

ABSTRACT
The genome RNA of human parainfluenza virus type 2 (hPIV2) that acts as the template for the polymerase complex is entirely encapsidated by the nucleoprotein (NP). Recently, the crystal structure of NP of PIV5, a virus closely related to hPIV2, was resolved in association with RNA. Ten amino acids that contact the bound RNA were identified and are strictly conserved between PIV5 and hPIV2 NP. Mutation of hPIV2 NP Q202 (which contacts a base rather than the RNA backbone) to various amino acids resulted in an over 30-fold increase of polymerase activity as evidenced by a minireplicon assay, even though the RNA-binding affinity was unaltered. Using various modified minireplicons, we found that the enhanced reporter gene expression could be accounted for by increased minigenome replication, whereas mRNA synthesis itself was not affected by Q202 mutation. Moreover, the enhanced activities were still observed in minigenomes partially lacking the leader sequence and which were not of hexamer genome length. Unexpectedly, recombinant hPIV2 possessing the NP Q202A mutation could not be recovered from cDNA.
IMPORTANCE We examined the importance of amino acids in the putative RNA-binding domain of hPIV2 NP for polymerase activity using minireplicons. Abnormally enhanced genome replication was observed upon substitution mutation of the NP Q202 position to various amino acids. Surprisingly, this mutation enabled polymerase to use minigenomes that were partially lacking the leader sequence and not of hexamer genome length. This mutation does not affect fundamental properties of NP, e.g., recognition of gene junctional and editing signals. However, the strongly enhanced polymerase activity may not be viable for the infectious life cycle. This report highlights the potential of the polymerase complex with point mutations in NP and helps our detailed understanding of the molecular basis of gene expression.
KEYWORDS: human parainfluenza virus type 2, encapsidation, nucleoprotein, polymerase
INTRODUCTION
Human parainfluenza virus type 2 (hPIV2) is a major respiratory pathogen and a member of the Rubulavirus genus of the family Paramyxoviridae, which includes simian virus 41, hPIV4, mumps virus, and PIV5. The hPIV2 genome RNA contains six tandemly linked genes (3′-NP-P/V-M-F-HN-L-5′) and is tightly coated by the nucleoprotein (NP) to form a helical nucleocapsid that serves as the template for viral RNA synthesis. The phosphoprotein (P) and large protein (L) together form the viral RNA-dependent RNA polymerase (RdRp), which is responsible for both transcription to produce mRNAs and replication to produce genomes and antigenomes (1). hPIV2 NP contains 543 amino acids, whose functional domains are partially identified. The N-terminal 294 amino acids are required for NP self-assembly, and two C-terminal regions (amino acids 295 to 402 and 403 to 494) are essential for binding to the C- and N-terminal regions of P, respectively (2). Moreover, NP interacts with L at amino acids 403 to 494 (3). However, the RNA-binding domain of this protein has not been determined.
NPs from different nonsegmented negative-strand RNA viruses (nsNSV) share common structural features despite low sequence identity across different families. Crystals of NP of respiratory syncytial virus (RSV), vesicular stomatitis virus (VSV), and rabies virus in association with RNA show various oligomeric nucleocapsid-ring structures, made by lateral contacts of NP protomers (4–7). The RNA is sequestered in a long groove between the N-terminal domain (NTD) and the C-terminal domain (CTD). In the Paramyxoviridae, each NP binds to six nucleotides (nt), and the genome obeys a “rule of six” (R/6); there is a strict requirement for their genomes to consist of multiples of 6 nt (8, 9).
Recently, the high-resolution crystal structure of the PIV5 NP-RNA ring complex assembled in Escherichia coli was determined (10). PIV5 NP encapsidates RNA in its positively charged groove between the NTD and CTD, similar to the case for other nsNSV. Each NP associates 6 nt, with a conformation of alternating three bases facing in (toward the protein core) and three bases facing out (toward the solvent). Amino acids from the NTD bind bases facing inward, and those from the CTD bind bases facing outward. Because of the strong similarity between hPIV2 and PIV5 NP genes, this report provides information regarding the relative importance of the RNA-binding residues in minigenome gene expression and has uncovered surprising properties of NPQ202.
RESULTS
Importance of NP amino acids that contact the RNA for minireplicon gene expression.
Ten amino acids of PIV5 NP directly contact the RNA, namely, K194, R195, Y260, Q202, M266, G267, Y350, A351, R354, and S355 (10), and are strictly conserved between hPIV2 and PIV5 NP (Fig. 1A). The RNA-binding pocket of PIV5 NP is subdivided into two subpockets. Pocket 1 includes K194, R195, and Y260, and pocket 2 includes M266, G267, Y350, A351, and R354, all of which bind phosphates of the ribose-phosphate backbone. Q202 and S355 are exceptional because they bind a base and a ribose, respectively (Fig. 1B).
FIG 1.

Mutagenesis analysis using the hPIV2 Rluc minireplicon assay for amino acids in the putative RNA-binding domain of NP. (A) Comparison of hPIV2 and PIV5 NP amino acid sequences. Bold numbers over the nucleotides indicate amino acids responsible for RNA binding. The accession numbers are BAE00051 and YP_138511 for hPIV2 and PIV5, respectively. (B) Schematic diagram representing the putative binding of amino acids in the NP with ribonucleotides. The pentagons with numerals and hexagons with “B” and “P” represent ribose, base, and phosphate, respectively. Amino acids included in two RNA-binding pockets estimated by the PIV5 NP structure are indicated. (C) Relative Rluc activity in the minireplicon assay with the hPIV2 polymerase complex including NP mutants. The Rluc expression from minigenomes is normalized to internal control Fluc expression, and relative values are shown (NPwt = 1). NP−, result for the Rluc minigenome without NP plasmid. Data represent means and standard deviations from triplicate experiments. Expression of NP, P, and L was detected by Western blotting using specific MAbs. *, P < 0.01; **, P < 0.05.
To examine the contribution of these amino acids to gene expression, we prepared a series of plasmids expressing NP mutants whose RNA-binding residues were replaced by alanine (A351 was omitted from this study). Using hPIV2 Renilla luciferase (Rluc) minireplicons, we analyzed the effects of alanine substitution on reporter gene expression. We first examined protein expression by Western blotting using a monoclonal antibody (MAb) for NP. Eight of the nine mutations did not affect the protein expression; that of the R354A mutant was clearly low, presumably indicating its importance for protein conformation (Fig. 1C). Analyses of Rluc expressions in these minireplicons revealed various patterns. All the residues which contact the phosphates of the bases pointing outward mostly eliminate gene expression when mutated to Ala (M266A is a partial exception here), indicating that these residues are important for gene expression. In contrast, all the residues contacting the ribose-phosphate backbone of the bases facing inward are essentially neutral when mutated to Ala, suggesting that these residues are not as important for gene expression. In further contrast, Q202, the only residue that directly contacts a base (Fig. 1B, base 1), has the most remarkable effect upon mutation to Ala; it enhances minigenome reporter gene expression by >30-fold.
To examine Q202 in more detail, we constructed plasmids expressing NPs whose Q202 was replaced by all other amino acids (except for Q202C, because E. coli would not replicate this plasmid). We first confirmed that all substitutions of NP did not affect protein expression (Fig. 2A). Various patterns of Rluc expression by these mutations were again observed (Fig. 2A and B). In two mutants, Q202P and -K, Rluc expression lower than that of the wild type (wt) was observed (Fig. 2B). In Q202T, a stronger loss of Rluc expression was observed (Fig. 2B). In contrast, strongly enhanced Rluc expression was observed in several other mutants (Fig. 2A). Q202V, -S, -L, and -M showed Rluc expression similar to that of Q202A. Although Q202G, -I, -F, -Y, -W, -D, -E, -N, -H, and -R showed higher Rluc expression than wt, the enhancements were lower than for Q202A. Although Q itself contains a side chain that is neither charged nor aromatic, the stronger enhancements seem to be due to mutation to noncharged and nonaromatic amino acids. Given the very disparate effects of mutating Q202 to S versus T and to R versus K, it is difficult to find a simple correlation. The loss of Q at position 202 seems to be important, since most mutations enhance gene expression. Because Q202A showed one of the strongest effects, we used this mutant for further studies.
FIG 2.

Mutagenesis analysis of hPIV2 NP Q202 using the Rluc minireplicon assay. (A) Relative Rluc activity in the minireplicon assay with the hPIV2 polymerase complex including NP mutants. The Rluc expression from minigenomes is normalized to internal control Fluc expression, and relative values are shown (NPwt = 1). NP−, result from the Rluc minigenome without NP plasmid. Data represent means and standard deviations from triplicate experiments. Expression of NP, P, and L was detected by Western blotting using specific MAbs. (B) Extension from panel A, displaying the relative Rluc activity in the minireplicon assay using NP Q202T, P, and K. (C) Relative amount of antigenome during the hPIV2 Rluc minireplicon assay. The NP-encapsidated antigenome was obtained by immunoprecipitation after performing the assay. The relative amounts of antigenome were measured by qRT-PCR. Data represent means and standard deviations from triplicate experiments. *, P < 0.01; **, P < 0.05.
We next examined antigenome levels during the Rluc minireplicon assay with wt NP (NPwt) or NPQ202 mutants. After performing the standard assay, NP-encapsidated RNA was collected by immunoprecipitation (IP) using a MAb against NP, and the relative amounts of antigenome RNA were measured by quantitative real-time reverse transcription-PCR (qRT-PCR) (Fig. 2C). In the presence of NPQ202A, NPQ202S, and NPQ202L, in which Rluc expression was enhanced approximately 30-fold over the wt level, there was an approximately 30-fold increase in antigenome levels. In the presence of NPwt and NPQ202K, where Rluc expression was low, antigenome levels were similarly low. Thus, at least for these mutants, enhanced and reduced gene expression can be accounted for by enhanced and reduced minigenome replication, respectively.
Contribution of Q202A to NP-P interaction, NP-NP self-interaction, and RNA binding.
We examined whether the Q202A mutation affected NP-P interaction, NP-NP self-interaction, and NP-RNA binding. Plasmid-driven NP and P were immunoprecipitated with anti-NP MAbs, and the precipitate was examined by Western blotting for P. P immunoprecipitated with NPQ202A was at a level comparable to that with NPwt, indicating that the Q202A mutation does not affect NP-P interaction (Fig. 3A). The NP-NP interaction was analyzed by IP using full-length NP and a C-terminally truncated mutant, NPΔC100, that conserves the potential to interact with NP (2). Cells expressing both proteins were subjected to IP using a MAb against amino acids 495 to 543 of NP, and precipitated proteins were examined by Western blotting using a MAb against amino acids 1 to 100 of NP (Fig. 3B). NP(wt)ΔC100 was immunoprecipitated with NPQ202A in the same amounts as with NPwt. Moreover, the interaction between NPQ202A and NP(Q202A)ΔC100 was also at the same levels with others, indicating that the Q202A mutation does not affect NP-NP self-interaction.
FIG 3.

Effects of NPQ202A on NP-P interaction, NP-NP self-interaction, and NP-RNA binding (A) Interaction between NP and P. BSR T7/5 cells transfected with NP- and P-expressing plasmids were subjected to IP assay using a mixture of anti-NP MAbs. Both NPwt and NPQ202A were detected by Western blotting using anti-NP MAb, and P was detected by anti-P/V MAb. (B) Interaction between NP and NPΔC100. BSR T7/5 cells transfected with NP- and NPΔC100-expressing plasmids were subjected to IP assay using a MAb against the C terminus of NP (MAb 159-1, which reacts with NP amino acids 495 to 543). NPwt, NPQ202A, NP(wt)ΔC100, and NP(Q202A)ΔC100 were detected by Western blotting using a MAb against N terminus of NP (MAb 330-1, which reacts with NP amino acids 1 to 100). (C) Interaction between NP mutants and the minigenome. BSR T7/5 cells transfected with NP- and minigenome-expressing plasmids were subjected to RNA IP assay using an anti-NP MAb. (D) Interaction between NP mutants and P in the form of the NP-RNA complex. BSR T7/5 cells transfected with P-, NP-, and minigenome-expressing plasmids were subjected to RNA IP assay using an anti-P/V MAb. The relative amounts of RNA were measured by qRT-PCR. Data represent means and standard deviations from triplicate experiments. Proteins were detected by Western blotting using anti-NP and -P/V MAbs. *, P < 0.01; **, P < 0.05.
To examine whether Q202A affects NP-RNA binding, we performed an RNA IP assay. nsNSV NP preferentially encapsidates viral RNA by recognizing the 3′ and/or 5′ cis-acting RNA elements (11). Therefore, we used the hPIV2 Rluc minigenome that encodes both 3′ and 5′ cis-acting RNA elements as a substrate for NP-RNA binding. Plasmid-driven NP mutant and wt minigenomes were immunoprecipitated with anti-NP MAbs, and their relative amounts of RNA were measured by qRT-PCR (Fig. 3C). The relative amount of minigenome immunoprecipitated by NPQ202A was comparable to that by NPwt. Moreover, we examined the binding of P to the NP-RNA complex. Cells expressing P and mutant NP as well as the hPIV2 minigenome were subjected to RNA IP assay using anti-P/V MAb. The relative amounts of immunoprecipitated minigenome forming the complex with NPQ202A and NPwt were at similar levels (Fig. 3D). These data indicate that the Q202A mutation does not affect RNA binding (efficiency of encapsidation).
Unsuccessful recovery of rPIV2 possessing the NPQ202A mutation.
To examine the effect of NPQ202A on the viral life cycle, we attempted to generate recombinant hPIV2 (rPIV2) with the NP Q202A point mutation by reverse genetics. According to the standard protocol, BSR T7/5 cells were transfected with plasmids expressing NPwt, P, L, and the full genome with either NPQ202A or NPwt. Three days posttransfection, the culture medium was transferred to Vero cells, which are highly sensitive for hPIV2 infection. At a further 2 days postinfection, cell fusion was observed in the Vero cells inoculated with rPIV2 NPwt, whereas no obvious cytopathic effect was observed in rPIV2 NPQ202A (Fig. 4A). We also confirmed the infection of rPIV2 NPwt by indirect immunofluorescence assay (IFA) using anti-P MAb, whereas no apparent viral protein was observed in rPIV2 NPQ202A infection (Fig. 4B). We could not find any cytopathic effect in the rPIV2 NPQ202A-infected Vero cells after observation for 10 days. Identical results were obtained in three independent trials. We were also unable to rescue rPIV2-NPQ202A using NPQ202A as the support plasmid (data not shown).
FIG 4.
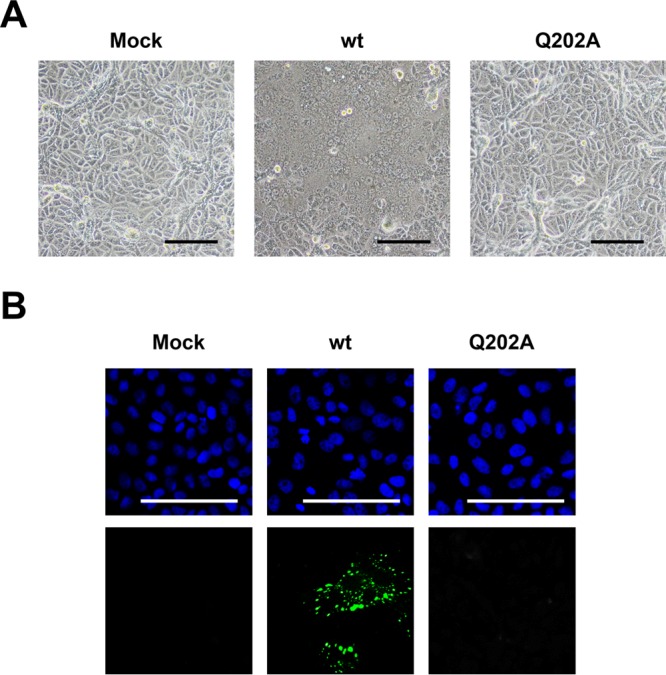
Rescue of recombinant hPIV2 with the NP Q202A mutation. The culture medium of BSR T7/5 cells transfected with plasmids encoding NP, P, L, and full genome from hPIV2 wt or hPIV2 NPQ202A were transferred to Vero cells. (A) After 48 h, cells were observed under a light microscope. (B) Cells were fixed and stained with 4′,6-diamidino-2-phenylindole (DAPI) (upper panel) and anti-P MAb (lower panel). The scale bars correspond to 100 μm.
The NP Q202A mutation maintains abnormally enhanced gene expression in the presence of junctional sequences.
Viral genes are separated by junctional sequences that enable transcription of multiple mRNAs from the nsNSV RNA genome (11). We examined whether NPQ202A could not only transcribe mRNAs but also accommodate gene junctions, i.e., could stop and restart mRNA synthesis from bicistronic minigenomes. We constructed an Rluc- and firefly luciferase (Fluc)-encoding dual minigenome containing the hPIV2 P-M junction (hPIV2-Rluc-Fluc). Upon examination, both Rluc and Fluc activities were found at 48 h posttransfection compared with the NP negative control (Fig. 5A). We subsequently examined the effect of NPQ202A in this assay and observed reporter expression over 30-fold-higher than that of NPwt for both Rluc and Fluc (Fig. 5A). Similar results were obtained using a minigenome containing 3 reporter genes and two junctions (Fig. 5B). mRNA synthesis from bi- and tricistronic minigenomes suggests that gene end and gene start signals are normally processed by RdRp in the presence of NPQ202A.
FIG 5.

Effects of NPQ202A on dual and triple minireplicon assays. Relative luciferase activities in the dual (A) and triple (B) minireplicon assays with the hPIV2 polymerase complex including NPwt or NPQ202A are shown. The Rluc, Fluc, and Gluc expression is shown as relative values (NPwt = 1). NP−, result from the assays without NP plasmid. Data represent means and standard deviations from triplicate experiments. *, P < 0.01.
The NP Q202A mutation does not affect mRNA editing.
The unedited rubulavirus P gene mRNAs code for the V protein; P protein expression requires mRNA editing in which two Gs are inserted into the mRNA in response to a cis-acting editing sequence (9, 12). We next examined whether mRNA editing occurs normally in the presence of Q202A, to determine whether this remaining property of rubulavirus mRNA synthesis is affected. We therefore added the hPIV2 P gene editing signal (3′-GGGUUGAAAUUCUCCCCCCC-5′) at position 12 from the start of the Rluc gene of the Rluc-Fluc dual minigenome (Fig. 6A). Translation of unedited mRNA results in the short peptide MTSKPNFKRGGL. When hPIV2 RdRp recognizes the editing signal during transcription, GG is added (5′-CCCAACUUUAAGAGGGGGGGGG-3′). This converts the edited mRNA into one that expresses functional Rluc (Fig. 6A, “Edited by hPIV2 RdRp”). We performed the hPIV2 Rluc/edit-Fluc minireplicon assay using NPwt and NPQ202A and applied the resultant Rluc and Fluc values to the mathematical formula shown in Materials and Methods. We computed the intrinsic Rluc editing efficiencies as 18.6% (±3.4%) for NPwt and 19.6% (±2.5%) for NPQ202A, which are not significantly different (Fig. 6B). Thus, all aspects of mRNA synthesis that we have examined, including the ability of RdRp to accommodate gene junctions and to respond to the P gene cis-acting editing signal, apparently take place normally in the presence of NPQ202A.
FIG 6.

Effect of NPQ202A on gene editing. (A) Schematic diagram of the minireplicon assay for examining gene editing efficiency. An hPIV2 editing signal was added in the Rluc gene of the hPIV2-Rluc-Fluc minigenome. The translated amino acid sequences are shown. After GG is added by hPIV2 RdRp, the resulting sequence can be converted to the Rluc open reading frame. (B) Intrinsic editing efficiency. The calculation method is described in the text. Data represent means and standard deviations from triplicate experiments.
NPQ202A enables RdRp to use incomplete minigenomes partially lacking leader sequences and which do not conform to the rule of 6.
hPIV2 minigenomes replicate to higher levels when equivalent amounts of NPQ202A rather than NPwt genes support minigenome replication. This enhanced replication is presumably due to some advantage that NPQ202A-containing replication complexes have in promoter recognition and initiation of RNA synthesis at the genome 3′ end. Since the cis-acting promoter sequences are recognized by RdRp in the context of the NP-RNA nucleocapsid (12), NP is well placed to affect promoter strength.
It is unusual for mutant proteins to confer an apparent advantage, even at the restricted level of minigenome replication in transfected cells. However, there is a somewhat parallel situation with Sendai virus (SeV). SeV minigenomes replicate to 5- to 10-fold-higher levels when a mutant P gene (that does not express C proteins) supports minigenome replication than when a wt P gene is used; C proteins are thought to act as promoter-specific inhibitors of genome replication (13). One indication that the SeV C proteins act at the level of promoter recognition concerns the paramyxovirus “rule of six” (R/6) and how it governs genome replication (see Discussion). SeV minigenomes that do not conform to the rule of six (R/6minus) do not replicate when C proteins are expressed, but they replicate relatively well in the absence of C expression. To gather further evidence that hPIV2 NPQ202A acts at the level of promoter recognition, we examined whether NPQ202A behaved similarly on R/6minus hPIV2 minigenomes.
We have previously described hPIV2 minigenomes with successive 6-nt deletions in the leader region, which are inactive when NPwt supports minigenome replication (14). As they are all of hexamer length, their inactivity is presumably due to missing cis-acting sequences and/or because the spacing between the two elements of the bipartite promoter, which is critical, has been altered (15, 16). We confirmed that successive deletions of six contiguous bases (d#0 to d#8) in the leader region completely disrupts Rluc expression (Fig. 7A). When the same experiment was carried out with NPQ202A, clear Rluc expression in d#3 to d#8 was found, which varied from 2- to 30-fold that of NPwt on R/6wt minigenomes. Thus, although highly variable, enhanced expression by NPQ202A is maintained even on these R/6minus minigenomes. We also examined the effect of NPQ202A on R/6minus minigenomes that were not of hexamer length by successively adding back the 6 nt that were originally deleted from d#5, generating minigenomes of 6n + 1 to 6n + 5 genome length (d#5 + 1 to d#5 + 5) (Fig. 7B). Partial restoration of the missing nucleotides did not restore replication supported by NPwt, presumably because only minigenomes of hexamer length are active. However, NPQ202A again restored replication to these minigenome that were not of hexamer length (Fig. 7B), even though their cis-acting sequences were no longer in the wt hexamer context. The effects of NPQ202A on R/6minus hPIV2 minigenome replication further suggest that these mutant NPs, like the SeV C proteins, operate at the level of promoter recognition.
FIG 7.

Effects of NPQ202A on unique minireplicon assays using incomplete minigenomes partially lacking leader sequences and which do not conform to the rule of six. (A) Relative Rluc expression in the minireplicon assay partially lacking the leader sequence determined by using the hPIV2 polymerase complex including NPwt or NPQ202A. (B) Relative luciferase expression in the minireplicon assay partially lacking 30 to 35 nucleotides from the 3′ terminus determined by using the hPIV2 polymerase complex including NPwt or NPQ202A. The Rluc expression from minigenomes is normalized to an internal control Fluc expression, and relative values are shown (NPwt = 1). NP−, result for the Rluc minigenome without NP plasmid. “NP wt” in the graph for NPQ202A indicates the result for NPwt using the normal minigenome. Data represent means and standard deviations from triplicate experiments. *, P < 0.01.
DISCUSSION
We examined the contribution of the 10 amino acids of hPIV2 NP that directly contact the genome RNA in NP-RNAs to reporter gene expression. Alanine mutagenesis using Rluc minireplicons identified point mutations, such as Y260, G267, and Y350, that strongly reduced reporter expression (Fig. 1C), suggesting that these residues that contact the ribose-phosphate backbone of the bases pointing toward the solvent are critical for gene expression. In contrast, we surprisingly found abnormally enhanced Rluc expression when NPQ202A was used to support minigenome replication (Fig. 1C). Moreover, the amount of antigenome produced in this assay was correspondingly much higher than that using NPwt (Fig. 2C). Antigenome levels presumably reflect those of genomes. For example, in trailer-trailer copyback minigenomes, where minigenomes and miniantigenomes are initiated from very similar if not identical promoters, the fact that NPQ202A also strongly increases miniantigenome levels relative to NPwt (data not shown) suggests that “classical” back-and-forth genome replication is indeed abnormally enhanced by NPQ202A.
The most interesting property of the Q202A mutation was its ability not only to abnormally enhance reporter expression in R/6wt minigenomes but also to recover the enhanced expression from R/6minus minigenomes (Fig. 7). Paramyxovirus replication promoters are bipartite in nature, being composed, at a minimum, of the first 12 nt at the 3′ genome end and a second component in the 5′ untranslated region (UTR) of the first (NP) gene, which for SeV and hPIV3 (genus Respirovirus) is simply a cytosine at hexamer position 1 of hexamers 14, 15, and 16 from the 3′ end [i.e., (3′CNNNNN)3] (17, 18) and for PIV5 (genus Rubulavirus) is CG at hexamer positions 5 and 6 of hexamers 13, 14, and 15 [(3′NNNNCG)3] (16). These two promoter elements are found on the same vertical face of the helical nucleocapsid, and their precise nucleotide spacing, as well as their hexamer phase or context, can be critical (15, 16, 19–21). R/6minus minigenomes are inactive when supported by NPwt, presumably because their promoter cannot be recognized and RNA synthesis does not initiate within these replication complexes. When supported by NPQ202A, the promoters of these minigenome NP-RNAs appear to regain activity.
The possible contributions of the Q202 mutation to genome encapsidation or other aspects of the replication machinery need to be further examined. However, our discussion focuses on initiation at replication promoters because (i) we were unable to find any aspect of the mechanics of mRNA synthesis per se that is affected by Q202A and (ii) our results are consistent with what is known about replication promoters. It is presumably not coincidental that Q202 is the only one of these 10 residues that contacts a base, and in particular, base 1 of the triplet that points toward the solvent. We do not know whether Q202 has a particular affinity for any base (in these NP-RNA ring structures assembled in E. coli, the bound RNA is heterogeneous and all bases are modeled as uracils). Given that Q202 is likely to be contacting every 6th base in the NP-RNA, Q202 must interact with all bases. Moreover, although it is the bases at hexamer positions 5 and 6 that form the essential elements of the PIV5 internal replication promoter element, the base at position 1 forms a 3-base stack with those at positions 6 and 5 (Fig. 1B). The interaction of Q202 with the base at position 1 can then presumably affect the stability of the 3-base stack and thus affect replication promoter activity. The presumed complexity of this interaction could explain the bewildering effects of mutating Q202 to all other amino acids (Fig. 2A), e.g., why mutation to some residues strongly enhances promoter activity relative to that of NPwt (e.g., Q202A), whereas the presence of other amino acids in place of Q202 inhibits promoter activity (e.g., Q202T).
hPIV2 NPQ202A has another unexpected property, one that presents an apparent paradox. For R/6wt minigenomes, as much as 35 times more reporter is expressed when NPQ202A supports minigenome replication as when NPwt is used, as if NPwt acts as an inhibitor of minigenome replication relative to NPQ202A. Although highly speculative, one way to reconcile these results is to propose that RdRp in R/6wt- and NPwt-containing replication complexes may be constrained in its interaction with the R/6wt promoter, because RdRp may need to adopt a particular conformation before it can initiate. Q202 is presumably important in maintaining this restriction. In contrast, RdRp in R/6wt and NPQ202A-containing replication complexes does not have to adopt this constrained conformation (because of the absence of Q202) and thus initiates more frequently. For the same reason, RdRp in NPQ202A-containing replication complexes can initiate RNA synthesis reasonably well even on R/6minus NP-RNAs. This more frequent (and illegitimate?) initiation is perfectly compatible with minigenome replication that requires only a subset of the NP functions needed for a complete viral life cycle. In this view, the constrained RdRp conformation is not needed for initiation at the genome 3′ end per se, but the precise manner in which RdRp initiates RNA synthesis here may be somehow critical for the successful completion of an infectious life cycle.
nsNSV RdRps carry out both transcription and replication. Given their very different modes of RNA synthesis, their commitment to one or the other must occur relatively early in each case and be maintained until the process is completed. For example, replicases do not respond to gene editing and junctional signals, unlike transcriptases. All nsNSV RdRps are thought to start RNA synthesis (or at least enter the NP-RNA template) at the genome 3′ end, guided by their interaction with the promoter. In contrast to the case for segmented negative-strand RNA viruses (sNSV), where RdRp commitment to transcription or replication is determined by whether RNA synthesis initiates with a capped host cell primer or ATP, nsNSV RdRp is thought to initiate at the genome 3′ end with ATP for both forms of RNA synthesis (22). The commitment is thought to come shortly thereafter. nsNSV RdRps whose nascent RNA chains begin being assembled with NP before these RdRps terminate leader RNA synthesis become committed to replication; they read through the leader/NP junction and continue to the very end of template. In contrast, RdRps that initiate RNA synthesis at the NP gene start site and then cap the nascent NP mRNA become committed to transcription (23, 24). Structural studies of both sNSV and nsNSV RdRps suggest that both can be found in a number of alternative states or conformations, each of which may be important during different steps of RNA synthesis (25–28).
In this view, nsNSV RdRps act as transcriptases or replicases because they have adopted particular conformations that commit them to each form of RNA synthesis and from which they cannot escape until each process is completed. Once the processes are completed, these RdRps are free to reinitiate RNA synthesis at the genome 3′ end as uncommitted RdRp. It is possible that they must adopt a hypothetical constrained conformation during promoter recognition so that they can switch to the conformation of a committed replicase or transcriptase and, most importantly, to do so such that this mode of RNA synthesis is fixed, i.e., maintained until the process is completed. R/6wt replication complexes containing NPQ202A that initiate RNA synthesis (albeit more frequently) without adopting the hypothetical constrained conformation may not be as able to effect these switches in conformation with high fidelity, i.e., such that each conformation maintains each mode of RNA synthesis until that process is completed. In the absence of this conformational fidelity to each mode of RNA synthesis, RNAs that are absent in wt infections and which compromise the production of infectious progeny may be produced. In this view, the lowered frequency of initiation on NPwt-containing replication complexes relative to NPQ202A–containing complexes can be considered the price the virus pays for ensuring the fidelity of each form of subsequent RNA synthesis.
MATERIALS AND METHODS
Cells and antibodies.
BSR T7/5 cells that constitutively express T7 RNA polymerases (29) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS). Vero cells were cultured in Eagle's minimal essential medium supplemented with 10% FCS. Monoclonal antibodies (MAbs) against hPIV2 NP (16-1A, 159-1, 306-1, 330-1, and 366-1), P/V (315-1), P (335-1), and L (70-6-2) were as described previously (2, 3, 30).
Plasmid construction.
Renilla luciferase expressing the minigenome plasmid of hPIV2 (hPIV2-Rluc) was constructed as described previously (14). Multiple luciferases, including Rluc, firefly luciferase (Fluc), and Gaussia secretory luciferase (Gluc), expressing minigenome plasmids of hPIV2 (hPIV2-Rluc-Fluc and hPIV2-Rluc-Fluc-Gluc) were constructed by a standard PCR method. In brief, each luciferase gene and junctional sequence of hPIV2 genome were amplified and connected. The P-M junction and M-F junction were arranged between Rluc and Fluc and between Fluc and Gluc, respectively. The hPIV2-Rluc-Fluc with P/V editing signal in the Rluc gene (hPIV2-Rluc/edit-Fluc) was constructed by adding the editing signal in the Rluc gene, and 5 nt were added directly after the Fluc gene to follow the hexamer genome length. A series of modified hPIV2-Rluc constructs possessing partially deleted leader sequences were constructed by a standard PCR mutagenesis method. All minigenomes were expressed as the negative-sense RNA under the control of a T7 RNA polymerase promoter. The hPIV2 NP wild-type (NPwt), P, L, and Fluc genes cloned into the pTM1 vector, which contains a T7 promoter and an encephalomyocarditis virus internal ribosome entry site, were as described previously (31, 32). A series of pTM1-NP mutants were constructed by a standard PCR mutagenesis method. A plasmid expressing NPwt with the C-terminal 100 amino acids truncated, pTM1-NP(wt)ΔC100, was prepared as described previously (2), and pTM1-NP(Q202A)ΔC100 was constructed by a standard PCR mutagenesis method. pPIV2, containing the full-length cDNA (15,654 nt; GenBank accession number ABI176531) of the hPIV2 Toshiba strain was constructed as described previously (33). A single point mutation at the NP Q202 position was introduced by a standard PCR mutagenesis method.
hPIV2 minireplicon assay.
The hPIV2 Rluc minireplicon assay was performed in BSR T7/5 cells cultured in 12-well plates. One hundred thousand BSR T7/5 cells were seeded in 12-well plate. Plasmids hPIV2-Rluc (0.5 μg), pTM1-L (0.375 μg), pTM1-P (0.2 μg), and pTM1-NP (0.375 μg) or empty vector and Fluc (0.1 μg) were transfected by using XtremeGENE HP (Roche, Basel, Switzerland). At 24 h posttransfection, the Rluc and Fluc activities were measured by using the dual-luciferase assay kit (Promega, Madison, WI, USA) according to the manufacture's instructions. All results obtained for Rluc were normalized by the expression levels of Fluc. The hPIV2 Rluc-Fluc, Rluc-Fluc-Gluc, and Rluc/edit-Fluc minireplicon assays were performed in BSR T7/5 cells as described above with some modifications. Plasmids hPIV2-Rluc-Fluc, Rluc-Fluc-Gluc, or Rluc/edit-Fluc (0.5 μg), pTM1-L (0.375 μg), pTM1-P (0.2 μg), and pTM1-NP (0.375 μg) or empty vector were transfected. At 48 h (for hPIV2-Rluc-Fluc and Rluc/edit-Fluc) or 72 h (for hPIV2-Rluc-Fluc-Gluc) posttransfection, Rluc and Fluc activities were measured as described above. Gluc activity in the culture medium was measured by using the BioLux Gaussia luciferase assay kit (New England BioLabs, Ipswich, MA, USA) according to the manufacturer's instructions as described previously (34).
IP analysis.
NP-P interaction was analyzed by immunoprecipitation (IP) assay performed in BSR T7/5 cells transfected with 0.2 μg of pTM1-P and 0.8 μg of pTM1-NP mutants or empty vector. At 48 h posttransfection, cells were lysed in lysis buffer (20 mM Tris-Cl [pH 8.0], 150 mM NaCl, 10% glycerol, and 1% Triton X-100) containing cOmplete protease inhibitor (Roche). The supernatants obtained by centrifugation were incubated with MAbs against hPIV2 NP (mixture of 16-1A, 159-1, 306-1, 330-1, and 366-1) and protein A-Sepharose (GE Healthcare, Little Chalfont, UK). The immunoprecipitated sample was separated by SDS-PAGE, and proteins were detected by Western blotting using specific MAbs. NP-NP self-interaction was also analyzed by IP assay performed in BSR T7/5 cells transfected with 0.5 μg of pTM1-NPΔC100 and 0.5 μg of pTM1-NP or empty vector. The procedure for IP sample preparation was same as described above, and MAb NP 159-1, which recognizes NP amino acids 495 to 543, was used to specifically immunoprecipitate the full-length NP. The NPΔC100 that was coimmunoprecipitated with full-length NP was detected by Western blotting using MAb NP 330-1, which recognizes NP amino acids 1 to 100.
Analysis of antigenome replication during hPIV2 Rluc minireplicon assay.
Quantification of the antigenome was performed by quantitative real-time reverse transcription-PCR (qRT-PCR) using an RNA sample immunoprecipitated by an anti-NP MAb. At 48 h posttransfection of hPIV2 Rluc minireplicon components, BSR T7/5 cells were lysed in lysis buffer as described above. The supernatants obtained by centrifugation were incubated with a MAb (159-1) against hPIV2 NP and protein A-Sepharose. The immunoprecipitated sample was subjected to RNA extraction using Isogen (Nippon Gene, Tokyo, Japan). To increase the efficiency of RNA extraction, Dr.GenTLE precipitation carrier (TaKaRa, Shiga, Japan) was used according to the manufacturer's instructions. The cDNA synthesis was carried out with the PrimeScript RT reagent kit (TaKaRa) with a specific primer for antigenome RNA (5′-ACCAAGGGGAAAATCAATATGTT-3′). qRT-PCR was performed by using SsoAdvanced Universal SYBR green Supermix (Bio-Rad, Hercules, CA, USA) with primers for the Rluc gene (forward, ATAACTGGTCCGCAGTGGTG; reverse, TAAGAAGAGGCCGCGTTACC).
RNA IP analysis.
RNA IP analysis was performed in BSR T7/5 cells transfected with 0.5 μg of hPIV2-Rluc and 0.4 μg of pTM1-NP mutants or empty vector. At 48 h posttransfection, cells were lysed in lysis buffer as described above. The supernatants obtained by centrifugation were incubated with MAbs against hPIV2 NP (a mixture of 16-1A, 159-1, 306-1, 330-1, and 366-1) and protein A-Sepharose. Half of the immunoprecipitated sample was separated by SDS-PAGE, and then proteins were detected by Western blotting using specific MAbs. The other half was subjected to RNA extraction using Isogen with Dr.GenTLE precipitation carrier for qRT-PCR. The cDNA synthesis was carried out with the PrimeScript RT reagent kit with a specific primer for genome RNA (5′-ACCAAGGGGAGAATCAGATGGCATCG-3′). qRT-PCR for the Rluc gene was performed as described above. Interaction of P with NP-RNA was analyzed by RNA IP assay using BSR T7/5 cells transfected with 0.5 μg of hPIV2-Rluc, 0.2 μg of pTM1-P, and 0.4 μg of pTM1-NP mutants or empty vector. An Anti-P/V MAb (315-1) was used for precipitation by the method described above.
Generation of recombinant hPIV2.
Two hundred thousand BSR T7/5 cells were seeded in a 6-well plate. Cells were transfected with wt or NPQ202A pPIV2 (2.5 μg) together with the expression plasmids pTM1-NP (0.5 μg), pTM1-P (0.25 μg), and pTM1-L (0.5 μg) by using XtremeGENE HP. At 3 days posttransfection, the released viruses in the supernatant of the transfected cells were filtered with a 0.45-μm-pore-size filter and transferred to Vero cells. Virus rescue was confirmed by the appearance of cell fusion that is a characteristic of hPIV2 infection and by indirect immunofluorescence assay (IFA) using a MAb against hPIV2 P (335-1).
Calculation of gene editing efficiency.
The hPIV2-Rluc/edit-Fluc minireplicon assay with NPwt and NPQ202A was performed. Simultaneously, same assay using the hPIV2-Rluc-Fluc minigenome was performed to compare the editing efficiency. The Fluc expression by both minigenomes should be the internal control. The Rluc editing efficiency is calculated as follows: (i) the basic Rluc editing efficiency (bRed) = A/B × 100 (%), where A is the Rluc value from Rluc edit and B is the Rluc value from Rluc normal; (ii) the basic Fluc editing efficiency (bFed) = C/D × 100 (%), where C is the Fluc value from Fluc edit and D is the Fluc value from Fluc normal. Because the value of bFed is regarded as the internal control (100%), the intrinsic Rluc editing efficiency = bRed/bFed × 100 (%).
ACKNOWLEDGMENTS
We thank Ryo Kanehisa and Yuki Naka, Wakayama Medical University, for supporting the experiments.
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (16K19143).
We declare no conflicts of interest.
REFERENCES
- 1.Lamb RA, Parks GD. 2013. Paramyxoviridae: the viruses and their replication, p 957–995. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Nishio M, Tsurudome M, Ito M, Kawano M, Kusagawa S, Komada H, Ito Y. 1999. Mapping of domains on the human parainfluenza virus type 2 nucleocapsid protein (NP) required for NP-phosphoprotein or NP-NP interaction. J Gen Virol 80:2017–2022. doi: 10.1099/0022-1317-80-8-2017. [DOI] [PubMed] [Google Scholar]
- 3.Nishio M, Tsurudome M, Ito M, Ito Y. 2000. Mapping of domains on the human parainfluenza type 2 virus P and NP proteins that are involved in the interaction with the L protein. Virology 273:241–247. doi: 10.1006/viro.2000.0429. [DOI] [PubMed] [Google Scholar]
- 4.Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagné N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eléouët J-F, Rey FA. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283. doi: 10.1126/science.1177634. [DOI] [PubMed] [Google Scholar]
- 5.Green TJ, Zhang X, Wertz GW, Luo M. 2006. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 313:357–360. doi: 10.1126/science.1126953. [DOI] [PubMed] [Google Scholar]
- 6.Albertini AA, Wernimont AK, Muziol T, Ravelli RBG, Clapier CR, Schoehn G, Weissenhorn W, Ruigrok RWH. 2006. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 313:360–363. doi: 10.1126/science.1125280. [DOI] [PubMed] [Google Scholar]
- 7.Luo M, Green TJ, Zhang X, Tsao J, Qiu S. 2007. Conserved characteristics of the rhabdovirus nucleoprotein. Virus Res 129:246–251. doi: 10.1016/j.virusres.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolakofsky D, Pelet T, Garcin D, Hausmann S, Curran J, Roux L. 1998. Paramyxovirus RNA synthesis and the requirement for hexamer genome length: the rule of six revisited. J Virol 72:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolakofsky D, Roux L, Garcin D, Ruigrok RWH. 2005. Paramyxovirus mRNA editing, the “rule of six” and error catastrophe: A hypothesis. J Gen Virol 86:1869–1877. doi: 10.1099/vir.0.80986-0. [DOI] [PubMed] [Google Scholar]
- 10.Alayyoubi M, Leser GP, Kors CA, Lamb RA. 2015. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein–RNA complex. Proc Natl Acad Sci U S A 112:E1792–E1799. doi: 10.1073/pnas.1503941112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol 283:61–119. [DOI] [PubMed] [Google Scholar]
- 12.Kolakofsky D. 2016. Paramyxovirus RNA synthesis, mRNA editing, and genome hexamer phase: a review. Virology 498:94–98. doi: 10.1016/j.virol.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Tapparel C, Hausmann S, Pelet T, Curran J, Kolakofsky D, Roux L. 1997. Inhibition of Sendai virus genome replication due to promoter-increased selectivity: a possible role for the accessory C proteins. J Virol 71:9588–9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsumoto Y, Ohta K, Goto H, Nishio M. 2016. Parainfluenza virus chimeric mini-replicons indicate a novel regulatory element in the leader promoter. J Gen Virol 97:1520–1530. doi: 10.1099/jgv.0.000479. [DOI] [PubMed] [Google Scholar]
- 15.Murphy SK, Ito Y, Parks GD. 1998. A functional antigenomic promoter for the paramyxovirus simian virus 5 requires proper spacing between an essential internal segment and the 3′ terminus. J Virol 72:10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy SK, Parks GD. 1999. RNA replication for the paramyxovirus simian virus 5 requires an internal repeated (CGNNNN) sequence motif. J Virol 73:805–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tapparel C, Maurice D, Roux L. 1998. The activity of Sendai virus genomic and antigenomic promoters requires a second element past the leader template regions: a motif (GNNNNN)3 is essential for replication. J Virol 72:3117–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffman MA, Banerjee AK. 2000. Precise mapping of the replication and transcription promoters of human parainfluenza virus type 3. Virology 269:201–211. doi: 10.1006/viro.2000.0223. [DOI] [PubMed] [Google Scholar]
- 19.Ruigrok RWH, Crepin T, Kolakofsky D. 2011. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr Opin Microbiol 14:504–510. doi: 10.1016/j.mib.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Rassa JC, Wilson GM, Brewer G a, Parks GD. 2000. Spacing constraints on reinitiation of paramyxovirus transcription: the gene end U tract acts as a spacer to separate gene end from gene start sites. Virology 274:438–449. doi: 10.1006/viro.2000.0494. [DOI] [PubMed] [Google Scholar]
- 21.Keller MA, Murphy SK, Parks GD. 2001. RNA replication from the simian virus 5 antigenomic promoter requires three sequence-dependent elements separated by sequence-independent spacer regions. J Virol 75:3993–3998. doi: 10.1128/JVI.75.8.3993-3998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reguera J, Gerlach P, Cusack S. 2016. Towards a structural understanding of RNA synthesis by negative strand RNA viral polymerases. Curr Opin Struct Biol 36:75–84. doi: 10.1016/j.sbi.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Tekes G, Rahmeh AA, Whelan SP.. 2011. A freeze frame view of vesicular stomatitis virus transcription defines a minimal length of RNA for 5′ processing. Plos Pathog 7:e1002073. doi: 10.1371/journal.ppat.1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogino T, Banerjee AK. 2011. An unconventional pathway of mRNA cap formation by vesiculoviruses. Virus Res 162:100–109. doi: 10.1016/j.virusres.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rahmeh AA, Schenk AD, Danek EI, Kranzusch PJ, Liang B, Walz T, Whelan SP.. 2010. Molecular architecture of the vesicular stomatitis virus RNA polymerase. Proc Natl Acad Sci U S A 107:20075–20080. doi: 10.1073/pnas.1013559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang B, Li Z, Jenni S, Rahmeh AA, Morin BM, Grant T, Grigorieff N, Harrison SC, Whelan SP.. 2015. Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell 162:314–327. doi: 10.1016/j.cell.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thierry E, Guilligay D, Kosinski J, Bock T, Gaudon S, Round A, Pflug A, Hengrung Naren Omari Kamel EI, Florence B, Hart DJ, Beck M, Cusack S. 2016. Influenza polymerase can adopt an alternative configuration involving a radical repacking of PB2 domains. Mol Cell 61:125–137. doi: 10.1016/j.molcel.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerlach P, Malet H, Cusack S, Reguera J. 2015. Structural insights into bunyavirus replication and its regulation by the vRNA promoter. Cell 161:1267–1279. doi: 10.1016/j.cell.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buchholz UJ, Finke S, Conzelmann K-K. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishio M, Tsurudome M, Ito M, Watanabe N, Kawano M, Komada H, Ito Y. 1997. Human parainfluenza virus type 2 phosphoprotein: mapping of monoclonal antibody epitopes and location of the multimerization domain. J Gen Virol 78:1303–1308. doi: 10.1099/0022-1317-78-6-1303. [DOI] [PubMed] [Google Scholar]
- 31.Nishio M, Ohtsuka J, Tsurudome M, Nosaka T, Kolakofsky D. 2008. Human parainfluenza virus type 2 V protein inhibits genome replication by binding to the L protein: possible role in promoting viral fitness. J Virol 82:6130–6138. doi: 10.1128/JVI.02635-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishio M, Tsurudome M, Garcin D, Komada H, Ito M, Le Mercier P, Nosaka T, Kolakofsky D. 2011. Human parainfluenza virus type 2 L protein regions required for interaction with other viral proteins and mRNA capping. J Virol 85:725–732. doi: 10.1128/JVI.01226-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishio M, Tsurudome M, Ito M, Garcin D, Kolakofsky D, Ito Y. 2005. Identification of paramyxovirus V protein residues essential for STAT protein degradation and promotion of virus replication. J Virol 79:8591–8601. doi: 10.1128/JVI.79.13.8591-8601.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsumoto Y, Ohta K, Yumine N, Goto H, Nishio M. 2015. Identification of two essential aspartates for polymerase activity in parainfluenza virus L protein by a minireplicon system expressing secretory luciferase. Microbiol Immunol 59:676–683. doi: 10.1111/1348-0421.12329. [DOI] [PubMed] [Google Scholar]