

ABSTRACT
Human herpes simplex virus 1 (HSV-1) is a widespread pathogen, with 80% of the population being latently infected. To successfully evade the host, the virus has evolved strategies to counteract antiviral responses, including the gene-silencing and innate immunity machineries. The immediately early protein of the virus, infected cell protein 0 (ICP0), plays a central role in these processes. ICP0 blocks innate immunity, and one mechanism is by degrading hostile factors with its intrinsic E3 ligase activity. ICP0 also functions as a promiscuous transactivator, and it blocks repressor complexes to enable viral gene transcription. For these reasons, the growth of a ΔICP0 virus is impaired in most cells, except cells of the human osteosarcoma cell line U2OS, and it is only partially impaired in cells of the human osteosarcoma cell line Saos-2. We found that the two human osteosarcoma cell lines that supported the growth of the ΔICP0 virus failed to activate innate immune responses upon treatment with 2′3′-cyclic GAMP (2′3′-cGAMP), the natural agonist of STING (i.e., stimulator of interferon genes) or after infection with the ΔICP0 mutant virus. Innate immune responses were restored in these cells by transient expression of the STING protein but not after overexpression of interferon-inducible protein 16 (IFI16). Restoration of STING expression resulted in suppression of ΔICP0 virus gene expression and a decrease in viral yields. Overexpression of IFI16 also suppressed ΔICP0 virus gene expression, albeit to a lesser extent than STING. These data suggest that the susceptibility of U2OS and Saos-2 cells to the ΔICP0 HSV-1 is in part due to an impaired STING pathway.
IMPORTANCE The DNA sensor STING plays pivotal role in controlling HSV-1 infection both in cell culture and in mice. The HSV-1 genome encodes numerous proteins that are dedicated to combat host antiviral responses. The immediate early protein of the virus ICP0 plays major role in this process as it targets hostile host proteins for degradation with its E3 ligase activity, and it disrupts repressor complexes via protein-protein interaction to enable viral gene transcription. Therefore, the ΔICP0 HSV-1 virus is defective for growth in most cells, except the human osteosarcoma cell lines U2OS and Saos-2. We found that both cell lines that support ΔICP0 virus infection have defects in the STING DNA-sensing pathway, which partially accounts for the rescue of the ΔICP0 virus growth. Restoration of STING expression in these cells rescued innate immunity and suppressed ΔICP0 virus infection. This study underscores the importance of STING in the control of HSV-1.
KEYWORDS: STING, IFI16, ICP0, herpes simplex virus, innate immunity
INTRODUCTION
Herpes simplex virus (HSV) causes several pathologies, from benign cold sore to neonatal herpes infection and viral encephalitis (1). After primary infection in mucosal epithelial cells, the virus establishes a lifelong latent infection in sensory neurons. Periodically the virus is reactivated in sensory neurons due to weakened immune responses, stress, or hormonal changes to cause recurrent disease (1).
To counteract host antiviral responses, the virus encodes multiple products. Among them, the immediate early protein of the virus, infected cell protein 0 (ICP0), has two major functions: it is a promiscuous transactivator of genes introduced into the cells by infection or transfection, and it blocks innate immunity (2–13). Initially, ICP0 localizes in the nucleus at the ND10 bodies, where the viral genome is localized and silenced (14–16). ICP0 induces degradation of ND10 body components such as promyelocytic leukemia protein (PML) and SP100 through its E3 ubiquitin ligase activity (10–13). The disruption of ND10 bodies allows viral gene transcription and genome replication but also interferes with interferon gene expression induced by constituents of the ND10 bodies (10–13). ICP0 tandemly disrupts the DNA repressor complex CoREST/REST/HDAC1/2 (i.e., corepressor of repressor 1 silencing transcription/repressor 1 silencing transcription/histone deacetylase 1/2), while it recruits chromatin-remodeling enzymes to the viral genome such as the histone acetyltransferase CLOCK to enable viral gene expression (7, 17–21). Failure of ICP0 to execute either of these functions results in delays in viral gene expression (7). Later in infection, ICP0 translocates to the cytoplasm, where it acquires a new set of binding partners and exerts different functions (22). For instance, through its interaction with CIN85, ICP0 alters the abundance of surface receptors, such as epidermal growth factor receptor (EGFR), to alter surface signaling responses (23). In addition, cytoplasmic ICP0 blocks the activation of the transcriptional factor interferon regulatory factor 3 (IRF3) to inhibit downstream antiviral responses, although the exact mechanism remains unknown (24, 25).
The ICP0 gene is considered a nonessential gene for the growth of the virus in cell cultures. However, the growth of a ΔICP0 mutant virus in cell cultures at low multiplicity of infection is largely impaired due to the actions of the innate immunity and gene-silencing machineries (7, 26–28). At a higher multiplicity of infection, some copies of the viral genome escape the gene-silencing machinery and allow for viral gene transcription and replication (1, 7). Several years ago, it was demonstrated that the human osteosarcoma cell line U2OS could support the growth of the ΔICP0 virus up to high titers, but the mechanism responsible for the rescue of ICP0-deficient mutants has yet to be determined (29).
The DNA sensor STING restricts a broad spectrum of pathogens (30–32). STING is a transmembrane protein on the endoplasmic reticulum; it senses foreign nucleic acids and activates type I interferon and proinflammatory cytokines through the IRF3 or NF-κB pathways (30–33). The mechanism of STING activation following HSV-1 infection remains to be determined. In the cytoplasm, double-stranded DNA (dsDNA) can be recognized and processed by the cyclic GMP-AMP synthase (cGAS) that generates the noncanonical cyclic dinucleotide 2′3′-cGAMP, the natural ligand of STING (34, 35). STING is also associated with another microbial DNA sensor, interferon-inducible protein 16 (IFI16) (36). IFI16 resides mainly in the nucleus, contains domains involved in DNA binding and regulation of transcription, and interacts with p53, retinoblastoma 1, and BRCA-1 (37–39). On certain occasions, IFI16 translocates from the nucleus to the cytoplasm, where it interacts with STING, leading to an augmentation of STING activity (36). In the case of HSV-1, we and others have demonstrated that depletion of STING from cells results in increased yields of wild-type HSV-1 and the ΔICP0 mutant virus (30, 40). Moreover, infection of STING knockout mice with HSV-1 results in uncontrollable viral replication ending in premature death (30, 31, 41). HSV-1 has developed mechanisms to counteract innate immune responses induced by these DNA sensors. For instance, IFI16 is degraded during the early steps of HSV-1 infection, and thus its function is abrogated prior to the initiation of viral replication (42–44). Conversely, STING is stabilized during HSV-1 infection, and a fraction of STING is exported from cells within extracellular vesicles and delivered to uninfected cells to control virus dissemination (40). Christensen et al. showed in a macrophage model that ICP27 inhibits IRF3 phosphorylation by interacting with STING and TANK-binding kinase 1 (TBK1) (45).
Previously we demonstrated that the growth of the ΔICP0 mutant virus could be partially rescued in STING knockdown cells, in a cell-type-dependent manner (40). In this study, we investigated the ability of STING to activate innate immunity in the human osteosarcoma cell lines U2OS and Saos-2, which support the growth of ΔICP0 virus. We found that following infection with the ΔICP0 mutant virus, both the U2OS and the Saos-2 cells were unable to mount innate immune responses. Moreover, U2OS and Saos-2 did not activate innate immune responses after exposure to the ligand of STING, 2′3′-cGAMP. This defect was most likely due to very low levels of STING expressed in U2OS and Saos-2 cells. Transient expression of STING protein in U2OS and Saos-2 cells restored their ability to activate innate immunity after exposure to foreign nucleic acids and suppressed the growth of ΔICP0 virus. In contrast, IFI16 protein was constantly expressed in U2OS and Saos-2 cells, and a subsequent increase in IFI16 by overexpression suppressed, albeit to a lesser extent, the growth of ΔICP0 virus without restoring the ability of the cells to activate innate immunity through the STING pathway. We conclude that deficiencies in the STING pathway in U2OS cells and Saos-2 contribute partially to the susceptibility of these cells to ICP0-deficient viruses.
RESULTS
ΔICP0 mutant virus growth is impaired in HEL cells compared to U2OS and Saos-2 cells.
The growth of the ΔICP0 mutant virus is impaired at low multiplicity of infection in most laboratory cell lines. One cell line in which the ICP0 deletion mutant virus will replicate is the human osteosarcoma cell line U2OS, as previously reported (29). We analyzed the growth of the ΔICP0 mutant virus in two human osteosarcoma cell lines, U2OS and Saos-2, and in the human immortalized lung fibroblast line HEL. For this purpose, cells were infected with the ΔICP0 mutant virus at 0.01 PFU/cell, and the release of progeny virus was measured at 3, 24, and 48 h postinfection using titration assays in U2OS cells. As shown in Fig. 1, the HEL cells imposed a strong restriction on the ΔICP0 mutant virus, with progeny virus yields almost 2-log10 less than those of the U2OS cells at 24 and 48 h postinfection. The ΔICP0 mutant virus growth in Saos-2 displayed an intermediate phenotype, with progeny virus yields 10-fold higher than those of the HEL cells but 10-fold lower than those of U2OS cells at 48 h postinfection (Fig. 1). Thus, several host factors restrict or are required for optimum viral growth, suggesting that Saos-2 cells may share features of both U2OS and HEL cells regarding ΔICP0 mutant virus growth.
FIG 1.
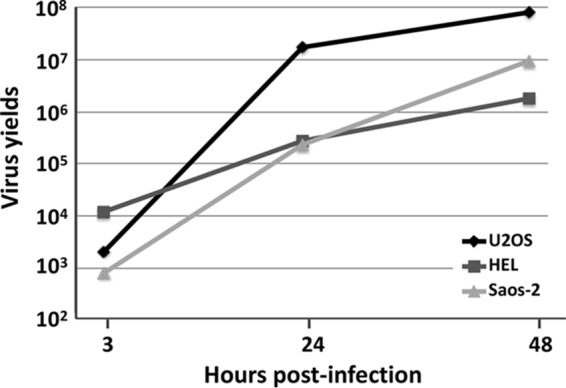
Rescue of ΔICP0 virus growth in U2OS and Saos-2 cells but not in HEL cells. HEL, U2OS, and Saos-2 cells were exposed to the ΔICP0 mutant virus at 0.01 PFU/cell. The cells were harvested at 3, 24, and 48 h postinfection, and progeny viruses were titrated in U2OS cells.
U2OS and Saos-2 cell lines failed to activate innate immune responses.
Innate immunity plays a pivotal role in restricting HSV-1 during the early steps of the infection. The STING pathway restricts HSV-1 by inducing type I interferon followed by the activation of other antiviral factors, such as interferon-stimulated gene 56 (ISG56) and ISG15. To investigate such responses, we exposed HEL, U2OS, Saos-2, and STING knockdown cells to the ΔICP0 mutant virus (1 PFU/cell) or to the natural agonist of STING, 2′3′-cGAMP (3 μM), for 9 h. Cells were then harvested, total RNA was extracted and converted to cDNA, and semiquantification of the ISG56 gene transcript was done by PCR analysis. As shown in Fig. 2A, HEL cells treated with the 2′3′-cGAMP or infected with the ΔICP0 mutant induced ISG56 transcription (compare lanes 3 and 4 to lane 2). In contrast, neither treatment of U2OS and Saos-2 cells with 2′3′-cGAMP (lanes 7 and 12) nor the infection of cultures with the ΔICP0 mutant virus (lanes 8 and 11) triggered ISG56 expression. In addition, no activation of innate immune responses by these two stimuli was detected in STING knockdown HEL cells (compare lanes 15 and 16 to lanes 3 and 4). The experiment was repeated two more independent times with similar results. Notably, treatment with 2′3′-cGAMP caused a reduction in the amounts of the STING transcripts, as shown in panel A (compare lane 4 to lane 2). A control dose-dependent assay demonstrated that doses of 2′3′-cGAMP ranging between 1 to 3 μM activated innate immunity and slightly increased the amounts of the STING protein, but higher doses of the STING agonist did not activate innate immunity and caused a reduction in the amounts of the STING transcripts, without necessarily affecting the amounts of the STING protein (data not shown here). Therefore, a negative-feedback loop may link the activity of the STING protein with the amounts of its transcripts, and this loop could account for the reduction in the amounts of the STING transcripts observed in panel A, lane 4.
FIG 2.
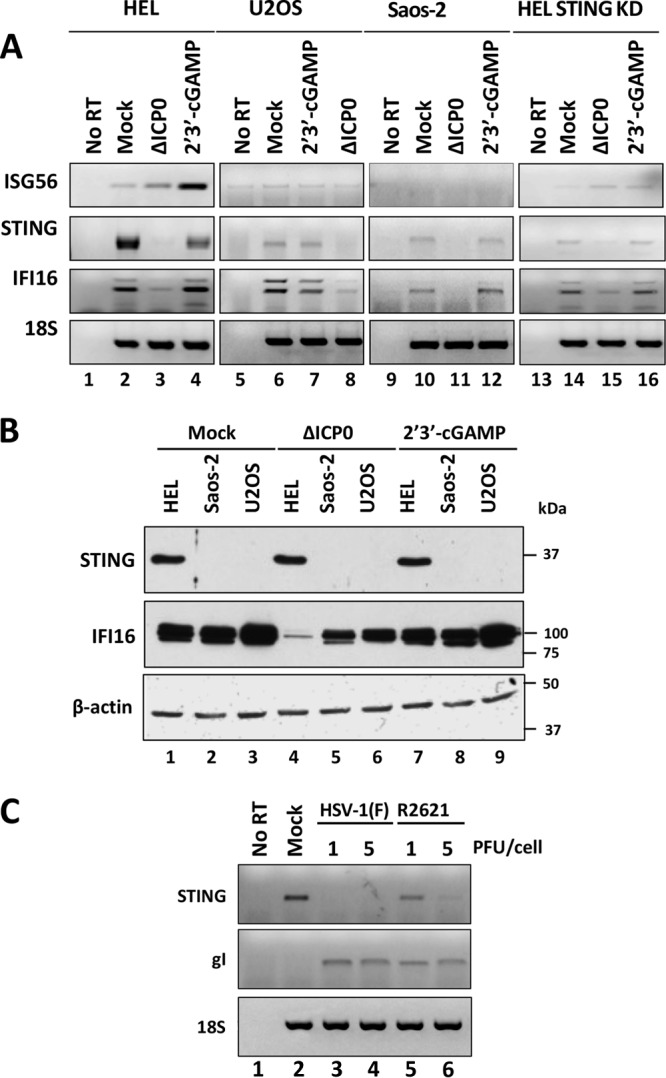
U2OS and Saos-2 cells do not mount innate immunity after treatment with 2′3′-cGAMP or infection with the ΔICP0 mutant virus. (A) HEL, U2OS, Saos-2, and STING knockdown HEL cells were exposed to the ΔICP0 mutant at 1 PFU/cell or treated with 2′3′cGAMP (3 μM). At 9 h postexposure, the cells were harvested, and semiquantitative PCR analysis was done using STING, IFI16, and ISG56 primers; 18S was used as a control. (B) Immunoblot analysis was performed using replicate cultures as in panel A. Membranes were reacted with antibodies against STING and IFI16. β-Actin was used as a loading control. (C) HEL cells were exposed to HSV-1(F) or to the R2621 mutant virus at 1 or 5 PFU/cell. The cells were harvested at 18 h after infection, and semiquantitative PCR analysis was done using STING and gI primers; 18S served as a control.
Since U2OS and Saos-2 did not activate interferon-stimulated gene expression following treatment with 2′3′-cGAMP, we assessed the expression of STING and IFI16 at both the mRNA and protein levels under the same conditions. As shown in Fig. 2B, the STING protein was present in HEL cells (Fig. 2B, lane 1). In contrast, STING protein was undetectable in U2OS and Saos-2 cells (Fig. 2B, lanes 2 and 3). Similarly, the transcript of STING was barely detectable by semiquantitative PCR analysis in U2OS and Saos-2 cells, while it was abundantly present in HEL cells (Fig. 2A, compare lanes 6 and 10 to lane 2). Notably, STING mRNA levels in HEL cells decreased after infection with the ΔICP0 mutant virus compared to mock- or 2′3′-cGAMP-treated cells, but no reductions in the protein levels were observed (Fig. 2A, compare lane 3 to lanes 1 and 4, and B, compare lane 4 to lanes 1 and 7). Reductions in the levels of STING transcripts were observed both in wild-type virus and in the virion host shutoff protein deletion mutant (ΔVHS) virus (R2621)-infected cells, at 5 PFU/cell (Fig. 2C, compare lanes 3 and 4 to lane 2), but the transcripts of STING were relatively stable in ΔVHS mutant virus-infected cells at a lower multiplicity of infection (1 PFU/cell) compared to the wild-type virus-infected cells (Fig. 2C, compare lane 5 to lanes 2 and 6). This could be due to other delays in the ΔVHS mutant virus-infected cells emanating from the presence of intact host transcripts. Previously we demonstrated that the STING protein is stable in wild-type virus and ΔICP0 mutant-infected HEL cells even up to 18 h postinfection (40). Moreover, the STING protein is stable in infected HEL cells after blocking protein synthesis by adding cycloheximide (CHX), even 18 h after the addition of CHX in infected cultures (40). Therefore, the reduction in the amounts of the STING transcripts during infection of the HEL cells by the ΔICP0 mutant (Fig. 2A) is not reflected in the protein levels (Fig. 2B). Treatment with 2′3′-cGAMP did not significantly alter the levels of the STING transcripts or protein (Fig. 2A and B).
In contrast to STING, the IFI16 protein is expressed in all of the cell lines at comparable levels (Fig. 2B, compare lanes 1 to 3). The transcripts for the IFI16 isoforms were also present in the three cell lines, and the levels were comparable between HEL and U2OS cells (Fig. 2A, compare lanes 2, 6, and 10). Infection with the ΔICP0 mutant virus at 1 PFU/cell, caused a decrease in the accumulation of the IFI16 protein and of its transcripts (Fig. 2A, compare lanes 3, 8, and 11 to lanes 2, 6, and 10), as has been previously described (40, 42). The decrease in the amounts of IFI16 protein by ΔICP0 virus was greater in HEL cells than in U2OS and Saos-2 cells (Fig. 2B, compare lanes 5 and 6 to lane 4 and lanes 2 and 3 to lane 1). Differences in the stability of IFI16 protein during HSV-1 infection between different cell types were previously reported (43). Treatment with 2′3′-cGAMP did not affect the accumulation of the IFI16 transcripts or the amounts of the protein. Taken together, the ability of the ΔICP0 virus to grow in U2OS and Saos-2 cells, even at a low multiplicity of infection, is in part due to lack of expression of the STING protein with concomitant deficiencies in activation of STING-dependent innate immune responses.
Rescue of STING expression in U2OS and Saos-2 cells restores innate immune responses.
We sought to determine whether transient expression of STING in U2OS and Saos-2 cells could restore innate immune responses and restrict ΔICP0 mutant virus infection. U2OS cells were transfected with a vector expressing STING or IFI16 or with the pUC19 control plasmid. At 36 h posttransfection, cells were exposed to 2′3′-cGAMP (3 μM) or to the ΔICP0 mutant virus (0.1 PFU/cell) for 8 h prior to RNA extraction and semiquantification of transcripts by PCR analysis. The results shown in Fig. 3A can be summarized as follows. First, the levels of the STING transcripts were negligible in U2OS cells except after the transfection with the STING-expressing plasmid (Fig. 3A, compare lanes 1 to 7 and 11 to 13 to lanes 8 to 10). Second, the three isoforms of IFI16 were detectable in untreated U2OS cells or after 2′3′-cGAMP treatment, but reductions in their levels were observed after infection with the ΔICP0 mutant virus (compare lanes 2, 4, 5, 7, 8, and 10 to lanes 3, 6, and 9). Reductions in the amounts of the IFI16 transcripts during HSV-1 infection were reported elsewhere too (43). Following transfection with the plasmid expressing IFI16, an increase in two splicing variants (b and c) was observed (compare lanes 11 to 13 to lanes 1 to 10). In IFI16-transfected cells, infection with the ΔICP0 mutant virus did not affect the accumulation of the overexpressed IFI16 transcripts (compare lane 12 to lanes 11 and 13). Third, U2OS cells transfected with the plasmid expressing STING exhibited a 7-fold induction in the ISG15 gene transcription compared to untransfected or pUC19-transfected U2OS cells (Fig. 3B). Activation of ISG15 expression following transfection with the plasmid expressing STING was probably due to the plasmid being recognized as foreign DNA by the newly synthesized STING. No induction of ISG15 transcription was noticed following transfection with the plasmid expressing IFI16 (Fig. 3B). Fourth, treatment with the agonist of STING, 2′3′-cGAMP, did not induce ISG15 transcription in untransfected, IFI16-transfected, or control DNA-transfected cells and did not have an additive effect on the activity of STING in the STING-transfected cells, most likely because the plasmid DNA itself had already activated STING. Fifth, infection with the ΔICP0 mutant virus reduced but did not abrogate the effects of overexpressed STING, as shown by the quantities of the ISG15 gene transcript, suggesting that the viral genes expressed by the ΔICP0 mutant virus could counteract the antiviral effects of STING, at a step downstream of its activation by the foreign DNA. Moreover, this downmodulation in the activity of STING was independent of ICP0. Finally, similar results were obtained from the analysis of proinflammatory genes, such as the gene coding for interleukin-6 (IL-6) (Fig. 3B). A similar experiment was performed in Saos-2 cells. As shown in Fig. 3C, transient expression of STING in Saos-2 cells activated ISG15 and ISG56 transcription by 30- and 20-fold, respectively, but no activation was observed in control-transfected cells. Infection with the ΔICP0 mutant virus did not moderate this time the innate immune responses activated by STING, most likely because innate immune responses were more robust in Saos-2 than in U2OS cells or because Saos-2 cells inhibit the virus at a step prior to the downmodulation of the STING activity. The entire experiment was repeated two more times with similar results. We conclude that restoration of STING expression in U2OS and Saos-2 cells could rescue antiviral responses.
FIG 3.

Restoration of STING expression in U2OS cells rescues innate immunity. (A) The U2OS cells were either mock transfected (lanes 2 to 4) or transfected with STING (lanes 8 to 10), IFI16-expressing plasmids (lanes 11 to 13), or pUC19 (lanes 5 to 7) as a control. At 36 h posttransfection, the cells were exposed to the ΔICP0 mutant virus at 0.1 PFU/cell (lanes 3, 6, 9, and 12) or to 2′3′-cGAMP (3 μM) (lanes 4, 7, 10, and 13). At 8 h postexposure, cells were harvested, total RNA was extracted, and the STING and IFI16 transcripts were semiquantified by PCR analysis. 18S was used as a control. (B) RNAs from panel A were used for quantification of the IL-6 and ISG15 transcripts by real-time PCR analysis. The experiment was repeated two more independent times with similar results. (C) The Saos-2 cells were transfected with the STING-expressing plasmid or with EGFP-expressing plasmid as a control or remained untransfected (NT). At 36 h posttransfection, the cells were exposed to the ΔICP0 mutant virus (0.1 PFU/cell). Analysis of ISG transcription was done at 8 h postinfection as in panel A.
Restoration of STING expression in U2OS and Saos-2 cells impaired the ΔICP0 mutant virus growth.
Next, we assessed whether restoration of STING expression could restrict ΔICP0 virus infection in U2OS and Saos-2 cells. In the first experiment, the human osteosarcoma cells were transfected as described above with the plasmids expressing STING or IFI16. Nontransfected cells (NT) or pUC19- or enhanced green fluorescent protein (EGFP)-transfected cells served as controls. At 36 h posttransfection, the cells were infected with the ΔICP0 mutant virus at 0.1 PFU/cell. The cells were harvested 8 h postinfection, and quantification of viral gene transcripts was done by real-time PCR analysis. Compared to ΔICP0 mutant virus-infected but nontransfected U2OS cells, the levels of transcripts for immediate early protein ICP22 and for the late protein glycoprotein I (gI) in control plasmid-transfected cells were 80% and 60%, respectively (Fig. 4A). Transient expression of STING imposed a strong suppression of viral gene transcription, as the amounts of the ICP22 and gI transcripts were approximately 15% of the control. IFI16 overexpression also had a suppressive effect, albeit to a lesser extent than STING, as the ICP22 and gI transcripts were 40% of the level of the control. In a replicate assay at 36 h after transfection with the STING- and IFI16-expressing plasmids, U2OS cells were exposed to the ΔICP0 mutant virus (0.1 PFU/cell) and harvested at 4 and 8 h postinfection. Viral gene expression and the expression of STING and IFI16 proteins were assessed by immunoblot analysis. As shown in Fig. 4B, the STING protein accumulated in the U2OS cells transfected with the STING-expressing plasmid (Fig. 4B, lanes 3, 7, and 11), but it was undetectable in the untransfected U2OS cells. IFI16 was expressed in U2OS cells and accumulated further in cells transfected with the IFI16-expressing plasmid (Fig. 4B, lanes 4, 8, and 12). The immediate early protein of the virus ICP4 was detectable at 4 h after infection in nontransfected or control-transfected cells (Fig. 4B, lanes 5 and 6), but a significant delay in the accumulation of ICP4 was observed in STING-transfected cells (lane 7), and only a small reduction in the amounts of ICP4 was noticed in IFI16-transfected cells (lane 8). The accumulation of ICP4 at 8 h after infection followed a similar pattern to that at 4 h after infection (compare lanes 9 to 12 to lanes 5 to 8). The accumulation of the late protein VP22 was apparent at 8 h after infection in the nontransfected U2OS cells (Fig. 4B, lane 9), but it was undetectable in the U2OS cells overexpressing STING protein (Fig. 4B, lane 11). IFI16 protein overexpression caused to a lesser extent reductions in the levels of the VP22 protein (Fig. 4B, compare lane 12 to lanes 9 and 11). Similarly, transient expression of STING in Saos-2 cells imposed a robust suppression of viral gene transcription compared to the controls (Fig. 4C). We conclude that transient expression of STING or IFI16 in U2OS and Saos-2 cells impairs ΔICP0 mutant virus gene transcription, which is reflected in the levels of the viral proteins. In a second experiment, we measured the effects of exogenous STING or IFI16 expressed in U2OS cells on ΔICP0 mutant virus growth. U2OS cells were transfected with a STING- or IFI16-expressing plasmid or with a control EGFP-expressing plasmid or the control plasmid pUC19. At 24 h posttransfection, the cells were infected with the ΔICP0 mutant virus (0.01 PFU/cell for panel D or 0.05 PFU/cell for panel E). The cells were harvested at 3, 24, or 48 h after infection, and the progeny virus was titrated using U2OS cells. As shown in Fig. 4D, the control-transfected cells had only a minor impact on ΔICP0 virus growth compared to the untransfected cells. In contrast, expression of STING in U2OS cells caused an approximately 10-fold reduction in the ΔICP0 mutant virus yields compare to the controls. The expression of STING in the U2OS cells is shown in Fig. 4D. Similar results were obtained in the assay shown in Fig. 4E. Overexpression of STING caused a 10-fold reduction in the ΔICP0 virus yields but not the control plasmids. Consistent with the viral gene expression assays, overexpression of IFI16 caused a reduction in the ΔICP0 virus yields but to a lesser extent than STING. We conclude that restoration of the STING DNA-sensing pathway in U2OS cells restricts the infection by the ΔICP0 mutant virus.
FIG 4.

STING expression in U2OS cells blocks viral gene expression and impairs ΔICP0 mutant virus growth. (A) RNAs from the experiment in Fig. 3A were used for quantification of the viral gene transcripts ICP22 and gI by real-time PCR analysis. (B) U2OS cells were transfected/infected as in Fig. 3A. At 4 and 8 h postinfection, the cells were harvested and lysed, equal amount of proteins were separated in replicate 10% denaturing polyacrylamide gels, and immunoblot analysis was done with the IFI16, STING, VP22, and ICP4 antibodies. β-Actin was used as a loading control. (C) RNAs from the experiment in Fig. 3C were used for quantification of the viral gene transcripts ICP22 and gI by real-time PCR analysis. (D) U2OS cells were transfected with an EGFP- or STING-expressing plasmid, as detailed in Materials and Methods. At 24 h posttransfection, the cells were infected with the ΔICP0 mutant at 0.01 PFU/cell. The cells were harvested 3, 24, and 48 h postinfection, and titration of progeny viruses was done in U2OS cells. Replicate cultures were analyzed for the expression of STING in U2OS-transfected cells. (E) Transfections of U2OS cells with STING, IFI16-expressing plasmids, or the control pUC19 were done as in panel D. At 24 h posttransfection, the cells were infected with the ΔICP0 mutant at 0.005 PFU/cell. The cells were harvested 3, 24, and 48 h postinfection, and titration of progeny viruses was done in U2OS cells.
DISCUSSION
During infection, HSV-1 effectively counteracts host antiviral mechanisms to ensure successful replication. Several viral proteins have dedicated functions to evade the host, such as ICP0, a viral E3 ligase that targets hostile proteins for degradation and disrupts repressor complexes (11–13), ICP27, which stimulates the transportation of intron-less mRNAs to the cytoplasm (46), VHS, the viral RNase that targets for degradation mainly AU-rich element (ARE)-containing mRNAs expressed to block the infection (47, 48), γ1 34.5, which prevents the translational shutoff through protein kinase R (PKR) activation (49, 50), and Us11, which downmodulates the Rig-like receptor (RLR) pathway by interacting with RIG-I and MDA-5 and others (11, 51). ICP0 remains one of the most studied HSV-1 proteins as it exerts essential functions immediately after the release of viral DNA in the nucleus, such as inhibition of innate immunity and of the gene-silencing machineries (1). Consequently, ICP0 mutant viruses fail to evade innate immunity, display defects in initiation of viral gene transcription, and hence produce less progeny viruses. Previously, it was demonstrated that ICP0 deletion mutant virus could replicate in the human osteosarcoma cell line U2OS, although the mechanism has remained unknown (29). Our work presented here was aimed at understanding the characteristics of the U2OS cells that supported the growth of ICP0 deletion mutant virus.
In this study, we compared the levels of growth of the ΔICP0 mutant virus in three cell lines: two human osteosarcoma cell lines (U2OS and Saos-2) and immortalized human lung fibroblasts (HEL). As previously reported, infection of U2OS cells with the ΔICP0 mutant virus at low multiplicity supports growth of the virus to high titers (29). Compared to the U2OS cells, the yields of the ΔICP0 mutant were decreased in Saos-2 cells by 10-fold and in HEL cells by 100-fold. Consistent with previous studies (29), the U2OS cells either complemented the lack of ICP0 protein or did not express a hostile protein, and that permitted better replication of ICP0 mutant virus, while the HEL cells restricted ΔICP0 virus infection. As ICP0 has a major role in blocking innate immunity during viral infection, we analyzed the ability of these cells to mount innate immune responses against viral infection or after exposure to the STING agonist 2′3′-cGAMP. While innate immune responses were induced in HEL cells in both cases, the two human osteosarcoma cell lines failed to mount an innate immune response. In STING knockdown HEL cells, neither treatment with 2′3′-cGAMP nor infection with the ΔICP0 mutant virus induced ISG expression, suggesting that STING plays central role in innate immune responses activated by these stimuli. The absence of responses in U2OS and Saos-2 cells after 2′3-cGAMP treatment suggested possible impairment in the STING pathway. Analysis of the expression of STING in U2OS and Saos-2 cells demonstrated that both cell lines have a deficit in STING expression as the amounts of the protein and transcripts were nearly undetectable. In addition to STING, the DNA sensor IFI16 also has a role in HSV-1 restriction. In contrast to STING, the amounts of the IFI16 protein were comparable among the three cell lines tested. Also, as has been described by others and us, we found that IFI16 expression was decreased following infection with the ΔICP0 mutant virus (40, 42). Interestingly, we noticed that the elimination of IFI16 protein in ΔICP0 mutant virus-infected cells was more efficient in the HEL cells, which have intact innate immunity, as opposed to the U2OS and Saos-2 cells, which have defects in the STING pathway. Defects in the IFI16 pathway as well or other changes in the two osteosarcoma cell lines cannot be excluded (40, 42–44). Differences in the stabilities of IFI16 protein during HSV-1 infection between various cell lines were recently reported, although the mechanistic details remain unknown (43).
The STING pathway plays an important role in restricting HSV-1 (30–32). Both STING-depleted cells and STING knockout mice present an increase in the severity of HSV-1 infection (30–32, 40). Previously, we reported that the growth of the ΔICP0 mutant virus was partially rescued in the HEL STING knockdown cells as they failed to mount innate immune responses (40). Thus, we hypothesized that the negligible amounts of the STING protein present in U2OS and Saos-2 cells could account for the lack of innate immunity upon infection. To test our hypothesis, we performed transient-transfection assays in the human osteosarcoma cells to rescue the expression of STING, using a STING-expressing plasmid. Similar analysis was done in U2OS cells transfected with a control-expressing plasmid or with an IFI16-expressing plasmid. Following transfection with the plasmid control or a plasmid expressing IFI16, we did not detect an induction of innate immunity genes after infection with the ΔICP0 mutant virus or exposure to the 2′3′-cGAMP. STING transfection induced similar levels of ISG15 and IL-6 expression between untreated and 2′3′-cGAMP-treated cells, and this induction was moderately reduced after ΔICP0 infection. The self-induction of innate immunity and inflammatory genes after transfecting U2OS cells with the plasmid expressing STING may be due to sensing of the transfected DNA per se by the newly synthetized STING. During transfection, the DNA delivered into the cells is in excess: thus, an additive stimulation of innate immunity genes by the 2′3′-cGAMP could not be observed. The reduction of innate immunity gene transcription upon infection of the STING-transfected U2OS cells with the ΔICP0 mutant virus suggests that viral genes can moderate to some extent the STING activity, at a step downstream of the activation of STING by the exogenous DNA. ICP0 is not required for this downmodulation process. This phenomenon was observed in U2OS cells but not in Saos-2 cells. One interpretation could be that in Saos-2 cells, the innate immunity gene transcription was induced to a greater extent than in U2OS cells, following transfection with the STING-expressing plasmid, and therefore ΔICP0 mutant virus failed to reduce ISG transcription. On the other hand, Saos-2 cells are less permissive than U2OS cells, as demonstrated in Fig. 1 and reported previously (29). Thus, an alternative interpretation for the failure of the ΔICP0 virus to downmodulate the STING activity in Saos-2 could be that the ΔICP0 virus fails to bypass a barrier that acts before STING. In U2OS cells, it may be the case that this barrier is missing. These data imply that U2OS cells allow for viral gene expression and virus replication but also permit inhibition of the STING antiviral responses by the virus in an ICP0-independent mechanism.
We also found that the expression of viral genes after transient expression of STING or IFI16 proteins was reduced by 90% and 60%, respectively. The overexpression of IFI16 allowed for accumulation of IFI16 protein during ΔICP0 virus infection; therefore, a negative impact on the ΔICP0 virus gene expression was observed. Nevertheless, the infection by the ΔICP0 mutant virus was suppressed more drastically after rescuing STING expression rather than after overexpressing IFI16. The inhibitory effect by STING might come from a dual effect—first by the presence of innate immunity factors induced by the transfected DNA prior to the infection and second by the responses triggered during the viral infection by the rescued STING pathway.
Another observation was that during infection either with the wild-type virus or with the ΔICP0 mutant virus, the amounts of the STING transcripts declined. The viral RNase VHS appears to have an indirect role in this process as in ΔVHS mutant virus-infected cells, the amounts of the STING transcripts still declined at high multiplicities of infection. At lower multiplicities of infection, the transcripts of STING were stable in the absence of the viral RNase VHS. Maybe delays in infection of the ΔVHS virus caused by the presence of intact hostile cellular mRNAs result in delays in elimination of the STING transcripts. Despite the reduction in the amounts of the STING transcripts, no alterations in the abundance of the STING protein were detected. The STING protein is stable throughout the course of the infection by either HSV-1 or the ΔICP0 mutant. Moreover, as we previously reported STING is stable during infection, even up to 18 h after blocking of protein synthesis in infected cells with cycloheximide (CHX) (40). One occasion that elimination of the STING protein was observed was after infection of cancer cells (e.g., HEp-2 or HeLa cells) with mutant viruses that have delays in the expression of late gene functions (e.g., ΔICP0 and ΔICP4 mutant viruses). However, these viruses in immortalized cells did not induce elimination of the STING protein (40). A fraction of STING is also excreted out of the infected cells in extracellular vesicles (EVs) (40, 52). A possible hypothesis is that reduction in the amounts of the STING transcripts along with excretion of the protein might contribute to the moderation of the activity of STING during HSV infection.
Previous reports characterized the effects of IFI16 during HSV-1 infection (36, 53). Small hairpin RNA against IFI16 led to a reduction in beta interferon expression upon infection and an increase in virus yields (50, 51). In other studies, recruitment of IFI16 to the PML nuclear bodies together with the viral genome was demonstrated, suggesting a role of IFI16 in transcriptional repression of the viral DNA (54, 62). Depletion of p204, the mouse functional ortholog of IFI16, from bone marrow-derived macrophages resulted in decreased IRF3 and NF-κB responses to HSV-1 infection, while depletion of p204 expression from mouse cornea resulted in increased HSV-1 replication in the cornea tissue (36, 53). In our study, we found that the overexpression of IFI16 reduced viral gene expression but did not induce innate immune responses following treatment with 2′3′-cGAMP or exposure to the ΔICP0 virus, while rescue of STING expression induced innate immunity and suppressed the ΔICP0 virus. Thus, the mechanism of inhibition of HSV by IFI16 remains to be determined.
In this study, we demonstrated a defect in the STING pathway in two osteosarcoma cell lines. This defect prevents both of the cell lines from triggering an innate immune response upon 2′3′-cGAMP treatment or after ΔICP0 mutant virus infection. The lack of innate immune responses upon infection represents a hallmark for the susceptibility of the U2OS cell line. However, the Saos-2 cell line presents the same defect but has moderate susceptibility to the infection; therefore, the high susceptibility of U2OS cannot be explained only by an absence of innate immunity. The U2OS and Saos-2 lines have been extensively used to study the characteristics of the p53 and retinoblastoma (pRb) proteins (29). Saos-2 cells encode a functionally inactive form of pRb truncated at its carboxy terminus and contain a deletion in the gene encoding p53, whereas U2OS cells encode functional pRb, but they carry only one copy of p53 and one copy of ATRX: hence the expression of these proteins is halved (29, 55). Another study demonstrated that ATRX (i.e., alpha-thalassemia/mental retardation syndrome X-linked protein), which contributes to transcriptional repression and chromatin assembly during herpes infection, is not expressed in U2OS cells (56). Therefore, U2OS cells appear to have defects in multiple hostile components that enable optimum virus growth. On the other hand, Saos-2 could have fewer defects or have defects in components utilized by the virus for optimum growth. For instance, p53, which is missing from Saos-2 cells, has an overall positive role in HSV-1 infection (63). ATRX, which is not expressed in U2OS cells, has a negative impact on HSV-1 infection; therefore, the virus represses its expression (56, 57).
In conclusion, we demonstrated that defects in the STING pathway contribute to the rescue of the ΔICP0 mutant virus growth. These data implied that ICP0 interferes with the STING pathway. The transcriptional factor IRF3 is a major component of the STING pathway, and it is already known to be blocked by ICP0, albeit the mechanism remains unknown (24, 25). Whether ICP0 has additional effects on the STING pathway remains to be identified. Also, the observation that two osteosarcoma cell lines have defects in the STING pathway suggests that maybe the lack of STING activity contributes to the immune resistance of these tumors.
MATERIALS AND METHODS
Cell lines and virus.
The HEL (immortalized human embryonic lung fibroblast) and Saos-2 (human osteosarcoma) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. U2OS cells (human osteosarcoma) were cultured in McCoy's 5A medium supplemented with 10% FBS and antibiotics. The HEL STING knockdown cell line was reported elsewhere (40). HSV-1(F) is a limited-passage isolate described before (58). The properties of the ΔICP0 and R2621 mutant viruses were described elsewhere (59, 60).
HSV titration assays.
HEL, Saos-2, and U2OS cells were infected with ΔICP0 virus at 0.01 PFU/cell. Cells were harvested at the indicated times after infection. U2OS cells were used for the titration, as before (18). At 48 h postinfection, the cells were fixed using methanol and stained with Giemsa stain. Plaques were counted under an Olympus stereomicroscope.
Immunoblot analysis.
The immunoblotting procedures have been described elsewhere (17). At the times indicated in the figure legends, the cells were lysed in a triple-detergent buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate supplemented with 50 mM NaF, 0.2 mM Na3VO4, and protease inhibitor cocktail). Equal amounts of proteins were electrophoretically separated, electrically transferred to nitrocellulose sheets, blocked with phosphate-buffered saline (PBS) supplemented with 0.1% (vol/vol) Tween 20 (PBST) and 5% nonfat milk, and reacted overnight at 4°C with appropriate primary antibodies diluted in PBST–5% nonfat milk. STING (R&D systems), IFI16 (Abcam), and β-actin (Sigma) mouse monoclonal antibodies were used in a 1:1,000 dilution. The rabbit polyclonal antibody against VP22 and the mouse monoclonal antibody against ICP4 were kindly provided by B. Roizman and used in a 1:1,000 dilution. Finally, protein bands were visualized with 5-bromo-4-chloro-3-indolylphosphate (BCIP)-nitroblue tetrazolium (NBT) (Denville Scientific) or with ECL enhanced chemiluminescence Western blotting detection reagents (Amersham Biosciences) according to the manufacturer's instructions.
Total RNA extraction and semiquantitative or real-time PCR analysis.
HEL, Saos-2 and U2OS cells, seeded in 6-well plates at a 80% confluence, were mock infected, infected with the ΔICP0 mutant virus, or treated with 2′3′-cGAMP (3 μM) (Sigma). At indicated times posttreatment, the cells were lysed in 1 ml TRIzol (Life Technologies) and total RNA was extracted using phenol-chloroform procedures. DNase treatment was performed using Turbo DNase enzyme (Ambion) according to the manufacturer's instructions. cDNA synthesis was performed using the SuperScript III first-strand kit (Invitrogen) according to the supplier's instructions. Semiquantitative PCR analysis was performed using the GoTaqG2 Hot Start polymerase (Promega) and an equal volume (1 μl) of cDNA that was generated from 1 μg of total RNA per sample. For the semiquantitative PCR analysis, we analyzed the products of the reactions between cycles 18 and 28. We found that at cycle 25, the amplifications were in the exponential range and the quantities of the products were adequate to be appreciated by ethidium bromide analysis in 2% agarose gels. Standard curves were performed to optimize the conditions for each primer set. The annealing temperature was set up at 4°C lower than the lowest melting temperature (Tm) between a primer set. Real-time PCR analyses were performed using SYBR green reagent (Invitrogen) or TaqMan (Applied Biosystems) according to the manufacturer's recommendations. The 18S rRNA (Ambion) was used for normalization. Predesigned probe (6-carboxyfluorescein [FAM]/MGB) for ISG15 was obtained through Thermo Fisher. The following primer sequences were used: STING, forward, 5′-TTCGAACTTACAATCAGCATTACAA-3′, and reverse, 5′-CTCATAGATGCTGTTGCTGTAAACC-3′; IFI16, forward, 5′-CCAGCACAACCTTCCCTGAGAGCCATCT-3′, and reverse, 5′-GAAACTGCTGCTTGGTGTTGSTGGAGGC-3′; gI, forward, 5′-CCCACGGTCAGTCTGGTATC-3′, and reverse, 5′-TTTGTGTCCCATGGGGTAGT-3′; and ISG56, forward, 5′-GGAAAAAAAGCCCACATTTGAGGT-3′, and reverse, 5′-CTTTTGAAATTCCTGAAACCGACCA-3′.
Transfection/infection assays.
U2OS and Saos-2 cells seeded in 6-well plates at a 80% confluence were transfected with 1 μg of pUC19 (New England BioLabs), pcDNA-Flag-STING (kindly provided by R. Weichselbaum's lab, University of Chicago), pcDNA-IFI16 (Addgene, [61]), or pEGFP-N3 (Invitrogen) plasmids using the Lipofectamine 3000 reagent (Invitrogen), according to the manufacturer's instructions. Briefly, for each reaction, 1 μg of plasmid (per well) was mixed in 200 μl Opti-MEM I (Fisher Scientific) with 3 μl of the P3000 reagent. In a separate tube, 200 μl of Opti-MEM I was mixed with 3 μl of the Lipofectamine 3000 reagent. After 5 min of incubation at room temperature, the contents of the two tubes were combined, and the mixture was incubated for another 20 min at room temperature and added to the cells in 10% McCoy's medium. The medium was replaced 3 h posttransfection with complete McCoy's medium. At 24 or 36 h posttransfection, the cells were mock infected or infected with the ΔICP0 mutant virus at 0.1 or 0.01 PFU/cell, in complete McCoy's medium, as indicated in the legends to the respective figures. The medium was replaced 2 h postinfection with complete McCoy's medium. At the times postinfection indicated in the legends to the figures, the cells were harvested in TRIzol reagent (Life Technologies), and total RNA was extracted and analyzed by real-time PCR analysis, as described above, or the cells were harvested for titration of progeny viruses.
ACKNOWLEDGMENTS
We thank Bernard Roizman, University of Chicago, for kindly providing the R7910 and the R2621 viruses, the rabbit polyclonal antibody against VP22, and the mouse monoclonal antibody against ICP4. We thank Edward Stephens, University of Kansas Medical Center, for editing the manuscript. The Saos-2 cells were kindly provided by Tomoo Iwakuma, University of Kansas Medical Center. Many thanks to K-INBRE student Alec Ebersole for technical support.
This work was partly funded by K-INBRE funds P20 GM103418. Maria Kalamvoki is funded through KUMC startup funds.
T.D. and M.K. designed and performed research, analyzed data, and wrote the manuscript.
REFERENCES
- 1.Knipe DM, Howley PM (ed). 2013. Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Cai WZ, Schaffer PA. 1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J Virol 63:4579–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Everett RD. 1984. Trans activation of transcription by herpes virus products: requirement for two HSV-1 immediate-early polypeptides for maximum activity. EMBO J 3:3135–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Everett RD. 1985. Activation of cellular promoters during herpes virus infection of biochemically transformed cells. EMBO J 4:1973–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gelman IH, Silverstein S. 1985. Identification of immediate early genes from herpes simplex virus that transactivate the virus thymidine kinase gene. Proc Natl Acad Sci U S A 82:5265–5269. doi: 10.1073/pnas.82.16.5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalamvoki M, Roizman B. 2010. Role of herpes simplex virus ICP0 in the transactivation of genes introduced by infection or transfection: a reappraisal. J Virol 84:4222–4228. doi: 10.1128/JVI.02585-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roizman B. 2011. The checkpoints of viral gene expression in productive and latent infection: the role of the HDAC/CoREST/LSD1/REST repressor complex. J Virol 85:7474–7482. doi: 10.1128/JVI.00180-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gelman IH, Silverstein S. 1986. Co-ordinate regulation of herpes simplex virus gene expression is mediated by the functional interaction of two immediate early gene products. J Mol Biol 191:395–409. doi: 10.1016/0022-2836(86)90135-X. [DOI] [PubMed] [Google Scholar]
- 9.Hagglund R, Roizman B. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J Virol 78:2169–2178. doi: 10.1128/JVI.78.5.2169-2178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chelbi-Alix M, de Thé H. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941. doi: 10.1038/sj.onc.1202366. [DOI] [PubMed] [Google Scholar]
- 11.Lanfranca MP, Mostafa HH, Davido DJ. 2014. HSV-1 ICP0: an E3 ubiquitin ligase that counteracts host intrinsic and innate immunity. Cells 3:438–454. doi: 10.3390/cells3020438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J Virol 72:6581–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu H, Roizman B. 2003. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc Natl Acad Sci U S A 100:8963–8968. doi: 10.1073/pnas.1533420100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sourvinos G, Everett RD. 2002. Visualization of parental HSV-1 genomes and replication compartments in association with ND10 in live infected cells. EMBO J 21:4989–4997. doi: 10.1093/emboj/cdf458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everett RD, Murray J, Orr A, Preston CM. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J Virol 81:10991–11004. doi: 10.1128/JVI.00705-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everett RD. 2013. The spatial organization of DNA virus genomes in the nucleus. PLoS Pathog 9:e1003386. doi: 10.1371/journal.ppat.1003386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalamvoki M, Roizman B. 2010. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc Natl Acad Sci U S A 107:17721–17726. doi: 10.1073/pnas.1012991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalamvoki M, Roizman B. 2011. The histone acetyltransferase CLOCK is an essential component of the herpes simplex virus 1 transcriptome that includes TFIID, ICP4, ICP27, and ICP22. J Virol 85:9472–9477. doi: 10.1128/JVI.00876-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci U S A 104:17134–17139. doi: 10.1073/pnas.0707266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu H, Roizman B. 2009. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J Virol 83:4376–4385. doi: 10.1128/JVI.02515-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roizman B, Gu H, Mandel G. 2005. The first 30 minutes in the life of a virus: unREST in the nucleus. Cell Cycle 4:1019–1021. doi: 10.4161/cc.4.8.1902. [DOI] [PubMed] [Google Scholar]
- 22.Lopez P, Sant CV, Roizman B. 2001. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J Virol 75:3832–3840. doi: 10.1128/JVI.75.8.3832-3840.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang Y, Kurakin A, Roizman B. 2005. Herpes simplex virus 1 infected cell protein 0 forms a complex with CIN85 and Cbl and mediates the degradation of EGF receptor from cell surfaces. Proc Natl Acad Sci U S A 102:5838–5843. doi: 10.1073/pnas.0501253102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor KE, Chew MV, Ashkar AA, Mossman KL. 2014. Novel roles of cytoplasmic ICP0: proteasome-independent functions of the RING finger are required to block interferon-stimulated gene production but not to promote viral replication. J Virol 88:8091–8101. doi: 10.1128/JVI.00944-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paladino P, Collins SE, Mossman KL. 2010. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS One 5:e10428. doi: 10.1371/journal.pone.0010428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sacks WR, Schaffer PA. 1987. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J Virol 61:829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai W, Schaffer PA. 1992. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J Virol 66:2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Panagiotidis C, Silverstein S. 1992. Multimerization of ICP0, a herpes simplex virus immediate-early protein. J Virol 66:5598–5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao F, Schaffer PA. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J Virol 69:6249–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paludan SR, Bowie AG. 2013. Immune sensing of DNA. Immunity 38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lio CW, McDonald B, Takahashi M, Dhanwani R, Sharma N, Huang J, Pham E, Benedict CA, Sharma S. 2016. cGAS-STING signaling regulates initial innate control of cytomegalovirus infection. J Virol 90:7789–7797. doi: 10.1128/JVI.01040-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type-I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dawson MJ, Trapani JA. 1995. The interferon inducible autoantigen, IFI 16: localization to the nucleolus and identification of a DNA-binding domain. Biochem Biophys Res Commun 214:152–162. doi: 10.1006/bbrc.1995.2269. [DOI] [PubMed] [Google Scholar]
- 38.Johnstone RW, Wei W, Greenway A, Trapani JA. 2000. Functional interaction between p53 and the interferon-inducible nucleoprotein IFI 16. Oncogene 19:6033–6042. doi: 10.1038/sj.onc.1204005. [DOI] [PubMed] [Google Scholar]
- 39.Hertel L, Rolle S, De Andrea M, Azzimonti B, Osello R, Gribaudo G, Gariglio M, Landolfo S. 2000. The retinoblastoma protein is an essential mediator that links the interferon-inducible 204 gene to cell-cycle regulation. Oncogene 19:3598–3608. doi: 10.1038/sj.onc.1203697. [DOI] [PubMed] [Google Scholar]
- 40.Kalamvoki M, Roizman B. 2014. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc Natl Acad Sci U S A 111:E611–E617. doi: 10.1073/pnas.1323414111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parker ZM, Murphy AA, Leib DA. 2015. The role of the DNA sensor STING in protection from lethal infection following corneal and intracerebral challenge with HSV-1. J Virol 89:11080–11091. doi: 10.1128/JVI.00954-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuchet-Lourenço D, Anderson G, Sloan E, Orr A, Everett RD. 2013. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J Virol 87:13422–13432. doi: 10.1128/JVI.02474-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orzalli MH, Broekema NM, Knipe DM. 2016. Relative contributions of herpes simplex virus 1 ICP0 and vhs to loss of cellular IFI16 vary in different human cell types. J Virol 90:8351–8359. doi: 10.1128/JVI.00939-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, Mettenleiter T, Chen ZJ, Knipe DM, Sandri-Goldin RM, Enquist LW, Hartmann R, Mogensen TH, Rice SA, Nyman TA, Matikainen S, Paludan SR. 2016. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J 35:1385–1399. doi: 10.15252/embj.201593458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sandri-Goldin RM. 2011. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol 6:1261–1277. doi: 10.2217/fmb.11.119. [DOI] [PubMed] [Google Scholar]
- 47.Taddeo B, Esclatine A, Roizman B. 2004. Post-transcriptional processing of cellular RNAs in herpes simplex virus-infected cells. Biochem Soc Trans 32:697–701. doi: 10.1042/BST0320697. [DOI] [PubMed] [Google Scholar]
- 48.Taddeo B, Zhang W, Roizman B. 2006. The UL41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc Natl Acad Sci U S A 103:2827–2832. doi: 10.1073/pnas.0510712103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 50.Chou J, Chen JJ, Gross M, Roizman B. 1995. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5− mutants of herpes simplex virus 1. Proc Natl Acad Sci U S A 92:10516–10520. doi: 10.1073/pnas.92.23.10516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xing J, Ni L, Wang S, Wang K, Lin R, Zheng C. 2013. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-κB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol 87:9788–9801. doi: 10.1128/JVI.01440-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kalamvoki M, Deschamps T. 2016. Extracellular vesicles during herpes simplex virus type 1 infection: an inquire. Virol J 13:63. doi: 10.1186/s12985-016-0518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJ. 2012. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol 5:173–183. doi: 10.1038/mi.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Everett RD. 2015. Dynamic response of IFI16 and promyelocytic leukemia nuclear body components to herpes simplex virus 1 infection. J Virol 90:167–179. doi: 10.1128/JVI.02249-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akan P, Alexeyenko A, Costea PI, Hedberg L, Solnestam BW, Lundin S, Hällman J, Lundberg E, Uhlén M, Lundeberg J. 2012. Comprehensive analysis of the genome transcriptome and proteome landscapes of three tumor cell lines. Genome Med 4:86. doi: 10.1186/gm387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Newhart A, Rafalska-Metcalf IU, Yang T, Negorev DG, Janicki SM. 2012. Single-cell analysis of Daxx and ATRX-dependent transcriptional repression. J Cell Sci 125:5489–5501. doi: 10.1242/jcs.110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jurak I, Silverstein LB, Sharma M, Coen DM. 2012. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J Virol 86:10093–10102. doi: 10.1128/JVI.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 59.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol 71:7328–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poon APW, Roizman B. 1997. Differentiation of the shutoff of protein synthesis by virion host shutoff and mutant γ134.5 genes of herpes simplex virus 1. Virology 229:98–105. doi: 10.1006/viro.1996.8425. [DOI] [PubMed] [Google Scholar]
- 61.Liao JC, Lam R, Brazda V, Duan S, Ravichandran M, Ma J, Xiao T, Tempel W, Zuo X, Wang Y-X, Chirgadze NY, Arrowsmith CH. 2011. Interferon-inducible protein 16: insight into the interaction with tumor suppressor p53. Structure 19:418–429. doi: 10.1016/j.str.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, Chandran B. 2014. IFI16 restricts HSV-1 replication by accumulating on the HSV-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog 10:e1004503. doi: 10.1371/journal.ppat.1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. 2013. Roles of p53 in herpes simplex virus 1 replication. J Virol 87:9323–9332. doi: 10.1128/JVI.01581-13. [DOI] [PMC free article] [PubMed] [Google Scholar]