

Abstract
Naturally produced polybrominated diphenyl ethers (PBDEs) pervade the marine environment and structurally resemble toxic man-made brominated flame retardants. PBDEs bioaccumulate in marine animals and are likely transferred to the human food chain. However, the biogenic basis for PBDE production in one of their most prolific sources, marine sponges of the order Dysideidae, remains unidentified. Here, we report the discovery of PBDE biosynthetic gene clusters within sponge microbiome-associated cyanobacterial endosymbionts by employing an unbiased metagenome mining approach. By expression of PBDE biosynthetic genes in heterologous cyanobacterial hosts, we correlate the structural diversity of naturally produced PBDEs to modifications within PBDE biosynthetic gene clusters in multiple sponge holobionts. Our results establish the genetic and molecular foundation for the production of PBDEs in one of the most abundant natural sources of these molecules, further setting the stage for a metagenomic-based inventory of other PBDE sources in the marine environment.
Keywords: PBDE, cyanobacteria, natural product, microbiome, metagenome, sponge
Halogenated organic compounds have a sordid record in history (http://chm.pops.int/). Once widely used in a variety of applications, notorious man-made polyhalogenated aromatic molecules such as DDT (insecticide), PCBs (industrial coolants), and polybrominated diphenyl ethers (PBDEs, fire retardants) have been restricted from commercial use owing to their toxicity and bioaccumulation in the environment (Figure 1A). Polyhalogenated dibenzo-p-dioxins like TCDD are widely recognized to be among the most toxic and environmentally persistent man-made pollutants1. Remarkably, nature is also a prolific producer of polyhalogenated organic compounds, including PBDEs that mimic and even surpass the toxicity associated with their anthropogenic counterparts, such as BDE47 and deca-BDE2.
Figure 1. Synthetic and naturally occurring polyhalogenated molecules.

Structural similarity between (A) synthetic and (B) naturally produced marine halogenated phenolic toxins and pollutants.
Distinguished from man-made PBDEs by the presence of additional hydroxyl and methoxyl moieties, naturally produced hydroxylated polybrominated diphenyl ethers (OH-BDEs) such as 2OH-BDE68 (1) and 6OH-BDE47 (2) and their respective methoxylated forms-2MeO-BDE68 (3) and 6MeO-BDE47 (4, Figure 1B), are ubiquitous natural product chemicals found across all trophic levels in marine biota, from cyanobacteria and macroalgae to whales and sea-birds3. While we recently reported the biosynthetic basis for the natural production of polybrominated phenolics, including PBDEs, by marine γ-proteobacteria4, the most prodigious natural sources of PBDEs in the marine environment are the benthic filter feeding marine sponges (Porifera) within the family Dysideidae5-8. In Dysideidae, PBDEs can exceed 10% of the sponge tissue by dry weight9,10. PBDE containing Dysideidae are ubiquitous in the Indo-Pacific subtropical region stretching from India to French Polynesia. In addition to OH-BDEs such as 1 and 2, Dysideidae sponges harbor methoxylated derivatives of dihydroxylated polybrominated diphenyl ethers (di-OH-BDEs) such as 2MeO,6’OH-BDE68 (5), thought to be derived via methylation of 2,6’OH-BDE68 (6), and polybrominated dioxins such as spongiadioxin C (7) with its corresponding methoxylated derivative 8 (Figure 1B)11,12. Naturally produced PBDEs and dioxins are moreover routinely detected in marine animals13-15, including human analytes16-18, indicating that sponge-derived polybrominated natural products, among other natural sources, bioaccumulate in the marine food web with potential for transfer to humans.
While the natural occurrence of PBDEs in the Dysideidae has been extensively documented over the last four decades, the genetic and molecular basis for their production has not been established. There are two principal factors that have precipitated this information gap. First, cell sorting and chemical localization studies revealed that Dysideidae-derived PBDEs localize to endosymbiotic filamentous cyanobacteria present in the sponge microbiome9. However, compound localization is not conclusive evidence of biogenic origin as natural products may be compartmentalized away from their biosynthetic source in complex communities19-22. The primary cyanobacterial symbiont of Dysideidae sponges has been described as Hormoscilla spongeliae (formerly Oscillatoria spongeliae23). Having thus far resisted laboratory cultivation24 and characterized only by morphology9, cell ultrastructure9,24, and 16S rRNA gene signatures25,26, genomic data was not previously available for H. spongeliae. Second, and in concert with our previous description of PBDE biogenesis from marine γ-proteobacteria4, chemical structures of naturally produced PBDEs and dioxins imply that genes encoding their biosynthesis cannot be expected to resemble modular natural product assembly lines that lend themselves to computational mining and discovery from genomic and metagenomic datasets27. Furthermore, in light of published literature28,29 invoking the contribution of an entirely different class of halogenating enzymes in the biosynthesis of polybrominated phenolics than what we had characterized previously from marine Pseudoalteromonas and Marinomonas bacteria4, it was not apparent that the biochemical logic for the production in PBDEs in marine γ-proteobacteria would be preserved in Dysideidae sponges.
In this report, we describe a chemical hypothesis-directed metagenome mining approach to identify genes responsible for the biosynthesis of naturally produced PBDEs and polybrominated dioxins in three phylogenetically distinct clades of Dysideidae sponges. Using an approach that does not bias the search for biosynthetic genes towards either the sponge host or the symbiotic microbial community, we demonstrate that the PBDE biosynthetic genes reside in H. spongeliae symbionts, while no candidate genes were found in the host or other associated organisms. Biosynthetic genes identified by whole tissue metagenomic sequencing were experimentally validated by heterologous expression in a cyanobacterial host. In addition, structural diversity of PBDEs present in different sponge metabolomes were linked to biosynthetic gene cluster variability among closely-related H. spongeliae strains in three different Dysideidae holobionts. Our results provide genetic and biochemical logic for the production of polybrominated toxins from one of the most prominent natural sources of PBDEs in the ocean, and now set the stage for metagenomic inventorying and discovery of other geographically dispersed producers.
RESULTS
Correlation of PBDE chemistry with sponge holobionts
We collected eighteen sponge specimens morphologically similar to previously described Lamellodysidea herbacea and Dysidea granulosa8 from multiple sites within the U.S. Territory of Guam in 2014 and 2015 (Supplementary Results, Supplementary Figure 1, Supplementary Table 1). A molecular phylogeny of sponge ribosomal Internal Transcribed Spacer (ITS-2) sequences derived from these samples along with sequences previously reported from other Indo-Pacific locations25,30,31 revealed four well-supported, monophyletic clades (Figure 2A, Supplementary Figure 2). Clades I–III encompass a taxonomically contested group of Lamellodysidea/Lendenfeldia/Dysidea genera32, whereas Clade IV corresponds to Dysidea granulosa. For clarity, in this report, we refer to each sponge group by the clades shown in Figure 2A rather than genus and species nomenclature. Multiple samples were obtained corresponding to Clades Ia, Ib, III, and IV, while no specimens belonging to the Dysideidae Clade II were identified in our collection.
Figure 2. Host and symbiont phylogenies.

(A) Left: Midpoint-rooted, maximum likelihood (ML) phylogeny based on sponge ribosomal ITS-2 sequences from the current study (in bold) along with representative literature examples. Identical sequences are listed at a single branch tip, with symbols denoting dominant natural product chemistry. Bootstrap values ≥70 are shown; scale bar indicates substitutions per site. Matching superscripts indicate literature ITS-2 and 16S rRNA sequences obtained from the same sponge specimen. Right: H. spongeliae ML tree of near-full length 16S rRNA gene sequences, including closest free-living relative Oscillatoria corallinae, and rooted on closest relative with a sequenced genome, Moorea producens 3L. One full-length 16S rRNA gene sequence (GUM098) was obtained from clone library (see Figure 3). Remaining Clade IV 16S rRNA sequences in this study, denoted in parentheses, are represented by 370 bp sequences obtained from high-throughput community data that are 100% identical to the GUM098 full-length sequence. (B) Chemical profiles, as monitored by absorbance at 280 nm by HPLC with in situ photographs of representative sponges for each clade.
Mass spectrometry and NMR analyses of sponge extracts revealed clear correlations between the sponge ITS-2 sequence phylogeny (Figure 2A) and polybrominated phenolic chemistry (Figure 2B). Consistent with our previous characterization of PBDEs from Dysideidae8, the most abundant natural product in Clade Ia specimens was 5, together with minor amounts of 1 as detected by LC/MS/MS (Supplementary Figure 3). In contrast, molecule 1 dominated the product profile of Clade Ib. The D. granulosa Clade IV also contained 1, but in lower abundance than 2. We recovered high quantities of polybrominated phenolics from each of the Clade Ia, Ib, and IV sponge specimens with PBDEs vastly dominating the small molecule sponge metabolomes (Figure 2B). In contrast, Clade III samples from this study contained neither PBDEs, nor polychlorinated peptides such as dysidenin previously reported in other Dysidea sponges with ITS-2 sequences falling in this clade25,26,33. Nevertheless, all sponge specimens collected in this study, including those from Clade III, contained cyanobacterial assemblages morphologically similar to those shown in Supplementary Figure 4. 16S rRNA gene sequence analysis revealed that cyanobacteria derived from closely related sponges always claded together with a high level of congruence between the H. spongeliae 16S rRNA and sponge ITS-2 phylogenies (Figure 2A).
Marine sponges are associated with complex microbiomes34,35. To gain insights into the microbial community composition of the different sponges, we inventoried the microbiomes of two specimens from each of Clades Ia, Ib, III, and IV by 16S rRNA gene amplification and deep Illumina sequencing, generating between 75,000-250,000 reads per sample (Supplementary Table 2). In each of sponge Clades Ia, Ib, and III, H. spongeliae constituted a large fraction of the sponge-associated bacterial community (Figure 3). In contrast, a lower abundance of H. spongeliae in Clade IV correlated with a greater overall microbial diversity than the other three sponge clades.
Figure 3. Representative 16S rRNA gene diversity profiles.
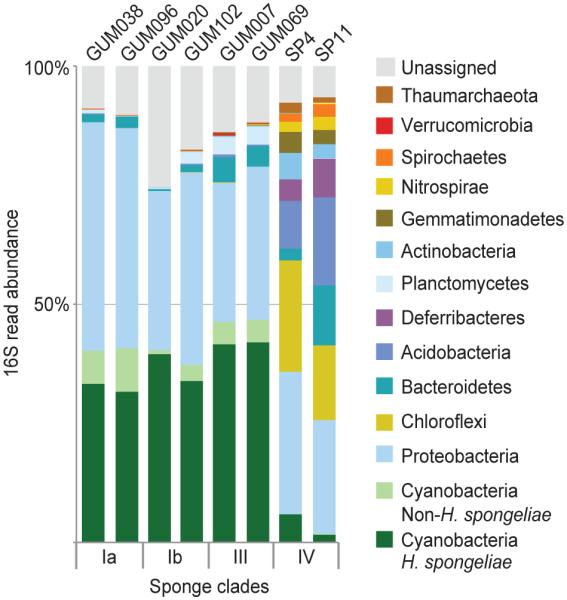
Genomic DNA for 16S amplification was isolated from cross sections of sponge tissues. Taxonomic groups are shown at the phylum level, with the exception of operational taxonomic units identified as H. spongeliae (dark green). For clarity, groups representing <0.1% of the data are not labeled.
Metagenomic identification of PBDE biosynthetic genes
DNA isolated from sponge tissues contained a complex mixture of sponge host plus commensal, symbiotic, parasitic, pathogenic, and/or prey microorganisms, providing us with an opportunity to identify genes encoding PBDE biosynthesis regardless of their species of origin. We used our previous description of the operon structures and chemical mechanisms responsible for producing polybrominated phenolics in marine γ-proteobacteria4,36 to guide our search for genes mediating biosynthesis of similar products in Dysideidae sponges. Briefly, biosynthesis of PBDEs in Pseudoalteromonas starts with conversion of chorismic acid to p-hydroxybenzoic acid (9, Figure 4A) by the chorismate lyase Bmp6, followed by decarboxylative bromination of 9 to 2,4-dibromophenol (10) by the flavin-dependent brominase Bmp5. Intermediate 10 then undergoes oxidative coupling by the cytochrome P450 enzyme (CYP450) Bmp7 to yield PBDEs such as 1, along with other regioisomeric products. Pseudoalteromonas genes bmp1–4, together with bmp8, have been shown to encode proteins involved in bromopyrrole synthesis37,38 and do not participate in the biosynthesis of polybrominated phenolics. Hence, we focused on a diagnostic pattern of Pseudoalteromonas bmp5–7 genes to query for PBDE biosynthetic genes in Dysideidae sponges.
Figure 4. Discovery of hs_bmp gene clusters.

(A) Previously identified bmp gene loci in the marine bacterium Pseudoalteromonas luteoviolacea 2ta16. Genes bmp1–4, together with bmp8, participate in the biosynthesis of bromopyrroles, while bmp9–10, encoding ferredoxin and ferredoxin reductase respectively, are electron transporters for the activity of CYP450-Bmp7. The catalytic roles of bmp5 gene products (yellow), bmp6 (blue), and bmp7 (red) are color-coded. In addition to ortho-OH-BDEs such as 1, catalytically promiscuous CYP450-Bmp7 also generates para-OH-BDE and biphenyl products as shown. (B) All Clade Ia (Sample SP12) scaffolds greater than 5 kb in length are displayed in grey. The scaffold containing the hs_bmp operon is designated by a gold circle. Other colored points represent scaffolds classified by amino acid sequence similarity of multiple predicted proteins to known sequences from the GenBank non-redundant database. (C) The color scheme from panel A is preserved for hs_bmp genes. Nucleotide identity ≥90% between different regions of the hs_bmp gene loci from different sponge clades is shown in grey shading.
In line with previous reports using sequence homologies of natural product biosynthetic enzymes to identify sponge-derived biosynthetic genes by degenerate primer design and PCR39,40, we initially attempted a PCR-based experimental approach to identify Pseudoalteromonas bmp5 homologs in the Dysideidae metagenomic DNA. However, no PCR amplicons could be generated. This prompted us to take a de novo metagenomic sequencing and assembly approach, enhancing computational detection of taxonomically distant orthologs by using amino acid rather than nucleic acid based sequence searches. Starting our analysis with Clade Ia sample SP12, assembled metagenomic scaffolds were classified taxonomically by comparing predicted proteins from all potential open reading frames (ORFs) to previously characterized proteins in the GenBank non-redundant (nr) database using the DarkHorse program to identify closest taxonomic relatives41 (Supplementary Table 3). Although not all scaffolds could be explicitly assigned to known taxa, in many cases, results were clear and unambiguous. Scaffolds greater than 5 kb containing five or more genes that all matched a single phylogenetic group were assigned to that group. When sequence similarity-based taxonomic results for the scaffolds from Clade Ia sample SP12 were combined with nucleotide composition (percent G+C) and depth of coverage (a measure of relative DNA abundance in the original sample), clusters of several dominant groups were clearly distinguishable (Figure 4B). These clusters included the sponge host, identified by matches to Porifera proteins, as well as representatives of Cyanobacteria, Bacteroidetes, and Alphaproteobacteria.
One scaffold of 15,853 nucleotides from the Clade Ia sample SP12 metagenome contained adjacent ORFs encoding homologs of the Pseudoalteromonas Bmp5–7 enzymes, as well as an additional CYP450 hydroxylase (hs_bmp12, see below). Three independent lines of evidence suggested that the consensus metagenomic sequences in this scaffold originated from cyanobacterial symbionts rather than the sponge host. First, when metagenomic scaffolds were clustered by nucleotide composition (percent G+C), depth of coverage, and predicted amino acid sequence matches to the GenBank non-redundant database, the scaffold containing bmp gene candidates clearly fell within the high abundance cyanobacterial cluster and well outside the range of identified sponge (Porifera) sequences (Figure 4B). Second, all sixteen predicted genes on this scaffold had closest GenBank matches to proteins from previously sequenced cyanobacteria (Supplementary Table 4), including a three-gene DevBCA transporter complex specific to cyanobacteria42. Finally, the depth of coverage for 16S rRNA genes assembled from the sample SP12 metagenome showed that sequences 99.6% identical to those previously published for H. spongeliae had the greatest representation of any bacterial taxa in this sample, at 113× coverage, while the next most abundant cyanobacterial 16S rRNA sequence had only 11× coverage (Supplementary Table 5). These relative abundances agree with independently obtained PCR amplification data (Figure 3), and are consistent with a single copy bmp operon originating from an H. spongeliae population genome containing two copies of the 16S rRNA gene, both of which were collapsed during metagenomic assembly into a single scaffold that had approximately double the depth of coverage of the bmp operon (68×, Figure 4B). Although 16S copy number has not been determined for H. spongeliae, two copies are found in the genome of Moorea producens, its closest sequenced relative, and many other members of cyanobacterial order Oscillatoriales. No other bacterial species were detected in sufficient abundance to support coverage of the bmp gene cluster observed in the metagenomic data.
We thus named the putative bmp gene cluster from the Clade Ia SP12 metagenome as hs_bmp (H. spongeliae derived bmp operon, Figure 4C), numbering individual genes by homology to the previously described Pseudoalteromonas bmp gene cluster4. The hs_bmp gene encoding a CYP450 hydroxylase with no analogous counterpart in the Pseudoalteromonas bmp gene cluster was designated hs_bmp12, preceded by a partial, 315 bp duplication of the 5’-region of the hs_bmp5 gene named hs_bmp11. The SP12 metagenomic nucleotide sequences for hs_bmp5-7 were only 35-38% identical to the corresponding Pseudoalteromonas Bmp5-7 genes, but 50-59% identical at the amino acid level (Supplementary Table 6), explaining why the SP12 hs_bmp locus was initially undetected by PCR screening, while subsequently identified via amino acid sequence-based similarity searches.
Functional characterization of hs_bmp genes
We next set out to functionally validate the Clade Ia SP12 hs_bmp genes in PBDE biosynthesis. Unfortunately, expression of the hs_bmp genes could not be achieved using Escherichia coli as a heterologous host, even when synthetic codon optimized sequences for hs_bmp genes were employed. We rationalized that a cyanobacterial host might be more applicable for expression as the hs_bmp gene cluster is cyanobacterial in origin. Using our versatile platform for the construction of vector systems43, we assembled plasmids for the integration of exogenous DNA in the chromosome of the cyanobacterial strain Synechococcus elongatus PCC 7942. We introduced Clade Ia SP12 hs_bmp7 driven by a synthetic promoter-riboswitch system into the S. elongatus PCC 7942 genome to generate Se7942-hs_bmp7 strain (see Online Methods for details, Supplementary Table 7 for strain description, Supplementary Figures 5–6). Upon induction of protein expression, we observed the specific bioconversion of exogenously added 10 to an OH-BDE product by LC/MS/MS (Figure 5A). NMR characterization of the isolated product established its identity as 1 (Supplementary Note). We similarly integrated the region spanning hs_bmp7 to hs_bmp12 as a single cassette into the S. elongatus PCC 7942 genome to generate the Se7942-hs_bmp7–12 strain (Supplementary Table 7, Supplementary Figures 6–7). Querying the product profile upon exogenous addition of 10 led to the identification of two isomeric di-OH-BDE products confirmed by NMR as 6 and 2,6’OH-BDE80 (11) (Figure 5A, Supplementary Note).
Figure 5. Functional characterization of Clade Ia hs_bmp genes.

(A) UV-absorption chromatograms monitored at 280 nm for organic extracts of the following strains supplemented with 50 μM 10: (bottom) control strain Se7942-C, (middle) Se7942-hs_bmp7, and (top) Se7942-hs_bmp7–12. Production of 1 was observed in Se7942-hs_bmp7, while the production of 6 and 11 was observed for Se7942-hs_bmp7–12. Together with 6 and 11, the production of a third tribrominated molecule was observed, the MS1 signature for which corresponded to the tribrominated dioxin 7 as shown in panel B.
In addition to the di-OH-BDEs 6 and 11, we observed the production of an additional tribrominated molecule (Figure 5A). Though the tribrominated product could not be isolated in sufficient quantities for NMR confirmation, using mass spectrometry, we identified its MS1 profile as corresponding to dioxin 7 (Figure 1), previously reported12 from Dysideidae sponges (Figure 5B). In that study, 7 was isolated along with its methoxylated derivative 8, as well as 3 and 5 (methoxylated derivatives of 1 and 6, respectively (Figure 1B)). Thus, the heterologous production of 7 in S. elongatus with PBDEs 1 and 6 successfully reconstitutes the natural product metabolome of Dysideidae sponges. The requirement of an additional hydroxylase, such as hs_bmp12, for the natural production of polybrominated dioxins and di-OH-BDEs mirrors our prior biomimetic studies in which a flavin-dependent hydroxylase was refactored in combination with bmp5 and bmp7 for the production of polybrominated dioxins and di-OH-BDEs36.
Identification of alternative hs_bmp operon structures
With the confirmation that Clade Ia SP12 hs_bmp genes encode the synthesis of PBDEs, we next turned to metagenomes from sponge specimens SP4 (Clade IV) and GUM007 (Clade III) to evaluate how their hs_bmp operon structures correlate with their respective polybrominated natural product variability (Figure 2). The Clade IV SP4 metagenome contained hs_bmp gene homologs on several small, incompletely assembled scaffolds of cyanobacterial origin (Supplementary Figure 8A). The scattered distribution and the reduced assembly coverage depth for the SP4 metagenome, as compared to the Clade Ia SP12 metagenome with comparable input reads (Figures 4B, Supplementary Figure 8A, Supplementary Table 3), is consistent with the higher microbiome diversity and reduced H. spongeliae relative abundance in Clade IV sponges (Figure 3). However, despite reduced H. spongeliae dominance, PBDE product abundance in Clade IV at 10.8% sponge dry weight is comparable to that of Clade Ia sponge specimens (12.0% sponge dry weight, see Supplementary Methods for details, Supplementary Figure 9). For specimen GUM007, in which no PBDEs were detected, we enriched the sponge tissue samples for cyanobacterial trichomes, as described previously24, prior to DNA extraction (see Supplementary Methods for details). The resulting GUM007 metagenomic assembly contained cyanobacterial scaffolds at greater than 2000× coverage, as well as a complete copy of the H. spongeliae 16S rRNA gene at 4000× coverage (Supplementary Figure 8B). Despite the high average coverage depth, we did not observe hs_bmp genes in metagenomic sequences from Clade III sponge specimen GUM007.
To obtain a more complete set of hs_bmp operon sequences from different sponge clades, we designed consensus primers based on the Clade Ia hs_bmp metagenomic sequence and partially assembled scaffolds from Clade IV (see Supplementary Methods for details, Supplementary Figure 10, Supplementary Table 8). Through several iterations of PCR amplification and Sanger sequencing, hs_bmp operon sequences were recovered from Clade Ib, for which no metagenomic sequences had been obtained, and gaps in the Clade IV hs_bmp operon were closed (Figure 4C). As expected, we did not detect hs_bmp gene amplification products in any Clade III sponge specimens. All PCR results were replicated in at least two sponge samples from each clade.
Consistent with their functional roles in the synthesis of PBDEs (Figure 4A), the hs_bmp gene clusters for Clades Ib and IV contained the core repertoire of hs_bmp5–7 genes and displayed high overall sequence similarity to Clade Ia across the regions spanning these operon elements (Figure 4C, Supplementary Table 9). We observed a variable region between hs_bmp6 and hs_bmp7 genes consistent with the expressed PBDE chemistry in Clades Ia, Ib, and IV. The hydroxylating CYP450 hs_bmp12 and hypothetical hs_bmp11 genes from Clade Ia present in the variable region were absent from Clade Ib. In the analogous variable region for Clade IV, two genes, hs_bmp13–14, encode peptides homologous to B12-dependent iron-sulfur cluster enzymes, followed by a disrupted repetition of the hs_bmp5 gene encoded by hs_bmp15–16, sharing 80% nucleotide identity with the hs_bmp5 gene. The Clade IV cluster also contains a hypothetical gene with no apparent functional role (hs_bmp17) upstream of hs_bmp6.
DISCUSSION
By integrating chemical characterization of secondary metabolites, molecular taxonomic classification of sponge hosts and their associated microbiomes, metagenomic sequencing and assembly, targeted PCR amplifications, and functional reconstruction by expression of candidate biosynthetic genes in a heterologous host, we have identified clusters of microbial genes encoding proteins responsible for the natural production of PBDEs in the Indo-Pacific Dysideidae sponges. Four distinct sponge/holobiont groups were observed based on the convergence of five factors: sponge phylogeny, cyanobacterial symbiont H. spongeliae phylogeny, microbial community composition, the presence or absence of specific polybrominated compounds, and the structure of biosynthetic gene clusters responsible for natural product synthesis. We established multiple lines of evidence that the biosynthetic operons identified in this study originate from cyanobacterial symbionts, rather than the sponge hosts or other host-associated microbial populations. A combination of nucleotide composition, metagenomic assembly coverage, 16S rRNA genes, and predicted protein similarity to previously sequenced database genomes support that the cyanobacterial species responsible for PBDE production is H. spongeliae.
Natural products synthesized by symbionts associated with marine invertebrate hosts have been postulated to serve numerous roles, among which chemical defense of the host is primary22,35,44,45. Instances of the symbiotic interaction being dependent on the natural product biosynthetic capacity of the symbiont have been reported, in that, once the symbiont loses the natural product biosynthetic capacity, it is also cleared from the host microbiome46. In light of this background, the absence of halogenated products in cyanobacterial symbionts from Clade III sponges suggests that PBDEs are not essential for host sponge viability, although potential roles at different host life stages have not been explored. The role of PBDEs in sponge-cyanobacteria symbiosis and their broader implications for marine chemical ecology remain to be elucidated.
Our results indicate that the structural diversity of polybrominated phenolic natural product molecules isolated from Dysideidae sponges is determined by symbiont genotype. The activity of hs_Bmp7 generates 1, the dominant PBDE present in Clade Ib sponges. The subsequent structural diversification of 1 is biosynthetically encoded within the variable region between the hs_bmp7 and hs_bmp6 genes in Clade Ia and Clade IV hs_bmp operons (Figure 4C). In this variable region, the Clade Ia operon possesses the CYP450 hydroxylase hs_bmp12 gene that participates in the synthesis of di-OH-BDEs such as 6 and 11, along with polybrominated dioxins (Figure 5). Correspondingly, the Clade IV locus encodes genes with homology to B12-dependent iron-sulfur cluster enzymes that could serve catalytic roles in the isomerization of 1 to 2. Together with our earlier proposal for the participation of promiscuous halogenases that further halogenate the ortho- and para-positions of phenoxyl-activated aromatic rings of 2 and 5 (ref 8), the three hs_bmp gene loci identified in this study unify the biosynthetic proposals for a vast majority of the naturally produced PBDEs detected in the marine environment7,8. However, gene candidates for the methylation of OH- and di-OH-BDEs, such as the postulated synthesis of 5 from 6, remain elusive. At present, we are unable to distinguish between the possibilities of the methyltransferase being present at a distant locus within the H. spongeliae genome versus methoxylation performed by other members of the microbiome or the sponge host itself.
Despite the conservation of the underlying PBDE biosynthetic mechanisms that we employed to identify the hs_bmp biosynthetic gene clusters in unbiased sponge metagenomes, the cyanobacterial hs_bmp5–7 and the proteobacterial bmp5–7 genes share limited nucleotide similarity. The evolutionary relationships and the mechanisms underlying the diversification of the hs_bmp and bmp gene clusters are presently not clear. One notable difference between the two pathways is that the H. spongeliae strains of the current study produce a much more limited repertoire of CYP450-coupled bromophenol dimers than Pseudoalteromonas, with the ortho-OH-BDE 1 being the predominant hs_Bmp7 product. Conversely, the Pseudoalteromonas Bmp7 is catalytically promiscuous in its oxidative bi-radical synthesis of a diverse series of products that include predominantly C–C coupled polybrominated biphenyls as well as minor C–O coupled para-OH-BDEs and ortho-OH-BDEs4 (Figure 4A). Natural biphenyls (produced by Pseudoalteromonas) and OH-BDEs and di-OH-BDEs (produced by both Pseudoalteromonas and Hormoscilla) are bioaccumulating at the highest trophic levels in the marine environment13-15,47, underscoring the ecological relevance of both bmp and hs_bmp biosynthetic gene clusters.
The natural production of marine PBDEs extends to macroalgal and other cyanobacterial sources with as yet unknown biosynthetic routes48,49. Using the hs_bmp genes as guides, we have identified homologs of individual PBDE biosynthetic genes within the incomplete draft genomes of other marine cyanobacteria, including sea grass-associated Moorea producens 3L, snail-associated Pleurocapsa sp. PCC 7319, and free-living Rivularia sp. PCC 7116 (Supplementary Table 4). While it remains to be determined whether these homologs indeed catalyze the construction of PBDEs, these findings support future genome-guided opportunities for discovering additional PBDE genetic signatures in other marine habitats, including host-associated microbiomes across multiple trophic levels. Without a doubt, the hs_bmp and bmp gene loci hold tremendous promise as genetic beacons to illuminate a comprehensive inventory of natural sources that underlie environmental and human exposure to these natural bioactive marine chemicals. Ecosystem dynamics in our oceans are rapidly changing in response to climate change, increasing eutrophication, and effects caused by the overexploitation of marine living resources. Genetic tools to inventory the natural producers of PBDEs will also allow us to monitor PBDE-producing source populations with far reaching implications for policy design and human health.
DATA AVAILABILITY
All sequence data associated with this project have been deposited under NCBI BioProject ID PRJNA320446. All other data supporting the findings of this study are available within the paper and its supplementary information files.
ONLINE METHODS
Extraction of polybrominated phenolics from sponge samples
Sponge material was frozen and lyophilized. Lyophilized sponges were extracted thrice with methanol (MeOH) (3×dry weight). Organic solvent was removed in vacuo and the resultant residue was separated between water and dichloromethane (CH2Cl2). The CH2Cl2 soluble fraction was collected, dried over magnesium sulfate (MgSO4), and solvent removed in vacuo. The resultant residue was dissolved in MeOH and the polybrominated phenolics were separated by reversed phase high performance liquid chromatography (HPLC) using water and MeCN as solvents after addition of 0.1% v/v trifloroacetic acid (TFA). Solvents for LC/MS/MS were supplemented with 0.1% v/v formic acid instead of TFA. All mass spectrometry data was collected in the negative ionization mode as has been reported previously8.
Quantification of PBDE abundance
Sponge specimens belonging to Clade Ia (L. herbacea) and Clade IV (D. granulosa) were frozen and lyophilized. Dried sponge tissue was crushed, and 300 mg of crushed sponge tissue was incubated with 9 mL MeOH for 2 days at room temperature. The organic layer was removed and centrifuged at 16,000×g for 10 min. The supernatant recovered after centrifugation was directly analyzed by HPLC as described above. Standard curves for area under PBDE peaks vs amount of PBDE injected were generated by injecting known amounts of 2 (isolated from Clade IV sponges as described previously8) under identical chromatographic conditions (Supplementary Figure 9). The standard curve was then used to dereplicate the amount of PBDEs present in sponge MeOH extracts. The extinction coefficient of 5 is assumed to be the same as 2.
Chemical extraction of heterologous Synechococcus elongatus PCC 7942 cultures
Cultures of S. elongatus PCC7942 were centrifuged. The supernatant was extracted twice with equal volumes of ethyl acetate (EtOAc). The pellets were extracted twice with 2:1 CH2Cl2:MeOH and extract clarified by filtration. The organic layers were combined, dried over MgSO4, and solvent removed in vacuo. The residue was dissolved in MeOH, centrifuged extensively, and used directly for LC/MS/MS analyses as has been reported previously8. For NMR analyses, PBDEs were isolated by preparative HPLC using water and MeCN as solvents supplemented with 0.1% v/v TFA. All NMR data were collected using MeOH-d4 as solvent.
Microscopy and visualization of cyanobacterial trichomes
Homogenate from fresh sponge was examined for the presence of H. spongeliae-like trichomes shortly after collection under a field microscope at approximately 200× magnification. For phase-contrast and fluorescence observations, homogenate was prepared from samples stored in RNAlater at −20 °C and imaged using a Zeiss Axioskop microscope.
Specimen preparation and extraction of metagenomic DNA
DNA was extracted from frozen whole tissue of sponge specimens as follows: sponge samples were lysed at 55 ºC for 30 min in buffer containing 4 M guanidine thiocyanate, 2% sarkosyl, 50 mM EDTA, 40 μg/ml proteinase K, and 15% β-mercaptoethanol. After incubation at 55 °C for 20 min, samples were homogenized by bead-beating with 0.1 mm silica beads for 20 sec. Lysed samples were centrifuged for 5 min at high speed. The supernatant was moved to a new tube, and extracted with one volume of phenol:chloroform:isoamyl alcohol (25:24:1). The resultant aqueous layer was cleaned using the Quick-gDNA MiniPrep kit (Zymo Research, Irvine CA), and eluted in Tris-EDTA buffer.
Given the lack of polybrominated phenolic chemistry for the GUM007 sponge specimen, an additional sample from this sponge was processed further to ensure high enrichment of cyanobacterial (H. spongeliae) trichomes. Enrichment was achieved by cutting the sponge to pieces of approximately 0.5 cm with sterile scissors, and firmly pressing the pieces between two sterile plates in phosphate buffered saline. The resulting homogenate containing sponge cells and bacteria was collected and centrifuged at 100×g for 5 min to pellet the cyanobacterial trichomes. The overlying supernatant containing mostly sponge cells and other bacteria was removed, and the presence of trichomes in the remaining pellet was verified by microscopy as described above. Metagenomic DNA was extracted from the GUM007 trichome-enriched pellet using a previously described protocol50.
Sequencing and phylogenetic analysis of sponge and H. spongeliae ribosomal genes
For each sponge, the Internal Transcribed Spacer (ITS-2) region of the ribosomal gene was amplified and sequenced from bulk metagenomic DNA as described previously31 using Q5 high fidelity DNA polymerase (New England Biolabs). The full-length bacterial 16S rRNA gene was amplified from each sample using the primers 27F (5’-AGAGTTTGATCCTGGCTCAG-3’) and 1492R (5’-GGTTACCTTGTTACGACTT-3’) for most samples. For sponge sample GUM098 in Clade IV, the cyano-specific forward primer CYA106F (5’-CGGACGGGTGAGTAACGCGTGA-3’)51 was used instead of 27F as this clade was found to contain a lower relative abundance of H. spongeliae. Conditions for all 16S PCRs were as follows: initial denaturation of 30 sec at 98 °C; 30 cycles of 10 sec at 98 °C, 30 sec at 55 °C, 1 min at 72 °C; with final extension of 2 min at 72 °C. The PCR products were gel-purified, and clone libraries were generated using the TOPO TA Cloning kit (Thermo Fisher Scientific) as per the manufacturer’s instructions. Clones from these libraries were screened by an initial sanger sequence for 16S rRNA gene inserts with high identity (98-100%) to published H. spongeliae sequences (deposited as Oscillatoria, with NCBI taxonomy as Hormoscilla). Eight clones were screened for all sponges, with the exception of GUM098, for which 56 clones were screened. A representative H. spongeliae sequence was chosen at random for each sponge and additionally sequenced to obtain 2-fold coverage of the approximately 1460 bp amplicon. All Sanger sequencing was performed by Eton Biosciences, San Diego, CA.
For both the ITS-2 and 16S gene sequence datasets, unique sequences were aligned using MUSCLE, and the alignment was manually examined. Maximum likelihood trees were constructed using RaxML version 8.0.26, using the GTR amino-acid substitution model with Gamma (Γ) correction for rate heterogeneity. The automatic ‘bootstopping criteria’ option was employed (-N autoMRE), resulting in 350 and 950 bootstrap replicates for the sponge ITS-2 and bacterial 16S phylogenetic reconstructions, respectively. The resulting trees were visualized in FigTree version 1.4.2 to produce the phylograms of sponges and symbionts. For the 16S tree Clade IV, one full-length 16S rRNA sequence (GUM098) was obtained from clone libraries of these high diversity samples (see Figure 3). Remaining Clade IV 16S rRNA sequences in this study, denoted in parentheses, are represented by 370 bp sequences obtained from high-throughput community data that are 100% identical to the GUM098 full-length sequence. Additional phylogenetic analysis was performed as above on a more extensive collection of published ITS-2 reference sequences to include broader geographic distribution and to evaluate sponge taxonomic placement, resulting in a reconstruction with 500 bootstrap replicates (Supplementary Figure 2).
Bacterial community profiling with 16S rRNA gene sequences
The V4 region (~410 bp) of the 16S rRNA gene was amplified from whole-tissue genomic DNA extractions, using a two-PCR protocol to create dual-barcoded amplicon pools as described in Illumina’s guide to “16S Metagenomic Sequencing Library Preparation”. The first reaction was performed in triplicate using the primer sequences 515F-Y (5'-GTGYCAGCMGCCGCGGTAA-3’) and 926R (5’-CCGYCAATTYMTTTRAGTTT-3’) as described previously52, which also included overhangs for attachment of Illumina-compatible indexes in a subsequent reaction. The initial reaction was performed using Q5 polymerase with the following conditions: initial denaturation of 30 sec at 98 °C; 25 cycles of 10 sec at 98 °C, 20 sec at 50 °C, 20 sec at 72 °C; with final extension of 2 min at 72 °C. Triplicate reactions were combined, and 5 μL of pooled sample was used as template in the second PCR with primers coding for the sequencing site and a unique set of indexes for each sample. For this second reaction, the following changes were made to the above program: reduction of cycles to 8, and change of annealing temperature to 56 °C. Barcoded amplicons were cleaned, pooled in equimolar concentrations, and multiplexed on a single run of 2×300 bp sequencing on Illumina’s MiSeq platform by the UC San Diego Institute for Genomic Medicine.
Reads were trimmed with Trimmomatic version 0.33, and merged using FLASH version 1.2.11. Successfully merged sequences from all samples were combined into a single file, and filtered to a minimum average quality score of q20 using scripts in Qiime version 1.9.1. Primers were removed with Cutadapt version 1.9.1, and were filtered again with Qiime scripts to exclude reads outside 200-600 bp in length or containing homopolymer runs greater than 6 bp. Sequences were checked for chimeras against the Ribosomal Database Project gold database (training database v9) using vsearch version 1.1.1 (https://github.com/torognes/vsearch). Sequences were clustered into OTUs of 99% similarity using UCLUST through the open-reference workflow in Qiime. This included PyNast alignment53 against the SILVA version 111 database54, and taxonomic assignment using a custom database composed of the SILVA v111 database plus the near full-length H. spongeliae sequences obtained in this study. Eukaryotic, chloroplast, and mitochondrial sequences were removed from the dataset. Singletons were discarded, and samples were normalized using the cumulative-sum scaling method in the R package MetagenomeSeq version 1.1255.
Metagenomic sequence assembly and bioinformatic analysis
Libraries were constructed from metagenomic DNA using the TruSeq Nano kit (Illumina, San Diego CA), and sequenced as either paired-end 100 bp or 150 bp reads on Illumina’s HiSeq 2500 platform. Preparation of metagenomic libraries and sequencing was performed by the UC San Diego Institute for Genomic Medicine.
Illumina paired end reads were quality filtered and trimmed using Trimmomatic version 0.33, then assembled using IDBA-UD version 1.1.1. Assembled scaffolds were mapped to input reads using the end-to-end option of Bowtie2 version 2.256. Depth of coverage was calculated using the idxstats module of samtools version 0.1.1957.
Assembled scaffolds were classified according to taxonomic origin as previously described58, first performing a blastx search against the GenBank nr reference database (downloaded October 16, 2015) using Diamond version 0.7.9, identifying the taxonomic classification of database match sequences using DarkHorse version 1.5 at a filter setting of zero41, then tallying the taxonomic distribution of database matches for each individual scaffold.
Identification of hs_bmp clusters from additional sponge clades
We used targeted amplification to capture the hs_bmp gene cluster in all host clades by using metagenomic sequence data from both SP12 (Clade Ia) and SP4 (Clade IV). Reads from the SP4 metagenomic dataset were recruited to the assembled hs_bmp cluster from SP12 and we then designed primers to target consensus locations (~99% identity) across the operon (Supplementary Figure 10). These primers were used in PCR from genomic DNA of representatives from each clade, and successfully amplified regions were sequenced and assembled per sample. The newly extended sequence was compared with the original SP12 assembly to identify additional consensus regions for design of more primers. We iterated this process, generating 39 primers (Supplementary Table 8), until the full operon was sequenced with at least 2-fold coverage for multiple specimens in Clades Ia, Ib, and IV. No specific amplification was achieved for Clade III specimens. All reactions were performed using Q5 high fidelity DNA polymerase, and cycling conditions followed recommended guidelines from New England Biolab’s Tm Calculator (http://tmcalculator.neb.com/) for primer annealing temperatures and extension times. Read recruitment and primer mapping was performed in Geneious, version 8.1.7. Pairwise comparison diagrams illustrating percent nucleotide identities between hs_bmp operons in Clades Ia, Ib, and IV were constructed using Artemis Comparison Tool (ACT) software, release 13.0.0.
Plasmid construction for hs_bmp gene expression in cyanobacterial hosts
We attempted to use Escherichia coli as a heterologous host to functionally express the hs_bmp genes. Using the pET and pETDuet vectors, we transformed E. coli with plasmids bearing the Clade Ia hs_bmp5–12 genes. Various expression conditions resulted in no product formation. Then, we individually expressed each gene alone, and as fusion constructs to the maltose binding protein (MBP). Again, we did not observe expression of the genes, or formation of products. Next, we generated E. coli codon-optimized genes for hs_bmp5 and hs_bmp7. Using codon optimized genes, expression of hs_bmp5 could still not be observed. Codon optimized hs_bmp7 demonstrated recombinant protein expression, but very low levels of conversion of 10 to 1 could be observed, and further interrogation revealed that the recombinant protein was expressed as insoluble inclusion bodies, even as MBP fusion constructs. At this stage, we rationalized that a cyanobacterial heterologous host might be more applicable for the functional expression of the hs_bmp genes.
The plasmid pCV0094 was used for chromosomal integration at Neutral Site 2 (NS2) and controlled heterologous expression in S. elongatus PCC 7942. pCV0094 was assembled using the GeneArt Seamless Cloning and Assembly Kit (Thermo Fisher Scientific) using standardized modular devices43,59 including S. elongatus NS2 homologous regions, a tetracycline resistance marker, an origin of replication for E. coli and bom site derived from pBR322, a tetracycline resistance marker, a kanamycin resistance marker for selection of S. elongatus recombinant strains and a functional module where gene expression is driven by PconII and protein translation is controlled with riboswitch F, which is induced upon addition of theophylline. ORFs were inserted into pCV0094 downstream of the riboswitch F using Gibson cloning methodology employing appropriately designed primers. All plasmids thus constructed were verified by restriction digestion and Sanger sequencing at Genewiz LLC (San Diego, CA).
Cyanobacterial strain construction and cultivation
Plasmid DNA was introduced into S. elongatus through natural transformation60. Briefly, growing cells from 2 to 5 mL of culture with an OD750 ranging from 0.2 to 0.5 were pelleted and washed once with 10 mM NaCl and once with BG11 medium, and then re-suspended with 200 μL of BG11 medium. About 0.5 μg of purified plasmid DNA was added to the cell suspension and incubated in the dark for 6 to 18 h under continuous shaking at 30 °C. The cells were then spread on BG11 selective plates (40 mL, 1.5% agarose, with 1 mM Na2S2O3) supplemented with antibiotic (5 μg/mL kanamycin). The plates were incubated at 30 °C under continuous illumination of 300 μmol photons m−2 s−1; isolated colonies became apparent 4 days later. For each engineered strain, 3 to 4 colonies were picked individually and streaked onto new selective plates; newly formed colonies were then used to draw small (2 cm2) patches on new selective plates; these patches were used to inoculate 50 or 100 mL flasks of BG11 medium supplemented with antibiotics and grown as liquid cultures at 30 °C with continuous shaking and continuous illumination of 200 μmol photons m−2 s−1.
Chromosomal integrations at the NS2 and strain segregation were confirmed by PCR using the primers pairs S7942NS2-LA-F (5'-CTCCAGTAAAGTCTTCGCCCGTAAC-3') S7942NS2-RA-R (5'-TTGGTGCTGTTCAGTCTGGATGC-3'). PCR amplifications were carried out with the Q5 polymerase (New England Biolabs) according to the manufacturer's instructions directly on 0.5 μL of culture diluted beforehand with sterile H2O to obtain a very light pale green cell suspension and boiled for 3 min (Supplementary Figure 6).
Cyanobacterial culture induction and feeding
According to our previous report59, prior to induction with theophylline and feeding the cultures with the appropriate substrates, new 50 or 100 mL liquid cultures were inoculated to an OD750 of 0.1 and grown for a few days at 30 °C with continuous shaking and continuous illumination of 75 μmol photons m−2 s−1 until they reached an OD750 of 0.3 to 0.4. The cultures were then induced with 2 mM theophylline (using a stock solution of 200 mM in DMSO) and supplemented with 10 at a final concentration of 50 μM. The cultures were then maintained in the same condition for 2 d, after which they were collected for extraction and chemical analyses. Each of the experiments were performed in duplicate and included a control strain carrying only the antibiotic marker at NS2. For preparative isolation and NMR characterization of 1,6, and 11, strains Se7942-hs_bmp7 and Se7942-hs_bmp7–12 were grown as 2 L cultures in BG11 supplemented with 5 μg/mL kanamycin, 1% CO2, and 5 mM HEPES. The cultures were mixed continuously with a magnetic stir bar, and were grown under continuous illumination of 75 μmol photons m−2 s−1 at 30 ºC. When the cultures reached an OD750 of 0.3 to 0.4, the cells were induced with theophylline and supplemented with 10 as described above and grown in the same condition for another 2 d.
Supplementary Material
ACKNOWLEDGEMENTS
We thank our colleague B.M. Duggan at the University of California, San Diego for assistance in acquiring NMR data. This work was supported by the US National Science Foundation (DGE-1144086 Graduate Research Fellowship to J.M.B., OCE-1313747 to P.R.J., E.E.A., and B.S.M., IOS-1120113 to J.S.B., MCB-1149552 to E.E.A.); the US National Institutes of Health (K99ES026620 to V.A., R01-GM107557 to E.W.S, P01-ES021921 to P.R.J., E.E.A., and B.S.M., R01-CA172310 to V.J.P., instrument grant S10-OD010640); the US Department of Energy (DE-EE0003373 to J.W.G.); and the Helen Hay Whitney Foundation postdoctoral fellowship to V.A.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
V.A., S.P., A.T., E.W.S., V.J.P., E.E.A., and B.S.M. designed the study. V.A. performed chemical characterization, V.A., A.T., J.W.G. performed cyanobacterial expression experiments, J.M.B., M.A.S., J.B., P.R.J performed phylogenetic analyses, S.P. performed metagenomic analyses. Z.L., V.J.P., J.S.B. provided sponge samples, analytical tools and reagents. V.A., J.M.B., S.P., E.E.A., and B.S.M. wrote the manuscript with input from all authors.
REFERENCES
- 1.Hites RA. Dioxins: an overview and history. Environ. Sci. Technol. 2011;45:16–20. doi: 10.1021/es1013664. [DOI] [PubMed] [Google Scholar]
- 2.Wiseman SB, et al. Polybrominated diphenyl ethers and their hydroxylated/methoxylated analogs: environmental sources, metabolic relationships, and relative toxicities. Mar. Pollut. Bull. 2011;63:179–188. doi: 10.1016/j.marpolbul.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Gribble GW. Naturally Occurring Organohalogen Compounds - A Comprehensive Update. Vol. 91. Springer; Vienna: 2010. [Google Scholar]
- 4.Agarwal V, et al. Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat. Chem. Biol. 2014;10:640–647. doi: 10.1038/nchembio.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma GM, Vig B. Studies on the antimicrobial substances of sponges. VI. Structures of two antibacterial substances isolated from the marine sponge Dysidea herbacea. Tetrahedron Lett. 1972;13:1715–1718. [Google Scholar]
- 6.Carte B, Faulkner DJ. Polybrominated diphenyl ethers from Dysidea herbacea, Dysidea chlorea and Phyllospongia foliascens. Tetrahedron. 1981;37:2335–2339. [Google Scholar]
- 7.Calcul L, et al. NMR strategy for unraveling structures of bioactive sponge-derived oxy-polyhalogenated diphenyl ethers. J. Nat. Prod. 2009;72:443–449. doi: 10.1021/np800737z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agarwal V, et al. Complexity of naturally produced polybrominated diphenyl ethers revealed via mass spectrometry. Environ. Sci. Technol. 2015;49:1339–1346. doi: 10.1021/es505440j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unson MD, Holland ND, Faulkner DJ. A brominated secondary metabolite synthesized by the cyanobacterial symbiont of a marine sponge and accumulation of the crystalline metabolite in the sponge tissue. Mar. Biol. 1994;119:1–11. [Google Scholar]
- 10.Becerro MA, Paul VJ. Effects of depth and light on secondary metabolites and cyanobacterial symbionts of the sponge Dysidea granulosa. Mar. Ecol. Prog. Ser. 2004;280:115–128. [Google Scholar]
- 11.Utkina NK, et al. Spongiadioxins A and B, two new polybrominated dibenzo-p-dioxins from an Australian marine sponge Dysidea dendyi. J. Nat. Prod. 2001;64:151–153. doi: 10.1021/np0003544. [DOI] [PubMed] [Google Scholar]
- 12.Utkina NK, Denisenko VA, Virovaya MV, Scholokova OV, Prokof'eva NG. Two new minor polybrominated dibenzo-p-dioxins from the marine sponge Dysidea dendyi. J. Nat. Prod. 2002;65:1213–1215. doi: 10.1021/np0106438. [DOI] [PubMed] [Google Scholar]
- 13.Vetter W, et al. Sponge halogenated natural products found at parts-per-million levels in marine mammals. Environ. Toxicol. Chem. 2002;21:2014–2019. [PubMed] [Google Scholar]
- 14.Teuten EL, Xu L, Reddy CM. Two abundant bioaccumulated halogenated compounds are natural products. Science. 2005;307:917–920. doi: 10.1126/science.1106882. [DOI] [PubMed] [Google Scholar]
- 15.Shaul NJ, et al. Nontargeted biomonitoring of halogenated organic compounds in two ecotypes of bottlenose dolphins (Tursiops truncatus) from the Southern California Bight. Environ. Sci. Technol. 2015;49:1328–1338. doi: 10.1021/es505156q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wan Y, et al. Hydroxylated polybrominated diphenyl ethers and bisphenol A in pregnant women and their matching fetuses: placental transfer and potential risks. Environ. Sci. Technol. 2010;44:5233–5239. doi: 10.1021/es1002764. [DOI] [PubMed] [Google Scholar]
- 17.Wang HS, et al. Hydroxylated and methoxylated polybrominated diphenyl ethers in blood plasma of humans in Hong Kong. Environ. Int. 2012;47:66–72. doi: 10.1016/j.envint.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Chen A, et al. Hydroxylated polybrominated diphenyl ethers in paired maternal and cord sera. Environ. Sci. Technol. 2013;47:3902–3908. doi: 10.1021/es3046839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salomon CE, Faulkner DJ. Localization studies of bioactive cyclic peptides in the ascidian Lissoclinum patella. J. Nat. Prod. 2002;65:689–692. doi: 10.1021/np010556f. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt EW, et al. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc. Natl. Acad. Sci. USA. 2005;102:7315–7320. doi: 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hildebrand M, et al. Approaches to identify, clone, and express symbiont bioactive metabolite genes. Nat. Prod. Rep. 2004;21:122–142. doi: 10.1039/b302336m. [DOI] [PubMed] [Google Scholar]
- 22.Simmons TL, et al. Biosynthetic origin of natural products isolated from marine microorganism-invertebrate assemblages. Proc. Natl. Acad. Sci. USA. 2008;105:4587–4594. doi: 10.1073/pnas.0709851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anagnostidis KK, Jiří Modern approach to the classification system of cyanophytes. 3 - Oscillatoriales. Algological Studies/Archiv für Hydrobiologie. 1988;50-53:327–472. [Google Scholar]
- 24.Hinde R, Pironet F, Borowitzka MA. Isolation of Oscillatoria spongeliae, the filamentous cyanobacterial symbiont of the marine sponge Dysidea herbacea. Mar. Biol. 1994;119:99–104. [Google Scholar]
- 25.Ridley CP, et al. Speciation and biosynthetic variation in four dictyoceratid sponges and their cyanobacterial symbiont, Oscillatoria spongeliae. Chem. Biol. 2005;12:397–406. doi: 10.1016/j.chembiol.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Ridley CP, Faulkner DJ, Haygood MG. Investigation of Oscillatoria spongeliae-dominated bacterial communities in four dictyoceratid sponges. Appl. Environ. Microbiol. 2005;71:7366–7375. doi: 10.1128/AEM.71.11.7366-7375.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medema MH, Fischbach MA. Computational approaches to natural product discovery. Nat. Chem. Biol. 2015;11:639–648. doi: 10.1038/nchembio.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wischang D, Hartung J. Bromination of phenols in bromoperoxidase-catalyzed oxidations. Tetrahedron. 2012;68:9456–9463. [Google Scholar]
- 29.Wischang D, Radlow M, Hartung J. Vanadate-dependent bromoperoxidases from Ascophyllum nodosum in the synthesis of brominated phenols and pyrroles. Dalton. Trans. 2013;42:11926–11940. doi: 10.1039/c3dt51582f. [DOI] [PubMed] [Google Scholar]
- 30.Erpenbeck D, et al. Evolution, radiation and chemotaxonomy of Lamellodysidea, a demosponge genus with anti-plasmodial metabolites. Mar. Biol. 2012;159:1119–1127. [Google Scholar]
- 31.Thacker RW, Starnes S. Host specificity of the symbiotic cyanobacterium Oscillatoria spongeliae in marine sponges, Dysidea spp. Mar. Biol. 2003;142:643–648. [Google Scholar]
- 32.Redmond NE, et al. Phylogeny and systematics of demospongiae in light of new small-subunit ribosomal DNA (18S) sequences. Integr. Comp. Biol. 2013;53:388–415. doi: 10.1093/icb/ict078. [DOI] [PubMed] [Google Scholar]
- 33.Unson MD, et al. New polychlorinated amino acid derivatives from the marine sponge Dysidea herbacea. J. Org. Chem. 1993;58:6336–6343. [Google Scholar]
- 34.Hentschel U, et al. Molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbiol. 2002;68:4431–4440. doi: 10.1128/AEM.68.9.4431-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hentschel U, Piel J, Degnan SM, Taylor MW. Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 2012;10:641–654. doi: 10.1038/nrmicro2839. [DOI] [PubMed] [Google Scholar]
- 36.Agarwal V, Moore BS. Enzymatic synthesis of polybrominated dioxins from the marine environment. ACS Chem. Biol. 2014;9:1980–1984. doi: 10.1021/cb5004338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El Gamal A, et al. Biosynthesis of coral settlement cue tetrabromopyrrole in marine bacteria by a uniquely adapted brominase-thioesterase enzyme pair. Proc. Natl. Acad. Sci. USA. 2016;113:3797–3802. doi: 10.1073/pnas.1519695113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Gamal A, Agarwal V, Rahman I, Moore BS. Enzymatic reductive dehalogenation controls the biosynthesis of marine bacterial pyrroles. J. Am. Chem. Soc. 2016;138:13167–13170. doi: 10.1021/jacs.6b08512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flatt P, et al. Identification of the cellular site of polychlorinated peptide biosynthesis in the marine sponge Dysidea (Lamellodysidea) herbacea and symbiotic cyanobacterium Oscillatoria spongeliae by CARD-FISH analysis. Mar. Biol. 2005;147:761–774. [Google Scholar]
- 40.Fisch KM, et al. Polyketide assembly lines of uncultivated sponge symbionts from structure-based gene targeting. Nat Chem Biol. 2009;5:494–501. doi: 10.1038/nchembio.176. [DOI] [PubMed] [Google Scholar]
- 41.Podell S, Gaasterland T. DarkHorse: a method for genome-wide prediction of horizontal gene transfer. Genome Biol. 2007;8:R16. doi: 10.1186/gb-2007-8-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fiedler G, Arnold M, Maldener I. Sequence and mutational analysis of the devBCA gene cluster encoding a putative ABC transporter in the cyanobacterium Anabaena variabilis ATCC 29413. Biochim. Biophys. Acta. 1998;1375:140–143. doi: 10.1016/s0005-2736(98)00147-3. [DOI] [PubMed] [Google Scholar]
- 43.Taton A, et al. Broad-host-range vector system for synthetic biology and biotechnology in cyanobacteria. Nucleic Acids Res. 2014;42:e136. doi: 10.1093/nar/gku673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Florez LV, Biedermann PH, Engl T, Kaltenpoth M. Defensive symbioses of animals with prokaryotic and eukaryotic microorganisms. Nat. Prod. Rep. 2015;32:904–936. doi: 10.1039/c5np00010f. [DOI] [PubMed] [Google Scholar]
- 45.Pennings SC, Pablo SR, Paul VJ, Duffy JE. Effects of sponge secondary metabolites in different diets on feeding by 3 groups of consumers. J Exper. Mar. Biol. Ecol. 1994;180:137–149. [Google Scholar]
- 46.Kwan JC, et al. Genome streamlining and chemical defense in a coral reef symbiosis. Proc. Natl. Acad. Sci. USA. 2012;109:20655–20660. doi: 10.1073/pnas.1213820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marsh G, et al. Identification, quantification, and synthesis of a novel dimethoxylated polybrominated biphenyl in marine mammals caught off the coast of Japan. Environ. Sci. Technol. 2005;39:8684–8690. doi: 10.1021/es051153v. [DOI] [PubMed] [Google Scholar]
- 48.Malmvarn A, Zebuhr Y, Kautsky L, Bergman K, Asplund L. Hydroxylated and methoxylated polybrominated diphenyl ethers and polybrominated dibenzo-p-dioxins in red alga and cyanobacteria living in the Baltic Sea. Chemosphere. 2008;72:910–916. doi: 10.1016/j.chemosphere.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 49.Suyama TL, Cao Z, Murray TF, Gerwick WH. Ichthyotoxic brominated diphenyl ethers from a mixed assemblage of a red alga and cyanobacterium: structure clarification and biological properties. Toxicon. 2010;55:204–210. doi: 10.1016/j.toxicon.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidt EW, Donia MS. Chapter 23. Cyanobactin ribosomally synthesized peptides--a case of deep metagenome mining. Methods Enzymol. 2009;458:575–596. doi: 10.1016/S0076-6879(09)04823-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nubel U, Garcia-Pichel F, Muyzer G. PCR primers to amplify 16S rRNA genes from cyanobacteria. Appl. Environ. Microbiol. 1997;63:3327–3332. doi: 10.1128/aem.63.8.3327-3332.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016;18:1403–1414. doi: 10.1111/1462-2920.13023. [DOI] [PubMed] [Google Scholar]
- 53.Caporaso JG, et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quast C, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods. 2013;10:1200–1202. doi: 10.1038/nmeth.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Podell S, et al. Assembly-driven community genomics of a hypersaline microbial ecosystem. PLoS One. 2013;8:e61692. doi: 10.1371/journal.pone.0061692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma AT, Schmidt CM, Golden JW. Regulation of gene expression in diverse cyanobacterial species by using theophylline-responsive riboswitches. Appl. Environ. Microbiol. 2014;80:6704–6713. doi: 10.1128/AEM.01697-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clerico EM, Ditty JL, Golden SS. Specialized techniques for site-directed mutagenesis in cyanobacteria. Methods Mol. Biol. 2007;362:155–171. doi: 10.1007/978-1-59745-257-1_11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data associated with this project have been deposited under NCBI BioProject ID PRJNA320446. All other data supporting the findings of this study are available within the paper and its supplementary information files.