonvesicular transport plays an important role in maintaining the correct distribution of cholesterol among organelles (Hao et al., 2002; Maxfield and Wustner, 2002; Maxfield and Menon, 2006; Mesmin et al., 2011). Cholesterol can be incorporated into transport vesicles that carry membrane components from one organelle to another. However, this is not the sole pathway of sterol and lipid transport because newly synthesized sterols can be transported from the ER to the plasma membrane when vesicular trafficking is inhibited by either genetic or pharmacological intervention (Urbani and Simoni, 1990). This strongly suggests that a large fraction of cholesterol transport is mediated by nonvesicular, carrier-mediated transport mechanisms, but the mechanisms are poorly understood (Hao et al., 2002).
Several proteins are recognized as soluble sterol transfer proteins (STPs; Prinz, 2007; Beh et al., 2012; Iaea et al., 2014). STPs can directly interact with membrane compartments to extract sterol from one membrane and deliver it to another (Prinz, 2007; Beh et al., 2012; Clark, 2012). However, in many cases, the precise role and function of these proteins in cellular sterol homeostasis are undetermined (Mesmin and Maxfield, 2009; Iaea and Maxfield, 2015). Among STPs, the steroidogenic acute regulatory protein (StAR)–related lipid-transfer (START) domain (STARD) family is involved in several aspects of intracellular lipid trafficking (Soccio et al., 2002; Iaea et al., 2014). STARD4 has been implicated as an important sterol transport protein involved in maintaining cholesterol homeostasis (Soccio et al., 2005; Mesmin et al., 2011). Overexpression of STARD4 increases cholesteryl ester accumulation in lipid droplets in an acyl-CoA:cholesterol acyl-transferase (ACAT)–dependent manner (Rodriguez-Agudo et al., 2011; Garbarino et al., 2012), and its expression is controlled at the transcriptional level by sterols (Soccio et al., 2002).
Cholesterol transport can be studied by introduction of radioactive sterols or precursors into cells, but this method is indirect and requires stringent purification of organelles, as well as demonstration that cholesterol does not redistribute during the purification process (Frolov et al., 1996; Hao et al., 2002; Iaea and Maxfield, 2015). Alternatively, fluorescent sterols provide an opportunity for directly imaging sterol trafficking in live cells (Fischer et al., 1984; Maxfield and Wustner, 2012; Iaea and Maxfield, 2015). Unlike synthetic cholesterol derivatives that have large fluorophores that can influence their interaction with other membrane components, dehydroergosterol (DHE) is a naturally occurring fluorescent sterol whose chemical structure is similar to that of cholesterol (Maxfield and Wustner, 2012; Iaea and Maxfield, 2015). Many studies have shown that DHE is generally a faithful mimic of cholesterol in model and biological membranes (Mukherjee et al., 1998; Hao et al., 2002; Iaea et al., 2015).
Previous studies from our laboratory used DHE and fluorescence recovery after photobleaching (FRAP) to measure sterol delivery from the plasma membrane to the ERC (Hao et al., 2002). After photobleaching, the DHE recovery in the ERC was reported to have a half-time of ∼2–3 min (Hao et al., 2002; Garbarino et al., 2012). However, only 40–50% of the ERC fluorescence was recovered, for unknown reasons (Hao et al., 2002; Garbarino et al., 2012). The DHE delivered to the ERC was mainly from the plasma membrane, which is the largest pool of cellular cholesterol. The recovery rate of DHE to the ERC was not greatly affected by ATP depletion, indicating that the transport was mainly nonvesicular (Hao et al., 2002). In agreement with this hypothesis, the rate of sterol delivery to the ERC was increased due to increasing cellular cholesterol carriers, either by microinjection of methyl-β-cyclodextrin (MCD) or overexpression of STARD4 (Mesmin et al., 2011). Under both of these conditions, the relative distribution of sterol in the plasma membrane and ERC was not greatly altered, suggesting that these organelles are near equilibrium in their cholesterol content (Mesmin and Maxfield, 2009; Mesmin et al., 2011; Iaea and Maxfield, 2015).
The rate of sterol efflux from the ERC to the plasma membrane was previously studied using excess extracellular MCD (Hao et al., 2002). Of interest, this study showed that transport of DHE from the ERC to plasma membrane was inhibited by ATP depletion, indicating that vesicular transport out of, but not into, the ERC is a major contributor to sterol transport kinetics (Hao and Maxfield, 2000). Of note, the study also found that the entire cholesterol pool was mobile, but the rate of transfer from the ERC to the plasma membrane was nearly 10-fold slower than transfer to the ERC (Hao et al., 2002).
Although previous studies of sterol trafficking were insightful, they had several limitations and shortfalls. For instance, DHE is weakly fluorescent and is susceptible to photobleaching (Maxfield and Wustner, 2012). This limits the number of time points that can be sampled before photobleaching of the probe (Hao et al., 2002). In addition, in previous FRAP studies, incomplete recovery prevented accurate and complete modeling of sterol transport from the plasma membrane to the ERC (Hao et al., 2002; Mesmin et al., 2011; Garbarino et al., 2012). Finally, efflux studies were limited due to cellular toxicity when using high concentrations of MCD (Hao et al., 2002).
In this study, we report detailed kinetics of sterol transport between the plasma membrane and the ERC by addressing many of the limitations of previous studies. With access to sensitive imaging cameras, we imaged DHE-labeled living cells with low illumination, which minimizes photobleaching and allows repeated measurements for up to 1 h. Compared with previous studies (Hao et al., 2002; Mesmin et al., 2011; Garbarino et al., 2012), we redesigned the FRAP method to enable full recovery of DHE fluorescence. We implemented an efflux method that uses a large exogenous cholesterol pool with hydroxypropyl cyclodextrin (HPCD)–cholesterol complexes to rapidly exchange with DHE at the plasma membrane. These new methods allow us to obtain detailed kinetic information describing sterol transport between the plasma membrane and the ERC because for the first time sterol transport from the plasma membrane to the ERC and efflux from the ERC to the plasma membrane can be modeled as single-rate processes. For the first time, we report bidirectional sterol transport kinetics between the ERC and plasma membrane and show that STARD4 is critical for these processes.
RESULTS
Cholesterol transport between ERC and plasma membrane
To measure the rate of cholesterol transport between the ERC and the plasma membrane, we assessed the trafficking kinetics of the fluorescent sterol DHE (Fischer et al., 1984) in human osteosarcoma (U2OS) cells stably expressing the scavenger receptor A (U20S-SRA; Majumdar et al., 2011). These cell were used previously to study sterol transport (Mesmin et al., 2011) and allow cholesterol loading through SRA. To measure the bidirectional transport of DHE between the plasma membrane and the ERC, shown schematically in Figure 1, we used FRAP of the ERC and developed a novel DHE efflux assay to measure export from the ERC based on rapid exchange of DHE with cholesterol at the plasma membrane.
FIGURE 1:

Schematic of fluorescence method to study sterol transport between plasma membrane and ERC. U2OS-SRA cells are loaded with DHE complexed to MCD for 1 min. DHE is allowed to equilibrate among cellular membranes for 120 min. At steady state, DHE is enriched in the ERC and plasma membrane (yellow). The ERC is identified using fluorescently labeled Tf (red outline). (A) To monitor transport to the ERC, the ERC is photobleached, and the FRAP of DHE is recorded. (B) To monitor transport from the ERC, cellular medium is exchanged with medium containing red blood cells (RBCs) and HPCD–cholesterol complex. As fluorescent DHE is exchanged at the plasma membrane for nonfluorescent cholesterol, the fluorescence signal of the ERC decreases over time.
MCD–DHE complexes were prepared as described previously (Sheets et al., 1999). Cells were labeled for 1 min with 25 mM MCD–5 mM DHE complex. In contrast to previous studies (Hao et al., 2002; Mesmin et al., 2011), cells were chased for 2 h in chase medium (see Materials and Methods) to allow the sterol to equilibrate among membranes. An ACAT inhibitor, 30 µg/ml Sandoz 58–035, was used during this period to prevent esterification and ensure that the entire DHE pool remained mobile during the experiment. In the last 20 min, the cells were labeled with fluorescent transferrin to identify the ERC.
To measure the sterol transport from the plasma membrane to the ERC, we used FRAP and monitored the recovery of DHE fluorescence in the ERC. Images were analyzed as described previously (Hao et al., 2002; Mesmin et al., 2011; Garbarino et al., 2012), with minor alterations (see Materials and Methods for details). In addition to measuring the DHE fluorescence in ERC and the entire cell fluorescence, a region of the plasma membrane adjacent to the ERC that was photobleached was also analyzed to account for diffusion of DHE in the plasma membrane into the photobleached region containing both plasma membrane and ERC. Taking the plasma membrane recovery rate into account, we can accurately measure DHE fluorescence recovery in the ERC.
In control U2OS-SRA cells, photobleaching of DHE in the ERC resulted in ∼30% fluorescence being recovered in 10 min, with a half-time for full recovery (t1/2) of 14.5 ± 1.5 min (Figure 2, A and C, and Supplemental Figure S1). All kinetics results are summarized in Table 1. All of the kinetic studies in this article were modeled as single first-order rate processes with 100% recovery. In some cases, deviations from the fit to a single rate process can be seen, but we have not attempted to fit the data to more complex models. A composite set of control data from multiple experiments is shown in all kinetics graphs.
FIGURE 2:

Bidirectional sterol transport. For both FRAP and efflux experiments, U2OS-SRA cells were labeled with DHE for 1 min and equilibrated for 120 min. In the last 20 min, cells were incubated with 20 μg/ml Tf–Alexa 546 at 37°C in medium 2/glucose to identify the ERC. Cells were maintained at 37°C. (A) For FRAP recovery to the ERC, an image was taken before photobleaching. DHE in the ERC was photobleached (yellow arrow), and images were taken every 30 s. (B) FRAP measurements for control, LPDS, and cholesterol-overloaded cells. Control, t1/2 = 14.5 ± 1.5 min; LPDS, t1/2 = 10.4 ± 1.3 min; overloading, t1/2 = 3.8 ± 0.6 min. (C) To record the loss of DHE from the ERC, an image was taken before exchanging with efflux medium. Images were analyzed as described in Materials and Methods. Cells were maintained at 37°C. Scale bar, 10 μm. Where indicated, cellular cholesterol levels were decreased or increased by overnight incubation with LPDS medium or acute treatment with HPCD–cholesterol complex, respectively. (D) DHE efflux curves for control, LPDS, and cholesterol-overloaded cells. Control, t1/2 = 14.9 ± 0.9 min; LPDS, t1/2 = 14.3 ± 1.3 min; overloading, t1/2 = 5.8 ± 0.3 min. Each data point is derived from an average of at least eight experiments (±SE). Data are fitted to single-exponential curves.
TABLE 1:
Half-times (t1/2) of FRAP and efflux experiments.
| FRAP t1/2 (min ± SE) | R-square | Efflux t1/2 (min ± SE) | R-square | |
|---|---|---|---|---|
| Control, 10 min | 14.5 ± 1.5 | 0.85 | 14.9 ± 0.9 | 0.99 |
| Control, 30 min | 12.5 ± 3.4 | 0.99 | ||
| LPDS | 10.4 ± 1.3 | 0.99 | 14.3 ± 1.3 | 0.98 |
| Overload | 3.8 ± 0.6 | 0.98 | 5.8 ± 0.3 | 0.99 |
| Energy depletion | 21.2 ± 3.2 | 0.99 | 18.3 ± 1.1 | 0.98 |
| Energy depletion, 30 min | 24.2 ± 1.8 | 0.94 | ||
| HPCD | 1.3 ± 0.3 | 0.98 | 4.1 ± 0.4 | 0.99 |
| STARD4 | 4.3 ± 0.5 | 0.97 | 4.9 ± 0.6 | 0.99 |
| CRISPR SD4 KD | 22.4 ± 2.1 | 0.98 | 20.2 ± 0.4 | 0.96 |
| CRISPR SD4 + mC-FKBP-SD4 | 11.0 ± 1.6 | 0.98 | 12.5 ± 0.4 | 0.98 |
| CRISPR SD4 + mC-FKBP-SD4 + MitoTrap | 19.7 ± 1.5 | 0.96 | 19.1 ± 0.5 | 0.96 |
| CRISPR SD4 + W108D mC-FKBP-SD4 | 21.9 ± 1.5 | 0.96 | 19.0 ± 0.2 | 0.95 |
| CRISPR SD4 + energy depletion | 24.1 ± 1.3 | 0.99 | 24.8 ± 0.3 | 0.94 |
Half-times (t1/2) for FRAP and efflux experiments (±SE) were determined by fitting curves to a single exponential. The R-square of the fit was determined using GraphPad Prism software.
To determine the rate of cholesterol transport from the ERC to the plasma membrane, we incubated U2OS-SRA cells in exchange medium that contained ∼109 sheep red blood cells, which acts as an exogenous cholesterol pool, plus partially cholesterol loaded HPCD. This allows for rapid exchange of fluorescent DHE for nonfluorescent cholesterol at the plasma membrane, resulting in reduction of DHE fluorescence in the cell. Incubation of cells with exchange medium did not result in toxicity as monitored by propidium iodide staining (unpublished data) or alterations to cellular cholesterol levels (Supplemental Figure S2). Similar to the analysis of FRAP experiments, efflux images were analyzed by examining loss of DHE fluorescence from the ERC and an adjacent plasma membrane region. This analysis ensures that we measure the loss of fluorescence in the ERC and that the plasma membrane fluorescence is accounted for in the analysis.
After placement of cells into exchange medium, DHE in the plasma membrane is exchanged with nonfluorescent cholesterol with t1/2 ≈ 2 min (Supplemental Figures S5, S7, S11, S13, S18, and S20). Because the plasma membrane pool of DHE exchanges rapidly, there is very little DHE transport from the plasma membrane back to the ERC during efflux experiments. Under control conditions, DHE fluorescence in the ERC was reduced with a t1/2 = 14.9 ± 0.9 min after the start of exchange with cholesterol at the plasma membrane (Figure 2, C and D, blue, and Supplemental Figure S1). Using both the FRAP and efflux methods, we were able to accurately determine the t1/2 of sterol transport to and from the ERC to be ∼12–15 min.
Intracellular cholesterol levels affect cholesterol transport
To evaluate the role of intracellular cholesterol levels in modulating cholesterol transport, we determined the DHE trafficking kinetics in U2OS-SRA cells after cholesterol depletion or transient overloading (Figure 2). After metabolic depletion by overnight incubation in medium with lipoprotein-depleted serum (LPDS) and the HMG-CoA reductase inhibitor mevastatin, cellular cholesterol levels were reduced by ∼25% compared with control cells (Supplemental Figure S3). Of interest, after metabolic depletion of cellular cholesterol, DHE was trafficked to and from the ERC with half-times similar to or slightly faster than in controls, 10.4 ± 1.3 and 14.3 ± 1.3 min (Figure 2 and Supplemental Figure S1, red), respectively. Cholesterol overloading was accomplished by transient treatment with HPCD–cholesterol complex for 5 min. This treatment increased cholesterol levels by ∼20% (Supplemental Figure S3). The increased cellular cholesterol levels resulted in rapid DHE transport both to and from the ERC with t1/2 = 3.8 ± 0.6 and 5.8 ± 0.3 min, respectively (Figure 2 and Supplemental Figure S1, green). These data indicate that moderate increases in cholesterol levels increase the rate constant for sterol transport, but moderate reductions in cholesterol do not greatly alter the transport rate constant.
Role of ATP-dependent transport in sterol transport rates
Intracellular transport of cholesterol between organelles is mediated by both vesicular and nonvesicular transport mechanisms (Maxfield and Menon, 2006; Mesmin and Maxfield, 2009; Iaea and Maxfield, 2015). Because vesicular transport is ATP dependent, we used ATP depletion by treatment with sodium azide and 2-deoxyglucose to inhibit vesicular transport between the ERC and the plasma membrane (Figure 3 and Supplemental Figures S4 and S5). We verified that endocytosis of fluorescently labeled transferrin (Tf) had been stopped by this treatment (unpublished data). After energy depletion, DHE trafficked to the ERC with a t1/2 = 21.2 ± 3.2 min and out of the ERC with a t1/2 = 18.3 ± 1.1 min (Figure 3 and Supplemental Figure S4).
FIGURE 3:

Role of vesicular sterol transport. U2OS-SRA cells were treated with energy depletion (ED) medium to deplete cellular ATP levels. (A) FRAP measurements for control and ED. Control, t1/2 = 14.5 ± 1.5 min; ED, t1/2 = 21.2 ± 3.2 min. (B) DHE efflux curves for control and ED. Control, t1/2 = 14.9 ± 0.9 min; ED, t1/2 = 18.3 ± 1.1 min. (C) Extended FRAP measurement for control and ED U2OS-SRA cells. Control, t1/2 = 12.5 ± 3.4 min; ED, t1/2 = 24.2 ± 1.8 min. Each data point is derived from an average of at least eight experiments (±SE). Data are fitted to single-exponential curves.
Because the recovery t1/2 exceeded the duration of the fluorescence imaging, we extended the measured recovery time from 10 to 30 min to ensure that the recovery time was measured accurately. After 30 min, ∼90% of the DHE in the ERC of control cells had recovered, compared with only ∼40% recovered in energy-depleted cells. Consistent with the 10-min FRAP data, in control cells, DHE trafficked to the ERC with a t1/2 = 12.5 ± 3.4 min (Figure 3C and Supplemental Figure S4, blue), and in ATP-depleted cells, t1/2 = 24.2 ± 1.8 (Figure 3C and Supplemental Figure S4, red).
Increasing levels of sterol transporters cause faster sterol transport
Previous work demonstrated that sterol transport is maintained in large part by nonvesicular mechanisms (Maxfield and Menon, 2006; Garbarino et al., 2012; Iaea and Maxfield, 2015). However, it has been impossible to quantify accurately the portion of sterol transport that is maintained by specific nonvesicular mechanisms, due to incomplete DHE recovery in previous experiments (Hao et al., 2002; Mesmin et al., 2011; Garbarino et al., 2012). Using the improved methods described here, we examined the role of soluble sterol transporters in moving sterol between the ERC and the plasma membrane (Figure 4). As a first test, we increased the number of soluble sterol carrier levels by microinjection of a relatively sterol-specific β-cyclodextrin (Kilsdonk et al., 1995; Yancey et al., 1996), HPCD. When U2OS-SRA cells were microinjected with (1–3) × 109 molecules of HPCD, nearly equal to the number of cholesterol molecules in a cell (Maxfield and Mondal, 2006), there was a substantial increase in the rate of sterol transport both to and from the ERC, with t1/2 = 1.3 ± 0.3 and 4.1 ± 0.4 min, respectively (Figure 4, A and B, and Supplemental Figures S6 and S7). This is in general agreement with accelerated sterol transport observed when cells were microinjected with MCD (Mesmin et al., 2011).
FIGURE 4:

Microinjected HPCD and STARD4 overexpression enhances bidirectional sterol transport. HPCD and rhodamine–dextran were microinjected immediately before labeling of cells with DHE. U2OS-SRA cells were transfected with GFP-STARD4 for 18 h before the experiment. (A) FRAP measurements for single cells microinjected with HPCD or transfected with GFP-STARD4. Control, t1/2 = 14.5 ± 1.5 min; HPCD (1 × 109 molecules/cell), t1/2 = 1.3 ± 0.3 min; GFP-STARD4, t1/2 = 4.3 ± 0.5 min. (B) DHE efflux curves for cells microinjected with HPCD or transfected with GFP-STARD4. Control, t1/2 = 14.9 ± 0.9 min; HPCD (0.5 × 109 molecules/cell), t1/2 = 4.1 ± 0.4 min; GFP-STARD4, t1/2 = 4.9 ± 0.6 min. Each data point is derived from an average of at least 10 experiments (±SE). Data are fitted to single-exponential curves.
To evaluate quantitatively the specific role of STARD4, we overexpressed it transiently. In control U2OS-SRA cells, there are ∼250,000 STARD4 copies per cell (Supplemental Figure S8). Transient overexpression of GFP-STARD4 increases the number of STARD4 molecules by fourfold over endogenous levels (Supplemental Figure S8), and this accelerates the rate of sterol transport to and from the ERC with t1/2 = 4.3 ± 0.5 and 4.9 ± 0.6 min, respectively (Figure 4, A and B, and Supplemental Figures S6 and S7). This is consistent with a report (Mesmin et al., 2011) in which overexpression of STARD4 resulted in accelerated sterol transport to the ERC compared with control cells. These results indicate that 106 STARD4 molecules per cell are nearly as effective as 109 HPCD molecules in transporting cholesterol to and from the ERC.
Reduction of STARD4 slows sterol transport between ERC and plasma membrane
STARD4 is transcriptionally regulated by the sterol-responsive element–binding protein-2 (SREBP-2) and has been implicated as an important sterol carrier (Mesmin et al., 2011; Garbarino et al., 2012; Iaea et al., 2014). However, the exact function of STARD4 in cholesterol transport and homeostasis is not well understood (Riegelhaupt et al., 2010). To address this, we used clustered regularly interspaced short palindromic repeats (CRISPR) to disrupt the genomic copy of STARD4 in U2OS-SRA cells (Supplemental Table S1). To minimize the complications that occur after stable knockdown of STARD4 (Mesmin et al., 2011), we transiently transfected U2OS-SRA with CRISPR constructs to disrupt the STARD4 gene and examined the effect 18 h later (Figure 5). Under these conditions, cells expressing the SD4 CRISPR constructs were viable and had comparable cholesterol levels to control cells but had ∼95% lower STARD4 protein levels (Supplemental Figure S9). Genomic disruption of STARD4 slowed sterol transport both to and from the ERC with t1/2 = 22.4 ± 2.1 and 20.2 ± 0.4 min, respectively (Figure 5, A and B, and Supplemental Figures S10 and S11). Of importance, the loss of STARD4 did not alter general vesicular transport, as the rate of Tf efflux from the ERC in both SD4 CRISPR knockdown and control cells was similar (Supplemental Figure S12).
FIGURE 5:

Depletion of STARD4 slows sterol transport between ERC and plasma membrane. U2OS-SRA cells were transfected with GFP-Cas9 (STARD4 CRISPR) constructs for 18 h before the experiment. (A) FRAP measurements for single cells expressing STARD4 CRISPR constructs. Control, t1/2 = 14.5 ± 1.5 min; SD4 CRISPR, t1/2 = 22.4 ± 2.1 min. (B) DHE efflux curves for cells expressing STARD4 CRISPR constructs. Control, t1/2 = 14.9 ± 0.9 min; SD4 CRISPR, t1/2 = 20.2 ± 0.4 min. Each data point is derived from an average of at least 11 experiments (±SE). Data are fitted to single-exponential curves.
To validate further the importance of STARD4 in sterol transport, we took advantage of a methodology that rapidly depletes soluble proteins by irreversible recruitment to organelles such as mitochondria (Kessels and Qualmann, 2002; Robinson et al., 2010). We expressed an mCherry-FKB-STARD4 in U2OS-SRA cells that also expressed a mitochondrial protein (Mito; Robinson et al., 2010) that contains an FRB domain. In the absence of rapamycin, FKBP-STARD4 is diffusely expressed throughout the cytosol. On rapamycin-induced FKBP-FRB dimerization, mCherry-FKB-STARD4 was rapidly depleted from the cytosol and targeted to mitochondria (Supplemental Figure S13). As a result of STARD4 recruitment to the mitochondria, it looks by conventional epifluorescence microscopy as though the mitochondria have become enriched in sterol (Supplemental Figures S13 and S14). However, confocal imaging of rapamycin-treated cells that have STARD4 recruited to the mitochondria shows that the enrichment of sterol is not in mitochondria but in another membrane compartment (Supplemental Figure S15).
The mCherry-FKBP-STARD4 was able to carry out sterol transport between the ERC and the plasma membrane. We measured FRAP and efflux t1/2 values of 11.0 ± 1.6 and 12.5 ± 0.4 min, respectively, in U2OS-SRA cells expressing mCherry-FKB-STARD4 in which endogenous STARD4 had been removed using CRISPR (Figure 6, A and B, and Supplemental Figures S13 andS16).
FIGURE 6:

Expression of wild-type, cytosolic-localized STARD4 restores sterol transport between ERC and plasma membrane. U2OS-SRA cells were transfected with mCherry-FKBP-STARD4 with or without MitoTrap 48 h before the experiment. STARD4 CRISPR constructs were transfected for 18 h before the experiment. To redistribute STARD4 from the cytosol to the mitochondria, cells expressing MitoTrap were treated with 1 μM rapamycin for 10 min before imaging. After rapamycin treatment, cells were washed, and fresh medium 2 was added. (A) FRAP measurements for single cells expressing mCherry-FKBP-STARD4 and MitoTrap in cells expressing STARD4 CRISPR constructs. Control, t1/2 = 14.5 ± 1.5 min; SD4 CRISPR, t1/2 = 11.0 ± 1.6 min; SD4 CRISPR + SD4 + Mito, t1/2 = 19.7 ± 1.5 min. (B) DHE efflux curves for cells expressing mCherry-FKBP-STARD4 and MitoTrap in cells expressing STARD4 CRISPR constructs Control, t1/2 = 14.9 ± 0.9 min; SD4 CRISPR, t1/2 = 12.5 ± 0.4 min; SD4 CRISPR + SD4 + Mito, t1/2 = 19.1 ± 0.3 min. Each data point is derived from an average of at least 10 experiments (±SE). Data are fitted to single-exponential curves.
U2OS-SRA cells expressing STARD4 CRISPR, mCherry-FKBP-STARD4, and MitoTrap were treated with rapamycin, leading to redistribution of STARD4 from the cytosol to the mitochondria. The depletion of STARD4 from the cytosol results in reduced sterol transfer between the ERC and plasma membrane, with FRAP and efflux t1/2 = 19.7 ± 1.5 and 19.1 ± 0.3 min, respectively (Figure 6, A and B, and Supplemental Figure S16).
Functional STARD4 is required for efficient sterol transport between the ERC and the plasma membrane
Previous work has shown that several regions of STARD4 are required for transfer of sterol between liposomes (Mesmin et al., 2011; Iaea et al., 2015). Specifically, introduction of an aspartic acid at the apex of the omega-1 loop resulted in attenuated sterol transfer between liposomes and reduced interaction with membranes (Iaea et al., 2015). We examined whether a W108D mutation in the omega-1 loop of STARD4 reduced the sterol transfer kinetics in cells (Figure 7). Expression of a STARD4 construct with a W108D mutation in mCherry-FKBP-STARD4 did not restore sterol transport to and from the ERC in cells lacking endogenous STARD4. We observed FRAP and efflux t1/2 = 21.9 ± 1.5 and 19.0 ± 0.2 min, respectively (Figure 7, A and B, and Supplemental Figures S17 and S18), showing that the described mechanism of STARD4 sterol transport between liposomes (Iaea et al., 2015) also occurs in cells.
FIGURE 7:

Expression of omega-1 loop mutant (W108D) of STARD4 does not restore sterol transport. (A) U2OS-SRA cells were transfected with either wild-type or W108D mCherry-FKBP-STARD4 48 h before the experiment. STARD4 CRISPR constructs were transfected for 18 h before the experiment. FRAP measurements for cells expressing wild-type or W108D mCherry-FKBP-STARD4 in cells expressing STARD4 CRISPR constructs. Control, t1/2 = 14.5 ± 1.5 min; SD4 CRISPR + SD4, t1/2 = 11.0 ± 1.6 min; SD4 CRISPR + W108D SD4, t1/2 = 21.9 ± 1.5 min. (B) DHE efflux curves for cells expressing wild-type or W108D mCherry-FKBP-STARD4 in cells expressing STARD4 CRISPR constructs. Control, t1/2 = 14.9 ± 0.9 min; SD4 CRISPR + SD4, t1/2 = 12.5 ± 0.4 min; SD4 CRISPR + W108D SD4, t1/2 = 19.0 ± 0.2 min. Each data point is derived from an average of at least 10 experiments (±SE). Data are fitted to single-exponential curves.
STARD4 is a major sterol transporter between the ERC and the plasma membrane
With the quantitative tools described here, we sought to determine the portion of sterol transport that is specifically mediated by STARD4. To accomplish this, we removed endogenous STARD4 using CRISPR and blocked vesicular transport by ATP depletion (Figure 8, A and B, and Supplemental Figures S19 and S20). We then measured the rate constants for delivery to the ERC by FRAP or transport out of the ERC by the efflux assay (Figure 8C) and plotted them against data described earlier for STARD4 CRISPR (Figure 5) and energy depletion (Figure 3). The combination of ATP depletion and knockout of STARD4 results in slow transport both to and from the ERC and plasma membrane, with t1/2 = 24.1 ± 1.3 and 24.8 ± 0.3 min, respectively (Figure 8, A and B, and Supplemental Figures S19 and S20) These half-times are slower than those for either ATP depletion alone (Figure 3) or STARD4 reduction by CRISPR (Figure 5). Comparing the normalized rate constants of treatments to control cells, we find that ATP-independent transport accounts for nearly 75% of sterol transport between the ERC and plasma membrane. STARD4 accounts for ∼25% of all sterol transport and ∼33% of nonvesicular transport between these organelles (Figure 8C).
FIGURE 8:

STARD4 is a major nonvesicular sterol transport protein. U2OS-SRA cells were transfected with STARD4 CRISPR constructs and/or treated with energy depletion (ED) medium. (A) FRAP measurements for cells expressing STARD4 CRISPR constructs with or without ED. Control, t1/2 = 14.5 ± 1.5 min; SD4 CRISPR, t1/2 = 22.4 ± 2.1 min; ED, t1/2 = 21.2 ± 3.2 min; SD4 CRISPR + ED, t1/2 = 24.1 ± 1.3 min. (B) DHE efflux measurements for cells expressing STARD4 CRISPR constructs with or without ED. Control, t1/2 = 14.9 ± 0.9 min; SD4 CRISPR, t1/2 = 20.2 ± 0.4 min; ED, t1/2 = 18.3 ± 1.1 min; SD4 CRISPR + ED, t1/2 = 24.8 ± 0.3 min. (C) Normalized sterol transfer rate constants for FRAP (red) and efflux (blue) experiments under control, ED, STARD4-CRISPR and CRISPR-ED conditions. Each data point is derived from an average of at least nine experiments (±SE). Data are fitted to single-exponential curves.
DISCUSSION
In this study, we developed greatly improved methods to monitor the trafficking of the fluorescent cholesterol analogue DHE (Fischer et al., 1984) between the ERC and plasma membrane of living cells. Although previous work reported sterol transfer kinetics to the ERC using FRAP (Hao et al., 2002; Mesmin et al., 2011; Garbarino et al., 2012), the kinetic modeling was seriously incomplete because there was only partial recovery of the DHE fluorescence in the ERC. We identified two main causes of the incomplete recovery. First, previous studies allowed ∼15 min after incorporation of DHE into the plasma membrane before starting kinetic measurements. This was based on imaging showing that the distribution of DHE in the cells was apparently unchanged after a 15-min chase (Hao et al., 2002; Mesmin et al., 2011; Garbarino et al., 2012). Allowing a longer equilibration time (2 h in this study) gave slower rate constants for transport but allowed for full recovery of DHE fluorescence into the ERC. The second issue was that with previous imaging equipment, only ∼5–10 measurements could be made before photobleaching made measurements unreliable. With improved optics and cameras, we reduced the illumination intensity so that greater than 20 measurements could be made on each cell. With these improvements, all of our FRAP kinetic studies were modeled as single first-order rate processes with 100% recovery.
We also developed a novel method for measuring the kinetics of efflux of DHE from the ERC to the plasma membrane. Again, there had been previous efflux measurements (Hao et al., 2002), but they used relatively low concentrations (5 mM) of cholesterol-free MCD to extract DHE from the plasma membrane. Here we used 80 mM HPCD that was preequilibrated with sheep erythrocytes so that it did not change the total sterol content when incubated with cells. This high concentration of HPCD rapidly exchanged DHE in the PM for cholesterol with t1/2 = 2 min, and this made DHE transport from the ERC to the plasma membrane effectively irreversible, allowing us to measure the transport rates in this direction. These efflux transport rates were also modeled as single first-order rate processes that went to completion. Thus we were able to measure sterol transfer rate constants in both directions between the ERC and the plasma membrane.
Using these methods, we found that transfer between the ERC and the plasma membrane has t1/2 ≈ 12–15 min in both directions. This transport is slower than previous reports, in which recovery of the ERC after photobleaching occurred with t1/2 ≈ 3 min (Hao et al., 2002; Mesmin et al., 2011; Garbarino et al., 2012). However, unlike previous reports (Hao et al., 2002; Mesmin et al., 2011; Garbarino et al., 2012), in which only ∼40–50% of the sterol pool in the ERC was recovered, we obtained 100% recovery. It is not clear why longer equilibration times lead to slower rates of transport and higher mobile fractions. One possibility is that the DHE labeling protocol, which uses MCD–DHE complexes, perturbs the cells. We showed previously that the loading protocol does not significantly alter total cell cholesterol (Supplemental Figure S2), but MCD is also known to interact with some other lipids (Zidovetzki and Levitan, 2007). It may take some time for the cells to recover from the perturbations due to loading with DHE.
Although multiple processes are responsible for sterol transport between the ERC and the plasma membrane, we found that transport kinetics could be modeled reasonably well in most cases by a single process, although there are likely several transport processes occurring simultaneously. This may be in part coincidental. Transport of a labeled sphingomyelin from the ERC to the plasma membrane of CHO cells by vesicle transport processes had a t1/2 ≈ 12 min (Mayor et al., 1993), indicating that bulk lipid flow by vesicle transport on the recycling pathway occurs at this rate, which is comparable to the overall t1/2 we observe for vesicular and nonvesicular transport of sterol. With the accuracy of the present measurements, it would be difficult to resolve multiple rate constants from our data, but with further methodological advances, more detailed analysis may become feasible.
We can now examine the effects of various manipulations on rates of sterol transport. Increasing cellular cholesterol by ∼20% by brief incubation with cholesterol-loaded HPCD caused a threefold to fourfold increase in rate constants for DHE transport in either direction between the plasma membrane and the ERC (Figure 2 and Table 1). A 25% reduction of cholesterol after overnight growth in lipoprotein-depleted serum and treatment with a statin had a more modest effect on DHE transport rates, with no significant change in efflux to the plasma membrane and a slight increase in transport rate to the ERC. The asymmetric effects of increasing and decreasing cholesterol levels would be consistent with studies indicating that plasma membrane cholesterol is held at a level at which small increases in cholesterol concentration lead to large changes in its chemical potential (i.e., the chemical activity coefficient increases rapidly when cholesterol concentration is increased; Radhakrishnan et al., 2008; Steck and Lange, 2010; Das et al., 2014). Previous studies showed that even small elevations of cellular cholesterol above a threshold lead to large increases in cholesterol esterification by ACAT, which requires transport to the endoplasmic reticulum (Xu and Tabas, 1991; Lange and Steck, 1997).
A substantial portion of sterol transport is mediated by nonvesicular transport mechanisms (Hao et al., 2002; Maxfield and Menon, 2006). Vesicle transport requires ATP, and in our cells, ATP depletion to levels that completely block Tf receptor endocytosis reduces the rate of transport between the ERC and the plasma membrane by only ∼25–33% (Figure 8C and Table 1). This suggests that ∼67–75% of the total sterol transport between these membranes is ATP independent and does not require vesicle transport.
We examined the role of a specific sterol transporter, STARD4, which has been implicated as an important sterol transporter in various cell types (Mesmin et al., 2011; Rodriguez-Agudo et al., 2011; Garbarino et al., 2012). Approximately fourfold increase in STARD4 expression led to approximately threefold increase in the rates of transport of DHE between the plasma membrane and the ERC (Figure 4 and Table 1). This confirms that STARD4 can transport sterol very effectively in cells. Of note, the cholesterol distribution was not altered when levels of soluble sterol transporters were increased, suggesting that cellular membranes are near or at chemical equilibrium (Mesmin et al., 2011; Iaea and Maxfield, 2015).
Previous work showed that transient knockdown of STARD4 resulted in altered cholesterol levels and sensing (Mesmin et al., 2011). We also found that long-term reduction of STARD4 levels to ∼20% of control levels by stable transfection with short hairpin RNA leads to large differences in cellular lipid content (D. B. Iaea, R. B. Chan, B. Zhou, R. Bareja, O. Elemento, G. Di Paolo, and F. R. Maxfield, unpublished data). To avoid these long-term effects, we used two approaches. First, we examined cells 18 h after disruption of the STARD4 gene with a CRISPR construct, which led to a 95% reduction in STARD4 protein. In these cells, sterol levels remained similar to control cells, but the distribution and transport between the ERC and the plasma membrane were perturbed. Comparing the rate constants for transport between the ERC and the plasma membrane indicates that STARD4 is a critical sterol transport protein and accounts for ∼25% of total and ∼33% of nonvesicular sterol transport (Figure 8C and Table 1). To achieve more rapid elimination of STARD4, we transfected SD4 CRISPR cells with a mCherry-FKB-STARD4 construct and a mitochondrial protein that contains an FRB domain. The transfection led to somewhat faster sterol transport than in control cells (Table 1), reflecting the expression levels of the STARD4 constructs. Addition of rapamycin (Figure 6) caused ∼35% reduction in the sterol transport rate constants, very close to the reduction obtained with CRISPR disruption of STARD4. These data suggest that STARD4 is responsible for a substantial amount of sterol transport between the ERC and the plasma membrane.
There is a complex interplay of vesicular and nonvesicular transport mechanisms that act to maintain cholesterol distribution (Maxfield and Menon, 2006; Mesmin and Maxfield, 2009; Iaea and Maxfield, 2015). This study reports bidirectional sterol transport kinetics between the ERC and the plasma membrane under various conditions. For the first time, we provide detailed sterol transfer kinetics between two major cholesterol-containing organelles and show that ∼66–75% of this sterol transport is performed by nonvesicular, soluble sterol transport proteins. Furthermore, we show that STARD4 accounts for ∼25% of total sterol transport and ∼33% of all nonvesicular transport between the plasma membrane and the ERC. Thus STARD4 is an important sterol transport protein involved in sterol transport between the ERC and plasma membrane.
MATERIALS AND METHODS
Materials
Sheep red blood cells were purchased from Innovative Research (Novi, MI). Alexa labeling kits were purchased from Invitrogen. Human Tf and DHE were purchased from Sigma-Aldrich. All tissue culture supplies were purchased from Invitrogen. All other chemicals were from Sigma-Aldrich. The media used are as follows: medium 2 (150 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2 and 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4); M2glucose (medium 2 containing 2 mg/ml glucose); chase medium (M2glucose containing 30 μg of Sandoz 58035); efflux medium (chase medium with 80 mM HPCD–cholesterol complex and ∼1 × 109 sheep red blood cells/ml); and energy depletion medium (medium 2 with 15 μM NaN3 and 15 μM 2-deoxyglucose). Rabbit anti-STARD4 monoclonal (EPR17847-32) antibody was purchased from Abcam. Rabbit anti–pan actin polyclonal (AANO1-A) antibody was purchased from Cytoskeleton.
Cell culture and transfection
U2OS-SRA is a modified human osteosarcoma cell line that expresses SRA (Majumdar et al., 2011). U2OS-SRA cells were grown at 37°C in a 5% CO2 humidified incubator in McCoy’s 5A medium supplemented with 10% fetal bovine serum (FBS), 1 mg/ml Geneticin as a selection for SRA, 100 U/ml penicillin, and 100 μg/ml streptomycin. For STARD4 expression, U2OS-SRA cells were transiently transfected with pEGFP-C1-hSTARD4 using FuGENE 6 transfection reagent (Promega, Madison, WI) according to the manufacturer’s instructions. To modulate STARD4 protein levels or localization, U2OS-SRA cells were transfected with STARD4 CRISPR-GFP (SD4 CRISPR), YFP-FRB-MitoTrap (Mito), and mCherry-FKBP-STARD4. For rapamycin-dependent redistribution of STARD4 to the mitochondria, cells were treated with 1 μM rapamycin for 10 min before FRAP and efflux experiments. All studies for transient transfection were performed 18 h after transfection unless stated otherwise. Cells for wide-field microscopy were plated on 35-mm plastic dishes, the bottoms of which were replaced with poly-d-lysine–coated coverslips.
Fluorescence labeling and immunofluorescence
Human Tf (Sigma-Aldrich) was iron loaded and purified by Sephacryl S-300 (Pharmacia LKB) gel-filtration chromatography and conjugated to Alexa 488, Alexa 546, or Alexa 633 according to the manufacturer’s instructions (Invitrogen). To label cells with Tf, cells were incubated with 20 µg/ml Alexa488/546/633–Tf for 20 min at 37°C to reach steady state.
Labeling cells with lipid analogues
MCD–DHE complexes were prepared similarly to a previously described procedure (Sheets et al., 1999), with minor alterations. In brief, DHE in ethanol was dried under argon and subsequently dissolved in 25 mM MCD in medium 2, making the initial ratio of MCD to DHE 5:1 (mol/mol). The resulting suspension was vortexed and sonicated until it clarified. It was then incubated in a rocking water bath overnight at 37°C and centrifuged at 15,000 × g for 10 min before labeling of cells.
Fluorescence recovery after photobleaching
Cells were labeled for 1 min with DHE-loaded MCD and chased for 2 h in chase medium to allow for the sterol to equilibrate among membranes. In the last 20 min, the cells were labeled with fluorescent Tf to identify the ERC. A prebleach image was acquired before bleaching. DHE in the ERC was photobleached for 30 s, and images were taken every 30 s for 10 min or every 60 s for 30 min.
DHE efflux assay
Cells were labeled for 1 min with DHE-loaded MCD and equilibrated for 2 h in chase medium to allow for the sterol to equilibrate among membranes. Cells being imaged were colabeled with 20 µg/ml Alexa 488/546/633–Tf for 20 min at 37°C in chase medium to identify the ERC. An image of the field was taken before the buffer was exchanged for efflux medium. DHE images were taken at 0, 0.5, 1, 3, 5, 10, 20, 30, 40, 50, and 60 min, and the fluorescence in the ERC was measured.
Fluorescence microscopy
Wide-field fluorescence microscopy and digital image acquisition were carried out using a Leica DMIRB microscope with ultraviolet-transmitting epi-illumination and equipped with an Andor iXonEM Blue electron-multiplying charge-coupled device (CCD) camera driven by MetaMorph Imaging System software (Universal Imaging/Molecular Devices, Sunnyvale, CA). All images were acquired using 63×/1.36 numerical aperture (NA) oil-immersion objectives with 2 × 2 pixel binning. DHE was imaged using a filter cube obtained from Chroma Technology (Bellows Falls, VT; 335-nm [20-nm band pass] excitation filter, 365-nm long-pass dichromatic filter, and 405-nm [40-nm band pass] emission filter). Standard tetramethylrhodamine isothiocyanate, fluorescein isothiocyanate, and Cy5 cubes were obtained from Chroma. Filipin staining was imaged using an A4 filter cube (Leica, Wetzlar, Germany).
Confocal microscopy
Cells were imaged on a Zeiss LSM 880 AxioObserver microscope equipped with a Plan-Apochromat oil 63×/1.4 NA differential interference contrast M27 objective. Z-stacks were obtained using a step size of 2 μm.
Microinjection
Cytosolic microinjections were performed using backloaded borosilicate glass capillaries and a Narishige micromanipulator. The microinjection solution was 10 mM HEPES (pH 6.9) and 140 mM KCl with the addition of 0.5 mg/ml rhodamine–dextran and, when indicated, 2.5 mM HPCD. To determine the number of HPCD molecules injected in a single cell, the cellular integrated fluorescence intensity of the coinjected dye, rhodamine–dextran, was compared with a standard curve obtained by imaging rhodamine–dextran in aqueous solutions of known concentrations placed in a hemocytometer chamber. In this way, the number of rhodamine–dextran molecules injected could be calculated, and the number of cellular HPCD molecules was determined based on this. Images of the standard solutions were acquired using imaging parameters identical to those used in microinjection experiments.
Measurement of STARD4 copy number per cell
To estimate the number of STARD4 molecules per cell, U2OS-SRA cells were counted and lysed. Proteins from a known fraction of the cell lysate were separated by 16% SDS–PAGE and immunoblotted with anti-STARD4 and anti-actin. Known amounts of purified hSTARD4 were loaded in parallel as standard references. Ten percent of the volume of a lysate of 2.0 × 106 cells was loaded. The band corresponding to STARD4 was quantified and compared with the protein standards. The same measurement was performed with cells expressing GFP-STARD4. In this case, 10% of the cells used for quantification were transfected with GFP-STARD4. The band corresponding to GFP-STARD4 was quantified, from which the number of GFP-STARD4 molecules per cell was calculated.
Free cholesterol measurement by gas chromatography–mass spectrometry
Cellular lipids were extracted twice with hexane/2-propanol (3:2). During the first extraction, β-sitosterol was added as an internal standard for quantification. Dried lipids were resuspended in hexane and separated on a Varian Factor Four capillary column using a Varian 400 gas chromatography/tandem mass spectrometry system (Rosenbaum et al., 2010). The protein concentration after solubilization with 0.5 M NaOH was determined by the bicinchoninic acid protein assay.
Transferrin recycling rates
Control and SD4 CRISPR U2OS-SRA cells were labeled with Alexa 488/546–Tf for 20 min. Labeled Tf was chased with 100-fold excess of unlabeled Tf in the presence of 20 μM deferoxamine and fixed at the indicated time points (Hao et al., 2002). Cells were imaged as described, and fluorescence intensity was measured for individual cells in multiple fields.
Cholesterol modulation
U2OS-SRA cells were grown for 24 h in metabolic depletion medium (McCoy’s 5A medium, similar to the growth medium, but with 5% lipoprotein-deficient serum in place of FBS, supplemented with 10 μM mevastatin) to block cholesterol synthesis and deplete cholesterol stores. To overload cholesterol by HPCD treatment, cells were incubated with 10 mM HPCD–cholesterol complex in chase medium for 5 min before labeling.
Energy depletion
U2OS-SRA cells were labeled with DHE and fluorescent Tf before energy depletion. Cells were incubated with 15 μM sodium azide and 15 μM 2-deoxyglucose for 15 min to deplete cellular ATP before and maintained throughout FRAP and efflux experiments (Hao et al., 2002).
STARD4 CRISPR
Paired single-guide RNAs targeting exon 4 of STARD4 were identified using Geneious software (www.geneious.com), and sequences encoding guide RNAs were cloned into the BbsI sites on PX461 (expressing the Cas9D10A, nickase mutant) according to the published protocol (www.genome-engineering.org). Genomic modifications in transfected cells were assessed using T7 endonuclease assay (M0302; New England Bioscience) after PCR amplification of the target region. Oligonucleotides used for cloning and PCR are listed in Table 1.
Mutation detection by SURVEYOR assay
Cas9-induced mutations were detected using the SURVEYOR Mutation Detection Kit (Transgenomic/IDT). Briefly, an ∼500–base pair region surrounding the expected mutation site was PCR amplified using Herculase II (600675; Agilent Technologies). PCR products were column purified (Qiagen) and subjected to a series of melt–anneal temperature cycles with annealing temperatures gradually lowered in each successive cycle. SURVEYOR nuclease was then added to selectively digest heteroduplex DNA. Digest products were visualized on a 2% agarose gel.
Molecular biology
mCherry-FKBP-STARD4 was generated by replacing MTM1 from mCherry-FKBP-MTM1 (Hammond et al., 2014) via Not1 and Sal1. Omega-1 loop mutation (W108D) was generated by site-directed mutagenesis. pMito-YFP-FRB (Robinson et al., 2010) was purchased from Addgene.
Data analysis for FRAP and efflux experiments
In both FRAP and efflux experiments, DHE images were analyzed using MetaMorph Discovery-1 image analysis software (Pipalia et al., 2007; Mesmin et al., 2011). The cell outlines were traced out manually in DHE images. The ERC was also manually outlined for each cell using the Tf-labeled images. We measured the DHE fluorescence intensity from the outlined ERC region (F’ERC) and the entire cell (Fcell), as well as the area of the ERC (AERC). Because DHE images were wide-field images, F’ERC included not only the fluorescence in the ERC but also the fluorescence contributed by the regions of the plasma membrane that were directly above and below the ERC. To calculate the fluorescence contributed by the plasma membrane (FPM in ERC) in the ERC region, a small region next to the ERC was selected manually. The integrated fluorescence (FPM) and the area (APM) in this region were measured. We used
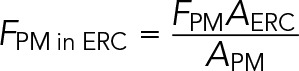 (1) (1)
|
This was based on the assumption that the DHE fluorescence present on the plasma membrane directly above and below the ERC was similar to that on the plasma membrane next to the ERC. The DHE fluorescence contributed only from the ERC (FERC) was calculated using
 (2) (2)
|
In FRAP experiments, FERC/Fcell was used to determine the fluorescence recovery ratio. FRAP data were fitted to a single exponential.
In efflux experiments, autofluorescence in the ERC (Fauto in ERC) was also considered. To calculate Fauto in ERC, cells without DHE labeling were imaged in parallel experiments. The images were first background corrected for using the fifth percentile of brightness in the image. The ERC was manually outlined for each cell using the Tf-labeled images. Fluorescence power from the outlined ERC region (Fauto) and the area of the ERC (Aauto) was measured. The autofluorescence intensity in the ERC (Iauto) was measured in each cell using
|
|
The averaged autofluorescence intensity in the ERC (Iaver auto) was calculated from >20 cells. The DHE fluorescence in the ERC without autofluorescence (FERC-auto) was calculated from
|
|
FERC-auto at each time point was normalized to FERC-auto at 0 min in each efflux experiment. Efflux data were then fitted to a single-exponential decay. In FRAP and efflux experiments, the half-time in each condition was averaged from multiple cells.
Supplementary Material
Acknowledgments
We are grateful to Lukas Dow and Ashlesha Muley (Weill Cornell Medicine) for assistance with implementing the CRISPR technology. We also thank Mingming Hao (Weill Cornell Medicine) for critical reading of the manuscript, Robert D. Phair (Integrative Bioinformatics) for extensive discussion of experimental results and design, as well as critical reading of the manuscript, and Xiaohui Zha (Ottawa Hospital Research Institute) for advice on the efflux assay. This work was supported by National Institutes of Health Grants F31-DK104631 (D.B.I.) and R37-DK27083 (F.R.M.).
Abbreviations used:
- ACAT
Acyl-CoA:cholesterol acyltransferase
- ATP
adenosine triphosphate
- CRISPR
clustered regularly interspaced short palindromic repeats
- DHE
dehydroergosterol
- ER
endoplasmic reticulum
- ERC
endocytic recycling compartment
- FRAP
fluorescence recovery after photobleaching
- HPCD
hydroxypropyl cyclodextrin
- LPDS
lipoprotein-depleted serum
- MCD
methyl-β-cyclodextrin
- SRA
scavenger receptor A
- SREBP-2
sterol-responsive element–binding protein-2
- StAR
steroidogenic acute regulatory protein
- STARD
START domain
- START
StAR–related lipid-transfer
- STPs
sterol transfer proteins
- Tf
transferrin
- U2OS
human osteosarcoma
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-07-0499) on February 16, 2017.
REFERENCES
- Beh CT, McMaster CR, Kozminski KG, Menon AK. A detour for yeast oxysterol binding proteins. J Biol Chem. 2012;287:11481–11488. doi: 10.1074/jbc.R111.338400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BJ. The mammalian START domain protein family in lipid transport in health and disease. J Endocrinol. 2012;212:257–275. doi: 10.1530/JOE-11-0313. [DOI] [PubMed] [Google Scholar]
- Das A, Brown MS, Anderson DD, Goldstein JL, Radhakrishnan A. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. Elife. 2014;3 doi: 10.7554/eLife.02882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer RT, Stephenson FA, Shafiee A, Schroeder F. delta 5,7,9(11)-Cholestatrien-3 beta-ol: a fluorescent cholesterol analogue. Chem Phys Lipids. 1984;36:1–14. doi: 10.1016/0009-3084(84)90086-0. [DOI] [PubMed] [Google Scholar]
- Frolov A, Woodford JK, Murphy EJ, Billheimer JT, Schroeder F. Spontaneous and protein-mediated sterol transfer between intracellular membranes. J Biol Chem. 1996;271:16075–16083. doi: 10.1074/jbc.271.27.16075. [DOI] [PubMed] [Google Scholar]
- Galan C, Woodard GE, Dionisio N, Salido GM, Rosado JA. Lipid rafts modulate the activation but not the maintenance of store-operated Ca(2+) entry. Biochim Biophys Acta. 2010;1803:1083–1093. doi: 10.1016/j.bbamcr.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Garbarino J, Pan M, Chin HF, Lund FW, Maxfield FR, Breslow JL. STARD4 knockdown in HepG2 cells disrupts cholesterol trafficking associated with the plasma membrane, ER, and ERC. J Lipid Res. 2012;53:2716–2725. doi: 10.1194/jlr.M032227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GR, Machner MP, Balla T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J Cell Biol. 2014;205:113–126. doi: 10.1083/jcb.201312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M, Lin SX, Karylowski OJ, Wustner D, McGraw TE, Maxfield FR. Vesicular and non-vesicular sterol transport in living cells. The endocytic recycling compartment is a major sterol storage organelle. J Biol Chem. 2002;277:609–617. doi: 10.1074/jbc.M108861200. [DOI] [PubMed] [Google Scholar]
- Hao M, Maxfield FR. Characterization of rapid membrane internalization and recycling. J Biol Chem. 2000;275:15279–15286. doi: 10.1074/jbc.275.20.15279. [DOI] [PubMed] [Google Scholar]
- Huang J, Feigenson GW. A microscopic interaction model of maximum solubility of cholesterol in lipid bilayers. Biophys J. 1999;76:2142–2157. doi: 10.1016/S0006-3495(99)77369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaea DB, Dikiy I, Kiburu I, Eliezer D, Maxfield FR. STARD4 membrane interactions and sterol binding. Biochemistry. 2015;54:4623–4636. doi: 10.1021/acs.biochem.5b00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaea DB, Mao S, Maxfield FR. Steroidogenic acute regulatory protein-related lipid transfer (START) proteins in non-vesicular cholesterol transport. In: Clark BJ, Stocco DM, editors. Cholesterol Transporters of the START Domain Protein Family in Health and Disease. New York: Springer; 2014. pp. 173–188. [Google Scholar]
- Iaea DB, Maxfield FR. Cholesterol trafficking and distribution. Essays Biochem. 2015;57:43–55. doi: 10.1042/bse0570043. [DOI] [PubMed] [Google Scholar]
- Kessels MM, Qualmann B. Syndapins integrate N-WASP in receptor-mediated endocytosis. EMBO J. 2002;21:6083–6094. doi: 10.1093/emboj/cdf604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilsdonk EP, Yancey PG, Stoudt GW, Bangerter FW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem. 1995;270:17250–17256. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- Lange Y, Dolde J, Steck TL. The rate of transmembrane movement of cholesterol in the human erythrocyte. J Biol Chem. 1981;256:5321–5323. [PubMed] [Google Scholar]
- Lange Y, Steck TL. Quantitation of the pool of cholesterol associated with acyl-CoA:cholesterol acyltransferase in human fibroblasts. J Biol Chem. 1997;272:13103–13108. doi: 10.1074/jbc.272.20.13103. [DOI] [PubMed] [Google Scholar]
- Liscum L, Munn NJ. Intracellular cholesterol transport. Biochim Biophys Acta. 1999;1438:19–37. doi: 10.1016/s1388-1981(99)00043-8. [DOI] [PubMed] [Google Scholar]
- Majumdar A, Capetillo-Zarate E, Cruz D, Gouras GK, Maxfield FR. Degradation of Alzheimer’s amyloid fibrils by microglia requires delivery of ClC-7 to lysosomes. Mol Biol Cell. 2011;22:1664–1676. doi: 10.1091/mbc.E10-09-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, Menon AK. Intracellular sterol transport and distribution. Curr Opin Cell Biol. 2006;18:379–385. doi: 10.1016/j.ceb.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Maxfield FR, Mondal M. Sterol and lipid trafficking in mammalian cells. Biochem Soc Trans. 2006;34:335–339. doi: 10.1042/BST0340335. [DOI] [PubMed] [Google Scholar]
- Maxfield FR, Wustner D. Intracellular cholesterol transport. J Clin Invest. 2002;110:891–898. doi: 10.1172/JCI16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, Wustner D. Analysis of cholesterol trafficking with fluorescent probes. Methods Cell Biol. 2012;108:367–393. doi: 10.1016/B978-0-12-386487-1.00017-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S, Presley JF, Maxfield FR. Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J Cell Biol. 1993;121:1257–1269. doi: 10.1083/jcb.121.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B, Maxfield FR. Intracellular sterol dynamics. Biochim Biophys Acta. 2009;1791:636–645. doi: 10.1016/j.bbalip.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B, Pipalia NH, Lund FW, Ramlall TF, Sokolov A, Eliezer D, Maxfield FR. STARD4 abundance regulates sterol transport and sensing. Mol Biol Cell. 2011;22:4004–4015. doi: 10.1091/mbc.E11-04-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Zha X, Tabas I, Maxfield FR. Cholesterol distribution in living cells: fluorescence imaging using dehydroergosterol as a fluorescent cholesterol analog. Biophys J. 1998;75:1915–1925. doi: 10.1016/S0006-3495(98)77632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani B, Ong HL, Liu X, Rauser K, Ambudkar IS, Singh BB. Lipid rafts determine clustering of STIM1 in endoplasmic reticulum-plasma membrane junctions and regulation of store-operated Ca2+ entry (SOCE) J Biol Chem. 2008;283:17333–17340. doi: 10.1074/jbc.M800107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MC, Johnson WJ, Rothblat GH. Mechanisms and consequences of cellular cholesterol exchange and transfer. Biochim Biophys Acta. 1987;906:223–276. doi: 10.1016/0304-4157(87)90013-x. [DOI] [PubMed] [Google Scholar]
- Pipalia NH, Hao M, Mukherjee S, Maxfield FR. Sterol, protein and lipid trafficking in Chinese hamster ovary cells with Niemann-Pick type C1 defect. Traffic. 2007;8:130–141. doi: 10.1111/j.1600-0854.2006.00513.x. [DOI] [PubMed] [Google Scholar]
- Prinz WA. Non-vesicular sterol transport in cells. Prog Lipid Res. 2007;46:297–314. doi: 10.1016/j.plipres.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 2008;8:512–521. doi: 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegelhaupt JJ, Waase MP, Garbarino J, Cruz DE, Breslow JL. Targeted disruption of steroidogenic acute regulatory protein D4 leads to modest weight reduction and minor alterations in lipid metabolism. J Lipid Res. 2010;51:1134–1143. doi: 10.1194/jlr.M003095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS, Sahlender DA, Foster SD. Rapid inactivation of proteins by rapamycin-induced rerouting to mitochondria. Dev Cell. 2010;18:324–331. doi: 10.1016/j.devcel.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Agudo D, Calderon-Dominguez M, Ren S, Marques D, Redford K, Medina-Torres MA, Hylemon P, Gil G, Pandak WM. Subcellular localization and regulation of StarD4 protein in macrophages and fibroblasts. Biochim Biophys Acta. 2011;1811:597–606. doi: 10.1016/j.bbalip.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum AI, Zhang G, Warren JD, Maxfield FR. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc Natl Acad Sci USA. 2010;107:5477–5482. doi: 10.1073/pnas.0914309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets ED, Holowka D, Baird B. Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcepsilonRI and their association with detergent-resistant membranes. J Cell Biol. 1999;145:877–887. doi: 10.1083/jcb.145.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol. 2010;11:688–699. doi: 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- Soccio RE, Adams RM, Maxwell KN, Breslow JL. Differential gene regulation of StarD4 and StarD5 cholesterol transfer proteins. Activation of StarD4 by sterol regulatory element-binding protein-2 and StarD5 by endoplasmic reticulum stress. J Biol Chem. 2005;280:19410–19418. doi: 10.1074/jbc.M501778200. [DOI] [PubMed] [Google Scholar]
- Soccio RE, Adams RM, Romanowski MJ, Sehayek E, Burley SK, Breslow JL. The cholesterol-regulated StarD4 gene encodes a StAR-related lipid transfer protein with two closely related homologues, StarD5 and StarD6. Proc Natl Acad Sci USA. 2002;99:6943–6948. doi: 10.1073/pnas.052143799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck TL, Lange Y. Cell cholesterol homeostasis: mediation by active cholesterol. Trends Cell Biol. 2010;20:680–687. doi: 10.1016/j.tcb.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck TL, Ye J, Lange Y. Probing red cell membrane cholesterol movement with cyclodextrin. Biophys J. 2002;83:2118–2125. doi: 10.1016/S0006-3495(02)73972-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbani L, Simoni RD. Cholesterol and vesicular stomatitis virus G protein take separate routes from the endoplasmic reticulum to the plasma membrane. J Biol Chem. 1990;265:1919–1923. [PubMed] [Google Scholar]
- van Meer G. Cellular lipidomics. EMBO J. 2005;24:3159–3165. doi: 10.1038/sj.emboj.7600798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Sprong H. Membrane lipids and vesicular traffic. Curr Opin Cell Biol. 2004;16:373–378. doi: 10.1016/j.ceb.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Xu XX, Tabas I. Lipoproteins activate acyl-coenzyme A:cholesterol acyltransferase in macrophages only after cellular cholesterol pools are expanded to a critical threshold level. J Biol Chem. 1991;266:17040–17048. [PubMed] [Google Scholar]
- Yancey PG, Rodrigueza WV, Kilsdonk EP, Stoudt GW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins. Demonstration Of kinetic pools and mechanism of efflux. J Biol Chem. 1996;271:16026–16034. doi: 10.1074/jbc.271.27.16026. [DOI] [PubMed] [Google Scholar]
- Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta. 2007;1768:1311–1324. doi: 10.1016/j.bbamem.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.