

ABSTRACT
The human immunodeficiency virus type 1 (HIV-1) capsid protein is an attractive therapeutic target, owing to its multifunctionality in virus replication and the high fitness cost of amino acid substitutions in capsids to HIV-1 infectivity. To date, small-molecule inhibitors have been identified that inhibit HIV-1 capsid assembly and/or impair its function in target cells. Here, we describe the mechanism of action of the previously reported capsid-targeting HIV-1 inhibitor, Boehringer-Ingelheim compound 1 (C1). We show that C1 acts during HIV-1 maturation to prevent assembly of a mature viral capsid. However, unlike the maturation inhibitor bevirimat, C1 did not significantly affect the kinetics or fidelity of Gag processing. HIV-1 particles produced in the presence of C1 contained unstable capsids that lacked associated electron density and exhibited impairments in early postentry stages of infection, most notably reverse transcription. C1 inhibited assembly of recombinant HIV-1 CA in vitro and induced aberrant cross-links in mutant HIV-1 particles capable of spontaneous intersubunit disulfide bonds at the interhexamer interface in the capsid lattice. Resistance to C1 was conferred by a single amino acid substitution within the compound-binding site in the N-terminal domain of the CA protein. Our results demonstrate that the binding site for C1 represents a new pharmacological vulnerability in the capsid assembly stage of the HIV-1 life cycle.
IMPORTANCE The HIV-1 capsid protein is an attractive but unexploited target for clinical drug development. Prior studies have identified HIV-1 capsid-targeting compounds that display different mechanisms of action, which in part reflects the requirement for capsid function at both the efferent and afferent phases of viral replication. Here, we show that one such compound, compound 1, interferes with assembly of the conical viral capsid during virion maturation and results in perturbations at a specific protein-protein interface in the capsid lattice. We also identify and characterize a mutation in the capsid protein that confers resistance to the inhibitor. This study reveals a novel mechanism by which a capsid-targeting small molecule can inhibit HIV-1 replication.
KEYWORDS: HIV-1, capsid, inhibitor, maturation
INTRODUCTION
The human immunodeficiency virus type 1 (HIV-1) infects and kills CD4+ T cells. Left untreated, patients with HIV-1 infection normally exhibit CD4 decline and immunodeficiency, eventually resulting in AIDS. In the past 20 years, the development and application of novel antiviral therapies for HIV-1 infection has resulted in long-term pharmacologic control of virus replication in patients. As a result, HIV-1 infection can be managed effectively as a chronic disease (1). Through the judicious administration of combinations of drugs against multiple viral and cellular targets, the emergence of drug resistance can be minimized. Nonetheless, poor adherence to therapy can compromise therapeutic effectiveness and result in antiviral resistance. Therefore, the development of effective inhibitors against new drug targets would help preserve therapeutic options for patients harboring drug-resistant HIV-1.
The HIV-1 capsid represents an attractive yet clinically unexploited target. Within the virion, the viral capsid is the protein shell that encases the viral ribonucleoprotein complex, which consists of genomic RNA, nucleocapsid protein, reverse transcriptase (RT), and integrase. Being a lentivirus, the HIV-1 capsid is conical in shape and consists of a polymer of a single viral protein, the capsid protein (CA). HIV-1 assembles as an immature particle, wherein CA is released from the Gag polyprotein upon proteolytic cleavage during particle maturation and self-assembles to form the closed, conical structure (2). CA consists of two independently folded domains, the N-terminal domain (NTD) and the C-terminal domain (CTD), which are connected by a flexible linker (1). The capsid consists of a lattice of approximately 1,500 CA subunits arranged in hexamers, into which are inserted 12 CA pentamers that allow for curvature and closure of the capsid ends (3). Hexamers are stabilized by intersubunit interactions between CA NTDs and between NTDs and CTDs of adjacent subunits (4, 5). The lattice of hexamers is further stabilized by 3-fold CTD-CTD interactions between subunits in neighboring hexamers (6, 7).
The capsid plays a crucial role in early steps of HIV-1 infection, including reverse transcription, nuclear entry, and integration. Most amino acid substitutions in CA that perturb capsid structure and/or stability are deleterious to infection (8–16). Several host factors are known to interact with the HIV-1 capsid, and amino acid substitutions or chemical inhibitors that perturb these interactions can inhibit infection (17–22). For example, the small-molecule HIV-1 inhibitors PF74 and BI-2 bind to a pocket between the NTD and CTD and prevent interactions of the viral capsid with the host proteins CPSF6 and Nup153 (19, 23–28). Additionally, compounds and peptides that prevent formation of the mature, functional viral capsid can inhibit HIV-1 replication. Examples include CAP-1 and NYAD-1, among others (29–31). Unfortunately, none of these inhibitors has advanced to clinical studies. Nonetheless, owing to the high sequence conservation of CA and its high sensitivity to mutagenesis, it is thought that antivirals targeting the viral capsid could become relatively durable therapies (16, 32).
To further understand the vulnerability of HIV-1 CA to small-molecule inhibition, we studied the mechanism of action of compound 1 (C1), a benzimidazole-based HIV-1 inhibitor previously described by researchers from Boehringer-Ingelheim Ltd. (33, 34). C1 was reported to prevent HIV-1 replication and to facilitate crystallization of purified CANTD for structural analysis. C1 binds to a region of the CA NTD near the exposed loop to which the host protein cyclophilin A (CypA) binds (34). In the present report, we show that C1 acts during HIV-1 maturation to prevent proper HIV-1 capsid assembly. A mutation in the C1 binding site of CA conferred resistance to the inhibitor, demonstrating that the antiviral target is CA.
RESULTS
C1 acts during HIV-1 assembly to inhibit viral infectivity.
In a previous study, Goudreau and coworkers reported that C1 inhibits HIV-1 replication with an 50% effective concentration (EC50) of 6.1 μM and showed that the compound binds to CA and inhibits CA assembly in vitro (33). In principle, CA-binding small-molecule HIV-1 inhibitors may act at both early and late stages of the virus replication cycle. To better define the mechanism of action of C1, we performed experiments to determine whether the compound acts during HIV-1 assembly or when present during the early stages of virus infection. HIV-1 particles capable of expressing the green fluorescent protein (GFP) reporter gene were generated by plasmid DNA transfection of cells cultured in the presence or absence of C1, and the viruses were harvested and tested for infectivity on various target cell types in the absence of additional inhibitor. Generation of HIV-1 in the presence of 50 μM C1 did not affect virus production as determined by quantification of viral p24 antigen or RT activity (Fig. 1A and B). However, the resulting particles were markedly impaired for infectivity relative to control (Fig. 1A). Varying the concentration of C1 in HEK293T producer cells in dose-response experiments yielded an EC50 of approximately 20 μM (Fig. 1C), in reasonable agreement with the previously reported antiviral potency. Importantly, C1 did not exhibit marked cytotoxic effects at concentrations of up to 100 μM (Fig. 1D), consistent with the lack of an effect of the compound on the levels of virus production.
FIG 1.

Effects of compound 1 on HIV-1 production and infectivity. (A) VSV-G-pseudotyped GFP reporter virus produced in the presence of 50 μM C1 was quantified by p24 ELISA, and infection was assayed by virus titration on TZM-bl cells. Results are normalized to virus produced in the presence of DMSO vehicle control. (B) Results of exogenous RT activity in disrupted virions normalized to CA content. Black and gray bars are results of duplicate assays performed at the indicated C1 concentration. (C) Viruses made in the presence of the indicated C1 concentration were assayed for infection of HOS and CEM-SS cells by flow cytometry for GFP expression. (D) The indicated cell lines were cultured in the presence of the indicated concentrations of C1 for 48 h, and cell proliferation was determined by MTT assay. Values were normalized to those obtained with the cultures containing 0.1 μM C1. (E) The infectivity of HIV-1 and SIV produced in the presence or absence of 20 μM C1 was assayed in TZM-bl cells. Values were normalized to the levels of RT activity present in each virus stock. SIVmac is SIVmac239, and HIVm2 is an HIV-1 mutant containing substitutions conferring resistance to BVM. (F) Effect of C1 removal on HIV-1 infectivity. HIV-1 was produced in the absence (control and C1 added after transfection) or presence (C1 added during virus production) of 40 μM C1, and infectivity was assayed after three rounds of ultrafiltration through a 100-kDa-molecular-mass cutoff filter to remove unbound compound. For the added-after-transfection sample, the virus was incubated in the presence of 40 μM C1 for 1 h prior to ultrafiltration. Infection values shown are relative to the control sample. Results shown in panels B, C, and D are representative of two independent experiments. Error bars in panels A, E, and F depict the range in values obtained from two independent experiments.
We next probed the specificity of C1 antiviral activity by asking whether it would inhibit the infection of functionally related viruses. In contrast to the results obtained with HIV-1, production of SIVmac239 virions in the presence of C1 only modestly reduced virus infectivity (Fig. 1E). Because the observed producer cell infectivity inhibition effect is reminiscent of the action of the previously described HIV-1 maturation inhibitor bevirimat (BVM/PA-457/DSB; BVM) (35, 36), we also tested whether a BVM-resistant HIV-1 mutant (HIVm2) is sensitive to C1. The infectivity of HIVm2 was markedly reduced by C1 (Fig. 1E), suggesting that C1 and BVM act on distinct molecular targets.
To determine whether C1 might also act during the early stage of HIV-1 infection, we used untreated virus to infect cells that were either treated with C1 or left untreated. C1 did not inhibit viral infectivity at concentrations of up to 64 μM under these conditions (data not shown). We also tested whether the reduced infectivity of HIV-1 particles generated in the presence of 40 μM C1 would be restored by removing the compound from the virus sample. HIV-1 particles remained impaired for infectivity despite three rounds of ultrafiltration (Fig. 1F). Moreover, the treatment of cell-free virus supernatant with C1 did not deleteriously affect virus infection (Fig. 1F). We accordingly conclude that C1 must be present during virus production to inhibit infectivity and that the compound acts irreversibly during HIV-1 particle formation.
HIV-1 particles produced in the presence of C1 exhibit impaired reverse transcription in target cells.
The reporter-based assay of HIV-1 infectivity requires completion of all of the early stages of infection, including entry, reverse transcription, uncoating, nuclear entry, and integration, as well as reporter gene transcription and translation. We sought to localize the infectivity defect induced by C1 to one or more of these stages. HIV-1 entry was quantitatively assessed using the BlaM-Vpr reporter fusion assay, which depends on delivery of a virion-associated β-lactamase protein reporter into the cytoplasm of target cells. HIV-1 reporter particles produced in the presence of C1 exhibited fusion signals similar to that of the control virions, indicating that the fusion of the C1-produced virions is not substantially impaired (Fig. 2A). To examine reverse transcription, we used quantitative PCR to measure the accumulation of early (minus-strand strong stop) and late (second-strand transfer) HIV-1 DNA species. HIV-1 particles produced in the presence of 40 μM C1 were markedly impaired for both stages of reverse transcription (Fig. 2B and C). Because C1-produced HIV-1 particles exhibited normal levels of RT enzyme activity (Fig. 1B), this result suggested that C1 induces a defect in the viral core, leading to abortive reverse transcription in target cells.
FIG 2.
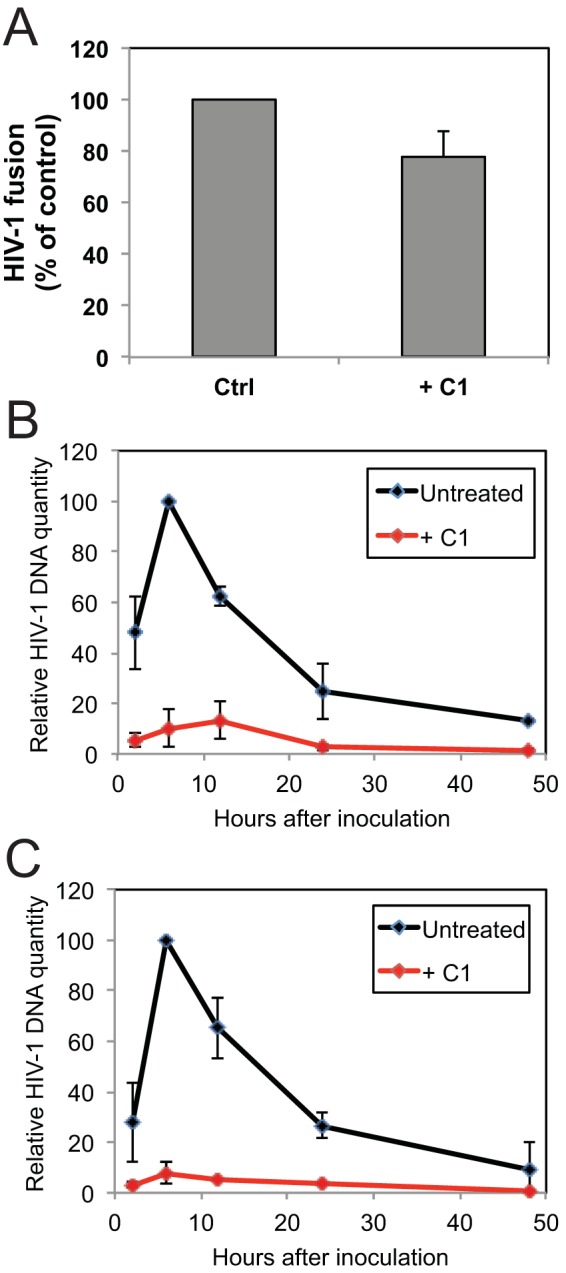
HIV-1 particles produced in the presence of C1 are impaired at an early postentry stage of infection. (A) HIV-1 particles containing BlaM-Vpr were produced in the presence or absence of 40 μM C1. Fusion with SupT1 target cells was quantified by flow cytometry by determining the number of cells in which CCF2-AM was cleaved by BlaM. Results shown are from two independent experiments, with error bars depicting the range in values. (B and C) Quantification of HIV-1 reverse transcription in SupT1 cells following inoculation with HIV-1 produced in the presence or absence of C1 (40 μM). Panels B and C show early and late reverse transcripts, respectively. Results shown are averages from three independent experiments, with error bars representing standard deviations.
C1 perturbs normal HIV-1 capsid assembly.
HIV-1 CA mutant particles that contain structurally aberrant cores frequently exhibit early postentry defects in infection. Therefore, we sought to identify structural and biochemical defects associated with C1-produced particles. Thin-section electron microscopic (EM) analysis of particles produced from cells treated with the dimethyl sulfoxide (DMSO) vehicle control revealed that ∼90% harbored cores with associated electron density from the viral ribonucleoprotein complex (37). Minor populations under these conditions included eccentric particles with electron density situated between the viral membrane and an often-seen electron-lucent core, as well as immature particles with a characteristic toroidal outer ring (Fig. 3). C1 treatment dramatically increased the population of eccentric particles, a pattern that was similar to that of the allosteric integrase inhibitor (ALLINI) BI-D, which was used as a control inhibitor (Fig. 3) (38). However, immunoblot analysis of pelleted particles produced in the presence of C1 revealed no major alterations of viral structural proteins, including CA, gp41, and gp120, although a slight reduction in the accumulation of the precursor Pr55gag protein was detected (Fig. 4A). Similarly, kinetic analysis of viral protein processing by pulse-chase labeling and immunoprecipitation failed to reveal a substantial effect of C1 on the kinetics of Pr55gag proteolysis (Fig. 4B), although a subtle delay in the accumulation of processed and unprocessed Gag proteins was observed in C1-treated cells. A slight delay in CA-SP1 cleavage in viral supernatants was also noted, reminiscent of the effect of the maturation inhibitor BVM on cleavage at this junction (39). However, the C1-induced delay was much less pronounced, and, unlike BVM, CA-SP1 failed to accumulate in particles produced in the presence of C1. The apparently slower CA-SP1 cleavage in particles released from C1-treated cells may also be related to the slower accumulation of particles into the supernatant. We conclude that C1 induces only subtle effects on viral Gag protein expression and processing.
FIG 3.

Electron microscopic analysis of the effects of C1 on the morphology of the HIV-1 core. HIV-1 particles were produced in the presence or absence of C1 (40 μM) and analyzed by thin-section electron microscopy. Representative images of mature, eccentric, and immature particles are shown in the two left, middle, and right pairs of images, respectively. Fractions of mature, immature, and eccentric particles were quantified and are shown in the right panel. Results are percentages normalized for the different morphologies from counting minimally 100 particles per experiment; error bars show the range in values across two independent experiments. The ALLINI BI-D (0.18 μM) was included in the experiment as a known compound that perturbs HIV-1 maturation (38).
FIG 4.
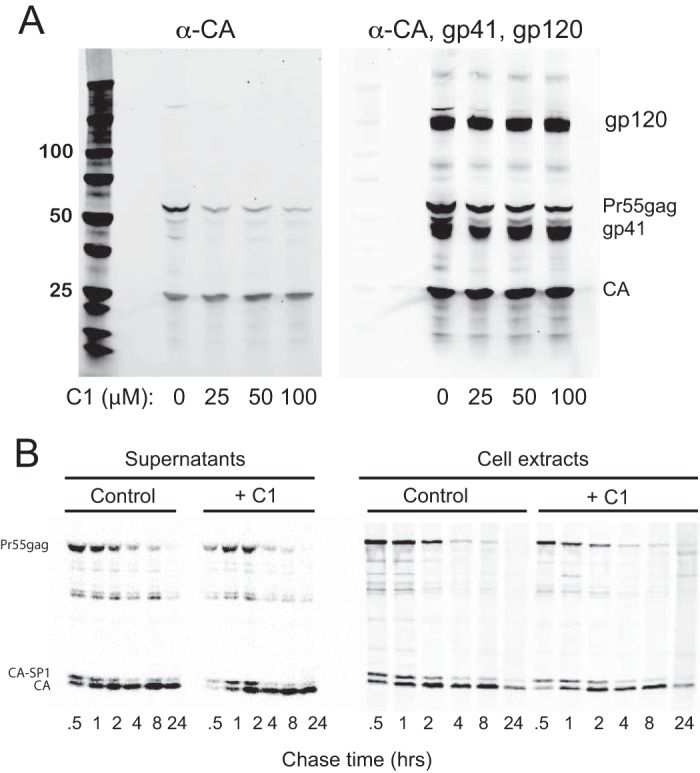
Compound 1 does not markedly affect processing of Pr55Gag during particle maturation. (A) Immunoblot analysis of HIV-1 particles produced in the presence of the indicated concentrations of C1. Pelleted particles were lysed and fractionated by SDS-PAGE, and proteins were detected by immunoblotting using a monoclonal antibody to HIV-1 CA (left) and a mixture of anti-CA and anti-Env (gp41 + gp120) antibodies. (B) Pulse-chase analysis of HIV-1 processing during particle maturation. HEK293T cells producing HIV-1 particles were cultured in the presence or absence of C1 (100 μM) and radiolabeled with [35S]methionine plus cysteine for 30 min, followed by culture in the presence of unlabeled medium. Supernatants and cells were collected at the indicated times of chase, and protein extracts were immunoprecipitated using a CA-specific monoclonal antibody and separated by SDS-PAGE. Bands were detected by phosphorimager analysis. The images shown in panels A and B are representative of results obtained from two and three independent experiments, respectively.
We also assayed the relative levels of viral genomic RNA incorporation in control and C1-produced virions. Viral RNA was extracted and subjected to cDNA synthesis, followed by quantitative PCR (qPCR) for HIV-1 cDNA. No significant alteration in relative viral RNA signal was observed in triplicate determinations (Fig. 5). We conclude that C1 does not affect the encapsidation of viral RNA during HIV-1 assembly.
FIG 5.
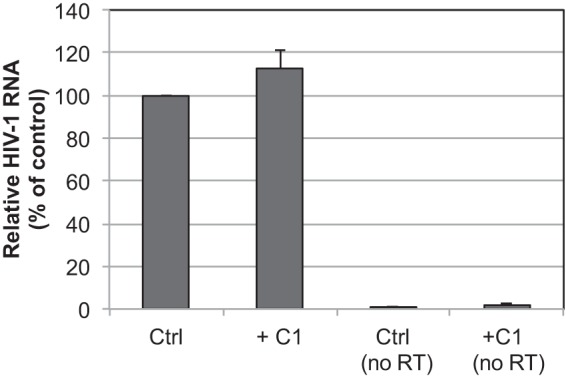
C1 does not inhibit HIV-1 particle encapsidation of viral RNA. RNA was extracted from DNase-treated HIV-1 particles produced in the presence or absence of C1 (40 μM) and analyzed by quantitative RT-PCR using primers specific for HIV-1 sequences. As controls, the RNA samples were subjected to PCR analysis without prior reverse transcription (no RT). Results shown are averages from three independent experiments, with error bars representing standard deviations.
Because the defect in reverse transcription of C1-produced particles is reminiscent of HIV-1 CA mutants containing unstable capsids, we sought to determine whether C1 affects capsid stability. To this end, we isolated core structures by a detergent spin-through centrifugation approach we developed to study capsid stability, and we quantified the levels of core-associated CA in the dense fractions of the gradient by p24 enzyme-linked immunosorbent assay (ELISA) (40, 41). Relative to untreated virions, which yielded approximately 11% of viral CA in the fractions corresponding to HIV-1 cores, C1-produced particles exhibited markedly lower levels of core-associated CA (∼1%) (Fig. 6). These results indicate that HIV-1 particles produced in the presence of C1 contain malformed and/or unstable cores. Notably, the vast majority of eccentric particles produced in the presence of ALLINIs harbored malformed capsids (37). Our interpretation is also consistent with the previous report that C1 inhibits CA assembly in vitro (33).
FIG 6.
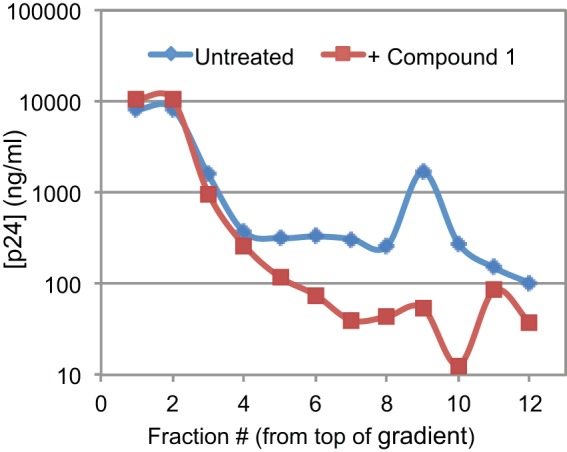
HIV-1 particles produced in the presence of C1 contain unstable capsids. HIV-1 particles were concentrated by ultracentrifugation and resuspended, and cores were released by ultracentrifugation of the virions through a layer of nonionic detergent into a sucrose density gradient. Shown is the level of CA present in each fraction, as quantified by p24 ELISA. The results shown are representative of three independent experiments. The fraction 10 dip in CA content was not observed in the other experiments.
Substitution of Thr at Arg132 confers resistance to C1.
Lemke and coworkers determined by X-ray crystallography that C1 binds to a novel binding pocket in the CA NTD and is contacted by several protein side chains, including Arg132, which forms a salt bridge with the compound (34). We previously showed that substitution of Thr for Arg132 can rescue the replication defect associated with the biochemically hyperstable CA mutant E45A (42) and speculated that the R132T substitution would be tolerated by the virus and confer resistance to C1. Indeed, we observed that HIV-1 containing the R132T substitution replicated in the presence of C1 concentrations of up to 100 μM, whereas as little as 25 μM C1 effectively inhibited replication of wild-type HIV-1 (Fig. 7A). Similarly, the infectivity of HIV-1 particles containing the R132T substitution was unaffected when the virus was produced in cell cultures containing concentrations of C1 as high as 100 μM (Fig. 7B). The R132T change accordingly conferred resistance to the morphological defect in virus core formation elicited by C1 treatment (Fig. 7C).
FIG 7.

R132T substitution in CA renders HIV-1 resistant to C1. (A) Cultures of CEM-SS cells were inoculated with HIV-1 (top) and HIV-1.CA.R132T (bottom), and cells were cultured in the presence of the indicated concentrations of C1. HIV-1 replication was monitored by quantifying p24 in the culture supernatants; the results are representative of those observed in two independent experiments. (B) Wild-type and R132T CA mutant GFP reporter viruses were produced in the presence and absence of C1 and assayed for infectivity by flow cytometry for GFP expression following infection of target cells in the absence of added inhibitor. Shown are the mean values from two independent experiments. (C) Quantification of wild-type and R132T mutant particle morphologies in the presence of DMSO or 40 μM C1 (means ± standard deviations for n = 2 experiments, with 65 to 424 particles counted per experiment). dpi, days postinfection.
C1 alters engineered disulfide cross-linking at the 3-fold interhexamer interface in the HIV-1 capsid.
To probe the capsid assembly defect induced by C1, we analyzed the effects of the compound on formation of engineered disulfide cross-links that can form spontaneously during HIV-1 capsid assembly. HIV-1 particles bearing paired Cys substitutions at positions 14 and 45, 68 and 212, 204 (single substitution), and 207 and 216 result in disulfide bonds forming between adjacent NTD and NTD (intrahexameric), NTD and CTD (intrahexameric), CTD and CTD (interhexameric), and CTD and CTD (interhexameric), respectively (6, 7, 15). The corresponding mutant viral particles were collected from transfected cells cultured in the presence or absence of C1 and pelleted, and proteins were separated by nonreducing SDS-PAGE and immunoblotted to detect the pattern of CA cross-linking. Particles produced in the presence of C1 exhibited normal CA banding patterns for the 14C/45C and 68C/212C mutants, which form cross-links (hexamer ladder) within CA hexamers (Fig. 8). In contrast, C1 treatment of the 204C and 207C/216C mutants, which cross-link at the 3-fold interhexamer interface, resulted in additional high-molecular-weight cross-linked CA forms (marked by asterisks in the figure). The corresponding mutant particles additionally containing the R132T substitution, which confers resistance to the antiviral action of C1, did not exhibit the higher cross-linked forms. These results suggest that the compound alters capsid assembly, leading to spurious cross-linked forms, and they link the altered cross-linking effect of the compound to its antiviral activity.
FIG 8.
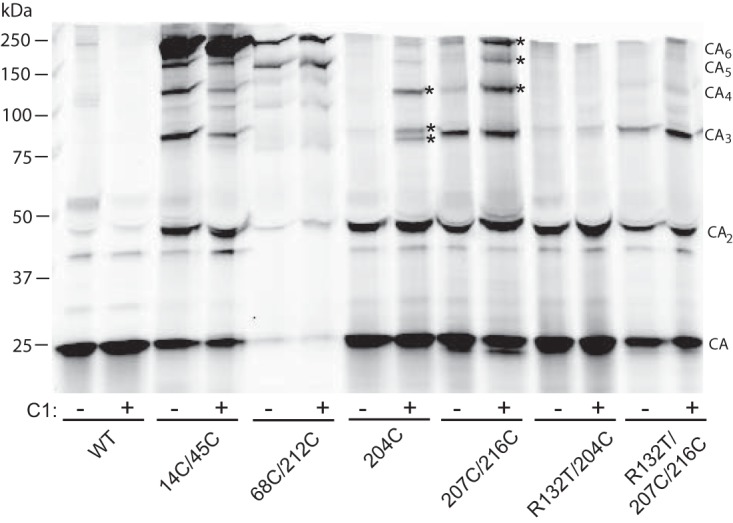
C1 induces aberrant cross-links in mutant particles capable of forming CA-CA disulfide bonds at the interhexamer interface. HIV-1 particles were produced by transfection of HIV-1 proviral constructs encoding the indicated Cys substitutions and cultured in the presence or absence of C1 (100 μM). Culture supernatants were harvested 2 days after transfection, and virions were pelleted and analyzed by nonreducing SDS-PAGE. The gel was immunoblotted with a probe for CA. Shown are representative results from one of three independent experiments. Asterisks indicate high-molecular-mass species that either formed or were enhanced by the presence of C1 and were dependent on Arg132 in CA.
DISCUSSION
Small molecules targeting the HIV-1 capsid have received much interest of late, as CA is an attractive therapeutic target (16, 32). Moreover, such compounds are useful as structural probes to answer questions regarding HIV-1 capsid function. CA-targeting compounds can function at both early and late stages of replication, owing to the involvement of CA surfaces in assembly, maturation, and postentry stages of replication, including reverse transcription and nuclear entry.
In the present study, we demonstrate that the small-molecule HIV-1 replication inhibitor C1 acts at a late stage of HIV-1 replication by specifically targeting CA. Previous studies had shown that C1 binds to CA and inhibits replication and had reported an X-ray crystal structure of C1 bound to the NTD of CA. In the structure, a carboxylate of C1 forms a salt bridge with Arg132 in CA. We observed that substitution of Thr for Arg132 rendered HIV-1 resistant to the antiviral effect of C1, thus identifying CA as the antiviral target. Thus, our studies support the functional relevance of the reported structure of the C1-CANTD complex. While we have not observed any deleterious effect of the R132T substitution on the infectivity of HIV-1 particles produced in the absence of inhibitors, the mutant virus exhibited a 3- to 4-day lag in replication in a T cell line (Fig. 7A). Among HIV-1 proteins, CA is relatively highly conserved and Arg132 is nearly invariant, being substituted for only by Lys, thus preserving the positive charge (43). Thus, while a single codon change (Arg to Thr) at this position is sufficient to confer resistance to C1, the high degree of conservation across circulating strains suggests that the Arg-to-Thr change is deleterious to HIV-1 fitness in vivo.
We also observed that C1 perturbs HIV-1 capsid assembly. Lemke and colleagues (34) previously showed that treatment of virus-producing cells with the benzimidazole inhibitor BM3 increased the percentage of eccentric particles, similar to our observation here with the chemically related C1 compound. We further showed that such aberrant cores are biochemically unstable and unable to promote reverse transcription in target cells, consistent with the phenotype of CA mutant viruses containing defective or unstable capsids. Although the present EM analysis lacked the sensitivity of the more in-depth cryoelectron tomography study that previously highlighted the formation of malformed capsids by ALLINIs (37), we nevertheless speculate that C1 treatment yielded similar defects. This interpretation is consistent with the biochemically unstable nature of HIV-1 cores that were formed in the presence of C1 (Fig. 6). While the effect of C1 on core morphology we observed was not absolute, it seems plausible that the morphological effects remain functionally related to the antiviral activity, since a CA substitution that rendered HIV-1 resistant to the antiviral activity also in large part prevented eccentric particle formation (Fig. 7C). C1 was originally identified in a screen for small molecules that inhibit the spontaneous assembly of HIV-1 CA in vitro (33), but inhibition of CA assembly was not clearly linked to its antiviral activity. Our results suggest that the assembly-inhibiting activity of C1 observed in vitro is also related to its antiviral activity.
We also observed that C1 induced aberrant CA-CA cross-links in particles containing engineered capsid intersubunit disulfide bonds. The aberrant cross-linking was observed with engineered disulfides at the interhexamer interface. This result is noteworthy, as interhexamer contacts are formed primarily between CA CTDs, while C1 binds the NTD. We postulate that the binding of the compound to the CA NTD allosterically modulates CTD orientation, thus preventing proper interactions between hexamers. This effect may be subtle, as the compound did not substantially inhibit the formation of the engineered disulfide cross-links at any of the intersubunit interfaces. It is also possible that C1 affects formation of the CA pentamers that are necessary for mature capsid formation or perturbs the interactions between pentamers and neighboring hexamers. Our new experimental approach involving analysis of intersubunit cross-links should prove useful in mechanistic analysis of other inhibitors that act during HIV-1 assembly and maturation.
The antiviral mechanism elucidated here contrasts with other known small-molecule CA-targeting HIV-1 inhibitors. Like the HIV-1 maturation inhibitor BVM and the ALLINI BI-D, C1 must be present during HIV-1 assembly and maturation to exhibit antiviral activity. However, BVM antiviral activity has been associated with a specific delay in cleavage of the CA-SP1 junction within the Gag precursor, thus resulting in accumulation of uncleaved CA-SP1 (35, 36). BVM binds specifically to immature HIV-1 particles, and resistance mutations occur in positions near and at the CA-SP1 junction (39, 44, 45). BVM also stabilizes the immature Gag lattice, an effect that may be either a cause or a consequence of delayed CA-SP1 cleavage (46). ALLINIs, in contrast, do not appear to alter capsid subunit interactions but act by disrupting IN-RNA interactions that are necessary for proper localization of the viral genomic ribonucleoprotein complex within the capsid (37, 47). At present, we have not excluded the possibility that C1 affects the intravirion localization of the ribonucleoprotein complex similarly to ALLINIs. Although the available evidence argues strongly that these compounds target different viral proteins (CA for C1 versus IN for ALLINIs), the effects of the compounds could be functionally related if an interaction between IN and CA is involved in maturation. The small-molecule inhibitor PF74 binds to the NTD-CTD interface within the hexamer and can act at both early and late stages in replication (19, 24, 48). PF74 can destabilize the viral capsid, but its major antiviral effect occurs between reverse transcription and integration (19, 27, 49). The potency of PF74 antiviral activity depends on expression of host cell factors CypA, Nup153, and TNPO3 in target cells (23, 28, 49, 50), and the PF74-binding pocket represents a binding site for host factor interactions, suggesting that the inhibitor's major effect is to alter virus-host cell interactions (19, 24).
While none of the currently reported CA-targeting compounds exhibits sufficient potency for clinical development, the variety of apparent antiviral mechanisms associated with such compounds suggests the intriguing possibility that the inhibitors act synergistically, and high potency may be achieved by creating bifunctional CA-targeting compounds.
MATERIALS AND METHODS
Plasmid DNAs.
Replication-competent HIV-1 molecular clones included pR9 (51) and pNL4-3 (52). NL4-3-based single-round reporter viruses included pNLENG1-IRES (53), which expresses GFP, and pNLX.Luc.R- (54), which expresses firefly luciferase. The R132T change in the CA region of gag was introduced into molecular clones by site-directed mutagenesis, and the mutation as well as absence of unwanted secondary changes throughout plasmid DNA coding regions was verified by dideoxy sequencing. Plasmids pMM310 (55) and pHCMV-VSV-G (56) were used to express beta-lactamase (BlaM)-Vpr fusion protein and vesicular stomatitis virus glycoprotein (VSV-G), respectively. SIVmac239 and HIVm2 (encoding two substitutions at the C terminus of CA) viruses were generated by transfection of the pBR239E and pNL4-3m2 plasmids, respectively (39). Plasmid pBR239E, a full-length clone of SIMmac239 in pBR322, was generously provided by Toshiaki Kodama (Oregon Regional Primate Research Center).
Small molecules.
C1 was synthesized and purified at the Vanderbilt Institute for Chemical Biology Chemical Synthesis Core or at Nanosyn (Santa Clara, CA) according to the previously reported procedure (33). The ALLINI BI-D (57) was synthesized at Haoyuan Chemexpress Co., Ltd. (Shanghai, People's Republic of China). Efavirenz (EFV) was obtained from the NIH Research and Reference Reagents program.
Cells and viruses.
HEK293T, HOS, and TZM-bl cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented to contain fetal bovine serum (FBS; 10%, vol/vol), penicillin (100 IU/ml), and streptomycin (100 μg/ml). SupT1 and CEM-SS cells were propagated in similarly supplemented RPMI 1640 medium. Viruses used for infectivity measures and transmission electron microscopy were generated by plasmid DNA transfection of HEK293T cells as previously described (58).
To study the effects of C1 on nascent HIV-1, the vehicle control DMSO or C1 was maintained in medium throughout the transfection procedure. The medium was changed 16 h after transfection, and virus particle-containing supernatants collected 48 h thereafter were clarified at 300 × g for 15 min, filtered through 0.45-μm filters (Nalgene), and either aliquoted and frozen at −80°C or concentrated by ultracentrifugation using an SW41Ti rotor at 24,000 rpm for 2 h at 4°C before freezing. Cell-free virus concentrations were determined using a commercial p24 ELISA kit (Advanced Biosciences Laboratories) or exogenous RT assay (59).
Assays of HIV-1 infectivity.
Approximately 20,000 HOS cells were seeded per well of a 24-well plate 1 day before infection. Cultures were inoculated with VSV-G-pseudotyped NLENG1-ES-IRES to achieve a multiplicity of infection in the range of 0.05 to 0.5. Where indicated, C1 or DMSO was added to target cells 1 h before infection. Percentages of GFP-positive cells were determined 48 h postinfection using a FACSCanto flow cytometer equipped with FACSDIVA software (BD Biosciences).
C1 activity against mature HIV-1 particles was analyzed after 1 h of incubation with drug or DMSO vector control at 37°C. The mixtures were subjected to three rounds of successive ultrafiltration in 100-kDa concentrators (Amicon) to remove the vast majority of unbound compound from the virus preparations prior to target cell infection.
Cell proliferation assay.
The effects of C1 on cell proliferation were measured with the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT)-based in vitro toxicology assay kit (TOX1; Sigma-Aldrich). HEK293T or TZM-bl cells were seeded in 96-well plates (6,000 cells per well in 100 μl complete medium). One day later, C1 was added at various final concentrations. Cultures contained a maximum DMSO concentration of 0.1% (vol/vol). Two days later, MTT solution (10 μl) was added to each well, and the plates were incubated for 2 h. After dissolution of the resulting MTT formazan crystals, the absorbance at 570 nm and 690 nm was measured, and the latter value was subtracted from the former value.
Capsid stability assay.
The intrinsic stability of HIV-1 cores was determined by quantifying the recovery of CA in dense fractions of a sucrose gradient following centrifugation of concentrated virus particles through a layer of 1% Triton X-100, as previously reported (40, 41). CA was quantified by p24 ELISA, and the results were reported as a percentage of the total CA detected within each gradient.
Quantification of HIV-1 reverse transcription in target cells.
DNA was extracted from HOS cells infected with DNase-treated (1 h at 37°C) VSV-G-pseudotyped NLENG1-ES-IRES virus using the DNeasy blood and tissue kit (Qiagen). Control infections were performed in the presence of 10 μM EFV to monitor residual plasmid after DNase treatment, and values obtained from control qPCR assays were subtracted from experimental samples. Duplicate PCR mixtures containing 25 ng DNA, 0.2 μM primers, 0.1 μM probe, and 1× QuantiTect probe PCR master mix were incubated at 50°C for 2 min and 95°C for 15 min, followed by 40 cycles of 94°C for 15 s, 58°C for 30 s, and 72°C for 30 s. Standards were prepared by endpoint diluting pNLENG1-ES-IRES plasmid in DNA from uninfected cells. Primers AE2963/AE4422 and probe AE2965 were used to detect late reverse transcripts, whereas primers AE989/AE990 with probe AE995 were used for early reverse transcripts (38).
Quantification of viral genomic RNA.
RNA was extracted from DNase-treated HIV-1 NL4-3 (250 ng p24) made in the presence of DMSO solvent control or 40 μM C1 using the viral RNA extraction kit (Qiagen). Triplicate PCR mixtures containing 25 ng RNA, 0.2 μM AE2963 and AE4422 primers, and 1× SYBR green master mix (Qiagen) with and without the supplied 1× RT mix were incubated at 50°C for 30 min and 95°C for 15 min, followed by 40 cycles of 96°C for 15 s, 55 to 90°C for 30 s, and 72°C for 30 s. To account for signal from plasmid DNA that potentially remained from transfection, values obtained in the absence of the RT mix were compared to those obtained in its presence.
Assay of HIV-1 particle fusion with target cells.
HIV-1 fusion with cells was quantified by the BlaM-Vpr assay essentially as previously described (38). SupT1 cells infected for 4 h at 37°C with 15 ng p24/ml of VSV-G-pseudotyped NLX.Luc.R- virus were washed and brought to room temperature before loading with the fluorogenic CCF2-AM substrate (Promega). BLaM activity was determined as the percentage of cells exhibiting blue fluorescence after 18 h by flow cytometry (BD Biosciences).
Pulse-chase kinetic assay of HIV-1 Gag processing.
HEK293T cells were transfected with the R9 molecular clone in 6-well dishes using the calcium phosphate method. Sixteen hours later, cells were washed to remove DNA precipitate, C1 (or DMSO as a solvent control) was added in complete medium to 100 μM, and cells were cultured for 1 h. Cells were then washed and cultured for 1 h in cysteine- and methionine-free DMEM with 2% dialyzed FBS. Proteins were metabolically labeled by addition of Tran-35S-label (MP Biomedicals) to 0.4 mCi/ml for 30 min. Labeling medium was removed, cultures were washed, and cells were redistributed into 24-well dishes and cultured in unlabeled medium for the indicated times. C1 was maintained at 100 μM in the treated cultures throughout the procedure. At various times during the chase period, the supernatants and cells were harvested. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, 0.15 M NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], pH 7.5), and HIV-1 proteins were immunoprecipitated with the CA-specific monoclonal antibody 183-H12-5C as previously described (39). Following SDS-PAGE, the proteins were fixed and the gels were soaked in Fluoro-Hance (Research Products International Corp.) and dried in a Bio-Rad vacuum gel dryer. Radioactive proteins were detected with a Bio-Rad phosphorimager.
Ultrastructural analysis of HIV-1 particles.
HIV-1 particles were generated by transfection of HEK293T cells with wild-type or R132T mutant pNL4-3. Viruses were pelleted at 30,000 rpm at 4°C for 1.5 h with a Beckman SW41 rotor. Pelleted virus was resuspended in a small volume of phosphate-buffered saline. An equal volume of 4% EM-grade paraformaldehyde (Electron Microscopy Sciences) was added, and samples were incubated at 4°C before embedding in Epon resin. Thin (75- to 100-nm) sections were applied to 200-mesh carbon-coated copper grids, stained with 0.2% lead citrate, and observed using a JEOL 1200EX microscope with an AMT 2k charge-coupled-device camera. Images were captured at 37,500× magnification and were visually inspected to classify viral particles as mature, eccentric, or immature.
ACKNOWLEDGMENTS
This work was supported by NIH grant P50 GM082251.
The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID: efavirenz (catalog number 4624) and HIV-1 p24 monoclonal antibody 183-H12-5C (catalog number 3537), from Bruce Chesebro and Kathy Wehrly. We thank David Levy for the generous gift of pNLENG1-IRES plasmid DNA. C1 was provided by the Vanderbilt Institute of Chemical Biology, Chemical Synthesis Core, Vanderbilt University, Nashville, Tennessee.
W.W., J.Z., A.N.E., and C.A. designed the study; W.W., J.Z., U.D.H., K.A.J., A.V.J., and Y.W. performed the experiments and interpreted the results; and W.W., A.N.E., and C.A. wrote the manuscript.
REFERENCES
- 1.Engelman A, Cherepanov P. 2012. The structural biology of HIV-1: mechanistic and therapeutic insights. Nat Rev Microbiol 10:279–290. doi: 10.1038/nrmicro2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sundquist WI, Krausslich HG. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2:a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283:80–83. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- 4.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganser-Pornillos BK, Cheng A, Yeager M. 2007. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131:70–79. doi: 10.1016/j.cell.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 6.Byeon IJ, Meng X, Jung J, Zhao G, Yang R, Ahn J, Shi J, Concel J, Aiken C, Zhang P, Gronenborn AM. 2009. Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139:780–790. doi: 10.1016/j.cell.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P. 2013. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Schwedler UK, Stemmler TL, Klishko VY, Li S, Albertine KH, Davis DR, Sundquist WI. 1998. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J 17:1555–1568. doi: 10.1093/emboj/17.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang S, Murakami T, Agresta BE, Campbell S, Freed EO, Levin JG. 2001. Human immunodeficiency virus type 1 N-terminal capsid mutants that exhibit aberrant core morphology and are blocked in initiation of reverse transcription in infected cells. J Virol 75:9357–9366. doi: 10.1128/JVI.75.19.9357-9366.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J Virol 77:5439–5450. doi: 10.1128/JVI.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scholz I, Arvidson B, Huseby D, Barklis E. 2005. Virus particle core defects caused by mutations in the human immunodeficiency virus capsid N-terminal domain. J Virol 79:1470–1479. doi: 10.1128/JVI.79.3.1470-1479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang S, Ablan S, Dueck M, Ayala-Lopez W, Soto B, Caplan M, Nagashima K, Hewlett IK, Freed EO, Levin JG. 2007. A second-site suppressor significantly improves the defective phenotype imposed by mutation of an aromatic residue in the N-terminal domain of the HIV-1 capsid protein. Virology 359:105–115. doi: 10.1016/j.virol.2006.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noviello CM, Lopez CS, Kukull B, McNett H, Still A, Eccles J, Sloan R, Barklis E. 2011. Second-site compensatory mutations of HIV-1 capsid mutations. J Virol 85:4730–4738. doi: 10.1128/JVI.00099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yufenyuy EL, Aiken C. 2013. The NTD-CTD intersubunit interface plays a critical role in assembly and stabilization of the HIV-1 capsid. Retrovirology 10:29. doi: 10.1186/1742-4690-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rihn SJ, Wilson SJ, Loman NJ, Alim M, Bakker SE, Bhella D, Gifford RJ, Rixon FJ, Bieniasz PD. 2013. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog 9:e1003461. doi: 10.1371/journal.ppat.1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, KewalRamani VN, Chin JW, Towers GJ, James LC. 2012. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog 8:e1002896. doi: 10.1371/journal.ppat.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, Halambage UD, Aiken C, James LC. 2014. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog 10:e1004459. doi: 10.1371/journal.ppat.1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. 1993. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73:1067–1078. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- 21.Franke EK, Yuan HE, Luban J. 1994. Specific incorporation of cyclophilin A into HIV-1 virions. Nature 372:359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- 22.Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh CT, Sodroski J, Gottlinger HG. 1994. Functional association of cyclophilin A with HIV-1 virions. Nature 372:363–365. doi: 10.1038/372363a0. [DOI] [PubMed] [Google Scholar]
- 23.Shi J, Zhou J, Shah VB, Aiken C, Whitby K. 2011. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J Virol 85:542–549. doi: 10.1128/JVI.01406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, Demeler B, Pornillos O, Ganser-Pornillos BK, Diaz-Griffero F, Ivanov DN, Yeager M. 2014. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci U S A 111:18625–18630. doi: 10.1073/pnas.1419945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fricke T, Buffone C, Opp S, Valle-Casuso J, Diaz-Griffero F. 2014. BI-2 destabilizes HIV-1 cores during infection and prevents binding of CPSF6 to the HIV-1 capsid. Retrovirology 11:120. doi: 10.1186/s12977-014-0120-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamorte L, Titolo S, Lemke CT, Goudreau N, Mercier JF, Wardrop E, Shah VB, von Schwedler UK, Langelier C, Banik SS, Aiken C, Sundquist WI, Mason SW. 2013. Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob Agents Chemother 57:4622–4631. doi: 10.1128/AAC.00985-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng K, Muranyi W, Glass B, Laketa V, Yant SR, Tsai L, Cihlar T, Muller B, Krausslich HG. 2014. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. eLife 3:e04114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matreyek KA, Yucel SS, Li X, Engelman A. 2013. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog 9:e1003693. doi: 10.1371/journal.ppat.1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF. 2003. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol 327:1013–1020. doi: 10.1016/S0022-2836(03)00289-4. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Zhao Q, Bhattacharya S, Waheed AA, Tong X, Hong A, Heck S, Curreli F, Goger M, Cowburn D, Freed EO, Debnath AK. 2008. A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J Mol Biol 378:565–580. doi: 10.1016/j.jmb.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kortagere S, Madani N, Mankowski MK, Schon A, Zentner I, Swaminathan G, Princiotto A, Anthony K, Oza A, Sierra LJ, Passic SR, Wang X, Jones DM, Stavale E, Krebs FC, Martin-Garcia J, Freire E, Ptak RG, Sodroski J, Cocklin S, Smith AB III. 2012. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J Virol 86:8472–8481. doi: 10.1128/JVI.05006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neira JL. 2009. The capsid protein of human immunodeficiency virus: molecular recognition and design of antiviral agents. FEBS J 276:6097. doi: 10.1111/j.1742-4658.2009.07312.x. [DOI] [PubMed] [Google Scholar]
- 33.Goudreau N, Lemke CT, Faucher AM, Grand-Maitre C, Goulet S, Lacoste JE, Rancourt J, Malenfant E, Mercier JF, Titolo S, Mason SW. 2013. Novel inhibitor binding site discovery on HIV-1 capsid N-terminal domain by NMR and X-ray crystallography. ACS Chem Biol 8:1074–1082. doi: 10.1021/cb400075f. [DOI] [PubMed] [Google Scholar]
- 34.Lemke CT, Titolo S, Goudreau N, Faucher AM, Mason SW, Bonneau P. 2013. A novel inhibitor-binding site on the HIV-1 capsid N-terminal domain leads to improved crystallization via compound-mediated dimerization. Acta Crystallogr D Biol Crystallogr 69:1115–1123. doi: 10.1107/S0907444913006409. [DOI] [PubMed] [Google Scholar]
- 35.Zhou J, Yuan X, Dismuke D, Forshey BM, Lundquist C, Lee KH, Aiken C, Chen CH. 2004. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J Virol 78:922–929. doi: 10.1128/JVI.78.2.922-929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, Freed EO, Wild CT. 2003. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc Natl Acad Sci U S A 100:13555–13560. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fontana J, Jurado KA, Cheng N, Ly NL, Fuchs JR, Gorelick RJ, Engelman AN, Steven AC. 2015. Distribution and redistribution of HIV-1 nucleocapsid protein in immature, mature, and integrase-inhibited virions: a role for integrase in maturation. J Virol 89:9765–9780. doi: 10.1128/JVI.01522-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, Kvaratskhelia M, Engelman A. 2013. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc Natl Acad Sci U S A 110:8690–8695. doi: 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou J, Chen CH, Aiken C. 2004. The sequence of the CA-SP1 junction accounts for the differential sensitivity of HIV-1 and SIV to the small molecule maturation inhibitor 3-O-}3′,3′-dimethylsuccinyl{-betulinic acid. Retrovirology 1:15. doi: 10.1186/1742-4690-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aiken C. 2009. Cell-free assays for HIV-1 uncoating. Methods Mol Biol 485:41–53. doi: 10.1007/978-1-59745-170-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah VB, Aiken C. 2011. In vitro uncoating of HIV-1 cores. J Vis Exp 2011:3384. doi: 10.3791/3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang R, Shi J, Byeon IJ, Ahn J, Sheehan JH, Meiler J, Gronenborn AM, Aiken C. 2012. Second-site suppressors of HIV-1 capsid mutations: restoration of intracellular activities without correction of intrinsic capsid stability defects. Retrovirology 9:30. doi: 10.1186/1742-4690-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li G, Verheyen J, Rhee SY, Voet A, Vandamme AM, Theys K. 2013. Functional conservation of HIV-1 Gag: implications for rational drug design. Retrovirology 10:126. doi: 10.1186/1742-4690-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou J, Huang L, Hachey DL, Chen CH, Aiken C. 2005. Inhibition of HIV-1 maturation via drug association with the viral Gag protein in immature HIV-1 particles. J Biol Chem 280:42149–42155. doi: 10.1074/jbc.M508951200. [DOI] [PubMed] [Google Scholar]
- 45.Adamson CS, Ablan SD, Boeras I, Goila-Gaur R, Soheilian F, Nagashima K, Li F, Salzwedel K, Sakalian M, Wild CT, Freed EO. 2006. In vitro resistance to the human immunodeficiency virus type 1 maturation inhibitor PA-457 (bevirimat). J Virol 80:10957–10971. doi: 10.1128/JVI.01369-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keller PW, Adamson CS, Heymann JB, Freed EO, Steven AC. 2011. HIV-1 maturation inhibitor bevirimat stabilizes the immature Gag lattice. J Virol 85:1420–1428. doi: 10.1128/JVI.01926-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kessl JJ, Kutluay SB, Townsend D, Rebensburg S, Slaughter A, Larue RC, Shkriabai N, Bakouche N, Fuchs JR, Bieniasz PD, Kvaratskhelia M. 2016. HIV-1 integrase binds the viral RNA genome and is essential during virion morphogenesis. Cell 166:1257–1268. doi: 10.1016/j.cell.2016.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL. 2010. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog 6:e1001220. doi: 10.1371/journal.ppat.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saito A, Ferhadian D, Sowd GA, Serrao E, Shi J, Halambage UD, Teng S, Soto J, Siddiqui MA, Engelman AN, Aiken C, Yamashita M. 2016. Roles of capsid-interacting host factors in multimodal inhibition of HIV-1 by PF74. J Virol 90:5808–5823. doi: 10.1128/JVI.03116-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah VB, Shi J, Hout DR, Oztop I, Krishnan L, Ahn J, Shotwell MS, Engelman A, Aiken C. 2013. The host proteins transportin SR2/TNPO3 and cyclophilin A exert opposing effects on HIV-1 uncoating. J Virol 87:422–432. doi: 10.1128/JVI.07177-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gallay P, Hope T, Chin D, Trono D. 1997. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc Natl Acad Sci U S A 94:9825–9830. doi: 10.1073/pnas.94.18.9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci U S A 101:4204–4209. doi: 10.1073/pnas.0306764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu R, Limon A, Devroe E, Silver PA, Cherepanov P, Engelman A. 2004. Class II integrase mutants with changes in putative nuclear localization signals are primarily blocked at a postnuclear entry step of human immunodeficiency virus type 1 replication. J Virol 78:12735–12746. doi: 10.1128/JVI.78.23.12735-12746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barnard RJ, Narayan S, Dornadula G, Miller MD, Young JA. 2004. Low pH is required for avian sarcoma and leukosis virus Env-dependent viral penetration into the cytosol and not for viral uncoating. J Virol 78:10433–10441. doi: 10.1128/JVI.78.19.10433-10441.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. 2007. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev 21:1767–1778. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang H, Jurado KA, Wu X, Shun MC, Li X, Ferris AL, Smith SJ, Patel PA, Fuchs JR, Cherepanov P, Kvaratskhelia M, Hughes SH, Engelman A. 2012. HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res 40:11518–11530. doi: 10.1093/nar/gks913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sowd GA, Serrao E, Wang H, Wang W, Fadel HJ, Poeschla EM, Engelman AN. 2016. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc Natl Acad Sci U S A 113:E1054–E1063. doi: 10.1073/pnas.1524213113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Willey RL, Smith DH, Lasky LA, Theodore TS, Earl PL, Moss B, Capon DJ, Martin MA. 1988. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J Virol 62:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]