

ABSTRACT
In this study, we describe the construction of the first genetically modified mutant of a halovirus infecting haloalkaliphilic Archaea. By random choice, we targeted ORF79, a currently uncharacterized viral gene of the haloalkaliphilic virus ϕCh1. We used a polyethylene glycol (PEG)-mediated transformation method to deliver a disruption cassette into a lysogenic strain of the haloalkaliphilic archaeon Natrialba magadii bearing ϕCh1 as a provirus. This approach yielded mutant virus particles carrying a disrupted version of ORF79. Disruption of ORF79 did not influence morphology of the mature virions. The mutant virus was able to infect cured strains of N. magadii, resulting in a lysogenic, ORF79-disrupted strain. Analysis of this strain carrying the mutant virus revealed a repressor function of ORF79. In the absence of gp79, onset of lysis and expression of viral proteins occurred prematurely compared to their timing in the wild-type strain. Constitutive expression of ORF79 in a cured strain of N. magadii reduced the plating efficiency of ϕCh1 by seven orders of magnitude. Overexpression of ORF79 in a lysogenic strain of N. magadii resulted in an inhibition of lysis and total absence of viral proteins as well as viral progeny. In further experiments, gp79 directly regulated the expression of the tail fiber protein ORF34 but did not influence the methyltransferase gene ORF94. Further, we describe the establishment of an inducible promoter for in vivo studies in N. magadii.
IMPORTANCE Genetic analyses of haloalkaliphilic Archaea or haloviruses are only rarely reported. Therefore, only little insight into the in vivo roles of proteins and their functions has been gained so far. We used a reverse genetics approach to identify the function of a yet undescribed gene of ϕCh1. We provide evidence that gp79, a currently unknown protein of ϕCh1, acts as a repressor protein of the viral life cycle, affecting the transition from the lysogenic to the lytic state of the virus. Thus, repressor genes in other haloviruses could be identified by sequence homologies to gp79 in the future. Moreover, we describe the use of an inducible promoter of N. magadii. Our work provides valuable tools for the identification of other unknown viral genes by our approach as well as for functional studies of proteins by inducible expression.
KEYWORDS: Archaea, Natrialba magadii, ϕCh1, halophiles, virus
INTRODUCTION
Halovirus ϕCh1 is a model for haloalkaliphilic viruses and infects the haloalkaliphilic archaeon Natrialba magadii (1). As typical for members of the Myoviridae family, ϕCh1 consists of an icosahedral head and a contractible tail and contains a double-stranded DNA (dsDNA) genome (2). The halovirus ϕCh1 is adapted to the extreme growth conditions of its host N. magadii, depending on high salt concentrations (4 to 5 M NaCl) and high pH values (8.5 to 11) (2). ϕCh1 was originally identified as a provirus integrated in the chromosome of the wild-type (wt) strain N. magadii L11. Induction of the lytic cycle occurs spontaneously at the transition of the late logarithmic to the stationary growth phase, upon which virus particles are produced and released by cell lysis. The cured strain N. magadii L13 can be reinfected by ϕCh1 (2).The genome of ϕCh1 is completely sequenced; it contains 58.5 kbp and consists of 98 open reading frames (ORFs) (2). A set of structural genes is organized in a gene cluster at the 5′ end of the viral genome. Several genes within this cluster show sequence homologies to other halovirus and bacteriophage genes (3), and the functions of 12 ORFs of ϕCh1 have been identified experimentally (4).
However, a vast majority of genes of archaeal viruses have no known homologues, and thus their function remains unknown (5). For ϕCh1, more than 60% of its ORFs seem be to genes with unique features (3, 5). In particular, genes organized in the 3′ region of ϕCh1 do not show sequence similarities to any known genes (3).
In this study, we describe the disruption and analysis of the arbitrarily chosen ORF79. ϕCh1 ORF79 was an uncharacterized gene for which previously no homologies to sequences with known functions or structural elements and no typical protein motifs or domains could be identified. ORF79 is located at the 3′ half (nucleotides [nt] 47669 to 48460) of the viral genome (3). We generated the first site-directed mutant of a haloalkaliphilic virus by disrupting ORF79 and performed phenotypic characterization of the mutant virus regarding structure and infection behavior. We investigated potential direct and indirect regulatory properties of the gene product of ORF79 (gp79) on expression of other viral proteins in in vivo assays and protein expression analyses. Our data show that gp79 is a novel repressor protein influencing the lytic life cycle of ϕCh1.
RESULTS
Disruption of ORF79 in the halovirus ϕCh1.
For the disruption of ORF79 of the halovirus ϕCh1, a homologous recombination approach was used. To this end, the lysogenic wild-type strain N. magadii L11 bearing ϕCh1 as a provirus was transformed with the suicide plasmid pΔORF79::novR (Fig. 1A). This plasmid contains the ORF79 of ϕCh1 disrupted by the gyrB gene, which confers resistance to novobiocin. By using PCR analysis with the primers 79-Nae and Nov-6, we screened for successfully transformed clones in which the integration of gyrB occurred at the locus of ORF79 (Fig. 1A and data not shown). As previously described, halophilic archaea are polyploid, containing up to 25 copies of their chromosomes (6). Therefore, the transformed clones contained both wild-type and disrupted versions of ORF79. In order to yield a genetically pure mutant strain, the transformed clones were grown in fluid cultures until lysis in order to generate virus particles. Only single copies of the virus genome are packed into the virus particles. Therefore, a mixture of wild-type virions and virions bearing the disrupted version of ORF79 was released. This pool of viruses was used for infection of the cured strain N. magadii L13. We used novobiocin to select for cells infected with the mutant viruses since the ORF79 gene is disrupted by the marker gene gyrB in these cells. Two single colonies were analyzed for the presence of the mutated provirus by PCR analysis with the primers 79-Nae and Nov-6 (Fig. 1A and B). By PCR analysis with the primers pKS-5 and pKS-3 specific for the backbone of pΔORF79::novR, we excluded the possibility of any remaining suicide plasmid DNA in the novobiocin-resistant clones. In one of the tested clones no plasmid DNA could be detected (Fig. 1C). In this clone, we confirmed the absence of wild-type ORF79 genes by PCR analysis with the primers 79-Nae and 79-Pst (Fig. 1D) and Southern blot analysis (Fig. 1E).
FIG 1.

Disruption of ORF79 of the virus ϕCh1 in the lysogenic strain N. magadii L11. (A) Schematic presentation of the locus of ORF79 of the virus ϕCh1 in the wild-type lysogenic strain N. magadii L11. The ORF79 disruption strain N. magadii L11-ΔORF79 is depicted. The ORF79 gene (gray arrow) is disrupted by the marker gene gyrB (black arrow) via homologous recombination with the suicide plasmid pΔORF79::novR. Small black arrows indicate positions of primers. Recognition sites of the restriction endonuclease AgeI are indicated. (B to D) PCR analyses with the indicated primers were performed using the indicated DNA samples. Screening for successful integration of the gyrB gene at the locus of ORF79 was performed (B). The absence of plasmid backbone DNA (C) and the absence of wild-type genes (D) were assessed. C1 and C2, candidates 1 and 2, resulting from the transformation of N. magadii L11 with the suicide plasmid pΔORF79::novR, followed by infection of N. magadii L13 with a pool of released viruses after lysis of a culture with disrupted ORF79; WT, wild-type strain N. magadii L11; P, suicide plasmid pΔORF79::novR. The pUC19/HaeIII size marker (pUC) or the Thermo Fisher Scientific GeneRuler 1-kb ladder was used as a size control. (E) A Southern blot analysis with AgeI-digested chromosomal DNA using the ORF79 as a probe (generated by PCR with the primers 79-Nae and 79-Pst) resulted in 2,554 bp for the wild-type strain N. magadii L11 (L11) and 5,332 bp for candidate 1 resulting from the transformation of N. magadii L11 with the suicide plasmid pΔORF79::novR (C2).
To characterize the ORF79 disruption in more detail, lysogenic strains of N. magadii L11 and N. magadii L11-ΔORF79 were compared (Fig. 2A to D). As a control, the cured strain N. magadii L13 was used. While the structural proteins E and gp34 could be detected in equal amounts in both strains at 91 h after inoculation (Fig. 2B and C), gp79 could be detected only in strain N. magadii L11 (Fig. 2D). For both the cured (L13) as well as the mutated (L11-ΔORF79) strain, no signal for gp79 could be detected (Fig. 2D).
FIG 2.

No gp79 can be detected within ϕCh1 virus particles and N. magadii L11-ΔORF79. (A to D) Analyses of lysogenic strains. The wild-type strain N. magadii L11 (L11), the ORF79 disruption strain N. magadii L11-ΔORF79 (Δ79), and the cured strain N. magadii L13 (L13) were grown in rich medium for 72 h. Total protein samples were taken and resolved by SDS-PAGE and stained with Coomassie (A) or used for Western blot analysis using polyclonal antibodies against protein E (B), protein gp34 (C), or gp79 (B). The Thermo Scientific unstained protein ladder (L) was used as a size control. (E to H) Analyses of virus particles. ϕCh1 wt as well as ϕCh1-ΔORF79 particles were isolated and compared for their total protein content by Coomassie staining (E) and for single protein content by Western blotting for protein E, gp34, and gp79, as indicated.
Stoichiometry and virus morphology of ϕCh1 wt and ϕCh1-ΔORF79.
Since the function of gp79 was unknown (3), we were interested whether the disruption of ORF79 had an effect on the morphology of ϕCh1. We did not detect differences in stoichiometry of the wt and mutant viruses when total proteins of both virus stocks were compared by Coomassie staining (Fig. 2E). This observation was also confirmed when structural components like protein E (Fig. 2F) and gp34 (Fig. 2G) were compared. However, the virus titers of the two preparations differed slightly. For ϕCh1 wt, we determined a titer of 2.9 × 1011 PFU; for ϕCh1-ΔORF79 we determined a titer of 1 × 109 PFU (data not shown). Next, we excluded the possibility of gp79 being a structural component. Using anti-gp79 antibodies, we detected no signal in either ϕCh1 wt or ϕCh1-ΔORF79 total viral protein by Western blotting (Fig. 2H). In contrast, gp79 was detected by anti-gp79 in the lysogenic strain (Fig. 2H). Additionally, we analyzed the viral particles resulting from lysis of N. magadii L11-ΔORF79 by negative staining electron microscopy and did not observe any morphological differences relative to the wild-type virus morphology (Fig. 3).
FIG 3.
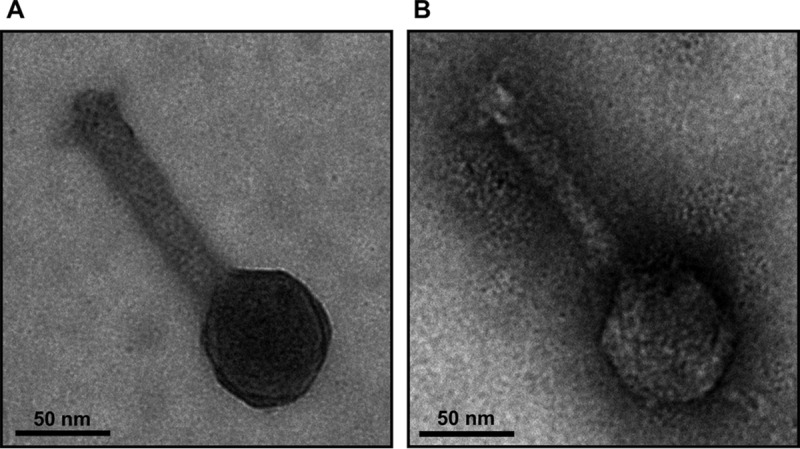
Electron micrographs of isolated ϕCh1 particles. Virions resulting from lysis of the wild-type strain N. magadii L11 (A) and the ORF79 disruption strain N. magadii L11-ΔORF79 (B) were isolated and negatively stained with uranyl formate for electron microscopy.
Impact of ORF79 on lysis behavior.
When next investigating the viral life cycle, we observed an earlier onset of lysis in the ORF79-disrupted strain than in the wild type (Fig. 4A). Lysis occurred 24 h earlier in N. magadii L11-ΔORF79 than in the wild-type N. magadii L11 strain. Complementation of the ORF79-deficient strain by transformation with the plasmid pNB102-ORF79 reestablished the normal growth and lysis behavior (Fig. 4A). Here, ORF79 was under the control of the ORF49 promoter (7) since the native promoter of ORF79 is unknown. The earlier onset of lysis and release of infectious virus particles from the ORF79-disrupted strain indicated a possible regulatory role of ORF79 in the lytic cycle of ϕCh1, which we investigated further.
FIG 4.
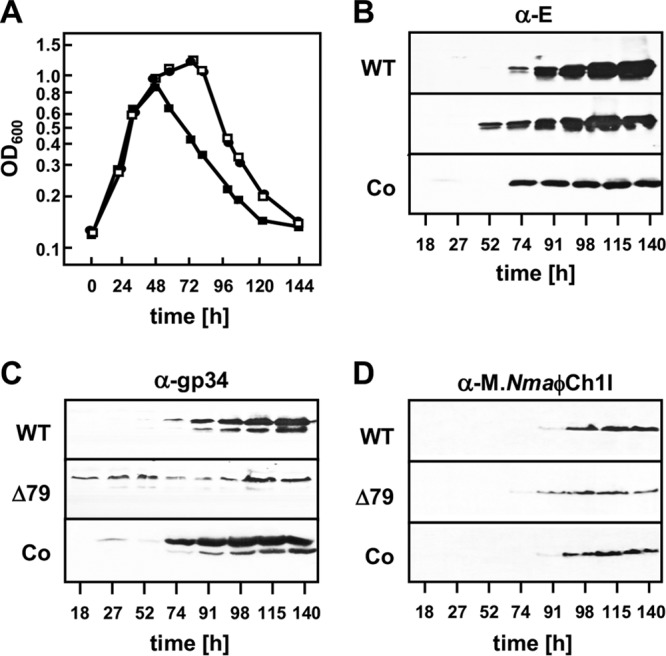
Influence of gp79 on growth of N. magadii and viral gene expression. The wild-type strain N. magadii L11 (circles; WT), the ORF79 disruption strain N. magadii L11-ΔORF79 (filled squares; Δ79), and the complemented strain N. magadii L11-ΔORF79 bearing pNB102-ORF79 (open squares; Co) were grown in rich medium (A). Samples were taken at indicated time points, optical density at 600 nm was measured, and total protein extracts were prepared. Protein samples were used for Western blot analysis using polyclonal antibodies against protein E, the tail fiber protein gp34, or the methyltransferase M.NmaϕCh1-I, as indicated.
Disruption of ORF79 results in deregulated expression of viral genes.
To study the potential regulatory influence of ORF79 on the lytic cycle in more detail, we compared the protein expression profiles of viral proteins in the wild-type strain, the ORF79 disruption strain, and the complemented strain. We chose three proteins encoded by genes on the 5′ end, the central part of the viral genome, and the 3′ end, respectively. Two genes encoded structural components of the ϕCh1 virion, the major capsid protein E encoded by ORF11 (3) and the tail fiber protein gp34 encoded by ORF34 (8). The third tested protein was the nonstructural N. magadii protein methyltransferase (M.NmaϕCh1-I), a late-gene product encoded ORF94 (9). The expression of these three proteins was analyzed by Western blot assays using polyclonal antibodies.
Protein E was detected in the cell lysate 24 h earlier in the ORF79 disruption strain than in the wild-type strain (Fig. 4B). This time period corresponded to the earlier onset of lysis in the mutant strain compared to that in the wild-type strain (Fig. 4A). An expression pattern similar to that of the wild type could be reestablished by the complementation of ORF79 (Fig. 4B). The expression of ORF34 in the wild-type strain N. magadii L11 was observed 74 h after inoculation, at the induction of lysis (Fig. 4C), confirming previous experiments (10). In contrast, the time course experiment performed with N. magadii L11-ΔORF79 showed constitutive expression of gp34 in the absence of ORF79 (Fig. 4C). Complementation of ORF79 restored the normal expression pattern (Fig. 4C). Slight expression of ORF34 was observed in one of the first three samples (27 h after inoculation). This phenomenon could not be seen in Western blotting using anti-E or anti-M.NmaϕCh1-I antibodies. For the late gene M.NmaϕCh1-I, a similar expression pattern as for protein E was observed in wild-type, mutant, and complemented strains (Fig. 4D). These results further strengthened our hypothesis for a possible role of ORF79 in regulation of the viral life cycle.
Since we detected an earlier onset of lysis in the N. magadii L11-ΔORF79 strain than in wt cells, we determined the eclipse in viral development and the burst size of viral particles. The eclipse was prolonged for 4 h (10 h for the ϕCh1-ΔORF79 virus compared to 6 h for the wt strain), and the burst size was reduced from 150 particles per cell (wt) to 50 particles per cell for ϕCh1-ΔORF79 (Table 1) while the number of particles released by the lysogenic mutant strain N. magadii L11-ΔORF79 was lower by a magnitude of 4. Together, these observations suggest that lysis occurred before the assembly of progeny particles was completed.
TABLE 1.
Characterization of mutant virus ϕCh1-ΔORF79
| Virus | Eclipse duration (h) | Burst size (no. of particles per cell) | No. of particles released after 144 h (PFU/ml) |
|---|---|---|---|
| ϕCh1 | 6 | 150 | 1 × 1011 |
| ϕCh1-ΔORF79 | 10 | 50 | 1 × 107 |
The presence of gp79 strongly reduces plating efficiency of the halovirus ϕCh1.
Since we suspected that gp79 acts as a repressor of lytic life cycle induction, we were interested in its effect on the plating efficiency of ϕCh1. Therefore, the cured wild-type strain N. magadii L13 was transformed with the plasmid pNB102-ORF79, bearing ORF79 under the control of the promoter of ORF49, or with an empty plasmid (pNB102) as a control. Subsequently, the plasmid-bearing strains and the wild-type strain were incubated with wild-type ϕCh1 virions, and their plating efficiencies were assessed. The presence of the empty plasmid had no effect on the plating efficiency of ϕCh1 (Table 2). However, the presence of gp79 in the cured strain N. magadii L13 reduced the plating efficiency of ϕCh1 by seven orders of magnitude (Table 2), demonstrating a drastic effect of the ORF79 gene product (gp79) on either the establishment of lysogeny or the induction of the lytic life cycle of ϕCh1.
TABLE 2.
Plating efficiency of N. magadii L13 strains with wild-type ϕCh1
| Strain | Titer (PFU/ml) |
|---|---|
| N. magadii L13 | 1 × 108 |
| N. magadii L13 (pNB102) | 1 × 108 |
| N. magadii L13 (pNB102-ORF79) | <1 × 101 |
gp79 represses the induction of the lytic life cycle in ϕCh1.
Next, we assessed whether overexpression of gp79 is able to prevent induction of lysis. To this end, the lysogenic strain N. magadii L11, bearing ϕCh1 as a provirus, was transformed with the plasmid pNB102-ORF79. In this gp79-overexpressing strain, lysis was completely impaired, but it was not in the control strain N. magadii L11 (pNB102) (Fig. 5A). In concordance, no infectious virions were present in the culture supernatant of the lysogenic N. magadii strain overexpressing gp79 (Fig. 5B).
FIG 5.

Influence of gp79 on the induction of the lytic life cycle of ϕCh1. The wild-type strain N. magadii L11 bearing pNB102 (circles; WT) and the ORF79-overexpressing strain N. magadii L11 bearing pNB102-ORF79 (squares; OE) were grown in rich medium. (A) Optical density at 600 nm was measured throughout growth at indicated time points. (B) The virus titer in the culture supernatant 115 h after inoculation was determined by infecting the cured strain N. magadii L13. (C to E) Total protein samples at the same time point were used for Western blot analysis using polyclonal antibodies against protein E, the tail fiber protein gp34, or the methyltransferase M.NmaϕCh1-I, as indicated. The respective purified proteins were used as positive control (+). (F) A Southern blot analysis with BglII-digested chromosomal DNA using a fragment of the ϕCh1 genome as a probe (generated by PCR with the primers Soj-5 and Soj-3) resulted in the expected signal at 3,130 bp for both strains. A New England BioLabs biotinylated DNA marker (L) was used as a size control.
Additionally, we did not detect the viral proteins E, gp34, and M.NmaϕCh1-I in the cells of the lysogenic N. magadii strain overexpressing gp79 (Fig. 5C to E) even though the genome of ϕCh1 was still integrated in the chromosome of N. magadii (Fig. 5F). This strongly suggested that gp79 functions as a repressor of induction of the lytic life cycle and, thus, viral protein expression.
gp79 directly regulates the expression of ORF34 but not of ORF94.
We observed earlier expression of the structural protein E and M.NmaϕCh1-I genes in the ORF79 disruption strain (Fig. 4B and D). Strikingly, expression of ORF34 was completely deregulated (Fig. 4C). The strong impact on the expression of ORF34 by the absence of gp79 prompted us to further investigate the potential role of gp79 as a repressor. First, we analyzed the transcription of ORF79 by reverse transcription-PCR (RT-PCR) in wt N. magadii L11. We detected the first signal 18 h after inoculation. The signal was constant throughout the experiment (Fig. 6A). When monitoring the synthesis of both proteins, gp79 and gp34, throughout growth in the wild-type lysogenic strain N. magadii L11 by Western blot analyses, we first detected gp79 expression 27 h after inoculation (Fig. 6B); 50 h later (74 h after inoculation) the signal decreased, and it disappeared completely 115 h after inoculation. Expression of ORF34 was first observed 74 h after inoculation and was constantly observed (Fig. 6B). We therefore conclude that gp79 is able to regulate the expression of ORF34. However, the regulation of gp79 itself is still unclear. Since the mRNA of ORF79 amplified and shown in Southern blot analysis (Fig. 6A) is continuously expressed in constant amounts compared to the protein synthesis shown in Western blotting (Fig. 6B), we conclude that the regulation occurs at a posttranslational level.
FIG 6.
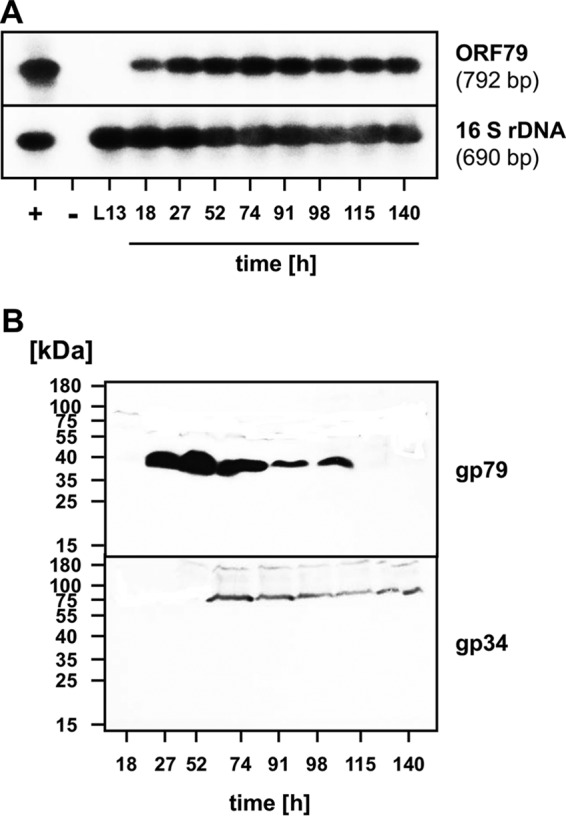
Gene expression of ORF79 and ORF34 and the occurrence of gp79 and gp34 in the wild-type lysogenic strain N. magadii L11. (A) Detection of ϕCh1 ORF79 transcripts during the life cycle of N. magadii L11 using RT-PCR. PCR and RT-PCR products from samples taken at various time points (18 to 140 h after inoculation) during the infection cycle of ϕCh1 in the lysogenic strain L11 were separated and used for Southern hybridizations. The upper panel represents results of RT-PCRs of samples taken at different time points using ORF79-specific primers 79-RT-1 and 79-RT-2. The lower panel represents results of control RT-PCRs using primers Nb-16F and Nb-16R for the detection of N. magadii 16S rRNA. As hybridization probes, the entire cloned ORF79 fragment and a fragment of the N. magadii 16S rRNA were used. The positive control (+) consisted of a PCR using ϕCh1 genomic DNA as a template (upper panel) or PCR using chromosomal DNA of N. magadii L11 (lower panel). The negative control (−) consisted of a PCR without a preceding RT reaction using total RNA isolated from N. magadii L11 at 120 h after inoculation. L13, RT-PCR using total RNA isolated from the virus-cured strain N. magadii L13. (B) The wild-type lysogenic strain N. magadii L11 was grown in rich medium for 140 h. Samples were taken at the indicated time points and used for Western blot analysis using polyclonal antibodies against gp79 or gp34.
In a next experiment, we further investigated the possible direct effect of gp79 on the expression of the tail fiber protein gp34 in a virus-free background. To this end, we transformed the cured strain N. magadii L13 with a plasmid bearing the tail fiber variant ORF3452 under the control of its own promoter (pNB102-ORF3452), together with a plasmid bearing ORF79 under the control of the inducible promoter of the tryptophanase tnaA (pRo-5-ptna-ORF79).
As reported earlier, the promoter of the Haloferax volcanii tryptophanase gene tnaA (11) is inducible with tryptophan and therefore tightly regulated. Homology searches identified the tryptophanase gene of N. magadii (Nmag_1937) (12), and its upstream region was analyzed to identify an inducible, tightly regulated promoter of N. magadii. The tryptophanase promoter is inducible by tryptophan on minimal medium, as determined with the reporter gene bgaH (Fig. 7A and B).
FIG 7.

Determination of the influence of gp79 on the expression of ORF3452 using the inducible tnaN promoter. (A and B) The cured strain N. magadii L13 bearing pRo-5-ptna-bgaH was grown in minimal medium and either induced by tryptophan addition every 24 h (open circles) or uninduced (filled squares). Samples were taken at the indicated time points, specific BgaH activity was measured (A), and the optical density at 700 nm was determined (B). (C and D) The cured strain N. magadii L13 bearing pNB102-ORF3452 and pRo-5-ptna-ORF79 was grown in minimal medium and either induced by tryptophan addition every 24 h (open circles; cont.) or uninduced (filled squares; un.) or induced only once 72 h after inoculation (filled circles; once). Samples were taken at the indicated time points, the optical density of the culture was measured at 700 nm (C), and total protein extracts were prepared. Protein samples were used for Western blot analysis using polyclonal antibodies against gp34 (D). (E) The cured strain N. magadii L13 bearing pNB102-Mtase and pRo-5-ptna-ORF79 was grown in minimal medium and either induced by tryptophan addition every 24 h (open circles; cont.) or uninduced (filled squares; un.). Total protein samples were taken at the indicated time points and used for Western blot analysis using a polyclonal antibody against M.NmaϕCh1-I.
The cured strain N. magadii L13 containing the two plasmids pNB102-ORF3452 and pRo-5-ptna-ORF79 was grown either without tryptophan addition, with tryptophan supplementation every 24 h, or with a single tryptophan supplement 72 h after inoculation (Fig. 7C). Without addition of tryptophan, ORF79 was not expressed, and ORF34 under the control of its own promoter was constitutively expressed in the stationary phase (Fig. 7D, upper panel). When ORF79 gene expression was induced continuously by daily tryptophan addition starting at day 3, gp34 expression diminished on day 4 and was not detectable at all after this time point (Fig. 7D, middle panel). A single induction event of ORF79 expression by addition of tryptophan at day 3 led to reduced expression of the tail fiber protein at days 4 and 5 (Fig. 7D, lower panel). In this culture, gp34 expression was restored to the levels before tryptophan addition on day 6, presumably the time point when the tryptophan was consumed and ORF79 was therefore no longer expressed. These findings demonstrate that gp79 directly acts as a repressor of the tail fiber protein ORF34 independently of other viral proteins in N. magadii.
In an analogous experiment, we tested the influence of gp79 on gene expression of M.NmaϕCh1-I. The cured strain N. magadii L13 was transformed with the plasmids pRo-5-ptna-ORF79 and pNB102-Mtase. Here, addition of tryptophan and therefore expression of gp79 did not change the expression pattern of the M.NmaϕCh1-I gene in the virus-free background (Fig. 7E). This demonstrates that the expression of ORF94 is not directly regulated by gp79. However, since M.NmaϕCh1-I was expressed earlier in the ORF79-disrupted strain N. magadii L11-ΔORF79 than in the wild-type strain, this strongly suggests that gp79 regulates the expression of additional regulatory factors. Based on these results together with the previous results of this study, gp79 seems to be a repressor directly regulating the expression of ORF34 and might additionally influence the whole lytic cell cycle by regulating expression of at least one further regulator.
DISCUSSION
Until recently, ϕCh1 was one of only four completely sequenced viruses infecting haloarchaea, along with the available genome data of viruses HF1, HF2, and BJ1 (3, 13–16). However, advanced sequencing techniques allow easier and faster studies, and thus the complete genome sequences of several other viruses have been described (17). So far, most sequenced genes of halophilic and haloalkaliphilic viruses do not match any genes in databases. This is most likely due to the fact that the great majority of prokaryotic sequences in today's databases are derived from bacteria, which are therefore not a good basis for sequence searches studying haloalkaliphilic genes in Archaea, a clearly distinct phylogenic group (18). Future basic research on archaea and their viruses will contribute to a better understanding of protein functions and will allow more substantial in silico analyses. Here, the randomly chosen ORF79 was demonstrated to encode a regulatory protein in ϕCh1, providing information for future bioinformatics approaches.
In this study, we describe the construction of the first disruption mutant of a halovirus by using a homologous recombination approach in its host organism. We exploited the fact that single copies of viral genomes are packed into the mature virions to yield a genetically uniform lysogenic strain by reinfecting a cured strain. This strategy proved to be faster and more efficient than the passaging procedure used previously (19) to obtain a genetically uniform N. magadii strain. We speculate that the reinfection strategy can also be used for manipulations of nearly all viral genes in haloviruses, provided that the packaging mechanisms as well as the viral machinery necessary for the establishment of lysogeny are not affected by the introduced mutation.
The disruption of ORF79 with the gyrB gene resulted in a lysogenic N. magadii strain which did not express gp79, and not even the N-terminal part could be detected by Western blotting. Here, we used polyclonal rabbit-raised antibodies targeting the whole gp79 protein produced in Escherichia coli. It is possible that the N-terminal part of gp79 is simply not immunogenic and could therefore not be detected. Indeed, the polyclonal antibody used against gp79 was not very potent, so the rabbit serum was diluted only 1:100 for the Western blot analyses performed. It is also possible that the N-terminal part of gp79 is unstable in N. magadii and therefore is immediately degraded after translation.
We observed an earlier onset of lysis and earlier expression of three tested proteins in the lysogenic, ORF79-disrupted strain. Mature, infectious virions were released upon lysis, proving that the whole lytic cycle occurs prematurely in this strain. Furthermore, expression of the tail fiber protein gp34 was completely deregulated in N. magadii L11-ΔORF79. These findings suggest that gp79 is a repressor of genes responsible for the induction of the lytic cycle of ϕCh1. This idea is supported by the finding that overexpression of gp79 completely prevented induction of the lytic cycle in the wild-type lysogenic strain N. magadii L11.
For the protein M.NmaϕCh1-I, we observed earlier expression in the ORF79-disrupted strain N. magadii L11-ΔORF79 but did not observe a direct effect on the protein in a virus-free background. Therefore, we propose that gp79 acts as a repressor of the lytic cycle by influencing the expression of at least one activator. At the same time, expression of ORF34 could be directly regulated by gp79 since we observed direct repression of the tail fiber protein-encoding gene by gp79 in a virus-free background.
However, in the wild-type strain N. magadii L11, gp79 can be detected also at late time points simultaneously with gp34, contradicting its role as a repressor. We hypothesize that at these late time points the repressor activity of gp79 is impaired since more copies of the viral genome are present in the cells, changing the ratio of gp79 and its target sequences. This would also explain why ORF34 was expressed in the ORF79 disruption strain at time points when no gp79 could be detected in the wild-type lysogenic strain. We speculate that gp79 is present in the wild-type lysogenic strain at low levels (undetected with the performed assays) and can act as a repressor because only few copies of the virus genome are present.
Further, we established the usage of the inducible promoter of the tryptophanase tnaA for in vivo studies in N. magadii in this study. The addition of tryptophan to minimal medium causes genes under the control of this promoter to be expressed. As tryptophan is consumed by N. magadii throughout growth, it has to be replenished by daily additions of tryptophan. This enables control over the expression behavior of the gene of interest since, without replenishment of tryptophan, gene expression will be terminated.
Newly performed bioinformatic analyses using the sequence of ORF79 revealed only very low homologies with other proteins, like the uncharacterized protein gp5 encoded by halovirus HHTV-2 (Uniprot accession number R4TKH2). However, motif searches using the Pfam (protein family) database revealed homologies to DNA binding domains like the adenovirus early E1A protein family (PF02703; http://pfam.xfam.org/family/PF02703) as well the DNA-binding domain of chromatin remodeling proteins and helicases (PF13891; http://pfam.xfam.org/family/PF13891) and primase zinc finger motifs (PF09329; http://pfam.xfam.org/family/PF09329). Again, these findings support our hypothesis that ORF79 encodes a regulatory element, most probably a repressor protein of virus ϕCh1.
MATERIALS AND METHODS
Strains, media, and growth conditions.
All strains used throughout this study are listed in Table 3. N. magadii was grown in rich medium (2) or in defined medium (3.5 M NaCl, 27 mM KCl, 2 mM Na2HPO4, 2 mM NaH2PO4, 12 mM NH4Cl, 5 mM leucine, 20 mM sodium acetate, 10 mM sodium pyruvate, 0.175 M Na2CO3, 1 mM MgSO4, 0.4 mM MnCl3, 0.3 mM CaCl2, 0.4 mM CuSo4 and 0.3 mM ZnSO4) at 37°C. Novobiocin and mevinolin were added to a final concentration of 3 to 4 μg/ml, respectively. Tryptophan was added to a final concentration of 2 mM to activate the tryptophanase promoter starting at an optical density at 700 nm (OD700) of 0.1. Liquid cultures were grown in Erlenmeyer flasks on a rotary shaker at 200 rpm. Plates were incubated in sealed plastic bags for 1 to 2 weeks.
TABLE 3.
Strains used throughout this study
| Strain name | Description | Reference or source |
|---|---|---|
| N. magadii L11 | Wild-type strain, ϕCh1 provirus | 2 |
| N. magadii L13 | Cured strain | 2 |
| N. magadii L11-ΔORF79 | Disruption of ORF79 of ϕCh1 by gyrB | This study |
| E. coli XL1-Blue | endA1 gyrA96 hsdR17 (rK− mK+) lac recA1 relA1 supE44 thi (F′ lacIq lacZΔM15 proAB+ tet) | Stratagene |
| E. coli Rosetta | F− ompT hsdSB (rB− mB−) gal dcm lacY1 (DE3) pRARE6 (Cmr) | Novagen |
E. coli was incubated in LB medium at 37°C as previously described (20). Ampicillin and tetracycline were added as appropriate to final concentrations of 100 μg/ml and 10 μg/ml, respectively.
Plasmid constructions.
A list of plasmids used for the transformation of N. magadii and all primers used throughout this study are listed in Tables 4 and 5, respectively. E. coli XL1-Blue (Agilent Technologies, Santa Clara, CA, USA) was used for all cloning purposes. All PCRs for cloning purposes were performed using the Pwo polymerase (Peqlab, VWR International GmbH, Erlangen, Germany) according to the manufacturer's instructions.
TABLE 4.
Plasmids used for transformations of N. magadii strains in this study
| Plasmid name | Description | Reference or source |
|---|---|---|
| pΔORF79::novR | Suicide plasmid for the disruption of ORF79 with gyrB (bla) | This study |
| pNB102 | Shuttle vector (bla Mevr) | 22 |
| pNB102-Mtase | ORF94 and its upstream region in pNB102 | This study |
| pNB102-ORF3452 | ORF3452 and its upstream region in pNB102 | This study |
| pNB102-ORF79 | ORF79 under the control of the promoter of ORF49 in pNB102 | This study |
| pRo-5 | Shuttle vector (bla gyrB) | 24 |
| pRo-5-ptna-bgaH | bgaH under the control of the tna promoter in pRo-5 | This study |
| pRo-5-ptna-ORF79 | ORF79 under the control of the tna promoter in pRo-5 | This study |
TABLE 5.
Primer used throughout this study
| Primer name | Sequence (5′–3′) |
|---|---|
| 34-Kpn | CAGCAGGGTACCCGGCGTTCGAGGTCA |
| 36-3 | CAGCAGAAGCTTATTCAGGTTTCATGTCGCTG |
| 49-Kpn | CAGCGGTACCTTGCGTTCAGTTCCG |
| 79-Bgl | GATAAGATCTATGGTCGAAGTGACGAACC |
| 79-KB | GACGAGATCTGGTACCTCAAGCATCGGTCGAGTCA |
| 79-Kpn | GACGGGTACCTCAAGCATCGGTCGAGTCA |
| 79-Nae | GATAGCCGGCGACTCTCACAAGATCTC |
| 79-Pst | GATACTGCAGCTCTTTGTACCGATGCGTC |
| 79-RT-1 | GACGAACCTCACCGACTT |
| 79-RT-2 | GCATCGGTCGAGTCAC |
| MT-Kpn-5 | GAATGGTACCGCGAGTCGGACAACGTTC |
| MT-Xba-3 | GATCTCTAGATCACTCATTATCACCGGCGT |
| Nb-16f | GGAGACCATTCCGG |
| Nb-16r | GGATCCGTCTTCCAG |
| Nov-6 | GGGATCGCAGAGGAGC |
| pKS-3 | GCCCTCCCGTATCGTAGT |
| pKS-5 | GTAGCTCTTGATCCGGCA |
| prom-2 | GACGACGGATCCTCCTGGGCCTCTTTG |
| RSET-Nde | GACGCATATGCGGGGTTCTCATCATC |
| Soj-5 | GCAGCAAGATCTATCGGAGTTACCAACCAGAAA |
| Soj-3 | CAGCAGCTGCAGCAGCAGTCAGCCATGGAATCCCT |
| Tna-N1 | GATTAAGCTTCTGAGGAATCGACCGGTT |
| Tna-N2 | GAACGGATCCCATATGTAGCAACTCGAGAACGCC |
For the construction of pRSET-ORF79, the ORF79 gene was amplified by PCR with the primers 79-Bgl and 79-Kpn using the ϕCh1 DNA as a template. The PCR product was digested with BglII and KpnI and ligated into pRSET-A (Invitrogen, Thermo Fisher Scientific Inc., Waltham, MA, USA), digested with BamHI and KpnI.
For the construction of pΔORF79::novR, a DNA fragment (nt 47649 to 48497) including the sequence of ORF79 was amplified by PCR with the primers 79-Nae and 79-Pst using the ϕCh1 DNA as a template. The PCR product was digested with NaeI and PstI and ligated into pKSII+ (Agilent Technologies). This plasmid was digested with SacI and treated with T4 DNA polymerase. The gyrB gene, released from the plasmid pMDS11 (21) by digestion with HindIII and treatment with the Klenow fragment polymerase, was inserted, resulting in pΔORF79::novR.
For the construction of plasmid pNB102-ORF79, the promoter region of ORF49 (nt 34321 to 34479 of ϕCh1) was amplified by PCR with the primers prom-2 and 49-Kpn using the ϕCh1 DNA as a template. The PCR product was digested with KpnI and BamHI and cloned into the plasmid pUC19 (PubMed Identifier [PMID] 2985470), resulting in pUC19-prom49. ORF79 was amplified by PCR with the primers 79-Bgl and 79-PstI using the ϕCh1 DNA as a template. The PCR product was digested with BglII and PstI and ligated into the plasmid pUC19-prom49, which was digested with BamHI and PstI. The promoter gene fusion was released from this plasmid by digestion with HindIII, followed by blunting with Klenow polymerase and subsequent digestion with KpnI and ligated into the plasmid pNB102 (22), which had been digested with XbaI, filled in with Klenow polymerase, and restricted with KpnI.
For the construction of pRo-5-ptna-bgaH, the upstream region of the tnaA gene (nt 1969653 to 1970184; GenBank accession number CP001932) (12) was amplified by PCR with the primers TnaN-1 and TnaN-2 using chromosomal DNA of N. magadii L13 as the template. The PCR product was digested with BamHI and HindIII and inserted into pBAD24 (23), restricted with the same enzymes, resulting in the plasmid pBAD24-tnaN. The bgaH gene was released from the plasmid pRV1-pTna (11) by digestion with NdeI and BamHI and introduced into pBAD24-tnaN, restricted with the same enzymes. The fusion of the tnaN promoter and bgaH was released from this plasmid by digestion with BamHI, treatment with Klenow polymerase, and digestion with HindIII and was finally introduced into pRo-5 (24), digested with KpnI, treated with T4 DNA polymerase, and digested with HindIII.
For the construction of pRo-5-ptna-ORF79, the ORF79 including a His tag was amplified by PCR with the primers RSET-Nde and 79-KB using the plasmid pRSET-ORF79 as a template. The PCR product was digested with NdeI and BlgII and ligated into the plasmid pBAD24-tnaN. The fusion of the tnaN promoter and ORF79 was released from this plasmid by digestion with KpnI and HindIII and ligated into the shuttle vector pRo-5 (24), restricted with the same enzymes.
For the construction of pNB102-ORF3452, ORF3452 as well as its own promoter was amplified by PCR with the primers 34-Kpn and 36-3 using the plasmid pBgb52 (10) as the template. The PCR product was digested with KpnI and HindIII and ligated into pNB102 (22), restricted with the same enzymes.
For the construction of pNB102-MTase, the ORF94 (M.NmaϕCh1-I) as well as its own promoter was amplified by PCR with the primers MT-Kpn-5 and MT-Xba-3 using the ϕCh1 DNA as a template. The PCR product was digested with KpnI and XbaI and ligated into pNB102 (22), restricted with the same enzymes.
Natrialba magadii protoplast transformation.
Transformation of N. magadii was performed by generation of protoplasts using proteinase K and polyethylene glycol 600 (PEG600) as previously described (24). Plasmid DNA was used at 5 μg for the transformation of vectors and 30 μg for gene disruptions. Plates were incubated at 37°C in sealed bags for up to 4 weeks until colonies were visible.
Screening of Natrialba magadii transformants.
To screen for transformants, culture supernatant was diluted 1 to 10 in sterile double-distilled H2O (ddH2O) and mixed vigorously. One microliter of the solution was used as the template in a 25-μl reaction mixture using GoTaq DNA polymerase (Promega Corporation, Madison, WI, USA) according to the manufacturer's instructions.
DNA isolation from halophilic Archaea.
Chromosomal DNA of N. magadii was isolated by phenol-chloroform extraction, followed by precipitation with isopropanol as previously described (2).
RT-PCR.
Isolation of RNA and reverse transcription-PCR (RT-PCR) were performed as described previously (9). Ten nanograms of total RNA was used for each reaction. The primers 79-RT-1 and 79-RT-2 were used to amplify ORF79 (Table 5). Each RNA sample was subjected to a control PCR using the same buffers, amounts of RNA and primers, and cycling conditions as for the combined RT-PCR. None of the reactions revealed a PCR product, indicating that the RNA samples were free of DNA. To ensure that equal amounts of RNA were used for all samples, a control PCR specific for 16S rRNA was performed with primers Nb-16f and Nb-16r. To verify that the correct fragments had been amplified, all PCR products were separated on agarose gels and hybridized with a cloned ORF79 DNA fragment.
Southern hybridization.
Southern hybridization was performed as previously described (4) using a Phototope-Star detection kit (New England BioLabs, Inc., Ipswich, MA, USA) according to the manufacturer's instructions.
Protein expression and Western blot analyses.
For the recombinant production of the gp79, E, gp3452, and M.NmaϕCh1-I proteins, the plasmids pRSET-ORF79, pET-E (4), pQE-cp3452 (10), and pMT14 (9)were transformed into E. coli Rosetta (Novagen, Merck KGaA, Darmstadt, Germany), respectively. Single colonies were grown in LB broth at 37°C with shaking at 200 rpm. Gene expression was induced by the addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an OD600 of 0.3. Cells were harvested after 3 h by centrifugation at 6,000 × g for 10 min at 4°C, resuspended in buffer B (8 M urea, 0.1 M NaH2PO4, 10 mM Tris-HCl, pH 8.0), and disrupted by sonication. The His-tagged protein was affinity purified under denaturing conditions using a nickel-agarose column (Qiagen N.V., Venlo, The Netherlands) according to the manufacturer's instructions. The purified proteins were used for the generation of polyclonal antibodies in rabbits (Moravian-Biotechnology, Ltd., Brno, Czech Republic).
Western blot analyses were performed essentially as described previously (25) using polyclonal rabbit antibodies followed by anti-rabbit IgG-horseradish peroxidase (HRP) (GE Healthcare, Buckinghamshire, United Kingdom). Detection was performed with SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions.
Cell extracts were prepared as described previously (4). Cellular proteins were precipitated from the supernatants by the addition of 5% trichloroacetic acid (TCA) and resuspended according to the optical density of the culture sample.
Negative staining and TEM.
Virus particles were stained with 2% uranyl formate according to standard protocols (26) and viewed under a transmission electron microscope (TEM) (EM-900; Carl Zeiss, Oberkochen, Germany).
Virus titer determination.
Virus titers were determined by plating appropriate dilutions prepared in rich medium as described previously (2).
Determination of burst size and eclipse.
The determination of the burst size as well as of the eclipse period was performed as described by Witte et al. (2).
β-Galactosidase assays.
β-Galactosidase (BgaH) activities in N. magadii L13 were determined as described previously (27). The protein concentrations were quantified using the Bradford method (28).
Bioinformatics analyses.
Bioinformatics analyses were performed using the tools at http://www.uniprot.org and http://pfam.xfam.org/.
REFERENCES
- 1.Tindall BJ, Mills AA, Grant WD. 1984. Natronobacterium gen nov. and Natronococcus gen. nov., two genera of haloalkaliphilic archaebacteria. Syst Appl Microbiol 5:41–57. doi: 10.1016/S0723-2020(84)80050-8. [DOI] [Google Scholar]
- 2.Witte A, Baranyi U, Klein R, Sulzner M, Luo C, Wanner G, Kruger DH, Lubitz W. 1997. Characterization of Natronobacterium magadii phage ϕCh1, a unique archaeal phage containing DNA and RNA. Mol Microbiol 23:603–616. doi: 10.1046/j.1365-2958.1997.d01-1879.x. [DOI] [PubMed] [Google Scholar]
- 3.Klein R, Baranyi U, Rossler N, Greineder B, Scholz H, Witte A. 2002. Natrialba magadii virus ϕCh1: first complete nucleotide sequence and functional organization of a virus infecting a haloalkaliphilic archaeon. Mol Microbiol 45:851–863. doi: 10.1046/j.1365-2958.2002.03064.x. [DOI] [PubMed] [Google Scholar]
- 4.Klein R, Greineder B, Baranyi U, Witte A. 2000. The structural protein E of the archaeal virus ϕCh1: evidence for processing in Natrialba magadii during virus maturation. Virology 276:376–387. doi: 10.1006/viro.2000.0565. [DOI] [PubMed] [Google Scholar]
- 5.Prangishvili D, Garrett RA, Koonin EV. 2006. Evolutionary genomics of archaeal viruses: unique viral genomes in the third domain of life. Virus Res 117:52–67. doi: 10.1016/j.virusres.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Breuert S, Allers T, Spohn G, Soppa J. 2006. Regulated polyploidy in halophilic archaea. PLoS One 1:e92. doi: 10.1371/journal.pone.0000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iro M, Klein R, Galos B, Baranyi U, Rössler N, Witte A. 2007. The lysogenic region of virus ϕCh1: identification of a repressor-operator system and determination of its activity in halophilic Archaea. Extremophiles 11:383–396. doi: 10.1007/s00792-006-0040-3. [DOI] [PubMed] [Google Scholar]
- 8.Klein R, Rössler N, Iro M, Scholz H, Witte A. 2012. Haloarchaeal myovirus ϕCh1 harbours a phase variation system for the production of protein variants with distinct cell surface adhesion specificities. Mol Microbiol 83:137–150. doi: 10.1111/j.1365-2958.2011.07921.x. [DOI] [PubMed] [Google Scholar]
- 9.Baranyi U, Klein R, Lubitz W, Krüger DH, Witte A. 2000. The archaeal halophilic virus-encoded Dam-like methyltransferase M.ϕCh1-I methylates adenine residues and complements dam mutants in the low salt environment of Escherichia coli. Mol Microbiol 35:1168–1179. doi: 10.1046/j.1365-2958.2000.01786.x. [DOI] [PubMed] [Google Scholar]
- 10.Rössler N, Klein R, Scholz H, Witte A. 2004. Inversion within the haloalkaliphilic virus ϕCh1 DNA results in differential expression of structural proteins. Mol Microbiol 52:413–426. doi: 10.1111/j.1365-2958.2003.03983.x. [DOI] [PubMed] [Google Scholar]
- 11.Large A, Stamme C, Lange C, Duan Z, Allers T, Soppa J, Lund PA. 2007. Characterization of a tightly controlled promoter of the halophilic archaeon Haloferax volcanii and its use in the analysis of the essential cct1 gene. Mol Microbiol 66:1092–1106. doi: 10.1111/j.1365-2958.2007.05980.x. [DOI] [PubMed] [Google Scholar]
- 12.Siddaramappa S, Challacombe JF, Decastro RE, Pfeiffer F, Sastre DE, Gimenez MI, Paggi RA, Detter JC, Davenport KW, Goodwin LA, Kyrpides N, Tapia R, Pitluck S, Lucas S, Woyke T, Maupin-Furlow JA. 2012. A comparative genomics perspective on the genetic content of the alkaliphilic haloarchaeon Natrialba magadii ATCC 43099T. BMC Genomics 13:165. doi: 10.1186/1471-2164-13-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bath C, Dyall-Smith ML. 1998. His1, an archaeal virus of the Fuselloviridae family that infects Haloarcula hispanica. J Virol 72:9392–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pagaling E, Haigh RD, Grant WD, Cowan DA, Jones BE, Ma Y, Ventosa A, Heaphy S. 2007. Sequence analysis of an archaeal virus isolated from a hypersaline lake in Inner Mongolia, China. BMC Genomics 8:410. doi: 10.1186/1471-2164-8-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang SL, Nuttall S, Ngui K, Fisher C, Lopez P, Dyall-Smith M. 2002. HF2: a double-stranded DNA tailed haloarchaeal virus with a mosaic genome. Mol Microbiol 44:283–296. doi: 10.1046/j.1365-2958.2002.02890.x. [DOI] [PubMed] [Google Scholar]
- 16.Tang SL, Nuttall S, Dyall-Smith M. 2004. Haloviruses HF1 and HF2: evidence for a recent and large recombination event. J Bacteriol 186:2810–2817. doi: 10.1128/JB.186.9.2810-2817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sencilo A, Jacobs-Sera D, Russell DA, Ko CC, Bowman CA, Atanasova NS, Osterlund E, Oksanen HM, Bamford DH, Hatfull GF, Roine E, Hendrix RW. 2013. Snapshot of haloarchaeal tailed virus genomes. RNA Biol 10:803–816. doi: 10.4161/rna.24045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woese CR, Kandler O, Wheelis ML. 1990. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A 87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derntl C, Selb R, Klein R, Alte B, Witte A. 2015. Genomic manipulations in alkaliphilic haloarchaea demonstrated by a gene disruption in Natrialba magadii. FEMS Microbiol Lett 362:fnv179. doi: 10.1093/femsle/fnv179. [DOI] [PubMed] [Google Scholar]
- 20.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 21.Holmes ML, Nuttall SD, Dyall-Smith ML. 1991. Construction and use of halobacterial shuttle vectors and further studies on Haloferax DNA gyrase. J Bacteriol 173:3807–3813. doi: 10.1128/jb.173.12.3807-3813.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou M, Xiang H, Sun C, Tan H. 2004. Construction of a novel shuttle vector based on an RCR-plasmid from a haloalkaliphilic archaeon and transformation into other haloarchaea. Biotechnol Lett 26:1107–1113. doi: 10.1023/B:BILE.0000035493.21986.20. [DOI] [PubMed] [Google Scholar]
- 23.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayrhofer-Iro M, Ladurner A, Meissner C, Derntl C, Reiter M, Haider F, Dimmel K, Rössler N, Klein R, Baranyi U, Scholz H, Witte A. 2013. Utilization of virus ϕCh1 elements to establish a shuttle vector system for halo(alkali)philic Archaea via transformation of Natrialba magadii. Appl Environ Microbiol 79:2741–2748. doi: 10.1128/AEM.03287-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harlow E, Lane D. 1988. Antibodies: a laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 26.De Carlo S, Harris JR. 2011. Negative staining and cryo-negative staining of macromolecules and viruses for TEM. Micron 42:117–131. doi: 10.1016/j.micron.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holmes ML, Dyall-Smith ML. 2000. Sequence and expression of a halobacterial beta-galactosidase gene. Mol Microbiol 36:114–122. doi: 10.1046/j.1365-2958.2000.01832.x. [DOI] [PubMed] [Google Scholar]
- 28.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]