

Abstract
Background
Anticytokine autoantibodies occur across a range of hematologic, pulmonary and infectious diseases, however, systematic investigation of their presence and significance in autoimmune diseases is lacking.
Methods
Serum samples from patients with systemic lupus erythematosus (SLE) (n=199), primary Sjögren’s syndrome (SS) (n=150), rheumatoid arthritis (RA) (n=149) and healthy controls (n=200) were screened for 24 anticytokine autoantibodies using a multiplex bead-based assay. To evaluate biological activity of anticytokine autoantibodies, their ability to block cytokine-induced signal transduction or protein expression was measured. RNA sequencing was performed on whole blood in subset of controls and SLE patients.
Results
SLE and SS patients had striking excess of autoantibodies against interferons and the interferon-responsive chemokine interferon-inducible-protein-10 (IP-10). Only autoantibodies against type I interferon, interleukin (IL)-12 and IL-22 exhibited neutralizing activity. In SLE, anti-interferon-γ autoantibodies tracked with more disease activity, anti-double-stranded-DNA antibodies, and elevated expression of interferon-α/β-inducible genes. Conversely, SLE patients with blocking anti-interferon-α autoantibodies normalized their type I interferon gene expression signature. Anti-type III interferons (λ2, λ3), and anti-IP-10 autoantibodies were newly recognized and autoantibodies against macrophage-colony stimulating factor, IL-4, IL-7, IL-17 and IL-22, that have not been previously identified in rheumatologic conditions, were discovered.
Conclusions
Anticytokine autoantibodies were associated with distinct patterns of SLE, SS and RA. Anti-interferon autoantibodies were overrepresented in SLE and SS and fall into distinct functional classes with only a subset of anti-type I interferon antibodies exhibiting neutralizing activity. Anti-interferon-γ autoantibodies correlated with increased disease activity and interferon-related gene expression, suggesting that they may contribute to the pathogenesis of SLE.
Keywords: Anticytokine autoantibodies, Systemic Lupus Erythematosus (SLE), Primary Sjögren’s Syndrome (SS), Rheumatoid Arthritis (RA)
Introduction
Anticytokine autoantibodies have been found to cause acquired immunodeficiency, pulmonary alveolar proteinosis, and hematologic syndromes (1–4) through neutralizing activities that create functional deficiencies of the cognate cytokines. Autoantibodies against more common autoimmune targets such as nuclear antigens, citrullinated peptides or immunoglobulin, are generally not found in these patients, nor do they suffer from other autoimmune symptoms. Both systemic autoimmunity and anticytokine autoantibodies are observed in autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) syndrome, by genetic defects to the autoimmune regulator (AIRE) gene, which facilitates negative selection of autoreactive T cells in the thymus (5). In addition to autoimmune-mediated endocrinopathies and a wide range of other systemic autoimmune phenomena (6), affected individuals commonly demonstrate neutralizing autoantibodies against type I interferons, interleukin (IL)-17 and IL-22, the latter two of which may explain the common tendency for chronic mucocutaneous candidiasis (7). In rheumatologic diseases, autoantibodies against one or a small group of cytokines have been reported (8–19), but their spectrum and clinical impact remain largely unknown. Autoantibodies against type I and II interferons have been reported in up to 27% of systemic lupus erythematosus (SLE) sera (8–11). Their impact on the interferon signature and the pathogenesis of SLE is unclear, but their potential to influence interferon signaling, disease activity, and response to biologic therapeutics could be great (11). Previous reports of anticytokine autoantibodies in rheumatologic diseases have been isolated, with variations in the detection techniques employed and the anticytokine activities sought, complicating the formulation of generalizable conclusions.
It remains largely unknown whether anticytokine autoantibodies in rheumatologic diseases are pathogenic, protective, or simple reflections of a general tendency towards autoreactivity. Given that anticytokine autoantibodies can have important physiological roles in health (20), and can be beneficial (21) or detrimental in various contexts (22), it is critical to define their roles and significance in rheumatologic disease. They may confer benefit or detriment, depending not only on the activity of the autoantibody itself but also on the intrinsic role of the target cytokine. Further, their presence might even help classify patients who currently carry similar diagnoses. Therefore, we constructed a multiplexed bead-based assay to detect and quantitate 24 different anticytokine antibodies and evaluated a total of 498 patients diagnosed with SLE, primary Sjögren’s syndrome (SS) and rheumatoid arthritis (RA).
Methods
Participants
Archived sera from patients with SLE, SS, RA and healthy controls stored at −80°C were identified through institutional review board-approved protocols or using appropriate Office of Human Subjects Research-approved waivers. Samples were obtained through collaborations across the United States and Greece (Table S1). SLE and RA patients fulfilled American College of Rheumatology classification criteria (23, 24); SS patients met the European-American criteria (25). Available clinical data were collected on the day of sample collection using standardized forms developed for clinical research, including Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) (26) scores for SLE; focus score from minor salivary gland biopsy (27) for SS; and Disease Activity Score including 28 joints with erythrocyte sedimentation rate or C-reactive protein score (DAS28-ESR or DAS28-CRP) (28) for RA.
Measurement of Anticytokine Autoantibodies
Sera were screened using multiplex particle-based technology as previously described (29). Anticytokine autoantibodies were quantified as a function of the fluorescence intensity and was linear over a range (0–25,000) (29). Data were analyzed using Prism, version 6.0 (Graphpad Software, Inc.).
Functional Evaluation of Anticytokine Autoantibodies
We developed assays to detect specific effects of anticytokine autoantibodies in isolated normal peripheral blood mononuclear cells (PBMC) (30) or cell lines known to express the receptor for each cytokine of interest. The impact of autoantibody-containing serum (10%) versus that of autoantibody-negative serum (10%) on cytokine-specific signal transduction or protein expression was assessed by flow cytometry (31, 32) or by commercial cytokine detection kits (Bio-Rad) with assay details outlined in Table S2. Flow cytometry data were collected using a FACSCalibur (BD Biosciences) and analyzed using FlowJo version 9.7 (TreeStar). To confirm that the blocking ability was due to Immunoglobulin G (IgG) subclass, total IgG was captured on protein G column (Ab Spin Trap, GE Healthcare) and the flow-through collected prior to eluting the IgG fraction for 7 samples (4 SLE patients and 1 SS patient with anti-type I interferon blocking antibodies and 2 healthy controls). The ability of each fraction to inhibit interferon signaling was determined by flow cytometry as above.
All samples containing anti-interferon and anti-IL-12 autoantibodies were evaluated for their neutralization ability. For all other anticytokine autoantibodies detected, the three highest fluorescence intensity values from each cohort were evaluated for functional activity relative to autoantibodies negative serum. If cytokine-binding activity was detected, functional assessment of subsequent positive sera was continued within each cohort in order of decreasing fluorescence intensity until at least three sequential samples with no blocking activity were reached.
RNA Sequencing and Analysis
RNA processing and sequencing was performed on whole blood samples from participants as described by Isaac et al (33). Reads of 94 bases were mapped to the human transcriptome and genome hg19 using TopHat 2.0.8. (34) For each patient, a gene signature score was derived using the median of fold change from healthy controls of a set of 21 interferon-α/β-inducible genes (35) or 7 interferon-γ-inducible genes (36–38) to assess for interferon activity (Table S3). Patients with interferon-α/β-inducible gene score less than 4 were considered to have low interferon signature (35).
Statistical Analysis
For each anticytokine autoantibody, concentrations above two standard deviations of the mean of healthy controls were classified as positive. Thus, approximately 2.5% or 5 out of 200 healthy controls were considered positive. Using unsupervised hierarchal clustering by Ward’s method (39) the prevalence of each anticytokine autoantibody across the 3 groups was compared and a colored heatmap generated. Group differences were examined with the use of Fisher’s, Wilcoxon or Mann-Whitney tests. Statistical tests were performed at the 0.05 level. Statistical analysis was performed with the use of SAS, version 9.4 (SAS Institute) and Prism, version 6.0 (Graphpad Software, Inc.).
Results
Distinct Spectra of Anticytokine Autoantibodies Across Rheumatologic Diseases
Clinical characteristics of each group are detailed in Table S4. Sera from 76 (38%) patients with SLE (Figure 1A), 63 (42%) with SS (Figure 1B), but only 30 (20%) with RA (Figure 1C) tested positive for at least one anticytokine autoantibody (Table S5). About half of these patients made two or more anticytokine autoantibodies (Figure 1D); fourteen (7%) SLE patients and 9 (6%) SS patients made autoantibodies to 4 or more cytokines compared to only one RA patient. A heat map comparing the frequency of each anticytokine autoantibody confirmed a ‘hot-spot’ of autoantibodies targeting interferons and interferon-inducible-protein-10 (IP-10) in SLE and SS. Unsupervised clustering identified a close relationship between the spectra of anticytokine autoantibodies produced in SS and SLE compared to RA (Figure 1E). Forty-three (22%) SLE and 25 (17%) SS patients made autoantibodies to one of the interferons, significantly more than the 9 (6%) RA patients (p=0.0001 and 0.0038) (Table 1). There was no significant difference in the prevalence of anti-interferon autoantibodies between the SLE and SS patients. Out of the 43 SLE patients who made autoantibodies to any interferon, 22 (11%) were against type I interferon, 14 (7%) to type II interferon and 12 (6%) to type III interferon. There was little overlap between the subtype of interferon targeted, with only 5 of 43 patients making autoantibodies against more than one interferon and none against all three classes (Figure 1F). In contrast to autoantibodies against type I and type II interferons, anti-type III interferon autoantibodies were equally distributed across the three patient groups. Intriguingly, 10 out of 17 SLE and 11 out of 15 SS patients who had anti-IP-10 autoantibodies also made autoantibodies to one of the interferons.
Figure 1. Spectrum of Anticytokine Autoantibodies.
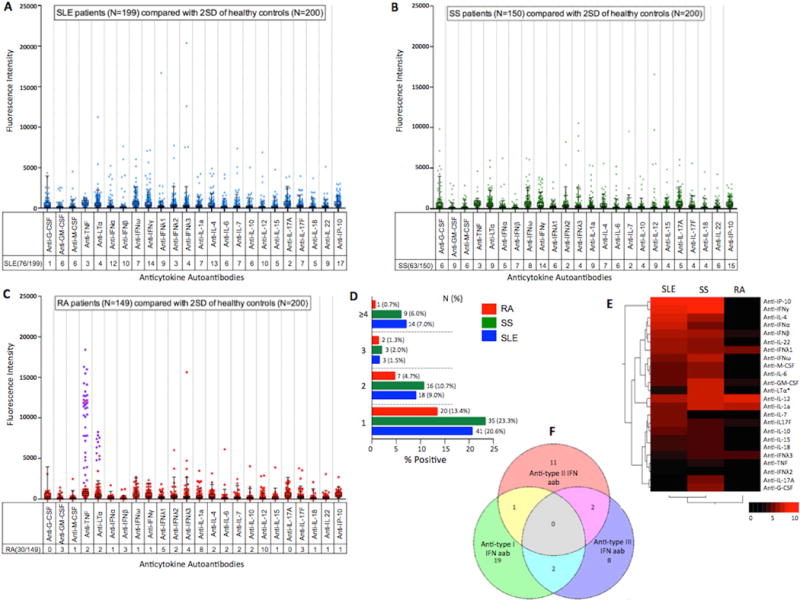
Dot plots of the distribution of all anticytokine autoantibodies measured by multiplex particle-based technology in patients with (A) Systemic Lupus Erythematosus (B) Primary Sjögren’s Syndrome and (C) Rheumatoid Arthritis and compared with two standard deviations (2SD) of the mean for healthy controls (box and whiskers). (D) Frequency of patients with 1, 2, 3 or ≥4 anticytokine autoantibodies. (E) Heatmap based on unsupervised hierarchal clustering of the prevalence of anticytokine autoantibodies in SLE, SS and RA patients. Key on bottom left indicates percent prevalence. (F) Distribution of anti-interferon autoantibodies based on type in SLE patients. Venn-diagram represents the overlap of autoantibodies against type I, II and III interferons in SLE patients with any anti-interferon autoantibody (N=43). Calculated using http://www.bioinformatics.lu/venn.php. Concentrations above two standard deviations (2SD) of mean of healthy controls were classified as positive. Anti-TNF*, anti-LTα*: patients who received anti-TNF biologics are shaded in purple in RA cohort dot plot and were analyzed as anti-TNF or anti-LTa negative for the heatmap.
G-CSF: granulocyte-colony stimulating factor; GM-CSF: granulocyte monocyte-colony stimulating factor; M-CSF: macrophage-colony stimulating factor; TNF: tumor necrosis factor; LTα: lymphotoxin-α; IFN: interferon; IL: interleukin; IP-10: interferon-inducible-protein-10
Table 1.
Prevalence of Anti-interferon Autoantibodies
| SLE (N=199) |
SS (N=150) |
RA (N=149) |
p-value# (SLE vs RA) |
p-value# (SS vs RA) |
p-value# (SLE vs SS) | |
|---|---|---|---|---|---|---|
| Any anti-interferon autoantibodies | 43 (21.6%) | 25 (16.7%) | 9 (6.0%) | 0.0001* | 0.0038* | 0.2485 |
| Anti-type I interferon autoantibodies | 22 (11.0%) | 13 (8.7%) | 3 (2.0%) | 0.0012* | 0.0105* | 0.4620 |
| Anti-type II interferon autoantibodies | 14 (7.0%) | 14 (9.3%) | 1 (0.7%) | 0.0038* | 0.0006* | 0.4339 |
| Anti-type III interferon autoantibodies | 12 (6.0%) | 11 (7.3%) | 7 (4.7%) | 0.5883 | 0.3381 | 0.6271 |
p-values calculated using Chi-square test.
By contrast, there was a striking paucity of anticytokine autoantibodies in RA (Figure 1C), most of which were against tumor necrosis factor (TNF) and lymphotoxin-α (LTα or TNFβ), in patients reported to be on TNF-blocking therapy. Etanercept is known to cross-react with LTα, and samples with autoantibodies against LTα were also positive for anti-TNF autoantibody. Interestingly, the majority of other anticytokine autoantibodies were directed against pro-inflammatory cytokines that may play a pathogenic role in RA: IL-12 (10 samples), IL-1α (8 samples), and IL-17F (3 samples).
Interestingly, all subjects who received rituximab, the anti-CD-20 monoclonal biologic, had low or absent levels of anticytokine autoantibodies. However, the causal role of this anti-B cell therapy on these autoantibody levels cannot be determined in this study and will require further exploration.
Functional Activity of Anticytokine Autoantibodies
As expected, type I interferon-induced phosphorylated signal transducer and activator of transcription-1 (STAT-1) in normal PBMC was unaffected when incubated with sera from patients and healthy controls that did not contain anti-type I interferon autoantibodies (Figure 2A). However, fifty percent (6 of 12) of SLE and 20% (1 of 5) of SS patients with anti-interferon-α autoantibodies prevented interferon-α-induced STAT-1 phosphorylation (Table S6). Fifty percent (5 of 10) of SLE and 14% (1 of 7) of SS patient sera with anti-interferon-β autoantibodies inhibited the action of their target cytokine, as well as 43% (3 of 7) SLE and 12% (1 of 8) of SS patient sera with autoantibodies against interferon-ω. Overall, nearly 50% of sera from SLE patients with anti-type I interferon autoantibodies were blocking, all of which were high-titer relative to other anti-type-I interferon-positive patients. IgG depletion experiments confirmed that the neutralizing activity was localized to the IgG subclass and not due to other immunoglobulin subclasses, serum factors, or extrinsic artifacts such as sodium heparin from the vacutainer (40) (Figure 3). In addition, the autoantibodies were specific, as no serum samples interfered with STAT-1 phosphorylation by an interferon for which they were autoantibody-negative. Only 3 of 149 patients with RA had anti-type I interferon autoantibodies, none of which were blocking, which is likely consistent with their observed low-titer (Figure 1C).
Figure 2. Examples of Neutralizing Anticytokine Autoantibodies.
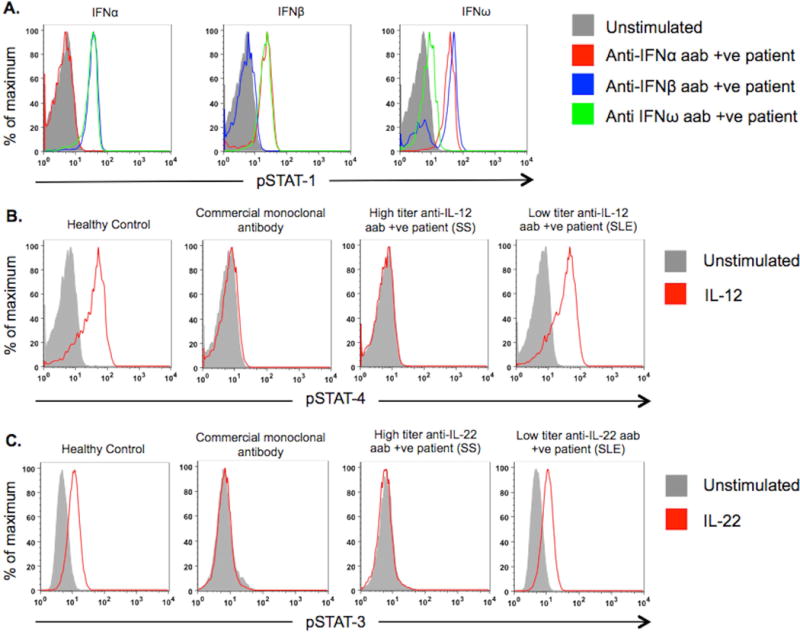
Representative examples of blocking antibodies demonstrated using (A) Normal PBMC incubated with healthy control or patient serum, left unstimulated or stimulated with interferon-α, interferon-β or interferon-ω. Interferon-induced STAT-1 phosphorylation was measured by flow cytometry. (B) Normal PBMC-derived lymphoblasts incubated with healthy control serum, healthy control serum spiked with 2μg/mL commercial monoclonal antibody against IL-12, high titer anti-IL-12 autoantibody patient serum, or serum from patient with low titer anti-IL-12-autoantibody, left unstimulated or stimulated with IL-12 and examined for phosphoSTAT-4 production. (C) A549 cells in presence of healthy control serum, healthy control serum spiked with commercial monoclonal antibody to IL-22 and sera from patients with high or low titer anti-IL-22 autoantibodies were left unstimulated or stimulated with IL-22.
aab: autoantibody; +ve: positive; IFN: interferon; IL: interleukin
Figure 3. Serum IgG Fraction Contains All the Interferon Neutralizing Activity.
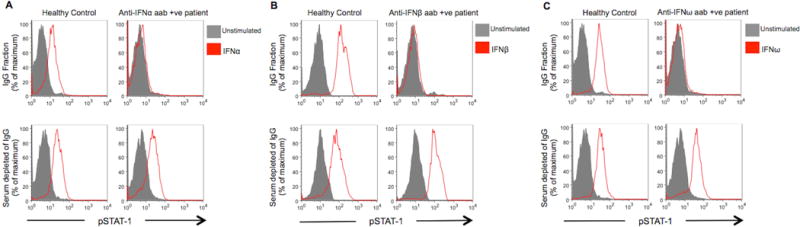
Total IgG, was captured on protein G (GE Healthcare) and the flow through collected prior to eluting the IgG fraction. Representative examples show the ability of each fraction to inhibit (A) interferon-α; (B) interferon-β or (C) interferon-ω induced STAT-1 phosphorylation in normal PBMC as measured by flow cytometry.
aab: autoantibody; +ve: positive; IFN: interferon
Other than type I interferons, only anticytokine autoantibodies directed against IL-12 and IL-22 exhibited blocking activity. Sera from 10% (1 in 10) of SLE patients and 55% (5 in 9) of SS patients with anti-IL-12 autoantibodies prevented IL-12-induced STAT-4 phosphorylation (Figure 2B). Serum from only one patient (with SS) with anti-IL-22 autoantibodies blocked IL-22-induced STAT-3 phosphorylation (Figure 2C) similar to that demonstrated in APECED (7) but was unassociated with candidiasis in our patient. Sera from 2 healthy controls, with high-titer autoantibodies, showed blocking activity, one against interferon-α, interferon-ω and IL-12 and the other to interferon-α only. None of the other anticytokine autoantibodies were active in functional assays (Figure S1A–O).
Anti-interferon Autoantibodies Track with Disease Activity and Affect Interferon-stimulated Gene Expression in SLE
Genes activated by type I interferons are upregulated in circulating leukocytes in SLE, and their degree of elevation correlates with higher disease activity (37). SLE patients with anti-interferon autoantibodies had higher SLEDAI at the time of sampling than those without (p=0.0075), were more likely to have depressed complement levels (p=0.0092) and had higher anti-double-stranded-DNA antibodies (p=0.0002) (Table 2) but no difference in the prevalence of nephritis. Remarkably, the elevation in SLEDAI and laboratory abnormalities was almost completely accounted for by anti-interferon-γ autoantibodies, rather than by anti-type I interferon autoantibodies (Table 2). This effect was specific to anti-interferon autoantibodies, as SLE patients with other anticytokine autoantibodies did not show any difference, including those autoantibodies that were identified with equally high frequency, (e.g., anti-IL-4 autoantibodies and anti-IL-12 autoantibodies) (Table S7). By contrast, anticytokine autoantibodies did not predict disease activity in either SS or RA patients based on the minor salivary gland biopsy focus score or Disease Activity Score including 28 joints (DAS28) scores (Table S7). None of the other subgroup analyses in SS or RA cohorts were significantly different (data not shown).
Table 2.
Association Between Disease Activity Indices and Presence of Anti-interferon Autoantibodies in SLE Patients
| Any anti-interferon aab negative (n=148) | Any anti-interferon aab positive (n=43) | p-value# | |
|---|---|---|---|
| SLEDAI Score, median (range) | 0 (0–33) | 2 (0–18) | 0.0075* |
| Low complements (C3, C4), n (%) | 35 (23.8) | 19 (44.2) | 0.0092* |
| Increased anti-dsDNA, n (%) | 41 (27.7) | 25 (58.1) | 0.0002* |
| Any anti-interferon-α aab negative (n=179) | Any anti-interferon-α aab positive (n=12) | p-value# | |
|---|---|---|---|
| SLEDAI Score, median (range) | 1 (0–33) | 2 (0–4) | 0.8334 |
| Low complements (C3, C4), n (%) | 29 (29.2) | 2 (16.7) | 0.5142 |
| Increased anti-dsDNA, n (%) | 35 (33.0) | 7 (41.7) | 0.0736 |
| Any anti-interferon-β aab negative (n=181) | Any anti-interferon-β aab positive (n=10) | p-value# | |
|---|---|---|---|
| SLEDAI Score, median (range) | 1 (0–33) | 2.5 (0–8) | 0.1597 |
| Low complements (C3, C4), n (%) | 49 (27.2) | 5 (50.0) | 0.1201 |
| Increased anti-dsDNA, n (%) | 60 (33.3) | 6 (60.0) | 0.0967 |
| Any anti-interferon-ω aab negative (n=184) | Any anti-interferon-ω aab positive (n=7) | p-value# | |
|---|---|---|---|
| SLEDAI Score, median (range) | 1.5 (0–33) | 2 (0–4) | 0.4868 |
| Low complements (C3, C4), n (%) | 53 (28.8) | 1 (14.3) | 0.6752 |
| Increased anti-dsDNA, n (%) | 62 (33.7) | 4 (57.1) | 0.2370 |
| Anti-interferon-γ aab negative (n=177) | Anti-interferon-γ aab positive (n=14) | p-value# | |
|---|---|---|---|
| SLEDAI Score, median (range) | 1 (0–33) | 4 (0–18) | 0.0022* |
| Low complements (C3, C4), n (%) | 47 (26.7) | 7 (50) | 0.0629 |
| Increased anti-dsDNA, n (%) | 55 (31.1) | 11 (78.6) | 0.0003* |
p-values calculated using Wilcoxon-Mann-Whitney test or Fisher’s exact test
SLEDAI: Systemic Lupus Erythematosus Disease Activity Index
anti-dsDNA: anti-double-stranded-DNA
aab: autoantibody
Low complements was defined as C3<80 mg/dl or C4<15 mg/dl and high anti-dsDNA antibody defined as >30 IU/mL.
To investigate whether anti-interferon autoantibodies in SLE patients altered the interferon signature associated with SLE, we correlated their presence or absence with RNA sequencing of the whole transcriptome of whole blood from 47 healthy controls and 95 SLE patients in the NIH cohort. Blood samples from SLE patients had a 4-fold overexpression of these interferon-α/β-inducible genes compared to healthy controls (Figure 4). Anti-interferon-α autoantibodies, particularly those with blocking activity, normalized expression of type-I interferon inducible genes, similar to the effects of therapeutic monoclonal anti-interferon-α (35). In fact, out of the 56 SLE patients with low interferon gene signature, 7 (12.5%) had anti-interferon-α autoantibodies. Conversely, SLE patients with anti-interferon-γ autoantibodies had 8-fold overexpression of interferon-α/β-inducible genes compared to patients with anti-interferon-α autoantibodies and to healthy controls. Consistent with these observations, increasing titers of anti-interferon-γ autoantibodies had a significant positive, though weak, correlation with interferon signature (r=0.3061, p=0.0027) and SLEDAI (r=0.3205, p=<0.0001) (Figure S2). Interestingly, 1 patient with anti-interferon-γ autoantibodies also had blocking anti-interferon-α autoantibodies and had normal type I gene expression, further confirming that these autoantibodies act similarly to therapeutic monoclonals. Analysis of 7 genes more specifically induced by interferon-γ (36–38) (Table S3) showed that they were not as elevated in SLE (Figure S3), similar to previous reports (37, 38). Taken together, these data suggest that anti-interferon autoantibodies in SLE are active in vivo and affect cellular aspects of disease expression. These data also suggest distinct functional roles for autoantibodies against type I and type II interferons in SLE that differ from those in patients with other anticytokine autoantibody-associated immunodeficiencies.
Figure 4. Interferon-α/β-inducible Genes and Anti-interferon Autoantibodies in SLE Patients.
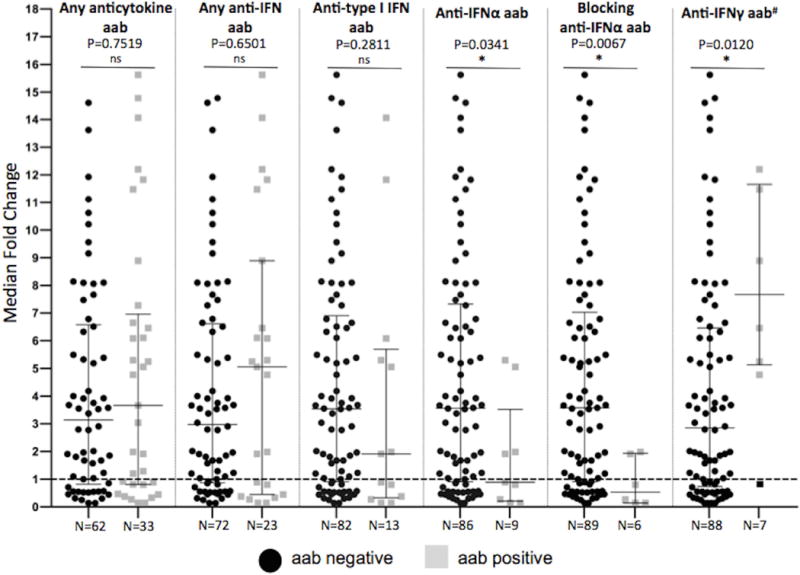
Median fold induction of 21 interferon-α/β-inducible genes in the NIH cohort of SLE patients (n=95) when compared to 47 healthy controls (dashed line). Patient subsets are compared based on presence or absence of specific autoantibodies. The horizontal bars represent the median with interquartile ranges. P-value determined by using Mann-Whitney test.
#: one patient with anti-interferon-γ autoantibody also had blocking anti-interferon-α autoantibody and is shaded black and excluded from the analysis.
aab: autoantibody; +ve: positive; IFN: interferon
Discussion
Using large international cohorts of carefully characterized patients we were able to identify distinct spectra of anticytokine autoantibodies, beyond only those directed at interferon-α, in SLE and SS that differed from those found in RA. Important to understanding the biology and management of these diseases, we found significant associations of anti-interferon autoantibodies with both clinical and in vitro aspects of disease. Whereas anti-type I interferon autoantibodies appeared to ameliorate the transcriptional profile but not the clinical aspects of SLE, autoantibodies to interferon-γ were associated with exacerbated disease activity and more pronounced in vitro signatures. SLE sera had the broadest range of autoantibodies in both number and activity, with higher frequency, more patients with more than one autoantibody, and more targeting of type I interferons.
The increased clinical severity and interferon signature seen in SLE patients with anti-interferon-γ autoantibodies is novel and appears specific, as no other anticytokine autoantibody we examined showed this activity. It is unexpected, insofar as previous studies have suggested that interferon-γ is an exacerbating factor in SLE, so it might have been anticipated that its neutralization would be ameliorative (41). It remains unclear whether the autoantibodies to interferons are a consequence of high levels of interferons seen in severe SLE or are independent disease modifiers. Although these anti-interferon-γ autoantibodies did not activate STAT-1 directly, in vivo they may interact with other factors to allow activation of STAT-1 and interferon-responsive genes. In the setting of interferon-γ receptor deficiency interferon-α signaling is increased for certain targets, which may explain part of the transcriptome effect we see in the presence of anti-interferon-γ autoantibodies (42).
Autoantibodies targeting types I and II interferons, granulocyte monocyte-colony stimulating factor, TNF, LTα, IL-1α, IL-6, IL-10, and IL-12 have been previously described in reports looking at small populations and one or few cytokine targets (8–16). However, autoantibodies to macrophage-colony stimulating factor, IL-4, IL-7, IL-17 and IL-22 have not previously been identified in rheumatologic conditions, nor have autoantibodies to type III interferons (λ2, λ3) or IP-10 been previously reported in humans. The few patients with RA who had circulating autoantibodies not attributable to therapy targeted IL-12, IL17F and IL-1α, distinct from those targeted in SLE and SS. The presence of blocking anticytokine autoantibodies in only 1% of the presumably healthy controls may reflect elicitation of antibodies for some other reason, subclinical disease or a predilection for future rheumatologic disease. Longitudinal follow up of asymptomatic normals would help address whether these are transient or permanent and whether they have prognostic importance. Alternatively, this may simply reflect that anticytokine autoantibodies can rarely occur in the absence of disease and are only one factor in complex syndromes.
The high incidence of anticytokine autoantibodies in SLE and SS may reflect their nature as systemic autoimmune diseases with multi-organ involvement, in which immunological self-tolerance is profoundly breached. In contrast, in RA the synovium is the main target of autoimmunity and extra-articular manifestations are rare. Immunization with cytokines complexed to protein carriers without adjuvant can elicit neutralizing and long-lasting anticytokine autoantibodies, suggesting that B cells harboring anticytokine specificities latently circulate, even in normal individuals (43). Presentation of cytokines by activated antigen presenting cells could contribute to providing T cell help to elicit anticytokine autoantibodies in the setting of ongoing autoimmune disease. Whether anticytokine autoantibodies, like anti-citrullinated protein autoantibodies and rheumatoid factor in RA, and anti-nuclear antibodies in SLE, precede or result from the onset of clinical disease is unknown. It is likely relevant that many of the cytokines most frequently targeted by anticytokine autoantibodies, such as interferons in SLE and SS, and IL-1α and IL-12 in RA, have been implicated in the pathogenesis of the diseases in which they are found. It will be important to determine whether anticytokine autoantibodies are seen in systemic autoinflammatory diseases such as familial Mediterranean fever, in which autoreactive B and T cells are lacking. These diseases may help distinguish between the development of anticytokine autoantibodies because of abundant cytokine exposure versus their arising as part of the underlying autoimmune diathesis.
While some anticytokine autoantibodies in rheumatologic disease may be due to generalized B cell hyperactivation and may not be disease modifying, the anti-interferon-γ autoantibodies we have identified are strongly associated with a distinct disease course. Anti-interferon-α autoantibodies appeared to be functional in vivo, since type I interferon-induced genes were reduced to baseline in individuals with neutralizing interferon-α autoantibodies. The frequency of anti-interferon-α autoantibodies and their association with reduced interferon transcriptional signature are consistent with the results of Morimoto et al (11), who found a reduction in laboratory and clinical parameters of SLE in those with reduced interferon signatures associated with anti-interferon-α autoantibodies. We did not find such a clinical effect, perhaps due to different recruitment criteria. In addition, while rituximab may be an important therapeutic agent in adult-onset immunodeficiency associated with anti-interferon-γ autoantibodies (44), its impact on anticytokine autoantibodies in SLE, SS, and RA patients will require analysis of treated patients over time with careful monitoring.
Understanding the role of anticytokine autoantibodies in disease pathogenesis remains limited by the diagnostic techniques and the broad range of potential targets. For example, the etiology of pulmonary alveolar proteinosis remained obscure for over 40 years after its initial description until autoantibodies to granulocyte monocyte-colony stimulating factor (GM-CSF) were proven to be causal (45, 46). This identification not only shed light on the biology of surfactant homeostasis but also led to therapeutic options including recombinant GM-CSF (47, 48) and rituximab (49) Similarly, the identification of anti-interferon-γ autoantibodies in Thai and Taiwanese patients with disseminated opportunistic infections came over a decade after their initial description and expanded our understanding of interferon-γ biology and possible therapies including rituximab (1, 44).
These data show that anticytokine autoantibodies are a common feature of systemic autoimmune diseases and extend to more cytokines than previously appreciated. Anti-interferon autoantibodies in SLE can have biological activity both in vitro and in vivo, possibly altering course and expression of clinical disease. Determining the influence that anti-interferon autoantibodies have on disease expression is imperative, since we are now in an age of cytokine and anticytokine therapeutics. Endogenous anti-interferon antibodies may affect the success of anti-interferon monoclonal antibodies. In addition to their roles in disease pathogenesis, better understanding of the nature and biological function of anticytokine autoantibodies may help inform the design of future therapeutic approaches. With the development of targeted agents and the promise of personalized medicine, the time to fully understand each patient’s disease, and the epigenetic modifiers such as anticytokine autoantibodies, is upon us.
Supplementary Material
Acknowledgments
We thank Dr. Peter K. Gregersen at the Feinstein Institute for Medical Research, Manhasset, New York for providing access to Autoimmune Biomarkers Collaborative Network (ABCoN) clinical samples.
Funding: This work was supported by the Divisions of Intramural Research, National Institute of Allergy and Infectious Diseases and National Institute of Arthritis and Musculoskeletal and Skin Diseases, both at the National Institutes of Health, Bethesda, Maryland. Dr. Gupta received a Rheumatology Research Foundation Scientist Development Award from the American College of Rheumatology.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Author Contributions:
S.G. guided study design, developed and performed the screening and functional assays, and analyzed all data. I.P.T. collected and collaborated in data analysis on the Greek cohort. L.B.R. developed, performed and analyzed functional assays. S.H. provided sera and clinical data from NIH SLE cohort and collaborated in data analysis. I.A. provided sera and clinical data from NIH SS cohort and collaborated in data analysis. Z.G.M. performed statistical analyses. J.R. and C.J. performed RNA sequencing and analysis. R.M.S. and S.M.H. guided study design and data analysis/interpretation. H.M.M. provided sera and clinical data from Greek cohort and data analysis/interpretation. S.K.B. guided study design and data analysis/interpretation. All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved final version.
References
- 1.Browne SK, Burbelo PD, Chetchotisakd P, Suputtamongkol Y, Kiertiburanakul S, Shaw PA, et al. Adult-onset immunodeficiency in Thailand and Taiwan. The New England journal of medicine. 2012;367(8):725–34. doi: 10.1056/NEJMoa1111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. The New England journal of medicine. 2003;349(26):2527–39. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 3.Casadevall N, Dupuy E, Molho-Sabatier P, Tobelem G, Varet B, Mayeux P. Autoantibodies against erythropoietin in a patient with pure red-cell aplasia. The New England journal of medicine. 1996;334(10):630–3. doi: 10.1056/NEJM199603073341004. [DOI] [PubMed] [Google Scholar]
- 4.Browne SK, Holland SM. Anticytokine autoantibodies in infectious diseases: pathogenesis and mechanisms. The Lancet infectious diseases. 2010;10(12):875–85. doi: 10.1016/S1473-3099(10)70196-1. [DOI] [PubMed] [Google Scholar]
- 5.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298(5597):1395–401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 6.Kisand K, Peterson P. Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy: known and novel aspects of the syndrome. Ann N Y Acad Sci. 2011;1246:77–91. doi: 10.1111/j.1749-6632.2011.06308.x. [DOI] [PubMed] [Google Scholar]
- 7.Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. The Journal of experimental medicine. 2010;207(2):299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Panem S, Check IJ, Henriksen D, Vilcek J. Antibodies to alpha-interferon in a patient with systemic lupus erythematosus. J Immunol. 1982;129(1):1–3. [PubMed] [Google Scholar]
- 9.Suit BE, Axelrod D, Moutsopoulos HM, Decker JL, Hooks JJ. Detection of anti-interferon antibodies in systemic lupus erythematosus. Clinical and experimental rheumatology. 1983;1(2):133–5. [PubMed] [Google Scholar]
- 10.Slavikova M, Schmeisser H, Kontsekova E, Mateicka F, Borecky L, Kontsek P. Incidence of autoantibodies against type I and type II interferons in a cohort of systemic lupus erythematosus patients in Slovakia. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2003;23(3):143–7. doi: 10.1089/107999003321532475. [DOI] [PubMed] [Google Scholar]
- 11.Morimoto AM, Flesher DT, Yang J, Wolslegel K, Wang X, Brady A, et al. Association of endogenous anti-interferon-alpha autoantibodies with decreased interferon-pathway and disease activity in patients with systemic lupus erythematosus. Arthritis and rheumatism. 2011;63(8):2407–15. doi: 10.1002/art.30399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meager A, Wadhwa M, Dilger P, Bird C, Thorpe R, Newsom-Davis J, et al. Anti-cytokine autoantibodies in autoimmunity: preponderance of neutralizing autoantibodies against interferon-alpha, interferon-omega and interleukin-12 in patients with thymoma and/or myasthenia gravis. Clinical and experimental immunology. 2003;132(1):128–36. doi: 10.1046/j.1365-2249.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graudal NA, Svenson M, Tarp U, Garred P, Jurik AG, Bendtzen K. Autoantibodies against interleukin 1alpha in rheumatoid arthritis: association with long term radiographic outcome. Annals of the rheumatic diseases. 2002;61(7):598–602. doi: 10.1136/ard.61.7.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans M, Abdou NI. Anti-interleukin-6 and soluble interleukin-6 receptor in systemic lupus erythematosus. Lupus. 1994;3(3):161–6. doi: 10.1177/096120339400300306. [DOI] [PubMed] [Google Scholar]
- 15.Sjowall C, Ernerudh J, Bengtsson AA, Sturfelt G, Skogh T. Reduced anti-TNFalpha autoantibody levels coincide with flare in systemic lupus erythematosus. Journal of autoimmunity. 2004;22(4):315–23. doi: 10.1016/j.jaut.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Takemura H, Suzuki H, Yoshizaki K, Ogata A, Yuhara T, Akama T, et al. Anti-interleukin-6 autoantibodies in rheumatic diseases. Increased frequency in the sera of patients with systemic sclerosis. Arthritis and rheumatism. 1992;35(8):940–3. doi: 10.1002/art.1780350814. [DOI] [PubMed] [Google Scholar]
- 17.Hellmich B, Csernok E, Schatz H, Gross WL, Schnabel A. Autoantibodies against granulocyte colony-stimulating factor in Felty’s syndrome and neutropenic systemic lupus erythematosus. Arthritis and rheumatism. 2002;46(9):2384–91. doi: 10.1002/art.10497. [DOI] [PubMed] [Google Scholar]
- 18.Schett G, Firbas U, Fureder W, Hiesberger H, Winkler S, Wachauer D, et al. Decreased serum erythropoietin and its relation to anti-erythropoietin antibodies in anaemia of systemic lupus erythematosus. Rheumatology (Oxford) 2001;40(4):424–31. doi: 10.1093/rheumatology/40.4.424. [DOI] [PubMed] [Google Scholar]
- 19.Price JV, Haddon DJ, Kemmer D, Delepine G, Mandelbaum G, Jarrell JA, et al. Protein microarray analysis reveals BAFF-binding autoantibodies in systemic lupus erythematosus. The Journal of clinical investigation. 2013;123(12):5135–45. doi: 10.1172/JCI70231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uchida K, Nakata K, Suzuki T, Luisetti M, Watanabe M, Koch DE, et al. Granulocyte/macrophage-colony-stimulating factor autoantibodies and myeloid cell immune functions in healthy subjects. Blood. 2009;113(11):2547–56. doi: 10.1182/blood-2009-05-155689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wildbaum G, Nahir MA, Karin N. Beneficial autoimmunity to proinflammatory mediators restrains the consequences of self-destructive immunity. Immunity. 2003;19(5):679–88. doi: 10.1016/s1074-7613(03)00291-7. [DOI] [PubMed] [Google Scholar]
- 22.Browne SK. Anticytokine autoantibody-associated immunodeficiency. Annual review of immunology. 2014;32:635–57. doi: 10.1146/annurev-immunol-032713-120222. [DOI] [PubMed] [Google Scholar]
- 23.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis and rheumatism. 1982;25(11):1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 24.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis and rheumatism. 1988;31(3):315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 25.Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjogren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Annals of the rheumatic diseases. 2002;61(6):554–8. doi: 10.1136/ard.61.6.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis and rheumatism. 1992;35(6):630–40. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- 27.Chisholm DM, Mason DK. Labial salivary gland biopsy in Sjogren’s disease. Journal of clinical pathology. 1968;21(5):656–60. doi: 10.1136/jcp.21.5.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prevoo ML, van ‘t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis and rheumatism. 1995;38(1):44–8. doi: 10.1002/art.1780380107. [DOI] [PubMed] [Google Scholar]
- 29.Ding L, Mo A, Jutivorakool K, Pancholi M, Holland SM, Browne SK. Determination of human anticytokine autoantibody profiles using a particle-based approach. Journal of clinical immunology. 2012;32(2):238–45. doi: 10.1007/s10875-011-9621-8. [DOI] [PubMed] [Google Scholar]
- 30.Holland SM, Dorman SE, Kwon A, Pitha-Rowe IF, Frucht DM, Gerstberger SM, et al. Abnormal regulation of interferon-gamma, interleukin-12, and tumor necrosis factor-alpha in human interferon-gamma receptor 1 deficiency. The Journal of infectious diseases. 1998;178(4):1095–104. doi: 10.1086/515670. [DOI] [PubMed] [Google Scholar]
- 31.Burbelo PD, Browne SK, Sampaio EP, Giaccone G, Zaman R, Kristosturyan E, et al. Anti-cytokine autoantibodies are associated with opportunistic infection in patients with thymic neoplasia. Blood. 2010;116(23):4848–58. doi: 10.1182/blood-2010-05-286161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He KL, Ting AT. A20 inhibits tumor necrosis factor (TNF) alpha-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Molecular and cellular biology. 2002;22(17):6034–45. doi: 10.1128/MCB.22.17.6034-6045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isaac J, Erthal J, Gordon J, Duverger O, Sun HW, Lichtler AC, et al. DLX3 regulates bone mass by targeting genes supporting osteoblast differentiation and mineral homeostasis in vivo. Cell death and differentiation. 2014;21(9):1365–76. doi: 10.1038/cdd.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao Y, Higgs BW, Morehouse C, de Los Reyes M, Trigona W, Brohawn P, et al. Development of Potential Pharmacodynamic and Diagnostic Markers for Anti-IFN-alpha Monoclonal Antibody Trials in Systemic Lupus Erythematosus. Human genomics and proteomics: HGP. 2009;2009 doi: 10.4061/2009/374312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crow MK, Wohlgemuth J. Microarray analysis of gene expression in lupus. Arthritis research & therapy. 2003;5(6):279–87. doi: 10.1186/ar1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis and rheumatism. 2005;52(5):1491–503. doi: 10.1002/art.21031. [DOI] [PubMed] [Google Scholar]
- 38.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2610–5. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward JH. Hierarchical Grouping to Optimize an Objective Function. Journal of the American Statistical Association. 1963;58(301):236–44. [Google Scholar]
- 40.Fritchley SJ, Kirby JA, Ali S. The antagonism of interferon-gamma (IFN-gamma) by heparin: examination of the blockade of class II MHC antigen and heat shock protein-70 expression. Clinical and experimental immunology. 2000;120(2):247–52. doi: 10.1046/j.1365-2249.2000.01178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Theofilopoulos AN, Koundouris S, Kono DH, Lawson BR. The role of IFN-gamma in systemic lupus erythematosus: a challenge to the Th1/Th2 paradigm in autoimmunity. Arthritis Res. 2001;3(3):136–41. doi: 10.1186/ar290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bax HI, Freeman AF, Ding L, Hsu AP, Marciano B, Kristosturyan E, et al. Interferon alpha treatment of patients with impaired interferon gamma signaling. Journal of clinical immunology. 2013;33(5):991–1001. doi: 10.1007/s10875-013-9882-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zagury D, Le Buanec H, Mathian A, Larcier P, Burnett R, Amoura Z, et al. IFNalpha kinoid vaccine-induced neutralizing antibodies prevent clinical manifestations in a lupus flare murine model. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(13):5294–9. doi: 10.1073/pnas.0900615106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Browne SK, Zaman R, Sampaio EP, Jutivorakool K, Rosen LB, Ding L, et al. Anti-CD20 (rituximab) therapy for anti-IFN-gamma autoantibody-associated nontuberculous mycobacterial infection. Blood. 2012;119(17):3933–9. doi: 10.1182/blood-2011-12-395707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitamura T, Tanaka N, Watanabe J, Uchida, Kanegasaki S, Yamada Y, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. The Journal of experimental medicine. 1999;190(6):875–80. doi: 10.1084/jem.190.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol. 2010;135(2):223–35. doi: 10.1016/j.clim.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kavuru MS, Sullivan EJ, Piccin R, Thomassen MJ, Stoller JK. Exogenous granulocyte-macrophage colony-stimulating factor administration for pulmonary alveolar proteinosis. American journal of respiratory and critical care medicine. 2000;161(4 Pt 1):1143–8. doi: 10.1164/ajrccm.161.4.9906044. [DOI] [PubMed] [Google Scholar]
- 48.Tazawa R, Trapnell BC, Inoue Y, Arai T, Takada T, Nasuhara Y, et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. American journal of respiratory and critical care medicine. 2010;181(12):1345–54. doi: 10.1164/rccm.200906-0978OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kavuru MS, Malur A, Marshall I, Barna BP, Meziane M, Huizar I, et al. An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. The European respiratory journal. 2011;38(6):1361–7. doi: 10.1183/09031936.00197710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.