

ABSTRACT
Several envelope glycoproteins are involved in herpesvirus entry into cells, direct cell-to-cell spread, and induction of cell fusion. The membrane fusion protein glycoprotein B (gB) and the presumably gB-activating heterodimer gH/gL are essential for these processes and conserved throughout the Herpesviridae. However, after extended cell culture passage of gL-negative mutants of the alphaherpesvirus pseudorabies virus (PrV), phenotypic revertants could be isolated which had acquired spontaneous mutations affecting the gL-interacting N-terminal part of the gH ectodomain (gDH and gHB4.1) (B. G. Klupp and T. C. Mettenleiter, J Virol 73:3014–3022, 1999; C. Schröter, M. Vallbracht, J. Altenschmidt, S. Kargoll, W. Fuchs, B. G. Klupp, and T. C. Mettenleiter, J Virol 90:2264–2272, 2016). To investigate the functional relevance of this part of gH in more detail, we introduced an in-frame deletion of 66 codons at the 5′ end of the plasmid-cloned gH gene (gH32/98). The N-terminal signal peptide was retained, and the deletion did not affect expression or processing of gH but abrogated its function in in vitro fusion assays. Insertion of the engineered gH gene into the PrV genome resulted in a defective mutant (pPrV-gH32/98K), which was incapable of entry and spread. Interestingly, in vitro activity of mutated gH32/98 was restored when it was coexpressed with hyperfusogenic gBB4.1, obtained from a passaged gL deletion mutant of PrV. Moreover, the entry and spread defects of pPrV-gH32/98K were compensated by the mutations in gBB4.1 in cis, as well as in trans, independent of gL. Thus, PrV gL and the gL-interacting domain of gH are not strictly required for function.
IMPORTANCE Membrane fusion is crucial for infectious entry and spread of enveloped viruses. While many enveloped viruses require only one or two proteins for receptor binding and membrane fusion, herpesvirus infection depends on several envelope glycoproteins. Besides subfamily-specific receptor binding proteins, the core fusion machinery consists of the conserved fusion protein gB and the gH/gL complex. The role of the latter is unclear, but it is hypothesized to interact with gB for fusion activation. Using isogenic virus recombinants, we demonstrate here that gL and the gL-binding domain of PrV gH are not strictly required for membrane fusion during virus entry and spread when concomitantly mutations in gB are present which increase its fusogenicity. Thus, our results strongly support the notion of a functional gB-gH interaction during the fusion process.
KEYWORDS: herpesvirus, pseudorabies virus, glycoproteins gH/gL, membrane fusion, virus entry, glycoprotein B
INTRODUCTION
Entry of enveloped viruses into host cells proceeds through fusion of the envelope with the host cell plasma membrane or with endosomal membranes. Whereas many viruses require only one or two receptor binding and/or fusion protein(s), herpesviruses are dependent on at least four viral envelope glycoproteins for this process. Subfamily-specific proteins like the alphaherpesvirus glycoprotein D (gD) mediate receptor binding, while the core fusion machinery conserved in all members of the Herpesviridae, comprising mammalian, avian, and reptilian herpesviruses, is composed of the homotrimeric glycoprotein B (gB) and a heterodimeric complex of the integral membrane glycoprotein H and the anchorless glycoprotein L (gH/gL) (reviewed in references 1 and 2).
Activation of the fusion machinery of pseudorabies virus ([PrV] Suid alphaherpesvirus 1) and of herpes simplex viruses 1 and 2 ([HSV-1/2] Human alphaherpesvirus 1 and 2) is initiated by binding of gD to specific cellular receptors like nectin-1 or herpesvirus entry mediator (HVEM) (3). Receptor binding leads to conformational changes in the C-terminal region of the HSV-1 gD ectodomain (4, 5). According to the current model, this conformational rearrangement of gD induces interaction with the gH/gL complex, which in turn is thought to activate the bona fide fusion protein gB to execute membrane fusion (6–9). While gD signaling is generally required for triggering membrane fusion in HSV, PrV gD is dispensable for direct cell-to-cell spread and in vitro cell fusion (10–12). Moreover, gD deletion mutants of PrV and bovine alphaherpesvirus 1 (BoHV-1) could be isolated, which were replication competent in cell culture in the presence of compensatory mutations in gB and gH (13–15), indicating that gD is not central to the fusion process.
Homologs of gB are found in all members of the Herpesviridae. The crystal structure of the ectodomain resembles that of a class III fusion protein, including a trimeric fold, a central alpha-helical coiled coil, and internal bipartite fusion loops (16). Several gB postfusion forms and a more condensed conformation of full-length HSV-1 gB, which is proposed to represent a prefusion or intermediate state, have been determined (17). Although gBs of HSV-1 (16) and of Epstein-Barr virus ([EBV] Human gammaherpesvirus 4) (18) share no sequence similarities with other class III fusion proteins, such as vesicular stomatitis virus (VSV) glycoprotein G (19) or baculovirus gp64 (20), their crystal structures are highly similar. However, unlike the latter proteins, gB is not able to merge membranes on its own but requires activation by the gH/gL complex (8, 21).
gH is a type I transmembrane protein with a short cytoplasmic tail and a large N-terminal ectodomain. It is associated with gL, which lacks a membrane anchor (2, 8, 22, 23). Although much work has gone into understanding the function of the gH/gL complex during membrane fusion, its role remains enigmatic. Despite experimental evidence that gH/gL may act as a fusion protein itself (24, 25), the recently determined crystal structures of soluble EBV gH/gL (22), HSV-2 gH/gL (8), varicella-zoster virus ([VZV] Human alphaherpesvirus 3) gH/gL (23), and of a core fragment of PrV gH (26) revealed no structural features resembling any known viral fusion protein. Furthermore, the structural analyses indicate that previously identified putative fusogenic and membrane-interacting peptides of HSV-1 gH are not exposed at the surface of gH and are unlikely to become available for interaction with membranes upon a conformational change (26). Interestingly, the structural analyses showed that the domain organization of the four gH homologs is strikingly similar although their amino acid sequences are only poorly conserved.
The most highly conserved region of gH is the membrane-proximal domain IV (designated H3 in HSV-2), which consists of a beta-sandwich comprising two opposed four-stranded beta-sheets, which are connected via a long crossover segment of the polypeptide chain, designated flap. Since the flap covers a conserved patch of hydrophobic amino acid residues, its movement during a receptor-triggered conformational change of gH is thought to enable interaction of the underlying hydrophobic surface with the viral envelope to promote the fusion process (26, 27). Functional conservation of this domain in PrV and HSV-1 could be demonstrated by analysis of chimeric gH consisting of PrV domains I to III and HSV-1 domain IV. This chimeric protein was not only capable of inducing membrane fusion when expressed together with HSV-1 or PrV gB and with PrV gD and gL but also supported replication of gH-deleted PrV (28). The less conserved domain III (H2) is composed of eight alpha-helices and contains a highly conserved amino acid stretch (serine-proline-cysteine) important for regulation of membrane fusion (29). Domain II (H1B) contains the “fence,” a sheet of antiparallel beta-chains, and a highly conserved bundle of three alpha-helices, designated syntaxin-like bundle (SLB) because of its structural similarities to a specific domain of cellular syntaxins. Recent studies indicate that the integrity and flexibility of the SLB are relevant for the function of PrV gH in membrane fusion (30). The N-terminal domain I of gH forms tight contacts with gL, and the presence of both proteins is critical for the secondary structure and the hydrophobic core of this domain in HSV and EBV (8, 22).
Due to a lack of a membrane anchor, gL is dependent on association with gH for virion incorporation (31–34). However, while gL is required for correct folding and transport of gH in most herpesviruses, it is not essential for gH virion incorporation in PrV, bovine gammaherpesvirus 4, and murid gammaherpesvirus 4 (35–37). More importantly, gL-deleted PrV mutants (PrV-ΔgL) are capable of limited cell-to-cell spread although they are defective in entry into target cells. However, after reversion analyses by serial cell culture passages, these gL-deleted PrV mutants regained wild-type-like replication properties by acquiring compensatory mutations in the gH, gB, and gD genes (38, 39).
Two independent reversion analyses resulted in two different revertants, but both contained compensatory mutations involving the predicted gL interaction domain of gH, which is located within the amino-terminal part of the gH ectodomain. The first passaging experiment selected for a chimeric gDH hybrid protein, which lacked the predicted gL interaction domain and consisted of the N-terminal 217 amino acids of gD, including the receptor binding domain, fused to the C-terminal 590 amino acids of gH (38). This hybrid protein was able to substitute not only for the lack of gL but also for the absence of wild-type gH and gD in complementation and in in vitro fusion assays (12). Recently, another infectious revertant, designated PrV-ΔgLPassB4.1, was isolated and characterized. As in gDH, compensatory mutations occurred in the gH gene of PrV-ΔgLPassB4.1 (gHB4.1) also located in the predicted gL-binding domain (39). Two point mutations in gHB4.1 (L70P and W103R) were found to be sufficient to compensate for the lack of gL in transfection-based fusion assays. However, mutations were also detected in gB (gBB4.1), which resulted in enhanced fusogenicity and gL-independent in vitro fusion also in the presence of wild-type gH, although further enhanced syncytium formation was observed after coexpression of gBB4.1 with the homologous gHB4.1 (39).
To investigate the functional relevance of the N-terminal part of the gH ectodomain in more detail and, in particular, in virus-infected cells, we deleted the predicted gL-binding domain (66 codons at the 5′ end, yielding gH32/98) in the plasmid-cloned PrV gH gene and tested the mutated gH in a virus-free transfection-based cell fusion assay. Furthermore, the mutated gH gene was inserted into the cloned PrV genome (27) in the presence or absence of gL and wild-type or mutated gB. Protein expression, as well as in vitro replication properties, including penetration, growth kinetics, and plaque formation of the obtained virus mutants, was investigated.
RESULTS
Deletion of the gL-binding domain of PrV gH does not affect protein expression and virion incorporation.
To investigate the functional relevance of the predicted gL-binding domain in PrV gH, we deleted codons 32 to 97 in the plasmid-cloned gH gene (gH32/98). The deletion excludes the signal peptide of gH, which is predicted to be cleaved behind amino acid 30 (Fig. 1B). To compare transient expression and processing of gH32/98 with those of the wild-type gH of PrV strain Kaplan (gHKa), RK13 cells were transfected with expression plasmids pcDNA-gH32/98KDE or pcDNA-gHKDE. (30) After 48 h cell lysates were analyzed by Western blotting. Targeted deletion of the predicted gL-binding domain led to expression of a truncated gH32/98 protein with a molecular mass of approximately 80 kDa, whereas the apparent mass of mature wild-type gH was approximately 90 kDa (Fig. 2). Smaller proteins representing immature gH precursors or degradation products were significantly less abundant, indicating that processing or stability of gH32/98 was not affected. Indirect immunofluorescence analyses of cells transfected with the expression plasmids for gHKa or gH32/98 demonstrated that the introduced in-frame deletion of the gL-binding domain had no apparent effect on expression level, cytoplasmic distribution, or surface localization of the protein (data not shown).
FIG 1.

Construction of virus mutants. (A) The wild-type PrV-Ka genome consists of unique long and short regions (UL and US, respectively), and inverted repeat sequences (IR, internal repeat; TR, terminal repeat). The positions of the relevant glycoprotein genes (gB, gD, gG, gH, and gL) are indicated. An EGFP expression cassette and a mini-F vector for BAC replication were inserted at the nonessential gG locus. (B) After deletion of the wild-type gH gene, modified forms were reinserted together with a kanamycin resistance gene (Kanr) by mutagenesis in E. coli. (C) In a similar manner, the gL gene was replaced by a zeocin resistance gene (Zeor). (D) Replacement of wild-type gB by mutated gBB4.1 was achieved by homologous recombination in BAC- and transfer plasmid-cotransfected RK13 cells and subsequent plaque purification of viable progeny viruses. Designations of virus recombinants, relevant restriction sites, regulatory sequences like promoters (PgX), polyadenylation signals (A+), an origin of DNA replication (ORIL), an Flp recombinase target site (FRT), and open reading frames (pointed rectangles) are indicated. The locations of signal peptides (S), transmembrane domains (TM), deletions (Δ), and amino acid substitutions in the deduced glycoproteins are highlighted. Vertical lines indicate tandem repeat sequences. TK, thymidine kinase. EP0, early protein 0.
FIG 2.
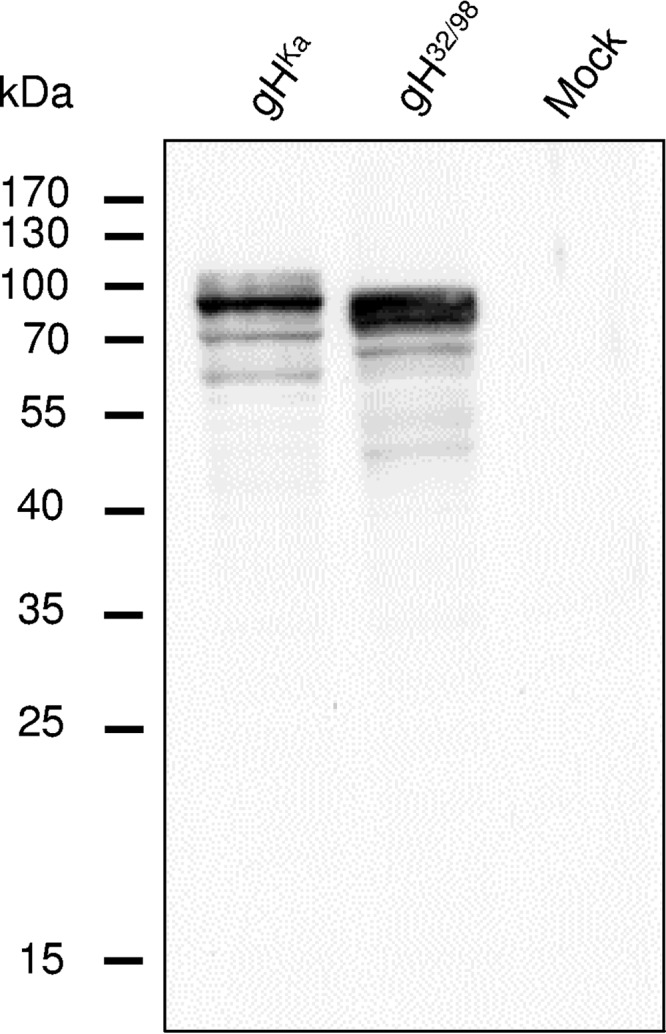
Western blot analyses of transfected RK13 cells. Lysates prepared 48 h after transfection with expression plasmids for wild-type gHKa, modified gH32/98, or the empty vector (Mock) were separated by SDS-PAGE. The blot was incubated with a PrV gH-specific rabbit antiserum. Molecular masses (kDa) of marker proteins are indicated.
After insertion of gH32/98 into the PrV genome, Western blot analyses of RK13 cells infected with phenotypically complemented virions of the resulting mutants pPrV-gH32/98K and pPrV-gH32/98KΔgLZ or with wild-type gH-containing mutants pPrV-gHK and pPrV-gHKΔgLZ (Fig. 1B and C) again revealed similar expression levels of truncated gH32/98 and of gHKa (Fig. 3A, top panel). Presence or absence of gL had no detectable effect on expression or processing of gH. However, only small amounts of immature 16- to 18-kDa forms of gL were detectable in cells infected with pPrVgH32/98K, in contrast to an abundant 20-kDa gL in pPrV-gHK-infected cells (Fig. 3A, second panel). This indicates that gH32/98 is unable to bind gL, which leads either to impaired gL processing and/or to rapid secretion of mature gL. Replacement of wild-type gBKa by the hyperfusogenic mutant gBB4.1 (39) in pPrVgH32/98KgBB4.1 and pPrVgH32/98KΔgLZgBB4.1 (Fig. 1D) did not alter the apparent molecular masses of the gB gene products nor the efficiency of furin cleavage of the precursor protein (Fig. 3A, third panel). Analysis of sucrose gradient-purified virions showed that gH32/98 is efficiently incorporated into virus particles irrespective of gL and of the presence of either gBKa or gBB4.1 (Fig. 3B, top panel). As expected, gL was only detectable in virions of pPrV-gHK which contained gHKa (Fig. 3B, second panel). Purity of the virion preparations was confirmed by the absence of pUL31, which is present only in infected cells and in primary enveloped particles in the perinuclear space (Fig. 3, bottom panels) (40).
FIG 3.

Western blot analyses of PrV-infected cells and purified virions. Lysates of RK13 cells prepared 20 h after infection (MOI of 5) with the indicated virus recombinants (A), and purified virion proteins were separated by SDS-PAGE (B). Blots were incubated with monospecific rabbit antiserum against PrV glycoproteins gH, gL, and gB and the nonstructural UL31 protein. Molecular masses of marker proteins are indicated.
Targeted deletion of the gL-binding domain leads to significantly reduced in vitro fusion activity of gH, which can be restored by compensatory mutations in gB.
Cell-cell fusion can be induced by coexpression of PrV or HSV glycoproteins gB, gD, and gH/gL in plasmid-transfected cells in vitro (12, 41). While the expression of all four HSV glycoproteins is necessary to induce membrane fusion, PrV gB and gH/gL are sufficient for this process, and the additional expression of PrV gD enhances in vitro fusion activity only moderately (12). However, omission of either gB or gH results in total abrogation of membrane fusion in either system.
To analyze the influence of the deletion of the predicted gL-binding domain of gH (gH32/98) on in vitro fusion activity, RK13 cells were cotransfected with expression plasmids encoding the mutated PrV gH32/98, wild-type gBKa, and optionally gL and gD. In addition, enhanced green fluorescent protein (EGFP) was coexpressed to facilitate evaluation of the assays by fluorescence microscopy. Assays with plasmids encoding wild-type gHKa, gBKa, gL, and gD served as positive controls, and results were set as 100%. Assays with the empty expression vector pcDNA3 served as negative controls. One day after transfection, the areas of cells containing three or more nuclei were measured and multiplied by the number of syncytia to quantify fusion activity. As expected, after cotransfection of wild-type gHKa, gBKa, and gL, formation of multiple syncytia was observed. However, fusion activity was completely abolished when either gL was omitted or gH32/98 was expressed instead of wild-type gHKa (Fig. 4). This suggested that the gL-binding domain of gH and/or the presence of gL in the fusion complex is essential for its function. As reported earlier, omission of gD had only minor effects on fusion induced by wild-type gBKa, gHKa, and gL (Fig. 4).
FIG 4.

In vitro fusion assays. RK13 cells were cotransfected with expression plasmids for EGFP, wild-type gB (gBKa), or mutated gBB4.1 and, optionally, gHKa or gH32/98, gDKa, and gLKa. One day posttransfection the areas of green fluorescing syncytia were measured, and total fusion activity was determined by multiplication of the mean syncytium area with the number of syncytia in 10 randomly selected fields. Fusion activities obtained with the four wild-type glycoproteins were set as 100%. Shown are the mean relative values and standard deviations of three independent experiments.
In recent studies, we identified an infectious gL-deleted PrV mutant, which carries compensatory mutations in the gH, gB, and gD (gHB4.1, gBB4.1, and gDB4.1) genes. While the mutations in gHB4.1 were found to be sufficient to compensate for the lack of gL in transient assays, the mutations present in gBB4.1 enhanced fusion activity (39). Therefore, it was investigated whether gH32/98 is able to trigger fusogenicity of the presumably more fusion prone gBB4.1 independent of gL. Interestingly, the fusion activity achieved with gH32/98 and gBB4.1 was much higher (approximately 550%) than that observed with the four wild-type proteins (Fig. 4). Moreover, fusion activity obtained with gH32/98 in combination with gBB4.1 reached approximately 80% of that obtained with wild-type gH in combination with gBB4.1, and in both cases this was independent of gL (Fig. 4). However, gBB4.1 alone was not sufficient to induce cell-cell fusion (39) (Fig. 4).
In summary, the wild-type-like in vitro syncytium formation mediated by gH32/98 together with gBB4.1 demonstrates that gL and the gL-binding domain of gH are dispensable for PrV glycoprotein-mediated membrane fusion if gB is modified by adaptive or compensatory mutations increasing its fusogenicity. This clearly differs from the situation in the gDH hybrid protein, which compensates the absence of gL without the requirement for modification in gB.
Deletion of the gL-binding domain in gH32/98 abrogates virus replication and spread.
After insertion of the mutated gH32/98 gene into the PrV genome by bacterial artificial chromosome (BAC) mutagenesis in Escherichia coli, no infectious virus of the resulting GFP-expressing recombinants pPrV-gH32/98K and pPrV-gH32/98KΔgLZ (Fig. 1) could be reconstituted in transfected RK13 cells, whereas an otherwise identical recombinant containing wild-type gH (pPrV-gHK) replicated efficiently. On trans-complementing RK13-gH/gL cells (42), the gH32/98-containing PrV mutants could, however, be isolated and propagated, indicating that they had no defects elsewhere in their genomes. After infection of RK13 cells with phenotypically complemented pPrV-gH32/98K (Fig. 5) or pPrV-gH32/98KΔgLZ (data not shown), only single GFP-expressing cells were observed, as described for gH-negative PrV mutants (11, 43). In growth kinetics studies performed on RK13 cells, no infectious progeny virus of pPrV-gH32/98K could be detected at any time (Fig. 6), even if titrations were performed on RK13-gH/gL cells (data not shown). Thus, gH32/98, although expressed and incorporated into virions, appeared to be nonfunctional in the viral context.
FIG 5.

Plaque assays on RK13-gBB4.1 (A) or RK13 cells (B). Cells were infected with the indicated PrV recombinants and incubated for 48 h under plaque assay conditions. Areas of 30 plaques per virus were measured, and percentages of wild-type (pPrV-gHK) sizes were calculated. Mean values and standard deviations of four independent experiments are shown. (C) Representative images of infected RK13 cells. Bar, 500 μm.
FIG 6.

In vitro growth kinetics of PrV mutants. RK13 cells were infected with pPrV-gHK, pPrV-gH32/98K, pPrV-gH32/98KgBB4.1, or pPrV-gH32/98KΔgLZgBB4.1 at an MOI of 0.01 (A) or 5 (B). Cells were harvested together with the supernatants after 0, 6, 12, 18, 24, 36, 48, and 72 h at 37°C and lysed by freeze-thawing, and progeny virus titers were determined on RK13 cells. Shown are mean results of four independent experiments with corresponding standard deviations.
Replication competence of pPrV-gH32/98K can be restored by compensatory mutations in gB.
Since transient in vitro fusion assays revealed that gH32/98 regained almost wild-type activity when coexpressed with the mutated gB of PrV-ΔgLPassB4.1 (see above), we assessed whether gBB4.1 is able to rescue the function of gH32/98 also in the viral context. A recombinant RK13 cell line which constitutively expressed gBB4.1 was used for replication studies. On RK13-gBB4.1 cells pPrV-gH32/98K and pPrV-gH32/98KΔgLZ spread to plaque areas of nearly 20% of the wild-type (pPrV-gHK) sizes (Fig. 5A). However, infectious titers remained 1,000-fold lower than the wild-type titer (results not shown). To exclude effects of inadequate gB expression or competition between gBB4.1 provided by the cells and the viral wild-type protein, the gB gene of the gH32/98-expressing PrV mutants was exchanged by homologous recombination after cotransfection of normal RK13 cells with BAC DNA of pPrV-gH32/98K or pPrV-gH32/98KΔgLZ and a transfer plasmid containing the PCR-amplified gB gene region of PrV-ΔgLPassB4.1 (Fig. 1D). This led to PrV mutants which could be efficiently propagated in noncomplementing cells. Sequence analyses of the gB genes of several plaque isolates revealed that all of them exhibited the two described point mutations leading to amino acid substitutions in the ectodomain of gBB4.1, E294G and G676R (Fig. 1D) (39). In contrast, two other described mutations of the gBB4.1 gene, leading to deletion of a lysine residue in the cytoplasmic tail (ΔK887) and of a duplicated stretch of 4 amino acids (AVRA) from positions 115 to 118 of wild-type gBKa, were not always introduced, indicating an inferior functional relevance. pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1, used for further characterization, exhibited the C-terminal deletion, but not the N-terminal mutation (Fig. 1D). In addition to the three mutations in gB and the desired deletions in gH/gL, no unwanted alterations were found in the gB, gD, gH, or gL genes of the investigated PrV mutants.
Whereas pPrV-gH32/98K and pPrV-gH32/98KΔgLZ were incapable of spread in RK13 cells, pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1 were able to form plaques although these were approximately 80% smaller than those formed by pPrV-gHK (Fig. 5B and C). Presence or absence of gL had no effect on plaque sizes of the gBB4.1-rescued gH32/98 mutants, and the size reduction compared to that of wild-type plaques was similar to that on gBB4.1-expressing cells (Fig. 5A).
To investigate the influence of the targeted deletion of the gL binding domain of gH in combination with the hyperfusogenic gBB4.1 on formation of infectious virus particles, RK13 cells were infected at multiplicities of infection (MOIs) of 0.01 (Fig. 6A) or 5 (Fig. 6B), and total progeny virus titers were determined 0, 6, 12, 18, 24, 36, 48, and 72 h postinfection (p.i.) on complementing RK13-gH/gL and noncomplementing RK13 cells. Since titers on both cell lines were almost identical, only those from RK13 cells are shown. Whereas no infectious pPrV-gH32/98K particles were formed in RK13 cells, pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1 grew to final titers of >106 PFU/ml, approximately 1 log10 unit lower than those of pPrV-gHK (Fig. 6). Thus, with respect to productive replication, cis-complementation of the gH32/98 mutants by virus-expressed gBB4.1 was much more efficient than trans-complementation on gBB4.1-expressing cells (see above). Plaque sizes, growth kinetics, and maximum titers of the gBB4.1-rescued viruses, pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1, were almost indistinguishable, indicating that in the absence of the gL-binding domain of gH, gL itself is no longer relevant for entry and viral spread. However, mutations in gBB4.1 were obviously essential for function of gH32/98.
Penetration of pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1 recombinants is significantly delayed.
Replication kinetics of pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1 after infection at both low and high MOIs revealed a markedly delayed titer increase compared to the titer of wild-type gH-expressing pPrV-gHK (Fig. 6). To investigate whether this effect was due to impaired virus entry, penetration studies were carried out. Whereas entry of pPrV-gHK into RK13 cells occurred very fast, with approximately 90% of infectious input virus protected from inactivation by low-pH treatment after 15 min at 37°C (Fig. 7), penetration of pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1 was significantly delayed. Even after 4 h, only 60 to 70% of the infectious particles of pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1 were internalized, and again no difference was observed between the gL-expressing and the gL-deleted mutants (Fig. 7). Penetration kinetics of pPrV-gH32/98K and PrV-gH32/98KΔgLZ could not be investigated since these mutants become infectious only after phenotypic complementation with wild-type gH. Nevertheless, our studies confirm that gL and the gL-binding domain of PrV gH are not strictly required for membrane fusion during virus entry and spread. However, in the viral context the minimum functional fusion complex consisting of N-terminally truncated gH32/98 and point-mutated gBB4.1 of PrV is less efficient with respect to speed or rate of fusion events than wild-type glycoproteins gB, gD, gH, and gL.
FIG 7.

Penetration kinetics of PrV mutants. RK13 cells were infected with approximately 250 PFU of pPrV-gHK, pPrV-gH32/98KgBB4.1, or pPrV-gH32/98KΔgLZgBB4.1. After adsorption at 4°C, the cells were incubated for 0, 15, 30, 60, 120, and 240 min at 37°C prior to acid inactivation of nonpenetrated virus. Numbers of plaques formed after 48 h were compared to those obtained without inactivation. Mean percentages of three independent experiments and standard deviations are shown.
DISCUSSION
Herpesviruses infect cells by fusion of the viral envelope with cellular membranes. While many other enveloped viruses use a single protein for attachment and fusion, herpesviruses require at least two conserved core fusion machinery components, gB and the heterodimeric gH/gL complex, and additional nonconserved subfamily-specific receptor binding proteins like gD (reviewed in references 2, 6, and 9).
In our previous work, we analyzed the relevance of the gH/gL complex by reversion analyses of gL-deleted PrV mutants (38, 39) and site-directed mutagenesis of PrV gH domains II, III, and IV (27, 30). In the present study, these approaches were extended to the structurally uncharacterized N-terminal gH domain I, which in HSV-2 and EBV was shown to tightly interact with gL (8, 22). In the previously described gL-independently replicating PrV mutants, this domain was either affected by point mutations (39) or replaced by the N-terminal part of gD, resulting in a gDH hybrid protein (38). Since chimeric gDH lacks gH amino acids 1 to 96, we artificially introduced a similar in-frame deletion of gH codons 32 to 97 (gH32/98) to investigate gD-independent function of truncated gH. The retained gH amino acids 1 to 30 are predicted to represent a cleaved N-terminal signal peptide required for translocation into the endoplasmic reticulum. Thus, the resulting N terminus of mature gH32/98 was similar to that of the gH core fragment (amino acids 107 to 639), whose crystal structure has been recently solved (26).
Deletion of the predicted gL-binding domain in gH32/98 did not impair expression and processing of gH in plasmid-transfected cells (Fig. 2) and showed no apparent differences from wild-type gH with respect to intracellular localization or cell surface expression. Furthermore, gH32/98 was efficiently incorporated into virions independent of gL (Fig. 3). This is in line with the previously described gL-independent virion incorporation of wild-type PrV gH (35), whereas in other herpesviruses like HSV-1 or VZV, gL is required for maturation and transport of gH to the future budding sites (31, 44–46). Several N-terminally truncated HSV-2 gH variants have been reported, which are transported without gL but still require gL for function (47). By amino acid substitutions in the corresponding part of HSV-2 gH, potential key residues were identified and hypothesized to form an endoplasmic reticulum (ER) retention signal, which is hidden in the presence of gL and then allows transport of the gH/gL complex to the future budding sites (47). However, these key residues are not conserved in PrV, VZV, or EBV. Furthermore, HSV-2 gL is considered to represent a scaffolding protein for gH (8) and was shown to be required for proper folding and function of the gH/gL complex (47). Although the N-terminal domain is not required for correct processing or virion targeting of PrV gH, its complex partner gL was incompletely processed in or secreted from cells infected with virus mutants expressing gH32/98 and was undetectable in extracellular virus particles. This demonstrates that gH32/98 is unable to bind and to target gL, which lacks a transmembrane domain, into virions. Thus, as hypothesized, amino acids 32 to 97 of PrV gH contain a region essential for gL interaction.
To investigate the impact of the deletion of the gL-binding domain of PrV gH on membrane fusion, we used a transient-transfection-based fusion assay (39) (Fig. 4). Unlike the wild-type protein, gH32/98 exhibited no detectable in vitro fusion activity when coexpressed with wild-type gB, gD, and gL. Omission of gD only slightly reduced activity of the wild-type glycoprotein set, whereas gL was indispensable (12, 39). Since PrV gD is obviously not required for in vitro fusion, the dysfunction of gH32/98 in our fusion assays was unlikely due to an impaired interaction with gD. Whereas the N-terminally mutated gHB4.1 and gDH of gL-independently replicating PrV mutants mediated considerable gL-independent in vitro fusion activity when they were coexpressed with wild-type gB (12, 38, 39), gH32/98 was inactive independent of gL. Thus, our results suggest that the deletion in gH domain I leads to downstream structural alterations which affect interaction with gB so that gH32/98 is not able to activate wild-type gB. In wild-type gH association with gL and in gDH, the gD part might induce or maintain a conformation of gH required for triggering gB-mediated fusion. In line with this hypothesis, the domain I-domain II interface of gH/gL has been considered to be relevant for gB activation in EBV and VZV (23, 48). However, when gH32/98 was used in combination with the mutated gBB4.1 obtained from the passaged, phenotypically reverted gL deletion mutant PrV-ΔgLPassB4.1 (39), fusion activity was rescued to a 5-fold higher level than observed with the wild-type set of glycoproteins, and this activity was independent of gD and gL (Fig. 4). Also in combination with wild-type gH, fusogenicity of gBB4.1 was gL independent and much higher than that of wild-type gB (39). This indicates that the conformational changes required for fusion might be facilitated by the mutations present in gBB4.1 and could be triggered even in the absence of gL and by suboptimal gH variants. Nevertheless, the gH trigger remained indispensable also for gBB4.1 (Fig. 4) (39).
In accordance with the missing in vitro fusion activity of wild-type gB together with gH32/98, engineered PrV recombinants expressing this protein combination were incapable of cell-to-cell spread and produced no infectious virus particles (Fig. 5 and 6). Thus, unlike gDH (38), gH32/98 is nonfunctional also in the viral context, confirming that the gD part of gDH is required for function of the hybrid protein in triggering fusion. However, on cells expressing gBB4.1, plaque formation of pPrV-gH32/98K and pPrV-gH32/98KΔgLZ was restored. The same effect was observed after replacement of the wild-type gB gene by gBB4.1, and the resulting mutants pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1 could be efficiently propagated on cells providing neither gB nor gH in trans (Fig. 5 and 6). Interestingly, the plaques produced by the gH32/98 mutants in the presence of gBB4.1 were approximately 80% smaller, and their maximum titers were one order of magnitude lower than those of isogenic PrV recombinants expressing wild-type gB, gD, gH, and gL, whereas in vitro fusion activity of gH32/98 and gBB4.1 was much higher than that of the complete wild-type glycoprotein set. These quantitative differences support the assumption that the mechanisms of membrane fusion during syncytium formation, virus entry, and virus spread are similar but not identical. Possibly, premature fusion events between cells and intracellular membranes induced by the hyperfusogenic gBB4.1 interfere with the efficiency of virion formation and spread. Furthermore, investigation of penetration kinetics revealed that entry of PrV mutants expressing truncated gH32/98 and gBB4.1 is considerably delayed compared to that in wild-type PrV (Fig. 7), which might contribute to the reduced plaque sizes and titers. Western blot analyses of purified virions of pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1 (Fig. 3B) provided no evidence that delayed penetration could be a consequence of less efficient virion incorporation of the two mutated glycoproteins. Thus, more likely, interaction of truncated gH32/98 with gD after receptor binding or with gBB4.1 prior to fusion might be less efficient than in wild-type particles. On the other hand, it is also conceivable that the gH32/98/gBB4.1-expressing PrV recombinants use endocytic/phagocytic pathways instead of direct fusion with the plasma membrane to enter cells.
For HSV-1 and -2 it has been shown recently that gH/gL rather than gB governs the speed and rate of fusion, with gH being the main contributor (49). The observed delay in penetration mediated by gH32/98 also emphasizes the importance of gH as a fusion regulator. In previous studies we detected similarly delayed penetration kinetics of PrV mutants which contained artificial disulfide bonds fixing the syntaxin-like bundle of alpha-helices in gH domain II, indicating that structural changes in gH are required to trigger fusion (30). Alterations in gH interaction with gD might play a role in this context since the N-terminal domain I, deleted in gH32/98, and domain II, comprising the syntaxin-like bundle, are the proposed interaction sites with gD in HSV-1 (50). Although PrV gD, unlike its HSV-1 homolog, is not generally required for membrane fusion, it is essential for infectious entry of extracellular virus particles (10, 11). Since, according to the current model, gD is thought to interact with gH/gL upon binding to one of its cellular receptors (4, 5), which then leads to activation of gH/gL to trigger gB fusogenicity (8), it is conceivable that activation of gH32/98 by gD is impaired due to the missing N-terminal domain. Conversion of gH/gL to an activated state by receptor-bound gD was recently suggested to be a key step in the fusion pathway. In a corresponding study, HSV-2 gH/gL with a deletion of 28 residues at the gH N terminus (gHΔ48/gL) was shown to induce low-level in vitro fusion without gD, suggesting that gHΔ48/gL resembled activated gH/gL (51). In contrast, the N-terminal deletion of 66 residues in PrV gH32/98 rendered the protein inactive in the presence of wild-type gB, irrespective of the presence or absence of gD and gL.
Remarkably, the gDH chimeric protein, which exhibits almost exactly the same deletion in the gH portion as gH32/98, showed wild-type-like function in transient fusion assays and during viral infection (12, 38). This suggests that the physical linkage in gDH might enable direct activation of the gH moiety after binding of the gD domain to a receptor. Furthermore, it is conceivable that the gD part of gDH adopts a stabilizing or modulating influence on gH structure which is normally executed by gL. Interestingly, unlike the PrV gDH protein (38), similar HSV-1 gDgH chimeras were not able to functionally substitute for gD, gH, or gL in either virus-free fusion assays of transfected cells or in complementation studies (52). This might reflect a larger dependence of the HSV-1 membrane fusion complex on gL and native gD. Concordantly, unlike the situation in HSV-1, gD is dispensable for cell-to-cell spread of PrV, gL is dispensable for virion incorporation of PrV gH, and fully replication competent gL and gD deletion mutants of PrV could be isolated after extended cell culture passage (10, 13, 38, 39).
Unlike deletion in PrV gDH, deletion of the gL-binding domain in gH32/98 results in a protein which is apparently unable to acquire the structure necessary for proper interaction with wild-type gB. This supports the idea that gH or gH/gL activates, rather than represses, gB fusion activity. In accordance with this it has been found that HSV-1 gH comes into close proximity to gB only after binding of gD to one of its receptors, suggesting that gB and gH/gL do not constitutively interact (9, 53). Furthermore, it is conceivable that activation of gH32/98 by receptor-bound gD is impaired, thereby preventing subsequent activation of wild-type gB, whereas mutated gBB4.1 can be activated by gH32/98 in a gD-independent manner. This hypothesis, however, remains to be verified.
In any case our present study shows that deletion of the gL-binding domain from gH (amino acids 32 to 97) led to a complete loss of in vitro fusion activity and function in the viral context in the presence of wild-type gB and that these defects were rescued to a considerable extent by mutated gBB4.1 isolated from the passaged gL deletion mutant, PrV-ΔgLPassB4.1 (39). gBB4.1 exhibits a replacement of glutamic acid by glycine at position 294 in domain I, which also contains the fusion loops required for function (16, 17). Furthermore, it has been hypothesized that replacement of the well-conserved glycine 676 by arginine in domain IV of gBB4.1 might decrease the energy required for the extensive conformational rearrangements of this protein part during the conversion of the pre- to the postfusion form (39).
In summary, our previous results from reversion analyses of gL-deleted PrV (38, 39) could now be confirmed and expanded by directed mutation of the gH gene in a cloned virus genome. Consistently, our findings reinforce the notion that gB and gH are the core players in herpesvirus-mediated membrane fusion. Although gL is usually also an essential component of the fusion machinery, its absence can be compensated by mutations in gH and/or gB. Our present study further demonstrates that the gL binding domain of PrV gH is dispensable for maturation and virion incorporation of gH, and, conditionally, also for membrane fusion in transient assays, as well as during viral entry and spread. However, compensatory mutations within gB are required to restore a functional fusion machinery. Thus, the core fragment of PrV gH analyzed by crystallography (26) indeed represents the core functional unit which can support membrane fusion without the need for gD and gL. Furthermore, this study emphasizes the importance of direct gH-gB interaction for the fusion process.
MATERIALS AND METHODS
Cells and viruses.
Rabbit kidney (RK13) cells were grown in Dulbecco's modified Eagle's minimum essential medium (MEM) supplemented with 10% fetal calf serum (FCS) at 37°C. The viruses described in this study were derived from pPrV-ΔgHABF (30), which is a gH-deleted mutant of PrV strain Kaplan (PrV-Ka) cloned as a bacterial artificial chromosome (BAC) (27). Viruses were propagated in normal RK13 cells, the gH- and gL-expressing cell line RK13-gH/gL (42), or RK13-gBB4.1 cells. The last cell line was obtained after stable transfection (X-tremeGENE HP reagent; Roche) of RK13 cells with pcDNA-gBB4.1 (39). For plaque assays, the virus inoculum was removed 2 h after infection, and cells were overlaid with semisolid medium containing 6 g/liter methylcellulose.
Mutagenesis of the cloned gH gene and generation of gH32/98-expressing PrV mutants.
Expression plasmid pcDNA-gHKDE (30) containing the gH open reading frame ([ORF] UL22) of PrV-Ka was used for in-frame deletion of the predicted gL-binding domain (codons 32 to 97) by site-directed mutagenesis (QuikChange II XL kit; Agilent) with the complementary pair of oligonucleotide primers PGH32/98-F and PGH32/98-R (Table 1). The resulting expression plasmid pcDNA-gH32/98KDE was digested with DrdI, and a 3.3-kbp fragment containing the modified gH gene together with a downstream kanamycin resistance (Kanr) gene was purified from an agarose gel and used for red-mediated mutagenesis of pPrV-ΔgHABF as described previously (27, 30) (Fig. 1). LB agar plates supplemented with kanamycin (50 μg/ml) and chloramphenicol (30 μg/ml) were used for selection of the desired BAC recombinants. DNA of the BAC recombinant pPrV-gH32/98K was obtained from overnight liquid cultures of single bacterial colonies by alkaline lysis, phenol-chloroform extraction, isopropanol precipitation, and RNase treatment. BAC DNA was used for transfection (FuGene HD transfection reagent; Promega) of RK13-gH/gL cells, and progeny virus isolated from single plaques was propagated and characterized.
TABLE 1.
Oligonucleotide primers for mutagenesis, PCR, and sequence analyses
| Primer name | Sequence (5′→ 3′)a | Nucleotide positionsa,b |
|---|---|---|
| PGH32/98-F | CTCGCCCGCGGCGCG/CCGCCCGTCTCCGCG | 60848–60862/61061–61075 |
| PGH32/98-R | CGCGGAGACGGGCGG/CGCGCCGCGGGCGAG | 60848–60862/61061–61075 (r) |
| UL1-For | CACAAAGCTTAGGATACACCAGCCGCGATG | 95482–95501 (r) |
| UL1-Rev | CACAGAATTCGGTCTCTTACTCGGCGGGGG | 95008–95027 |
| PgBR-F | ACAGGATCCCTTCGCGCACGACACGC | 19984–20000 (r) |
| PgBR-R | ACAGGATCCTCATCGTGGTCGCGGTC | 16250–16266 |
| gB-AssFW | TAACGGATCCATGCCCGCTGGTGGCGG | 19657–19673 (r) |
| gB-AssRV | CAGAATTCCTACAGGGCGTCGGGGTCC | 16911–16929 |
| gB-P3 | GATCTCCTGCACGGGGACGGGCACG | 19032–19056 (r) |
| gB-P7 | CCATCTACCGGCGGCGCTACAACA | 18329–18352 (r) |
| gD-For | CACAGAATTCACCTGCCAGCGCCATGC | 121129–121148 |
| gD-Rev | CACAGAATTCCATCGACGCCGGTACTGC | 122353–122370 (r) |
| PgH-PSF | TTCACGTCGGAGATGGGG | 60611–60628 |
| PgH-PSF2 | GGAAGCCCTTCGACCAG | 61875–61891 |
| PgHK-PSR | CGCGAGCCCATTTATACCC | (Kanr) |
| PgH-PSR2 | GTCGAGCAGGCTGAAGG | 62055–62071 (r) |
| PgH-WH3 | TGCACGAGAGCGACGACTACC | 61479–61499 |
| PgH-E555AR | CAGGTGCTGGGACGCGGGCATGCGATC | 62420–62446 (r) |
Nucleotide positions in the PrV Ka genome refer to GenBank accession number JQ809328 (57). Nonmatching nucleotides are shown in boldface, and restriction sites for cloning of PCR products are underlined.
Slashes in the first two primer sets denote the deletion of nucleotide positions 60863 through 61060. Reverse-strand PCR primers are indicated (r). PgHK-PSR binds to the inserted kanamycin resistance gene (Kanr) of the PrV recombinants.
Generation of gL-negative PrV mutants.
The gL ORF (UL1) of PrV-Ka was amplified from genomic DNA by PCR with primers UL1-For and UL1-Rev (Table 1) and inserted into expression vector pcDNA3 (Thermo Fisher Scientific) via engineered HindIII and EcoRI restriction sites. From pcDNA-UL1 a 0.2-kbp PshAI/SgrAI fragment containing gL codons 21 to 85 was deleted and replaced by a 0.45-kbp Eco72I fragment extracted from pcDNA3.1ZEO (Thermo Fisher Scientific). After cleavage of the resulting pcDNA-ΔgLZ with HindIII and EcoRI, the 0.75-kbp insert containing the zeocin resistance gene flanked by UL1 sequences was isolated and used for BAC mutagenesis of pPrV-gH32/98K and other mutants (Fig. 1C). The desired gL-negative BAC recombinants were selected on LB agar plates with zeocin (25 μg/ml) and chloramphenicol. Infectious virus of pPrV-gH32/98KΔgLZ was isolated and propagated on RK13-gH/gL cells.
Replacement of wild-type gB by mutated gBB4.1.
A 3.75-kbp fragment containing the gB gene region of PrV-ΔgLPassB4.1 (39) was amplified by PCR with primers PgBR-F and PgBR-R (Table 1) and cloned into BamHI-digested pUC19 (New England BioLabs) via engineered restriction sites. The obtained transfer plasmid pUC-PgBB4.1R, together with BAC DNA of pPrV-gH32/98K or pPrV-gH32/98KΔgLZ, was used for cotransfection of normal RK13 cells (Fig. 1D). Homologous recombination led to replication-competent progeny viruses, and two plaque isolates designated pPrV-gH32/98KgBB4.1 and pPrV-gH32/98KΔgLZgBB4.1 were further analyzed. All generated PrV mutants were characterized by restriction analysis and Southern blot hybridization of viral DNA (results not shown). Furthermore, the complete coding regions of gB, gD, gH, and gL were PCR amplified and sequenced using the primers indicated in Table 1, a BigDye Terminator, version 1.1, cycle sequencing kit, and a 3130 genetic analyzer (Applied Biosystems).
Western blot analyses.
RK13 cells were harvested 48 h after transfection (X-tremeGENE HP reagent; Roche) with gH expression plasmids or 20 h after infection with PrV recombinants at an MOI of 5. Virions were purified from culture supernatants by sucrose gradient centrifugation (35). Protein samples (5 μg of virion proteins or lysates of approximately 104 cells per lane) were separated by discontinuous sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and incubated with antibodies as described previously (54). Monospecific rabbit antiserum against PrV gH (38), gB (55), gL (35), and pUL31 (40) were used at a dilution of 1:10,000 (gH), 1:50,000 (gB, pUL31), or 1:1,000 (gL).
In vitro fusion assays.
To analyze the fusogenic potential of gH32/98, approximately 3 × 105 RK13 cells per well were seeded into 12-well cell culture plates. On the following day, cells were transfected with 400 ng each of expression plasmids for EGFP (pEGFP-N1; Clontech) and for PrV glycoprotein gBKa, gBB4.1, gDKa, gHKa (12, 30, 39), or gH32/98 (this study) in 100 μl of Opti-MEM using 2 μl of Lipofectamine 2000 (Thermo Fisher Scientific). Empty vector (pcDNA3) served as a negative control and was added to achieve a total DNA amount of 2 μg in all assays. The mixture was incubated for 20 min at room temperature and added to the cells. After 3 h cells were washed with phosphate-buffered saline (PBS) and incubated in MEM supplemented with 5% FCS for another 24 h at 37°C. Thereafter, cells were washed with PBS, fixed with 3% paraformaldehyde (PFA) for 20 min, and washed two times with PBS. Syncytium formation was analyzed using an Eclipse Ti-S fluorescence microscope and NIS-Elements imaging software (Nikon). Total fusion activity was determined by multiplication of the area of cells with three or more nuclei by the number of syncytia within 10 fields of view (5.5 mm2 each). The experiment was repeated four times, and average percent values of positive-control transfections as well as standard deviations were calculated.
In vitro replication studies.
For determination of growth kinetics, confluent monolayers of RK13 cells in 96-well plates were infected with the phenotypically complemented mutant pPrV-gH32/98K, pPrV-gH32/98KgBB4.1, pPrV-gH32/98KΔgLZgBB4.1, or the wild-type gH revertant pPrV-gHK (30) at an MOI of 0.01 or 5 and consecutively incubated on ice for 1 h and at 37°C for 2 h. Subsequently, the inoculum was removed, nonpenetrated virus was inactivated by low-pH treatment (56), and fresh medium was added. Immediately thereafter and after 6, 12, 18, 24, 36, 48, and 72 h at 37°C, the cells were harvested together with the supernatants and lysed by freeze-thawing (−70°C and 37°C). Progeny virus titers were determined by plaque assays on RK13 cells and RK13-gH/gL cells. After 48 h at 37°C under semisolid medium, 30 plaques or areas of cells infected with either of the GFP-expressing viruses were measured microscopically, and percentages of wild-type (pPrV-gHK) sizes were calculated. Mean results of three independent growth kinetics studies and comparative plaque assays, as well as standard deviations, were determined.
In vitro penetration kinetics.
For analysis of penetration kinetics, RK13 cells grown in six-well plates were infected on ice with approximately 250 PFU of PrV-gHK, pPrV-gH32/98KgBB4.1, or pPrV-gH32/98KΔgLZgBB4.1. After 1 h, the inoculum was replaced by prewarmed medium, and cells were incubated at 37°C. Before and 15, 30, 60, 120, and 240 min after the temperature shift, remaining extracellular virus particles were inactivated by low-pH treatment. Thereafter, cells were washed two times with PBS and overlaid with semisolid medium. For 100% penetration controls, infected cells were washed with PBS only and overlaid with semisolid medium. After 2 days at 37°C, plaques were counted, and the penetration rate was calculated by comparing the determined numbers with those in the corresponding controls. Mean values and standard deviations from three independent assays were determined.
ACKNOWLEDGMENTS
These studies were supported by the Deutsche Forschungsgemeinschaft (DFG grant Me 854/11-2).
We thank B. Wanner for providing plasmids for BAC mutagenesis and G. Strebelow for performing sequence analyses. The technical assistance of A. Landmesser, K. Günther, and K. Biebl is greatly appreciated.
REFERENCES
- 1.Harrison SC. 2015. Viral membrane fusion. Virology 479–480:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 65:1653–1668. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Giovine P, Settembre EC, Bhargava AK, Luftig MA, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. 2011. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog 7:e1002277. doi: 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lazear E, Whitbeck JC, Zuo Y, Carfi A, Cohen GH, Eisenberg RJ, Krummenacher C. 2014. Induction of conformational changes at the N terminus of herpes simplex virus glycoprotein D upon binding to HVEM and nectin-1. Virology 448:185–195. doi: 10.1016/j.virol.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J Biol Chem 284:17370–17382. doi: 10.1074/jbc.M109.005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper RS, Heldwein EE. 2015. Herpesvirus gB: a finely tuned fusion machine. Viruses 7:6552–6569. doi: 10.3390/v7122957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauh I, Mettenleiter TC. 1991. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J Virol 65:5348–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peeters B, de Wind N, Hooisma M, Wagenaar F, Gielkens A, Moormann R. 1992. Pseudorabies virus envelope glycoproteins gp50 and gII are essential for virus penetration, but only gII is involved in membrane fusion. J Virol 66:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klupp BG, Nixdorf R, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J Virol 74:6760–6768. doi: 10.1128/JVI.74.15.6760-6768.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt J, Klupp BG, Karger A, Mettenleiter TC. 1997. Adaptability in herpesviruses: glycoprotein D-independent infectivity of pseudorabies virus. J Virol 71:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schröder C, Linde G, Fehler F, Keil GM. 1997. From essential to beneficial: glycoprotein D loses importance for replication of bovine herpesvirus 1 in cell culture. J Virol 71:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt J, Gerdts V, Beyer J, Klupp BG, Mettenleiter TC. 2001. Glycoprotein D-independent infectivity of pseudorabies virus results in an alteration of in vivo host range and correlates with mutations in glycoproteins B and H. J Virol 75:10054–10064. doi: 10.1128/JVI.75.21.10054-10064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 17.Zeev-Ben-Mordehai T, Vasishtan D, Hernandez Duran A, Vollmer B, White P, Prasad Pandurangan A, Siebert CA, Topf M, Grunewald K. 2016. Two distinct trimeric conformations of natively membrane-anchored full-length herpes simplex virus 1 glycoprotein B. Proc Natl Acad Sci U S A 113:4176–4181. doi: 10.1073/pnas.1523234113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci U S A 106:2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roche S, Bressanelli S, Rey FA, Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- 20.Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat Struct Mol Biol 15:1024–1030. doi: 10.1038/nsmb.1484. [DOI] [PubMed] [Google Scholar]
- 21.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol 84:3825–3834. doi: 10.1128/JVI.02687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107:22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xing Y, Oliver SL, Nguyen T, Ciferri C, Nandi A, Hickman J, Giovani C, Yang E, Palladino G, Grose C, Uematsu Y, Lilja AE, Arvin AM, Carfi A. 2015. A site of varicella-zoster virus vulnerability identified by structural studies of neutralizing antibodies bound to the glycoprotein complex gHgL. Proc Natl Acad Sci U S A 112:6056–6061. doi: 10.1073/pnas.1501176112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galdiero S, Falanga A, Vitiello M, Browne H, Pedone C, Galdiero M. 2005. Fusogenic domains in herpes simplex virus type 1 glycoprotein H. J Biol Chem 280:28632–28643. doi: 10.1074/jbc.M505196200. [DOI] [PubMed] [Google Scholar]
- 25.Galdiero S, Falanga A, Vitiello M, D'Isanto M, Collins C, Orrei V, Browne H, Pedone C, Galdiero M. 2007. Evidence for a role of the membrane-proximal region of herpes simplex virus type 1 glycoprotein H in membrane fusion and virus inhibition. Chembiochem 8:885–895. doi: 10.1002/cbic.200700044. [DOI] [PubMed] [Google Scholar]
- 26.Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc Natl Acad Sci U S A 107:22635–22640. doi: 10.1073/pnas.1011507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuchs W, Backovic M, Klupp BG, Rey FA, Mettenleiter TC. 2012. Structure-based mutational analysis of the highly conserved domain IV of glycoprotein H of pseudorabies virus. J Virol 86:8002–8013. doi: 10.1128/JVI.00690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Böhm SW, Backovic M, Klupp BG, Rey FA, Mettenleiter TC, Fuchs W. 2015. Functional characterization of glycoprotein H chimeras composed of conserved domains of the pseudorabies virus and herpes simplex virus 1 homologs. J Virol 90:421–432. doi: 10.1128/JVI.01985-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schröter C, Klupp BG, Fuchs W, Gerhard M, Backovic M, Rey FA, Mettenleiter TC. 2014. The highly conserved proline at position 438 in pseudorabies virus gH is important for regulation of membrane fusion. J Virol 88:13064–13072. doi: 10.1128/JVI.01204-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Böhm SW, Eckroth E, Backovic M, Klupp BG, Rey FA, Mettenleiter TC, Fuchs W. 2015. Structure-based functional analyses of domains II and III of pseudorabies virus glycoprotein H. J Virol 89:1364–1376. doi: 10.1128/JVI.02765-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J Virol 66:2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaye JF, Gompels UA, Minson AC. 1992. Glycoprotein H of human cytomegalovirus (HCMV) forms a stable complex with the HCMV UL115 gene product. J Gen Virol 73:2693–2698. doi: 10.1099/0022-1317-73-10-2693. [DOI] [PubMed] [Google Scholar]
- 33.Klupp BG, Baumeister J, Karger A, Visser N, Mettenleiter TC. 1994. Identification and characterization of a novel structural glycoprotein in pseudorabies virus, gL. J Virol 68:3868–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu DX, Gompels UA, Nicholas J, Lelliott C. 1993. Identification and expression of the human herpesvirus 6 glycoprotein H and interaction with an accessory 40K glycoprotein. J Gen Virol 74:1847–1857. doi: 10.1099/0022-1317-74-9-1847. [DOI] [PubMed] [Google Scholar]
- 35.Klupp BG, Fuchs W, Weiland E, Mettenleiter TC. 1997. Pseudorabies virus glycoprotein L is necessary for virus infectivity but dispensable for virion localization of glycoprotein H. J Virol 71:7687–7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lete C, Machiels B, Stevenson PG, Vanderplasschen A, Gillet L. 2012. Bovine herpesvirus type 4 glycoprotein L is nonessential for infectivity but triggers virion endocytosis during entry. J Virol 86:2653–2664. doi: 10.1128/JVI.06238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gillet L, May JS, Colaco S, Stevenson PG. 2007. Glycoprotein L disruption reveals two functional forms of the murine gammaherpesvirus 68 glycoprotein H. J Virol 81:280–291. doi: 10.1128/JVI.01616-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klupp BG, Mettenleiter TC. 1999. Glycoprotein gL-independent infectivity of pseudorabies virus is mediated by a gD-gH fusion protein. J Virol 73:3014–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schröter C, Vallbracht M, Altenschmidt J, Kargoll S, Fuchs W, Klupp BG, Mettenleiter TC. 2015. Mutations in pseudorabies virus glycoproteins gB, gD, and gH functionally compensate for the absence of gL. J Virol 90:2264–2272. doi: 10.1128/JVI.02739-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J Virol 76:364–378. doi: 10.1128/JVI.76.1.364-378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J Virol 72:873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klupp B, Altenschmidt J, Granzow H, Fuchs W, Mettenleiter TC. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J Virol 82:6299–6309. doi: 10.1128/JVI.00386-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Babic N, Klupp BG, Makoschey B, Karger A, Flamand A, Mettenleiter TC. 1996. Glycoprotein gH of pseudorabies virus is essential for penetration and propagation in cell culture and in the nervous system of mice. J Gen Virol 77:2277–2285. doi: 10.1099/0022-1317-77-9-2277. [DOI] [PubMed] [Google Scholar]
- 44.Roop C, Hutchinson L, Johnson DC. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J Virol 67:2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klyachkin YM, Stoops KD, Geraghty RJ. 2006. Herpes simplex virus type 1 glycoprotein L mutants that fail to promote trafficking of glycoprotein H and fail to function in fusion can induce binding of glycoprotein L-dependent anti-glycoprotein H antibodies. J Gen Virol 87:759–767. doi: 10.1099/vir.0.81563-0. [DOI] [PubMed] [Google Scholar]
- 46.Duus KM, Grose C. 1996. Multiple regulatory effects of varicella-zoster virus (VZV) gL on trafficking patterns and fusogenic properties of VZV gH. J Virol 70:8961–8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cairns TM, Friedman LS, Lou H, Whitbeck JC, Shaner MS, Cohen GH, Eisenberg RJ. 2007. N-terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J Virol 81:5102–5111. doi: 10.1128/JVI.00097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Möhl BS, Chen J, Sathiyamoorthy K, Jardetzky TS, Longnecker R. 2016. Structural and mechanistic insights into the tropism of Epstein-Barr Virus. Mol Cells 39:286–291. doi: 10.14348/molcells.2016.0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Atanasiu D, Saw WT, Eisenberg RJ, Cohen GH. 2016. Regulation of herpes simplex virus glycoprotein-induced cascade of events governing cell-cell fusion. J Virol 90:10535–10544. doi: 10.1128/JVI.01501-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan Q, Longnecker R, Connolly SA. 2015. A Functional Interaction between herpes simplex virus 1 glycoprotein gH/gL domains I and II and gD is defined by using alphaherpesvirus gH and gL chimeras. J Virol 89:7159–7169. doi: 10.1128/JVI.00740-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atanasiu D, Cairns TM, Whitbeck JC, Saw WT, Rao S, Eisenberg RJ, Cohen GH. 2013. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. mBio 4:e00046-13. doi: 10.1128/mBio.00046-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cairns TM, Milne RS, Ponce-de-Leon M, Tobin DK, Cohen GH, Eisenberg RJ. 2003. Structure-function analysis of herpes simplex virus type 1 gD and gH-gL: clues from gDgH chimeras. J Virol 77:6731–6742. doi: 10.1128/JVI.77.12.6731-6742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci U S A 104:18718–18723. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pavlova SP, Veits J, Keil GM, Mettenleiter TC, Fuchs W. 2009. Protection of chickens against H5N1 highly pathogenic avian influenza virus infection by live vaccination with infectious laryngotracheitis virus recombinants expressing H5 hemagglutinin and N1 neuraminidase. Vaccine 27:773–785. doi: 10.1016/j.vaccine.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 55.Kopp M, Granzow H, Fuchs W, Klupp BG, Mundt E, Karger A, Mettenleiter TC. 2003. The pseudorabies virus UL11 protein is a virion component involved in secondary envelopment in the cytoplasm. J Virol 77:5339–5351. doi: 10.1128/JVI.77.9.5339-5351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mettenleiter TC. 1989. Glycoprotein gIII deletion mutants of pseudorabies virus are impaired in virus entry. Virology 171:623–625. doi: 10.1016/0042-6822(89)90635-1. [DOI] [PubMed] [Google Scholar]
- 57.Grimm KS, Klupp BG, Granzow H, Muller FM, Fuchs W, Mettenleiter TC. 2012. Analysis of viral and cellular factors influencing herpesvirus-induced nuclear envelope breakdown. J Virol 86:6512–6521. doi: 10.1128/JVI.00068-12. [DOI] [PMC free article] [PubMed] [Google Scholar]