

ABSTRACT
Although it has been known for over 40 years that eukaryotic mRNAs bear internal base modifications, it is only in the last 5 years that the importance of these modifications has begun to come into focus. The most common mRNA modification, the addition of a methyl group to the N6 position of adenosine (m6A), has been shown to affect splicing, translation, and stability, and m6A is also essential for embryonic development in organisms ranging from plants to mice. While all viral transcripts examined so far have been found to be extensively m6A modified, the role, if any, of m6A in regulating viral gene expression and replication was previously unknown. However, recent data generated using HIV-1 as a model system strongly suggest that sites of m6A addition not only are evolutionarily conserved but also enhance virus replication. It is therefore likely that the field of viral epitranscriptomics, which can be defined as the study of functionally relevant posttranscriptional modifications of viral RNA transcripts that do not change the nucleotide sequence of that RNA, is poised for a major expansion in scientific interest and may well fundamentally change our understanding of how viral replication is regulated.
KEYWORDS: Posttranscriptional gene regulation, RNA modification, N6-methyladenosine, mRNA function, mRNA stability, HIV-1
INTRODUCTION
While over 100 different modified bases have been identified on RNA transcripts in mammalian cells, the majority of these are restricted to noncoding RNAs, especially tRNAs. However, at least 10 distinct modified bases have now been reported to occur in mammalian mRNAs (1). In addition to the 7-methylguanosine cap that is added at the 5′ end of all cellular mRNAs, these include N6-methyladenosine (m6A), 2′-O-methyladenosine (Am), N6-2′-O-methyladenosine (m6Am), pseudouridine, and 5-methylcytosine. Of these, by far the most prevalent internal modified base found on mRNAs is m6A, and recent work has now begun to reveal how m6A affects mRNA function and how to precisely map the m6A residues present on mRNAs (1–3). m6A is also highly prevalent on a wide range of different viral RNA species (4–13), and recently, the first reports demonstrating a significant phenotypic effect of these m6A modifications have been published (10–14). Therefore, we will focus this review entirely on m6A and how this particular modification might affect different aspects of the viral life cycle.
m6A was first reported to be present on cellular mRNAs in 1975 with ∼3 internal m6A residues found on the average ∼2.2-kb transcript (15, 16). However, we now know that many cellular mRNAs, including mRNAs encoding housekeeping genes, lack any m6A residues, while highly regulated mRNAs may contain 10 or more (2, 3). The first demonstration of m6A residues on viral mRNAs soon followed and, using the biochemical approaches available at that time, a range of mRNAs encoded by several nuclear DNA and RNA viruses were then shown to bear fairly high levels of m6A, with the eight influenza A virus (IAV) mRNAs bearing an average of three m6A residues each (4–9). Subsequent work looking at each individual IAV mRNA revealed that IAV mRNAs actually contain from 1 to 8 m6A residues each (Table 1) (5). Investigators looking at transcripts encoded by the Rous sarcoma virus (RSV) also demonstrated that the RSV genomic RNA contained at least 8 m6A residues and were able to map two of these (7, 17). However, mutagenesis of these two m6A sites did not produce any phenotypic effect (17). In the absence of a more facile method to map the precise location of m6A on transcripts, and in the absence of information about which cellular factors produce and detect m6A residues, the field of viral epitranscriptomics, which can be defined as the study of functionally relevant posttranscriptional modifications of viral RNA transcripts that do not change the nucleotide sequence of that RNA, then became largely quiescent for almost 2 decades. During this time, researchers looking at aspects of gene regulation and development in a number of organisms were able to gradually identify several factors relevant to m6A addition and function and, perhaps most importantly, to develop techniques that map m6A sites with near single-nucleotide resolution.
TABLE 1.
Viruses encoding RNAs with reported m6A residues
| Virus | No. of m6A residues | References |
|---|---|---|
| RNA viruses | ||
| Influenza A virus | ∼24 | 4, 5 |
| Avian sarcoma virus | 13–15 | 6 |
| Rous sarcoma virus | 10–12 | 7 |
| Feline leukemia virus | NAa | 46 |
| HIV-1 | 10–14 | 10, 11 |
| Hepatitis C virus | ∼16 | 12 |
| Flavivirusesb | 5–12 | 12, 13 |
| DNA viruses | ||
| Adenovirus | NA | 8, 45 |
| SV40 | NA | 9, 43 |
| Herpes simplex virus 1 | NA | 44 |
NA, not available or not applicable.
Including Zika virus, yellow fever virus, Dengue virus, and West Nile virus, all of which were reported to contain multiple internal m6A residues.
The addition to m6A occurs predominantly in the nucleus and is mediated by the enzyme methyl transferase-like 3 (METTL3) together with several cofactors that have been reported to include METTL14, WTAP, KIAA1429, and RBM15/RBM15B (Fig. 1) (18–22). The human nucleus contains at least two proteins able to detect m6A residues, called YTHDC1 and YTHDC2 (23–26). YTHDC1, known as YT521-B in Drosophila, has been proposed to regulate mRNA splicing and is required for transcriptional repression by the long noncoding RNA XIST, which is heavily m6A modified (21, 24–26). Once exported from the nucleus, m6A residues on mRNAs are bound by three related cytoplasmic proteins, called YTHDF1, YTHDF2 and YTHDF3, which are believed to mediate the phenotypic effects of m6A on mRNA stability and translation (Fig. 1) (2, 3, 27).
FIG 1.

Overview of m6A addition to RNA transcripts. m6A addition to cellular mRNAs and to the majority of viral mRNAs occurs in the nucleus and is thought to be cotranscriptional. m6A addition is mediated by a complex consisting of METTL3 and several cofactors, including METTL14 and WTAP, which use SAM as a methyl donor. SAM is derived from SAC hydrolase (SAH) and this enzymatic step can be blocked by the drug DAA, resulting in a global inhibition of m6A addition. m6A can also be removed by the predominantly nuclear m6A demethylase ALKBH5, and can be detected in the nucleus by the m6A readers YTHDC1 and YTHDC2, which can modulate RNA. After nuclear export, m6A marks are bound by the cytoplasmic YTHDF1, YTHDF2, and/or YTHDF3 protein, which can regulate mRNA translation and/or stability. While m6A addition primarily occurs in the nucleus, METTL3 and other components of the m6A “writer” complex have been detected in the cytoplasm, possibly in response to stress; cytoplasmic RNA viruses also bear m6A marks.
In addition to METTL3 and its associated cofactors, referred to as m6A “writers,” and the various m6A-binding proteins, referred to as m6A “readers,” at least two proteins, ALKBH5 and FTO, have been proposed to function as m6A demethylases or “erasers” (28, 29). However, recent data suggest that FTO actually selectively demethylates the m6Am residues located at position 2 in many mRNAs and has a very limited ability to demethylate internal m6A residues (30). Nevertheless, the existence of at least one m6A eraser, the largely nuclear ALKBH5, means that m6A has the ability to function as a dynamic mRNA modification that can be added or removed in response to stress or other signals (2, 3).
A major reason why m6A has become a focus of research interest relates to the profound cellular phenotypes observed when m6A addition is perturbed. Loss of m6A addition is embryonic lethal in plants (31) and strongly perturbs development and sex determination in Drosophila (25, 26, 32). Moreover, loss of m6A addition blocks the differentiation of mammalian embryonic stem cells (33, 34). Importantly, the m6A addition machinery is evolutionarily conserved in all multicellular organisms examined thus far and is also present in fungi, including the yeast Saccharomyces cerevisiae (32), thus highlighting its potential importance.
The sequence specificity of the writer proteins that add m6A to mRNAs is not entirely clear, though it has been known for some time that the minimal sequence context is 5′-Rm6AC-3′ (where R is a purine) (6). A larger consensus sequence, 5′-RRm6ACH-3′ (where H is A, C, or U), has also been suggested (2, 3, 35), and evidence indicates that 5′-Gm6AC-3′ is generally preferred over 5′-Am6AC-3′ (6, 10). Yet, at most 10% of the consensus m6A sites found on mRNAs are actually modified and, despite the random distribution of consensus target sites, m6A residues are also, for currently unclear reasons, concentrated in the 3′ untranslated region (UTR) of cellular mRNAs (36, 37).
A major step forward in the study of m6A was the development of techniques to map the adenosine residues that are actually modified. The first reported technique, called Me-RIP-seq (37, 38), uses a commercially available antiserum that specifically recognizes m6A. With this protocol, mRNAs are first purified by poly(A) selection and are then fragmented to ∼100 to 200 nucleotide (nt) pieces. The fragmented RNA is then incubated with the m6A-specific antiserum, which enables the selective immunoprecipitation (IP) of m6A-containing RNA fragments. These are collected, subjected to deep sequencing, and then mapped onto the relevant genome or mRNA transcript using bioinformatics. The problems with Me-RIP-seq are 2-fold. First, because this technique is completely reliant on the relatively weak interaction between the antibody and m6A, the purification steps that can be performed are not that rigorous, resulting in significant nonspecific RNA background. Second, the precision of m6A site mapping that can be achieved is only 100 to 200 nt. As 5′-RAC-3′ sequences are expected to occur by chance every 32 nt, this technique cannot map m6A sites precisely and cannot distinguish between single m6A sites and m6A clusters.
The second technique used for m6A mapping, PA-m6A-seq, also relies on the same m6A-specific antiserum but uses poly(A)-containing mRNA derived from cells that have been pulsed with the highly photoactivatable uridine analog 4-thiouridine (4SU) (39). Once the antibody has been bound to the purified 4SU-labeled mRNA population, the antibody is cross-linked to the RNA by a pulse of UV light. The resultant RNA:protein complexes can then be rigorously purified prior to digestion with T1 RNase to remove RNA sequences that are not protected by the bound antibody. The antibody is then removed by proteinase K treatment, and the resultant ∼30-nt RNA fragments are deep sequenced. An additional advantage of this variation on the photoactivatable ribonucleoside-enhanced cross-linking and IP (PAR-CLIP) (40) protocol is that the cross-linked 4SU residue is misread by reverse transcriptase as a C, so that any residual contaminating RNA fragments can be discarded during bioinformatic analysis by including only reads bearing single U-to-C mutations. Other major advantages of PA-m6A-seq are the resultant extremely low background and the increased resolution of ∼30 nt. Despite claims that PA-m6A-seq can identify m6A residues at single-nucleotide resolution, the prevalence of the 5′-RAC-3′ motif means that there are quite often 2 or even 3 candidate A residues within the mapped m6A peak. This remains a problem, although targeted mutagenesis of individual 5′-RAC-3′ consensus sequences, followed by a repeat of the PA-m6A-seq analysis, represents one effective way to resolve this issue. Recently, another method for mapping m6A residues by cross-linking m6A-specific antibodies to RNA molecules, referred to as m6A individual-nucleotide-resolution cross-linking and IP (miCLIP), has been reported (41) that, as made clear by its name, claims single-nucleotide mapping of m6A sites. The key to this level of resolution is the authors' finding that UV cross-linking followed by reverse transcription specifically and uniquely results in the introduction of a C-to-T mutation at the cytosine present in the m6A consensus sequence 5′-Rm6AC-3′ in the ∼40-nt-long reads obtained, thus enabling the unequivocal bioinformatic identification of m6A residues on transcripts of interest.
A final method used to map m6A sites relies on the fact that the cytoplasmic YTHDF1, YTHDF2, and YTHDF3 reader proteins (Fig. 2) are all known to specifically bind to m6A (2, 3). A form of PAR-CLIP in which cells are pulsed with 4SU, cross-linked using UV light, and then subjected to immunoprecipitation of a YTHDF protein, followed by RNase T1 and proteinase K treatment and cDNA synthesis, can therefore identify precisely the YTHDF protein-binding sites on mRNAs (10). These sites should then define functionally relevant m6A residues. As in the case of PA-m6A-seq, YTHDF PAR-CLIP again maps m6A sites with ∼30-nt resolution and gives rise to almost no background. It remains theoretically possible that YTHDF1, YTHDF2, or YTHDF3 might also bind to RNA sites that lack m6A, though we have not so far observed this phenomenon.
FIG 2.
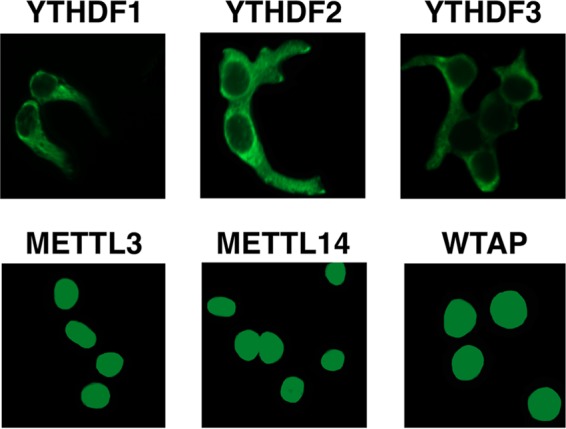
Subcellular locations of the m6A writers and readers. 293T cells were transfected with plasmids expressing FLAG-tagged versions of the m6A reader proteins YTHDF1, YTHDF2, and YTHDF3 (upper panels) and of the writer components METTL3, METTL14, and WTAP (lower panels), and were then subjected to immunofluorescence using an anti-FLAG antibody. These panels, which are intentionally slightly overexposed, reveal that the m6A writers are all tightly nuclear at steady state while the YTHDF readers are all cytoplasmic. Nevertheless, this result does not preclude the nucleocytoplasmic shuttling of any of these proteins, and the writers, in particular, have been proposed to enter the cytoplasm, possibly in response to stress.
While the techniques described above can accurately map m6A residues on all expressed RNA transcripts in a cell, they are at best semiquantitative and the actual level of m6A modification at any given site is therefore uncertain. One published technique called “site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography” (SCARLET) has been reported to enable the quantification of the level of m6A at specific sites on RNAs (42). However, as implied by its name, this procedure is technically complex, is expensive to perform, and assesses the level of m6A at one adenosine at a time. It is therefore clear that a simpler high-throughput approach that measures the level of m6A modification at multiple sites on the transcriptome simultaneously would represent an important technical advance.
Armed with the ability to inhibit m6A addition or function using RNA interference (RNAi) or gene editing and to map and mutate specific m6A residues on RNAs, it is now possible to begin to ask precisely how individual m6A residues, and the process of m6A addition in general, affect viral replication and gene expression. While only a few articles have appeared so far using this kind of approach, it appears likely that the emerging field of viral epitranscriptomics is not only poised for a major expansion but also has the potential to greatly influence our understanding of how viruses regulate their life cycle.
NUCLEAR RNA AND DNA VIRUSES
As the cellular proteins that add m6A to transcripts reside in the nucleus at steady state (2, 3) (Fig. 2), one might anticipate that, if viral RNAs are indeed m6A modified, this would primarily or exclusively occur for RNAs generated by nuclear DNA or RNA viruses. In fact, analyses of three DNA viruses (adenovirus, herpes simplex virus type 1, and SV40), four retroviruses (the closely related avian sarcoma virus and Rous sarcoma virus as well as HIV-1 and feline leukemia virus), and the orthomyxovirus influenza A virus (IAV) have revealed m6A residues present at levels that are at least as high as the number of m6A residues detected on cellular mRNAs (4–11, 43–46). Moreover, in HIV-1, where m6A residues have been mapped at near single-nucleotide resolution, the consensus m6A addition sites that are utilized are highly conserved across HIV-1 isolates (10). Given the plasticity of the HIV-1 genome, this conservation clearly implies that m6A facilitates some aspect of the replication cycle of HIV-1 and, by extension, of other nuclear viruses that express m6A-modified transcripts (Table 1). We note that m6A addition has been proposed to affect mRNA splicing (24–26), stability (27, 47), and translation (47–50), to modify RNA structure (51), and to inhibit the recognition of viral RNAs by Toll-like receptors and RIG-I (52, 53), and so m6A could positively regulate several aspects of the viral life cycle. Indeed, knockdown of the METTL3 and/or METTL14 m6A writers using RNA interference (RNAi) has been reported to inhibit HIV-1 replication up to 5-fold, while knockdown of the ALKBH5 m6A demethylase enhanced HIV-1 replication up to 8-fold (11, 14). Similarly, in CD4-positive T cells, overexpression of the predominant cytoplasmic reader protein YTHDF2 enhanced HIV-1 replication, while knockout of YTHDF2 by gene editing inhibited HIV-1 replication by 2-fold or more (10). We note that one group has reported, in contrast, that all of the YTHDF proteins can inhibit HIV-1 replication (14). However, this group exclusively analyzed replication of an HIV-1 variant bearing the firefly luciferase (FLuc) indicator gene in place of nef, and we have observed that FLuc actually contains prominent m6A modification sites that may well affect how YTHDF proteins affect the replication of this HIV-1-derived lentiviral vector (E. M. Kennedy and B. R. Cullen, unpublished results). We therefore believe it is essential that experiments addressing how m6A affects virus replication use wild-type viruses rather than viral mutants that have been modified to express an exogenous indicator gene. In conclusion, the prevalence and conservation of m6A residues on nuclear DNA and RNA viruses, combined with the limited number of reports looking at how m6A affects the replication of HIV-1, clearly suggest that m6A addition enhances viral gene expression and, hence, replication. However, the mechanistic basis for this positive effect currently remains unclear. We anticipate that ongoing efforts to precisely map m6A sites on viral transcripts, combined with the targeted mutagenesis of these m6A addition sites, will shed additional light on this question in the near future.
CYTOPLASMIC RNA VIRUSES
As noted above and demonstrated in Fig. 2, the cellular m6A writers METTL3, METTL14, and WTAP are all localized to the nucleus at steady state (2, 3). However, it has also been reported that METTL3 and METTL14 can be detected in the cytoplasm (12, 13, 54), suggesting that these proteins have the ability to shuttle between the nucleus and the cytoplasm and/or to enter the cytoplasm in response to stress.
If the m6A writers are indeed able to access the cytoplasm, then this raises the possibility that cytoplasmic viruses might also encode mRNAs bearing m6A residues. In fact, analyses of hepatitis C virus (HCV) and several different flaviviruses, including Zika virus, Dengue virus, yellow fever virus, and West Nile virus, have revealed at least 5 and to up to 16 m6A modification sites on the RNA genomes of these viruses (12, 13). In the case of HCV, the effect of m6A modifications has been analyzed in detail, and surprisingly and in marked contrast to HIV-1, knockdown of METTL3 and METTL14 mRNA using RNAi enhanced the production of infectious HCV virions, and knockdown of the mRNAs encoding the YTHDF proteins had a similar positive effect (12). Interestingly, HCV mRNA translation and RNA replication were both unaffected, thus suggesting that m6A on HCV RNAs might directly regulate the production of infectious HCV virions. Indeed, immunofluorescence analysis of HCV-infected cells showed that YTHDF proteins and the HCV structural proteins colocalize to the lipid droplets that function as sites of HCV virion morphogenesis, consistent with a direct role for m6A in regulating HCV virion production (12). Similarly, in the case of Zika virus, knockdown of METTL3 or METTL14 mRNA was also reported to enhance the production of Zika virions, while knockdown of ALKBH5 mRNA exerted an opposite inhibitory effect (13).
In general, viruses, especially RNA viruses that rely on virally encoded, error-prone RNA-dependent RNA polymerases, can rapidly evolve to inactivate sequences present on the viral RNA genome, such as targets for small interfering RNAs, that inhibit their replication in cis (55). Similarly, m6A addition to viral RNAs, which requires the consensus sequence 5′-RRm6ACH-3′, would also be easy for a virus to avoid if m6A indeed exerted an inhibitory effect in cis. It could be argued that for a virus that establishes long-term persistent infections, such as HCV, it might be advantageous to downregulate the rate of viral replication so as to mitigate host immune responses. However, this argument makes little sense in the case of Zika virus or the other flaviviruses listed in Table 1, which cause acute infections marked by high viremia, which are generally rapidly cleared by the host adaptive immune response. Thus, the fact that multiple m6A residues have been detected on all the flaviviruses analyzed so far (Table 1) argues that m6A addition has been selected for, rather than against, during flavivirus evolution. The observation that m6A can inhibit the release of infectious virions by Zika virus-infected cells is therefore difficult to understand. It is possible that m6A, as noted above, enables Zika virus to avoid the viral RNA-induced activation of innate antiviral immune responses, which might balance or enhance viral replication in vivo (52, 53). However, one would then have to argue that these antiviral responses have been lost in the cells used to analyze Zika virus growth in culture. Indeed, the Vero cells that were exclusively used by Linchinchi et al. (13) are known to be unable to mount an interferon response (56). Additional experiments using other cells that are fully competent to mount antiviral innate immune responses and cells in which m6A addition has been knocked out by gene editing, rather than knocked down using RNAi, are needed to resolve this conundrum.
m6A AS A TARGET FOR ANTIVIRAL THERAPY
If m6A indeed normally functions to enhance viral replication, as implied by the conservation of m6A on transcripts produced by diverse virus families (Table 1) and also supported by data generated using HIV-1 (10, 11), then m6A addition presents itself as a possible target for antiviral agents. The advantage of drugs that inhibit cellular proteins required for virus replication is that they make it very difficult for the virus to evolve resistance, while the disadvantage is that they can inhibit the normal physiological function of that protein and, hence, cause toxicity. So, is m6A addition a potential target for antiviral drug development? In fact, several lines of data suggest that this might be the case. Specifically, the S-adenosylhomocysteine (SAC) hydrolase inhibitor 3-deazaadenosine (DAA) has been shown to inhibit m6A addition and to act as a broad antiviral inhibitor (57–61).
The inhibition of SACH activity by DAA results in the accumulation of SAC in cells, which in turn results in depletion of S-adenosylmethionine (SAM), the methyl donor used by METTL3 to generate m6A (57) (Fig. 1). As SAM is used as a methyl donor by a wide range of cellular methylases, DAA is clearly not a specific inhibitor of m6A formation, though mRNA capping has been shown to be unaffected by DAA treatment (62). So, is DAA too toxic to use as an antiviral? In fact, several papers have reported using DAA to inhibit the replication of diverse viruses, including Rous sarcoma virus, HIV-1, respiratory syncytial virus, parainfluenza virus, vesicular stomatitis virus, measles virus, and reovirus (57–61), in cultured cells at concentrations that did not show any detectable cytopathic effects. Even more impressively, DAA was found to effectively block respiratory syncytial virus replication in cotton rats (58) and Ebola virus-induced fatality in mice (63, 64) at doses that did not give rise to any evident toxicity. Importantly, DAA is not incorporated into cellular nucleic acids (57) and does not have the structure expected for a nucleoside that can function as a chain terminator. Thus, it appears probable that it is indeed the inhibition of SAC hydrolase activity that underlies this inhibitory effect, although whether m6A addition is indeed the key target for DAA remains uncertain (57). Nevertheless, these observations are consistent with the hypothesis that m6A addition plays an important positive role in the life cycle of a wide range of viruses and suggest that an inhibitor that can specifically target METTL3 activity, rather than SAM-dependent methylation in general, might be well tolerated and could prove to be an effective broad-spectrum antiviral, especially for viruses that cause acute infections and disease. Given recent data suggesting that excessive m6A modification of cellular mRNAs might also contribute to the progression of some forms of cancer, such as acute myeloid leukemia (65), efforts to identify specific inhibitors of m6A addition would seem to be very timely.
CONCLUSIONS AND FUTURE DIRECTIONS
While the emerging field of viral epitranscriptomics is clearly in its infancy, we nevertheless feel that the limited data reported thus far are consistent with the hypothesis that m6A will emerge as a ubiquitous modification of viral RNA transcripts that profoundly influences several different aspects of the viral life cycle. Exactly how m6A exerts its phenotypic effects at a mechanistic level is still largely unclear in not only the viral but also cellular context, but there is no question that this area has now become the subject of an intense research effort that has begun to clarify aspects of this problem. Clearly, the next step will be to precisely map and then mutate m6A residues found on different viral genomes and then study the phenotypic consequences. Obviously, it will be critical to ensure that any observed inhibition of viral replication is indeed due to loss of m6A rather than to the inactivation of some other cis-acting RNA sequence. To control for this potential problem, and assuming that the observed phenotype is not too severe, one could test m6A-deficient viral mutants not only in wild-type cells but also in METTL3 knockout cells, where the mutant and parental viruses should replicate at equivalent levels. Such specific viral mutants should then enable a precise definition of how the addition of m6A to viral mRNAs regulates viral gene expression and replication.
ACKNOWLEDGMENTS
This work was supported in part by a National Institutes of Health grant (R21-AI130574) to B. R. Cullen. K. Tsai was supported by National Cancer Institute grant T32-CA009111.
We thank Joy Marshall for assistance with immunofluorescence analysis.
REFERENCES
- 1.Li S, Mason CE. 2014. The pivotal regulatory landscape of RNA modifications. Annu Rev Genomics Hum Genet 15:127–150. doi: 10.1146/annurev-genom-090413-025405. [DOI] [PubMed] [Google Scholar]
- 2.Meyer KD, Jaffrey SR. 2014. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol 15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yue Y, Liu J, He C. 2015. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev 29:1343–1355. doi: 10.1101/gad.262766.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krug RM, Morgan MA, Shatkin AJ. 1976. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J Virol 20:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Narayan P, Ayers DF, Rottman FM, Maroney PA, Nilsen TW. 1987. Unequal distribution of N6-methyladenosine in influenza virus mRNAs. Mol Cell Biol 7:1572–1575. doi: 10.1128/MCB.7.4.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dimock K, Stoltzfus CM. 1977. Sequence specificity of internal methylation in B77 avian sarcoma virus RNA subunits. Biochemistry 16:471–478. doi: 10.1021/bi00622a021. [DOI] [PubMed] [Google Scholar]
- 7.Kane SE, Beemon K. 1985. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol Cell Biol 5:2298–2306. doi: 10.1128/MCB.5.9.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sommer S, Salditt-Georgieff M, Bachenheimer S, Darnell JE, Furuichi Y, Morgan M, Shatkin AJ. 1976. The methylation of adenovirus-specific nuclear and cytoplasmic RNA. Nucleic Acids Res 3:749–765. doi: 10.1093/nar/3.3.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canaani D, Kahana C, Lavi S, Groner Y. 1979. Identification and mapping of N6-methyladenosine containing sequences in simian virus 40 RNA. Nucleic Acids Res 6:2879–2899. doi: 10.1093/nar/6.8.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kennedy EM, Bogerd HP, Kornepati AV, Kang D, Ghoshal D, Marshall JB, Poling BC, Tsai K, Gokhale NS, Horner SM, Cullen BR. 2016. Posttranscriptional m(6)A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe 19:675–685. doi: 10.1016/j.chom.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lichinchi G, Gao S, Saletore Y, Gonzalez GM, Bansal V, Wang Y, Mason CE, Rana TM. 2016. Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nature Microbiol 1:16011. doi: 10.1038/nmicrobiol.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gokhale NS, McIntyre AB, McFadden MJ, Roder AE, Kennedy EM, Gandara JA, Hopcraft SE, Quicke KM, Vazquez C, Willer J, Ilkayeva OR, Law BA, Holley CL, Garcia-Blanco MA, Evans MJ, Suthar MS, Bradrick SS, Mason CE, Horner SM. 2016. N6-Methyladenosine in Flaviviridae viral RNA genomes regulates infection. Cell Host Microbe 20:654–665. doi: 10.1016/j.chom.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lichinchi G, Zhao BS, Wu Y, Lu Z, Qin Y, He C, Rana TM. 2016. Dynamics of human and viral RNA methylation during Zika virus infection. Cell Host Microbe 20:666–673. doi: 10.1016/j.chom.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tirumuru N, Zhao BS, Lu W, Lu Z, He C, Wu L. 2016. N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. eLife 5:e15528. doi: 10.7554/eLife.15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desrosiers R, Friderici K, Rottman F. 1974. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A 71:3971–3975. doi: 10.1073/pnas.71.10.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desrosiers RC, Friderici KH, Rottman FM. 1975. Characterization of Novikoff hepatoma mRNA methylation and heterogeneity in the methylated 5′ terminus. Biochemistry 14:4367–4374. doi: 10.1021/bi00691a004. [DOI] [PubMed] [Google Scholar]
- 17.Kane SE, Beemon K. 1987. Inhibition of methylation at two internal N6-methyladenosine sites caused by GAC to GAU mutations. J Biol Chem 262:3422–3427. [PubMed] [Google Scholar]
- 18.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, Dai Q, Chen W, He C. 2014. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, Zhao X, Li A, Yang Y, Dahal U, Lou XM, Liu X, Huang J, Yuan WP, Zhu XF, Cheng T, Zhao YL, Wang X, Rendtlew Danielsen JM, Liu F, Yang YG. 2014. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24:177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, Sanjana NE, Freinkman E, Pacold ME, Satija R, Mikkelsen TS, Hacohen N, Zhang F, Carr SA, Lander ES, Regev A. 2014. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8:284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR. 2016. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537:369–373. doi: 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, Zou T, Yin P. 2016. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534:575–578. doi: 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- 23.Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, Lu Z, He C, Min J. 2014. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol 10:927–929. doi: 10.1038/nchembio.1654. [DOI] [PubMed] [Google Scholar]
- 24.Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, Wang X, Ma HL, Huang CM, Yang Y, Huang N, Jiang GB, Wang HL, Zhou Q, Wang XJ, Zhao YL, Yang YG. 2016. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell 61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG, Soller M. 2016. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 540:301–304. doi: 10.1038/nature20577. [DOI] [PubMed] [Google Scholar]
- 26.Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, Kreim N, Andrade-Navarro MA, Poeck B, Helm M, Roignant JY. 2016. m6A modulates neuronal functions and sex determination in Drosophila. Nature 540:242–247. doi: 10.1038/nature20568. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, Ren B, Pan T, He C. 2014. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, Lu Z, Bosmans RP, Dai Q, Hao YJ, Yang X, Zhao WM, Tong WM, Wang XJ, Bogdan F, Furu K, Fu Y, Jia G, Zhao X, Liu J, Krokan HE, Klungland A, Yang YG, He C. 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, He C. 2011. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, Gross SS, Elemento O, Debart F, Kiledjian M, Jaffrey SR. 2017. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 541:371–375. doi: 10.1038/nature21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, Fray RG. 2008. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 20:1278–1288. doi: 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hongay CF, Orr-Weaver TL. 2011. Drosophila Inducer of Meiosis 4 (IME4) is required for Notch signaling during oogenesis. Proc Natl Acad Sci U S A 108:14855–14860. doi: 10.1073/pnas.1111577108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, Ben-Haim MS, Eyal E, Yunger S, Pinto Y, Jaitin DA, Viukov S, Rais Y, Krupalnik V, Chomsky E, Zerbib M, Maza I, Rechavi Y, Massarwa R, Hanna S, Amit I, Levanon EY, Amariglio N, Stern-Ginossar N, Novershtern N, Rechavi G, Hanna JH. 2015. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 34.Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, Carter AC, Flynn RA, Zhou C, Lim KS, Dedon P, Wernig M, Mullen AC, Xing Y, Giallourakis CC, Chang HY. 2014. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15:707–719. doi: 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Csepany T, Lin A, Baldick CJ Jr, Beemon K. 1990. Sequence specificity of mRNA N6-adenosine methyltransferase. J Biol Chem 265:20117–20122. [PubMed] [Google Scholar]
- 36.Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, Haripal B, Zucker-Scharff I, Moore MJ, Park CY, Vagbo CB, Kussnierczyk A, Klungland A, Darnell JE Jr, Darnell RB. 2015. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev 29:2037–2053. doi: 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dominissini D, Moshitch-Moshkovitz S, Amariglio N, Rechavi G. 2015. Transcriptome-wide mapping of N6-methyladenosine by m6A-Seq. Methods Enzymol 560:131–147. doi: 10.1016/bs.mie.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, Han D, Dominissini D, Dai Q, Pan T, He C. 2015. High-resolution N(6)-methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew Chem Int Ed Engl 54:1587–1590. doi: 10.1002/anie.201410647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. 2015. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12:767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. 2013. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19:1848–1856. doi: 10.1261/rna.041178.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lavi S, Shatkin AJ. 1975. Methylated simian virus 40-specific RNA from nuclei and cytoplasm of infected BSC-1 cells. Proc Natl Acad Sci U S A 72:2012–2016. doi: 10.1073/pnas.72.6.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moss B, Gershowitz A, Stringer JR, Holland LE, Wagner EK. 1977. 5′-Terminal and internal methylated nucleosides in herpes simplex virus type 1 mRNA. J Virol 23:234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moss B, Koczot F. 1976. Sequence of methylated nucleotides at the 5′-terminus of adenovirus-specific RNA. J Virol 17:385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomason AR, Brian DA, Velicer LF, Rottman FM. 1976. Methylation of high-molecular-weight subunit RNA of feline leukemia virus. J Virol 20:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, He C. 20 January 2017. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. doi: 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. 2015. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 526:591–594. doi: 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. 2015. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 2015. 5′ UTR m(6)A promotes cap-independent translation. Cell 163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. 2015. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kariko K, Buckstein M, Ni H, Weissman D. 2005. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 53.Durbin AF, Wang C, Marcotrigiano J, Gehrke L. 2016. RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. mBio 7:e00833-16. doi: 10.1128/mBio.00833-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin S, Choe J, Du P, Triboulet R, Gregory RI. 2016. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell 62:335–345. doi: 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Westerhout EM, Ooms M, Vink M, Das AT, Berkhout B. 2005. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res 33:796–804. doi: 10.1093/nar/gki220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Desmyter J, Melnick JL, Rawls WE. 1968. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J Virol 2:955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bader JP, Brown NR, Chiang PK, Cantoni GL. 1978. 3-Deazaadenosine, an inhibitor of adenosylhomocysteine hydrolase, inhibits reproduction of Rous sarcoma virus and transformation of chick embryo cells. Virology 89:494–505. doi: 10.1016/0042-6822(78)90191-5. [DOI] [PubMed] [Google Scholar]
- 58.Wyde PR, Ambrose MW, Meyer HL, Zolinski CL, Gilbert BE. 1990. Evaluation of the toxicity and antiviral activity of carbocyclic 3-deazaadenosine against respiratory syncytial and parainfluenza type 3 viruses in tissue culture and in cotton rats. Antiviral Res 14:215–225. doi: 10.1016/0166-3542(90)90003-P. [DOI] [PubMed] [Google Scholar]
- 59.Gordon RK, Ginalski K, Rudnicki WR, Rychlewski L, Pankaskie MC, Bujnicki JM, Chiang PK. 2003. Anti-HIV-1 activity of 3-deaza-adenosine analogs. Inhibition of S-adenosylhomocysteine hydrolase and nucleotide congeners. Eur J Biochem 270:3507–3517. [DOI] [PubMed] [Google Scholar]
- 60.de Clercq E, Montgomery JA. 1983. Broad-spectrum antiviral activity of the carbocyclic analog of 3-deazaadenosine. Antiviral Res 3:17–24. doi: 10.1016/0166-3542(83)90011-6. [DOI] [PubMed] [Google Scholar]
- 61.Mayers DL, Mikovits JA, Joshi B, Hewlett IK, Estrada JS, Wolfe AD, Garcia GE, Doctor BP, Burke DS, Gordon RK, Lane JR, Chiang PK. 1995. Anti-human immunodeficiency virus 1 (HIV-1) activities of 3-deazaadenosine analogs: increased potency against 3′-azido-3′-deoxythymidine-resistant HIV-1 strains. Proc Natl Acad Sci U S A 92:215–219. doi: 10.1073/pnas.92.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, Okamura H. 2013. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155:793–806. doi: 10.1016/j.cell.2013.10.026. [DOI] [PubMed] [Google Scholar]
- 63.Bray M, Driscoll J, Huggins JW. 2000. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-l-homocysteine hydrolase inhibitor. Antiviral Res 45:135–147. doi: 10.1016/S0166-3542(00)00066-8. [DOI] [PubMed] [Google Scholar]
- 64.Huggins J, Zhang ZX, Bray M. 1999. Antiviral drug therapy of filovirus infections: S-adenosylhomocysteine hydrolase inhibitors inhibit Ebola virus in vitro and in a lethal mouse model. J Infect Dis 179 Suppl 1:S240–S247. doi: 10.1086/514316. [DOI] [PubMed] [Google Scholar]
- 65.Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, Uren PJ, Suresh U, Carew JS, Karnad AB, Weitman S, Tomlinson GE, Rao MK, Kornblau SM, Bansal S. 2014. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 28:1171–1174. doi: 10.1038/leu.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]