

ABSTRACT
In order to track the fate of HIV-1 particles from early entry events through productive infection, we developed a method to visualize HIV-1 DNA reverse transcription complexes by the incorporation and fluorescent labeling of the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) into nascent viral DNA during cellular entry. Monocyte-derived macrophages were chosen as natural targets of HIV-1, as they do not divide and therefore do not incorporate EdU into chromosomal DNA, which would obscure the detection of intranuclear HIV-1 genomes. Using this approach, we observed distinct EdU puncta in the cytoplasm of infected cells within 12 h postinfection and subsequent accumulation of puncta in the nucleus, which remained stable through 5 days. The depletion of the restriction factor SAMHD1 resulted in a markedly increased number of EdU puncta, allowing efficient quantification of HIV-1 reverse transcription events. Analysis of HIV-1 isolates bearing defined mutations in the capsid protein revealed differences in their cytoplasmic and nuclear accumulation, and data from quantitative PCR analysis closely recapitulated the EdU results. RNA fluorescence in situ hybridization identified actively transcribing, EdU-labeled HIV-1 genomes in productively infected cells, and immunofluorescence analysis confirmed that CDK9, phosphorylated at serine 175, was recruited to RNA-positive HIV-1 DNA, providing a means to directly observe transcriptionally active HIV-1 genomes in productively infected cells. Overall, this system allows stable labeling and monitoring of HIV genomic DNA within infected cells during cytoplasmic transit, nuclear import, and mRNA synthesis.
IMPORTANCE The fates of HIV-1 reverse transcription products within infected cells are not well understood. Although previous imaging approaches identified HIV-1 intermediates during early stages of infection, few have connected these events with the later stages that ultimately lead to proviral transcription and the production of progeny virus. Here we developed a technique to label HIV-1 genomes using modified nucleosides, allowing subsequent imaging of cytoplasmic and nuclear HIV-1 DNA in infected monocyte-derived macrophages. We used this technique to track the efficiency of nuclear entry as well as the fates of HIV-1 genomes in productively and nonproductively infected macrophages. We visualized transcriptionally active HIV-1 DNA, revealing that transcription occurs in a subset of HIV-1 genomes in productively infected cells. Collectively, this approach provides new insights into the nature of transcribing HIV-1 genomes and allows us to track the entire course of infection in macrophages, a key target of HIV-1 in infected individuals.
KEYWORDS: fluorescent-image analysis, human immunodeficiency virus, macrophages
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) infection can be viewed as occurring in early (entry) stages, including reverse transcription (RT), nuclear import, and the integration of the viral genome, and late (productive) stages, including viral transcription, protein synthesis, assembly, and release. After fusion with the cellular plasma membrane, the conical lattice core, composed of the genomic RNA and RT machinery encapsidated by the HIV-1 capsid (CA) protein, is delivered into the cytoplasm. While the virus transits toward the nucleus, it reverse transcribes its RNA genome into DNA and sheds the capsid lattice in a process known as uncoating. The majority of core uncoating appears to occur early following viral fusion; however, some CA is thought to remain associated with the reverse transcription complex (RTC), affecting its translocation through the nuclear pore and chromosomal targeting in the nucleus. Following viral integration, productive infection begins with the initiation of transcription mediated by host and viral proteins, resulting in the high-level expression of HIV-1 proteins and, ultimately, the budding of assembled virion particles. Understanding the connection of these early and late stages of HIV-1 infection is important to uncovering key biological aspects of the HIV-1 life cycle; however, there are few approaches that allow tracking of viral complexes through early and late stages of infection.
Although much is understood about the steps of viral membrane fusion to target cells in early HIV-1 infection, less is known about the sequence of events following the release of the viral particle into the target cell cytoplasm. It has become increasingly clear through both biochemical and imaging assays that reverse transcription and uncoating are closely linked, as specific CA mutations result in changes in the rates of uncoating and RT (1–3). Extensive studies examining the spatial and temporal aspects of RT and uncoating during infection have lent further evidence to this connection. A number of studies suggested that HIV-1 RT occurs during CA core disassembly in the cytoplasm during transit toward the nucleus (1, 4–7), whereas others suggested that that completion of HIV-1 RT signals the virus to uncoat while docked at the nuclear pore (8). Interestingly, in addition to modulating uncoating and RT, the interaction of CA with host proteins can dictate the nuclear import pathway and integration site selection, suggesting that CA may play a role in the nucleus of infected cells (9–12). Support for this finding has been observed in several recent imaging studies in which CA was shown to be associated with HIV-1 in the nuclei of infected cells (13–15).
Once in the nucleus, HIV-1 RT products can take a variety of forms (16), the most abundant of which in infected CD4+ T cells is linear DNA, the substrate for HIV-1 integration (17). While the majority of RT products stably integrate into the host genome, a proportion of HIV-1 genomes can adopt nonproductive, unintegrated forms like 1- and 2-long terminal repeat (LTR) circles (18, 19). Following integration, proviral transcription is initiated by cellular RNA polymerase II transcription factors, typically by NF-kappa B complexes. The synthesis of the viral transcriptional transactivator protein Tat dramatically increases transcription by recruiting the elongation complex P-TEFb to its promoter by directly binding cyclin T1 (CycT1), a subunit of P-TEFb (20). Additionally, the phosphorylation of serine 175 on the cyclin-dependent kinase 9 (CDK9) subunit of P-TEFb has been shown to assist in the recruitment of Tat/P-TEFb complexes to the HIV-1 promoter (21). While high-level HIV-1 transcription results in productive infection, the virus may also exist in a latent state, in which the provirus is integrated but not expressed.
Numerous refinements of imaging approaches have allowed the detection and localization of nuclear HIV-1 genomic structures, including the use of fluorescently tagged proteins associated with viral preintegration complexes (PICs) (22–25), DNA fluorescence in situ hybridization (FISH) (13, 26), staining of surrogate markers of DNA damage following the cleavage of a specific restriction site within the integrated provirus (27), and the incorporation of the thymidine nucleoside analog 5-ethynyl-2′-deoxyuridine (EdU) and subsequent fluorescent labeling (15). These approaches have provided valuable insights into intranuclear transport and integration site selection in infected cell nuclei. RNA FISH approaches have been utilized to monitor HIV-1 expression at the single-cell level in samples from infected patients, providing new insights in the tissue distribution of productively infected cells (28) and, when combined with DNA FISH, potential latent cell reservoirs in the body (29).
In this study, we developed an HIV genomic DNA labeling strategy combined with immunolabeling and RNA FISH to track HIV-1 genomes from early entry through integration and productive infection in infected cells. We utilized EdU incorporation into HIV-1 DNA followed by fluorescent click chemistry labeling (30) to track the early association of CA and HIV-1 DNA during infection of in vitro-cultured, primary monocyte-derived macrophages (MDM). We observed robust and specific labeling of HIV-1 RT products in the cytoplasm and nuclei of infected macrophages. EdU labeling revealed differences in reverse transcription and nuclear import phenotypes for defined HIV-1 CA mutants during early uncoating and nuclear entry events. Importantly, the EdU signal remained stable for several days after infection, allowing the identification of RT products in productively infected MDM. Remarkably, we were able to distinguish actively transcribing HIV-1 DNA from other inactive forms of intranuclear genomic structures by using a combination of EdU and RNA FISH techniques combined with immunofluorescence analysis of cellular transcription factors. This study for the first time connects fluorescence tracking of early and late stages of HIV-1 infection in MDM and directly identifies individual, actively transcribing HIV-1 genomes in productively infected cells. We demonstrate that a significant fraction of intranuclear genomic structures in both productively infected cells and nonproducing cells in cultures are not undergoing detectable transcription, suggesting that a significant number of integrated HIV-1 genomes in these cells either are in a latent state or are not competent for active transcription. These results provide new insights into the intracellular life cycle of HIV-1 and characterize a new technique that can aid in the further understanding of HIV infection.
RESULTS
EdU labels nascent, reverse-transcribed HIV-1 DNA in intracellular virion particles.
We first asked whether EdU could be incorporated into nascent HIV-1 DNA. Because dividing cells incorporate EdU into chromosomal DNA during cell division and therefore could mask the identification of labeled, intranuclear HIV-1 DNA, we decided to monitor EdU incorporation in in vitro-cultured MDM. The use of MDM provided three distinct advantages in our studies: (i) macrophages are natural targets of HIV-1 infection in vivo (31, 32), (ii) the cells are terminally differentiated and thus will not undergo cell division and consequent nuclear EdU incorporation, and (iii) we can control the level of deoxynucleoside triphosphates (dNTPs) in the cells by depleting sterile alpha motif and histidine/aspartic acid domain-containing protein 1 (SAMHD1) by the delivery of simian immunodeficiency virus (SIV) viral protein x (Vpx), which binds SAMHD1 and directs its proteolytic degradation (33, 34). In our experiments, Vpx is delivered by vesicular stomatitis virus G (VSV-G)-pseudotyped SIV virus-like particles (VLP) (subsequently referred to as SIV-VLP) that are defective for SIV genome packaging and efficiently deliver Vpx into target cells upon fusion and cytosolic delivery of the VLP contents (35).
We infected MDM with or without SIV-VLP for 16 h and washed and then infected the cells with a single-round HIV-1 strain (HIVLAI∂env), pseudotyped with the VSV-G glycoprotein, in the presence of EdU for 24 h. The cells were then fixed, EdU was fluorescently labeled (30), and the samples were subsequently immunostained for HIV-1 CA and nuclear envelope lamin proteins (Fig. 1). At 24 h postinfection (p.i.), we observed distinct, bright EdU puncta in HIV-1-infected MDM cultured without or with SIV-VLP (Fig. 1A and B). In MDM cultured without SIV-VLP, we found on average 1.6 total EdU puncta per cell and 0.7 nuclear puncta. As expected, MDM cultured with SIV-VLP prior to HIV-1 infection had significantly higher levels of total cellular and nuclear HIV: 6.4 and 4.4 puncta, respectively (Fig. 1C and D). Control samples in which MDM cultured with SIV-VLP were not infected with HIV or in which MDM cultured with SIV-VLP were infected with HIV without EdU had no detectable fluorescence signal (Fig. 1C and D), indicating that background incorporation of EdU into cellular (nuclear/mitochondrial) DNA was undetectable, allowing the unambiguous identification of EdU-labeled HIV-1 DNA in cytoplasmic and intranuclear compartments.
FIG 1.

Incorporation of EdU into HIV-1 particles in infected MDM. MDM were cultured without SIV-VLP (−Vpx) or with SIV-VLP (+Vpx) for 16 h and subsequently infected with VSV-G-pseudotyped HIVLAI∂env (HIV) in the presence of 10 μM EdU. (A and B) Cells were fixed after 24 h, labeled for EdU, stained for HIV-1 CA and nuclear lamin A/C, and imaged. (A) Representative z-sections of individual cells. Arrows indicate colocalized CA and EdU. (B, left) Representative z-sections of cells within a single field. (Right) Boxed cells (i and ii) showing enlarged images the same single z-sections of specified nuclei. Arrows denote colocalized intranuclear EdU and CA, and arrowheads denote EdU not colocalized with CA. (C and D) Numbers of total (C) and nuclear (D) EdU puncta per cell were quantified by using Imaris software. Mean values are indicated above the plots. No EdU, SIV-VLP-treated, HIV-1-infected cultures without EdU; No HIV, SIV-VLP-treated, EdU-containing cultures without HIV-1. (E) Percentage of EdU colocalized with CA in the nuclei of infected MDM. Fifty to seventy-five cells were analyzed under each condition in panels C to E. Data are representative of results from at least 3 independent experiments. A Mann-Whitney test was used to test for statistical significance in panel E (*, P < 0.05). Bars, 2.5 μm (A), 10 μm (B, left), and 5 μm (B, right).
We found approximately half of EdU puncta within the cytoplasm of HIVLAI∂env-infected MDM colocalized with the HIV-1 CA protein (Fig. 1A). This result is consistent with data from other studies that suggested that reverse transcription might occur within incompletely disassembled viral cores (1, 5, 6). Notably, a significant fraction of EdU puncta within the nuclei of infected MDM also colocalized with the HIV-1 CA protein (Fig. 1B). Quantification of EdU and CA colocalization demonstrated that 35% of nuclear EdU puncta colocalized with HIV-1 CA in MDM not cultured with SIV-VLP prior to HIV-1 infection, while only 15% colocalized with CA in MDM that had Vpx delivered prior to infection (Fig. 1E). The decreased colocalization of EdU and CA that we observed in MDM cultured with SIV-VLP may be a consequence of accelerated reverse transcription due to increased dNTP pools and thus a faster uncoating of the RTCs. These data demonstrating the association of HIV-1 DNA and CA in the nucleus of HIV-1-infected MDM are consistent with results from previous studies (13, 15) and further validate the model that capsid fragments assist the HIV RTC in entering the nucleus during primary cell infection.
To confirm that EdU incorporation was dependent on reverse transcription, MDM were infected with HIVLAI∂env encoding green fluorescent protein (GFP) in the nef open reading frame (HIVLAI∂env-GFP) in the presence of reverse transcriptase and integrase inhibitors. Figure 2 shows that the HIV-1 reverse transcriptase inhibitor nevirapine (NVP) potently blocks the accumulation of EdU puncta, whereas the HIV-1 integrase inhibitor raltegravir (RAL) does not. In these experiments, RAL and NVP blocked >90% of productive infection, and NVP treatment resulted in nearly undetectable levels of total and nuclear EdU per cell, whereas RAL had virtually no effect on EdU incorporation (Fig. 2A to D). These data confirm that we can equally detect integrated as well as unintegrated HIV-1 genomes in the nuclei of infected MDM, unlike a previous study that labeled intranuclear HIV-1 DNA by using FISH, in which unintegrated DNA was detected more efficiently than integrated provirus (13). In additional experiments, we confirmed that MDM cultured with SIV-VLP lacking Vpx prior to HIV-1 infection resulted in EdU incorporation similar to that in MDM not treated with SIV-VLP, indicating that SIV-VLP infection alone does not result in EdU incorporation (Fig. 2E).
FIG 2.

EdU incorporation is dependent upon RT but not integration and is not the result of Vpr or SIV-VLP. MDM were cultured with SIV-VLP for 16 h and subsequently infected with VSV-G-pseudotyped HIVLAI∂env that expresses GFP in place of the nef gene (HIV-GFP). (A) MDM were washed at 16 h p.i., and cells were labeled for EdU, stained for lamin A/C, and imaged at 48 h p.i. (A) Representative z-sections of nuclei with no drug (HIV-GFP), 10 μM RAL, or 1 μM NVP. (B) Percentage of GFP+ cells (n = ∼200 under each condition). (C and D) Numbers of total (C) and nuclear (D) EdU puncta per cell quantified by using Imaris software (n = ∼30 cells under each condition). Graphs in panels B to D are representative of results from 3 independent experiments. (E) MDM were cultured for 16 h without (No SIV) or with (SIV-Vpx−) SIV-VLP containing a null vpx gene. MDM were infected and imaged as described above for panel A, and the total number of EdU puncta per cell was quantified. (F and G) MDM were cultured for 16 h with SIV-VLP containing a null vpr gene, infected with HIV-GFP containing a functional (Vpr+) or null (Vpr−) vpr gene in the presence of EdU, washed at 16 h, and cultured for a total of 4 days. Cells were quantitatively imaged for the percentage of GFP+ cells (F) and the number of intranuclear EdU puncta per cell (G). Bar, 2.5 μm (A). Error bars represent standard errors of the means from 3 random fields in panels B and F.
Recent studies suggest that HIV-1 viral accessory protein R (Vpr) induces chromosomal damage and the cellular DNA damage response (DDR) (36–38). In our experiments, Vpr is incorporated into both SIV-VLP and HIV-1 virions. To rule out the possibility that the intranuclear EdU signal is a result of the Vpr-triggered incorporation of EdU into newly repaired chromosomal DNA, we cultured MDM with SIV-VLP produced from an SIV strain containing functional Vpx and a null vpr gene prior to HIV-1 infection. MDM were then infected with HIVLAI∂env-GFP strains carrying either a wild-type (WT) (Vpr+) or a null (Vpr−) vpr gene. MDM infected with Vpr− or Vpr+ HIVLAI∂env-GFP had similar levels of productive infection and identical amounts of nuclear EdU puncta at 4 days p.i. (Fig. 2F and G). Moreover, Vpr+ and Vpx+ SIV-VLP alone did not induce detectable EdU incorporation (Fig. 2C and D), confirming that EdU incorporation is not a consequence of Vpr expression or incorporation into incoming VLP. We used an SIV-VLP strain containing both Vpx and Vpr in subsequent experiments, as Vpr had no effect on EdU incorporation.
Fluorescence in situ hybridization confirms that EdU puncta identify HIV genomic DNA.
To further verify that EdU puncta in infected MDM during early infection accurately identify HIV-1 reverse transcription products, we subjected EdU-labeled, HIV-1-infected cells to DNA FISH to detect HIV-1 DNA. At 24 h p.i., approximately 75% of the cytoplasmic EdU puncta were colocalized with the DNA FISH signal, confirming that EdU puncta consisted of HIV-1 DNA (Fig. 3A). However, few intranuclear EdU puncta colocalized with the DNA FISH signal. We presume that this infrequent nuclear signal might have been due to the inefficient denaturation and hybridization of integrated proviral DNA in these experiments, as other studies have had similar difficulties in labeling integrated proviruses by DNA FISH (13). Together with the data illustrating the colocalization of specific HIV-1 DNA sequences with EdU, our data demonstrate that EdU puncta specifically identify locations of HIV-1 DNA in infected MDM, as EdU incorporation is dependent on only HIV-1 infection and RT and is not dependent on integration, culture with SIV-VLP, or Vpr delivery.
FIG 3.
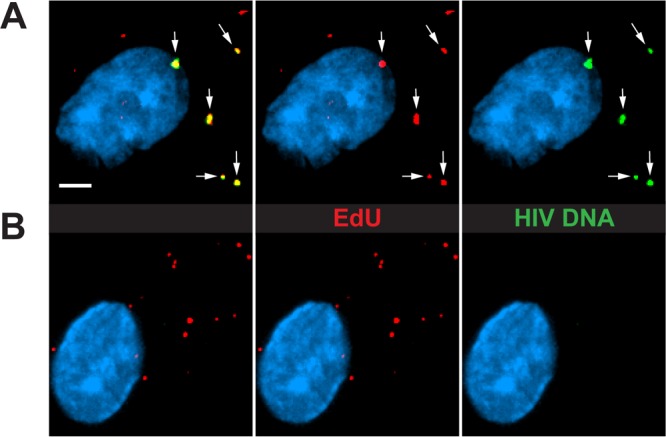
EdU puncta specifically identify sites of reverse-transcribed HIV DNA. MDM were cultured with SIV-VLP for 16 h, washed, and infected with HIV. At 24 h p.i., cells were fixed, labeled for EdU, stained for nuclear DNA (cyan), and hybridized with fluorescent HIV-1 DNA probes. Cells were then imaged at a high magnification to detect the colocalization of EdU and DNA FISH probes. (A) Representative MDM that were hybridized with DNA FISH probes. White arrows indicate the colocalization of EdU and DNA FISH probes. (B) Representative image of an MDM not hybridized with DNA probes. All images are three-dimensionally rendered whole-cell volumes. Bar, 2.5 μm.
Imaging of HIV-1 DNA localization for detection of reverse transcription and nuclear translocation defects conferred by HIV-1 CA mutants.
Previous studies have shown that certain CA mutations impact HIV-1 reverse transcription and/or nuclear import functions. The N74D and Q63A/67A mutations are of particular interest, in that they have been shown to be defective in reverse transcription or nuclear translocation (2, 9, 39, 40). Figure 4 shows data from a series of experiments in which MDM cultured with SIV-VLP were infected with HIVLAI∂env-GFP containing WT or mutant (Q63A/67A or N74D) CA proteins. At 4 days p.i., approximately 60% of MDM infected with WT CA-containing HIV-1 were GFP positive (GFP+), whereas HIV-1 containing either CA mutation was significantly less infectious (Q63A/67A, 23%; N74D, 14%) (Fig. 4A). Quantification of EdU-labeled HIV-1 DNA revealed that while HIV-1 strains with WT and Q63A/67A CA proteins had similar levels of total cellular EdU at 24 h p.i., the level for HIV-1 containing N74D CA was significantly reduced (7.2 and 6.1 EdU puncta/cell for the WT and the Q63A/67A mutant, respectively, versus 4.2 EdU puncta/cell for the N74D mutant) (Fig. 4B). The level of nuclear EdU, on the other hand, was significantly reduced in both mutants and closely mirrored the infectivity defects in the respective mutants. The Q63A/67A CA mutant had 2.5-fold reduced EdU and 2.6-fold reduced GFP levels, whereas the N74D CA mutant had 5.4-fold reduced EdU and 4.2-fold reduced GFP levels. To analyze the efficiency with which each CA mutant entered the nucleus, we quantified the percentage of HIV-1 genomes in each individual cell that were located in the nucleus. Thus, each point in Fig. 4D represents the percentage of EdU that is nuclear in an individual cell, and the means of these percentages are also indicated in Fig. 4D. HIV-1 containing WT CA was much more efficient in nuclear entry, with 70% of the total EdU being intranuclear on average, compared to only 19% and 4.5% nuclear EdU in HIV-1 containing Q63A/67A and N74D CAs, respectively (Fig. 4D). Taken together, these data indicate that although the Q63A/67A HIV-1 CA mutant reverse transcribes to the same extent as HIV-1 containing WT CA, it is defective for nuclear entry in comparison to virus with WT CA. On the other hand, HIV-1 containing N74D CA appeared to be significantly defective for both RT and nuclear entry based on the EdU labeling approach.
FIG 4.

CA mutations impact reverse transcription, nuclear import, and productive infection of MDM. (A) MDM were cultured with SIV-VLP for 16 h, washed, and infected with equivalent amounts [75 ng CA(p24)/ml] of HIV-GFP (WT) or HIV-GFP carrying the CA point mutant Q63A/67A or N74D in the presence of 10 μM EdU. WT CA virus in the presence of 1 μM nevirapine (WT +NVP) was added as a control. (A) MDM were washed at 24 h p.i., and the percentage of GFP+ MDM was quantified at 5 days p.i. Approximately 1,000 cells were counted under each condition. Error bars represent standard errors of the means from 3 random fields. (B and C) MDM were infected in parallel under the same conditions as those described above for panel A, and at 24 h p.i., MDM were fixed, labeled for EdU, stained for lamin A/C, and imaged. Numbers of total (B) and nuclear (C) EdU puncta per cell were quantified. (D) Using data shown in panels B and C, the percentage of total EdU puncta per cell that was nuclear was calculated. Each point indicates the percentage within a single cell. The means for all of the cells were quantified as indicated above. Approximately 70 cells were imaged under each condition in panels B to D. (E and F) MDM were infected in parallel under conditions identical to those described above for panel A but without EdU, and at 24 h p.i., total DNA was isolated from cells. Levels of HIV-1 late RT products (E) and 2-LTR circles (F) per human genome were quantified by qPCR by normalization to a standard curve of an HIV-GFP plasmid diluted in total MDM DNA. Error bars represent standard errors of the means from triplicate samples. All experiments are representative of data from 3 independent experiments. A Mann-Whitney test was used to test for statistical significance. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Quantitative PCR of HIV-1 DNA from infected MDM using previously defined amplicons (Late RT and 2-LTR) (19) showed a strong correlation with the EdU results (Fig. 4E and F). While the levels of total HIV-1 DNA were similar in wild-type and Q63A/67A CA mutant infections, they were significantly decreased following infection with HIV-1 containing N74D CA. 2-LTR circular DNA levels, which are used as a surrogate for RTC nuclear import, were significantly decreased in both CA mutants, reflecting the respective defects in the accumulation of reverse transcription products (N74D) and nuclear import (Q63A/67A). Of note, the total amount of HIV-1 DNA per cell measured by the total amount of EdU per cell was within approximately 2-fold of the total amount of HIV-1 DNA approximated by the number of late RT copies per human genome, indicating that the sensitivity of the EdU labeling approach is comparable to that of quantitative PCR (qPCR) in detecting HIV-1 DNA copies in infected MDM (Fig. 4B and E). For example, there were 7.2 EdU puncta per cell (Fig. 4B) and 17.9 late RT copies per genome (Fig. 4E) when MDM were infected with the WT CA virus, giving an EdU labeling efficiency of approximately 40% (7.2/17.9). Using qPCR to quantify the amount of 2-LTR circles per human genome, there was a 3.8-fold decrease in the level of the Q63A/67A CA mutant virus and an 8-fold decrease in N74D CA mutant HIV-1 compared to HIV-1 containing WT CA (Fig. 4F). Interestingly, the fold differences between the numbers of nuclear EdU puncta per cell were much lower (2.5-fold lower in the Q63A/67A mutant and 5.4-fold lower in the N74D mutant) (Fig. 4C). These data suggest that while 2-LTR qPCR is a useful surrogate to measure the extent of nuclear entry, direct observation by EdU labeling allows the detection of HIV-1 DNA regardless of its particular topology and can be used to quantify cytoplasmic and nuclear forms of HIV RTCs.
EdU-labeled HIV-1 is present in both productively and nonproductively infected MDM.
In the above-described experiments, we used EdU labeling to track HIV-1 DNA from the cytoplasm into the nucleus of infected MDM following HIV-1 entry. We next used this technique to determine whether the EdU signal persisted in cells containing transcriptionally active HIV-1 DNA. We quantified cytoplasmic and nuclear EdU accumulation over several days in culture. In these experiments, we found that productive infection could be detected at 2 days p.i. and leveled off after 4 to 5 days (Fig. 5A), consistent with the delayed infection rate observed in macrophages compared to cell lines and CD4 T cells (41, 42). The number of cytoplasmic EdU puncta increased initially between 12 and 24 h p.i. and decreased over time from 24 to 96 h p.i. (Fig. 5B). While the level of cytoplasmic EdU trended downwards, the number nuclear EdU puncta remained mostly stable within the nucleus, only slightly increasing over time between 12 and 96 h p.i. (Fig. 5C). This decrease in cytoplasmic HIV-1 DNA and the concomitant increase in nuclear HIV-1 DNA levels may reflect progressive nuclear entry and integration over time.
FIG 5.

Tracking HIV-1 nuclear import over time in productively and nonproductively infected MDM. (A) MDM were cultured with SIV-VLP for 16 h, infected with HIV-GFP for 12 h, washed, either fixed or cultured for the indicated times prior to fixation, and imaged for GFP expression. No HIV, SIV-VLP-treated and mock-infected cells. (B and C) MDM infected as described above for panel A were labeled for EdU, stained for lamin A/C (cyan), and imaged. The numbers of cytoplasmic (B) and nuclear (C) EdU puncta per cell were quantified at the indicated time points. (D, left) Representative z-section of cells in a single field at 96 h p.i. (Right) Boxed cells showing enlarged images of the same z-sections from the specified GFP+ (i) and GFP− (ii) cells. (E and F) The numbers of cytoplasmic (E) and nuclear (F) EdU puncta per cell stratified into GFP− and GFP+ MDM were extracted from the data in panels B and C by separating MDM based on their expression of GFP. (G) The percentage of GFP− and GFP+ MDM that contained any EdU was quantified. Cells under all conditions were quantified by using 25 to 30 cells except for panel A, where ∼1,000 cells were quantified for GFP expression. Error bars represent the standard errors from 6 to 9 random fields in panel A or from ∼30 cells in panels E and F. All data are representative of results from three independent experiments. A Mann-Whitney test was used to test for statistical significance in panels B and C, and Student's t test was used to determine statistical significance in panel F. *, P < 0.05; **, P < 0.01; ****, P < 0.0001. Bars, 5 μm (B, left) and 2.5 μm (B, right).
The analysis shown in Fig. 5B and C quantified all MDM regardless of whether they were productively infected (GFP+) or not (GFP negative [GFP−]). When we imaged cells for EdU and GFP simultaneously, we found many EdU puncta in both GFP-positive and -negative cells (Fig. 5D), indicating that we can detect HIV-1 DNA in productively (GFP+) and nonproductively (GFP−) infected MDM. Because we detected nuclear HIV-1 DNA in GFP+ and GFP− MDM, we next quantified the amounts of cytoplasmic (Fig. 5E) and nuclear (Fig. 5F) EdU puncta in GFP+ versus GFP− MDM. Because MDM did not express GFP until after 48 h, the numbers of EdU puncta in GFP+ and GFP− MDM could be compared only after this time. The number of cytoplasmic HIV-1 DNA copies as marked by EdU fell from approximately 1.8 puncta per cell at 48 h to nearly undetectable levels at the end of the time course in both GFP+ and GFP− MDM (Fig. 5E, black and gray bars). Interestingly, at each time point after 48 h, the number of intranuclear EdU puncta remained significantly higher in GFP+ MDM than in GFP− MDM (Fig. 5F).
We next analyzed the data shown in Fig. 5D and E to determine the percentage of productively infected cells containing at least one intranuclear EdU punctum as a measure of the sensitivity of the assay (Fig. 5F). After 48 h, essentially all GFP+ MDM had at least one intranuclear EdU punctum (Fig. 5F, gray bars). Somewhat unexpectedly, however, we found that 50% to 60% of GFP− MDM contained at least some nuclear HIV DNA (Fig. 5F, black bars), indicating that nonproductively infected cells can harbor transcriptionally silent nuclear HIV-1 DNA. Taken altogether, these data indicate that (i) EdU labeling of HIV-1 DNA sensitively detects intranuclear HIV DNA prior to and during productive infection, (ii) productively infected MDM have on average more intranuclear HIV DNA than do nonproductively infected MDM despite both MDM types losing cytoplasmic HIV DNA over time, and (iii) about half of the nonproductively infected MDM contain some amount of intranuclear HIV-1 DNA.
RNA FISH detects a subset of EdU-labeled HIV-1 genomes that are transcriptionally active.
We were interested in whether we could image transcriptionally active HIV-1 DNA. By using fluorescently labeled antisense RNA probes that hybridized to 5′- and 3′-LTR sequences, HIV-1 mRNA was found abundantly in the nucleus and cytoplasm of MDM that were productively infected (GFP+) but not in nonproductively infected MDM (GFP−) (Fig. 6A, top). Cells treated with RAL during infection did not show any GFP or HIV RNA expression, and they notably lacked any HIV-1 mRNA that may have been associated with incoming HIV virions (Fig. 6A, bottom). We found a striking colocalization of a subset of nuclear EdU with HIV RNA in productively infected, GFP+ MDM (Fig. 6B). The colocalized HIV DNA showed an almost complete overlap of robust and discrete HIV RNA signals within the nuclei of productively infected MDM. These RNA foci represented sites of intense, ongoing transcription, as their signal was much brighter than the surrounding cytoplasmic HIV-1 RNA signal, suggesting that we can more readily detect areas of high-level transcription.
FIG 6.

HIV RNA associates with a subset of nuclear HIV DNA. MDM were cultured with SIV-VLP for 16 h and infected without (−RAL) or with (+RAL) the integrase inhibitor RAL in the presence of 10 μM EdU. (A) MDM were washed at 16 h p.i., and at 5 days p.i., cells were fixed, hybridized to Stellaris RNA FISH probes, and imaged for GFP. Images are three-dimensionally rendered whole-cell volumes. (B) GFP+ cells were imaged at high magnification to detect the intranuclear colocalization of EdU and HIV-1 RNA. Top and bottom rows show z-sections of two separate GFP+ cells. Arrows indicate colocalized EdU and HIV RNA. (C) The number of nuclear EdU puncta per cell for RAL-untreated (HIV-GFP) and RAL-treated (RAL) cells was quantified at 5 days. (D) Percentage of nuclear EdU per cell that colocalized with HIV RNA. Data in panels C and D represent values for GFP+ MDM under RAL-untreated (HIV-GFP) conditions and GFP− MDM under RAL-treated conditions. A Mann-Whitney test was used to test for statistical significance in panel D. ****, P < 0.0001. Bars, 25 μm (A) and 2.5 μm (B).
The numbers of nuclear EdU puncta per cell in RAL-untreated and -treated MDM were not significantly different, confirming what we found in the above-described experiments (Fig. 2C and D and 6C). Nuclear HIV-1 DNA was defined by its colocalization with Hoechst nuclear staining, as this analysis showed results similar to those of lamin protein immunostaining when used to separate nuclear and cytoplasmic compartments (data not shown). We found that approximately 34% of the nuclear HIV-1 DNA per cell colocalized with HIV-1 mRNA in productively infected MDM, while only 2% of nuclear HIV-1 DNA was colocalized with HIV-1 mRNA in RAL-treated cells (Fig. 6D). Our colocalization data demonstrate that approximately one-third of intranuclear HIV-1 DNA is actively transcribing detectable HIV-1 mRNA during productive infection of MDM. However, as the mRNA foci primarily represent areas of intense transcription, we may not be able to detect HIV-1 DNA transcribing at a low level. This leaves the possibility that some EdU-labeled HIV-1 DNA not colocalized with RNA may represent viral genomes that are transcribing at a level below the limit of detection of our assay.
We identified a subset of EdU-labeled HIV-1 DNA that colocalized with CycT1 in the nuclei of GFP+, productively infected MDM (Fig. 7A). CycT1 is a C-type regulatory cyclin that comprises part of the heterodimer of the positive transcriptional elongation factor (P-TEFb), a transcription factor complex recruited to elongating HIV-1 transcripts by Tat (43). Thus, the colocalization of CycT1 and EdU suggested that P-TEFb was recruited to sites of HIV-1 DNA. However, as CycT1 was fairly ubiquitous throughout the nucleus of productively infected MDM, we sought a different immunostain that would display more specific and robust colocalization with HIV-1 DNA. Phosphorylation of serine 175 (pSer175) of the CDK9 serine/threonine kinase, the other monomer, along with CycT1, composing the heterodimer P-TEFb, was previously shown to enhance the interaction of P-TEFb with Tat, resulting in Tat-dependent activation of HIV-1 transcription (21). The colocalization of EdU-labeled HIV-1 DNA and pSer175 was observed for a subset of EdU in the nuclei of GFP+ MDM (Fig. 7B). This staining was more robust and less ubiquitous than with CycT1 and gave a more pronounced colocalization with HIV-1 DNA, demonstrating that the pSer175 modification on P-TEFb is a more specific marker for transcriptionally active P-TEFb.
FIG 7.

Phosphorylated P-TEFb associates with actively transcribing HIV-1 DNA. MDM were cultured with SIV-VLP for 16 h and infected with HIV-GFP in the presence of 10 μM EdU. (A) Cells were washed at 16 h p.i., and at 5 days p.i., cells were EdU labeled, stained for CycT1 and nuclear DNA (cyan), and imaged. The image is a three-dimensionally rendered whole-cell volume. Arrows indicate colocalized cyclin T1 and EdU. (B) In a parallel experiment, infected MDM were fixed at 5 days p.i., EdU labeled, and stained for phosphorylated serine 175 of P-TEFb (pSer175) and nuclear DNA (cyan). Single z-sections of two separate cells are shown, with arrows indicating colocalized EdU and pSer175. (C) The percentage of nuclear EdU puncta per cell which colocalized with pSer175 foci was quantified under both GFP+ RAL-untreated (HIV) and GFP− RAL-treated (RAL) conditions from multiple images. (D) MDM were infected as described above for panel A but with HIV lacking GFP, and at 5 days p.i., cells were fixed and labeled for EdU. Following EdU labeling, MDM were hybridized overnight to Stellaris RNA FISH probes as described in the legend of Fig. 6A and subsequently stained for pSer175 and nuclei (cyan). The image shows a single z-section of an RNA-expressing MDM, with arrows indicating the location of EdU that colocalized with both RNA and pSer175. (E and F) The percentages of intranuclear EdU puncta per cell that colocalized with RNA alone (E) or colocalized with both pSer175 and HIV RNA (F) were quantified. A Mann-Whitney test was used to determine statistical significance in panels E and F. ****, P < 0.0001. Bars, 2.5 μm (A, B, and D).
Interestingly, approximately one-third of the nuclear HIV-1 DNA per cell colocalized with pSer175 in productively infected MDM, a number similar to that for the colocalization of EdU with HIV RNA (Fig. 7C). This similarity between the colocalizations of pSer175 and RNA with EdU suggests that we can detect the fraction of transcriptionally active HIV in the nuclei of productively infected MDM. However, we also observed colocalization of 21% of nuclear EdU and pSer175 in RAL-treated MDM, suggesting that P-TEFb localized to HIV-1 DNA even in the absence of integration or detectable transcription (Fig. 7C). To identify HIV-1 DNA that was actively transcribing and associated with pSer175-modified P-TEFb, infected MDM were hybridized to RNA probes targeting the 5′ and 3′ LTRs of transcribed mRNAs following EdU click labeling. Following RNA FISH, we immunostained for pSer175 and looked for triple colocalization of HIV-1 DNA with both HIV-1 RNA and pSer175. We observed the specific colocalization of a subset of nuclear EdU with both HIV RNA and pSer175 in productively infected MDM (identified by RNA FISH-positive cells, as illustrated in Fig. 6A) (Fig. 7D). Initial quantification revealed results similar to those seen in Fig. 6C: 30.2% of the nuclear EdU per cell colocalized with RNA alone in productively infected MDM (Fig. 7E). Further analysis revealed that 17.2% of the nuclear EdU per cell colocalized with both pSer175 and HIV-1 mRNA in productively infected MDM, as opposed to the only 1.6% triple colocalization in RAL-treated MDM (Fig. 7F). Taken together, these data demonstrate that fewer than half of the nuclear HIV-1 DNAs in productively infected MDM are actively transcribing viral mRNA, as detected by RNA FISH.
DISCUSSION
In this study, we developed a novel imaging-based approach to quantitatively track HIV-1 genomic DNA through early and late stages of infection in macrophages and identified actively transcribing HIV-1 genomes in productively infected cells. We demonstrated that EdU was incorporated into HIV-1 DNA shortly after entry, and the depletion of the restriction factor SAMHD1 allowed the visualization of EdU-labeled HIV with higher efficiency. Importantly, EdU incorporation was dependent on RT and represented sites of HIV-1 DNA as revealed by DNA FISH hybridization, and EdU incorporation was not a consequence of Vpr, integration, or SIV-VLP treatment (Fig. 2 and 3). Thus, our EdU labeling approach provides a robust and highly specific signal to quantitatively detect HIV-1 genomes within infected MDM during cytoplasmic reverse transcription, nuclear entry, and active transcription from HIV-1 genomes.
HIV-1 CA colocalized with HIV-1 RT products in the cytoplasm of infected MDM early in infection, consistent with previous reports suggesting that reverse transcription of HIV-1 can occur within incompletely disassembled viral cores (1, 5–7, 15). We also observed CA-associated HIV-1 DNA within the nuclei of infected MDM, in line with more recent reports utilizing cell lines as well as MDM (13–15). Together, these data demonstrate that encapsidated virion particles enter the nucleus associated with HIV-1 DNA. These data also lend support to the numerous genetic studies that have found that CA is a key contributor to integration site selection within the nucleus (11, 12, 44–46). We further demonstrated that the colocalization of CA and HIV-1 DNA in the nucleus was significantly decreased in MDM that had Vpx delivered prior to infection. It is thought that the progression of reverse transcription results in instability and, ultimately, the uncoating of HIV-1 CA cores within infected cells (1, 3, 7). We hypothesize that the decreased amount of nuclear CA-containing HIV-1 DNA complexes early in infection of SAMHD1-depleted MDM reflects faster HIV-1 reverse transcription and, subsequently, faster core uncoating. Thus, slow reverse transcription in SAMHD1-containing MDM may result in incomplete uncoating and may lead to the nuclear import of more CA-containing HIV-1 complexes.
Although NVP effectively blocked the incorporation of EdU into reverse-transcribing HIV-1, the use of the HIV-1 integrase inhibitor RAL resulted in levels of EdU puncta per cell similar to those in MDM not treated with any drug (Fig. 2). This is in contrast to the HIV DNA FISH technique used previously by Chin et al. (13), which detected an increase in nuclear HIV-1 DNA levels in MDM treated with an HIV-1 integrase inhibitor compared to those in untreated cells. As noted by those authors, those data may reflect the inefficient detection of integrated HIV-1 DNA by FISH, consistent with our ability to detect more cytoplasmic than nuclear HIV-1 DNA using FISH (Fig. 3). Thus, EdU labeling of HIV-1 DNA provides a more effective technique than DNA FISH for detecting all of the unintegrated and integrated HIV-1 DNA products from entry through integration. Furthermore, by comparing the amount of total cellular EdU (Fig. 4B) to the number of late RT products per genome (Fig. 4E), we estimate that 40% of the products measured by qPCR were labeled by using EdU (7.2 EdU puncta per cell/17.9 late RT products per genome by using the WT CA virus), suggesting that this technique labels approximately half of the completed HIV-1 RT products within infected MDM. Additionally, because we are approximating the number of late RT products per genome by using a standard curve that approximates the human genome size as 3 billion base pairs, there is an inherent error built into the qPCR quantification. This combined with sampling error may help to explain why data from our EdU labeling technique do not exactly match the qPCR results but nevertheless suggests that EdU labeling is nearly as sensitive as qPCR.
EdU labeling of mutant CA-containing viruses confirmed that HIV-1 containing N74D CA is defective for reverse transcription in MDM (40, 47), while HIV-1 containing Q63A/67A CA can reverse transcribe efficiently but is defective for nuclear entry (39) (Fig. 4). While current qPCR techniques use 2-LTR circle levels as a surrogate marker of nuclear entry, EdU labeling likely identifies all forms of HIV-1 RT products within the nucleus, including 1-LTR and linear HIV-1 DNA species. This helped us resolve the efficiency of nuclear entry of all HIV-1 DNA species regardless of their topology. Importantly, because we quantified the percentage of EdU that was nuclear for each individual cell imaged, our data illustrated the decreased efficiency of nuclear entry for N74D and Q63A/67A CA viruses compared to the WT CA virus (Fig. 4D), a finding more specific than what could be achieved by using only raw numbers for late RT products and 2-LTR circles measured by qPCR. These CA mutant studies also revealed a subtler nuclear entry phenotype of HIV-1 containing Q63A/67A CA by using EdU labeling rather than 2-LTR circle analysis (Fig. 4D and F). Together, these data indicate that although Q63A/67A CA mutant HIV-1 RT products are less efficient at nuclear entry than WT CA HIV-1 RT products, they may enter the nucleus in forms that are not able to generate 2-LTR circles, an important detail that is not revealed by qPCR analysis.
Our studies monitoring EdU-labeled HIV-1 within the cytoplasm and nuclei of MDM during early HIV infection exclusively looked at VSV-G-pseudotyped HIV-1. We also successfully labeled CCR5-tropic HIV-1 in MDM using EdU (data not shown), but it was difficult to quantitatively track virions in these cells, as CCR5-tropic viruses fused less frequently and were much less infectious than VSV-G-pseudotyped viruses. Thus, we were unable to quantitatively compare levels of nuclear and cytoplasmic EdU accumulation in CCR5-tropic and VSV-G-pseudotyped HIV-1. However, we predict that entry through endosomes should not affect the quantification of cytoplasmic and nuclear EdU puncta, as VSV-G-pseudotyped HIV-1 would not be expected to have access to EdU within endosomes.
EdU labeling of HIV-1 DNA was also effective in tracking the loss of cytoplasmic EdU over a course of 4 days of infection. Interestingly, HIV-1 RT products were found in nuclei by as soon as 12 h p.i.; however, there was no detectable productive infection until 2 days p.i., demonstrating that productive infection progresses slowly over several days despite HIV-1 DNA already being within the nucleus. In contrast, CD4 T cells progress through integration and productive infection within a much shorter time frame (41, 42). Quantification of intranuclear HIV-1 DNA in productively (GFP+) versus nonproductively (GFP−) infected MDM demonstrated that after the start of productive infection, GFP+ MDM had significantly more intranuclear HIV-1 DNA than did GFP− nuclei. The ability to track EdU in GFP+ and GFP− MDM also demonstrated the strong sensitivity of our assay, as almost 100% of productively infected MDM had some intranuclear EdU. We also observed that around 50% of GFP− MDM harbored some amount of intranuclear HIV-1 DNA. A recent study suggested that a subset of MDM may be resistant to HIV-1 infection due to increased dUTP incorporation into HIV-1 RT products and their subsequent degradation (48). It is therefore possible that the fewer EdU puncta within the GFP− MDM subset may result from the degradation of uracilated HIV-1 RT products. The presence of some nuclear EdU in these nonproductively infected MDM may represent latent proviruses, but further studies are warranted to determine whether these nonproductively infected cells may harbor latent proviruses that can be reactivated, for example, by physiological stimuli or by newly described latency-reversing agents (49). These EdU puncta could also represent either episomal forms of HIV-1 cDNA or integrated, defective proviruses found in other studies (50).
Our subsequent RNA FISH studies demonstrated that a subset of nuclear EdU puncta were strongly associated with HIV-1 RNA. Approximately one-third of the genomes appeared to be actively transcribing, suggesting that there is a large amount of defective, nonproductive HIV-1 DNA in the nucleus. As we were able to detect only high-level HIV-1 transcription, some of the nuclear HIV-1 genomes that did not colocalize with RNA may be transcribing at a level below the limit of detection using RNA FISH. This large amount of what appeared to be nonproductive HIV-1 DNA may also represent MDM harboring increased amounts of abortive HIV-1 DNA compared to those in other cell types, as CD4 T cells have been shown to be killed by pyroptosis following incomplete reverse transcription during abortive infection (51, 52). Consistent with our RNA FISH results, we found that critical components of the active P-TEFb complex were recruited to a subset of HIV-1 DNA. Phosphorylated serine 175 of CDK9 (pSer175) demonstrated a similar colocalization frequency, with RNA FISH hybridization and pSer175 staining strongly staining approximately one-third of EdU puncta. However, in RAL-treated controls, 21% of EdU-labeled HIV-1 DNA was found to be associated with pSer175. We hypothesize that this may be due to the recruitment of transcription factors to HIV-1 DNA even in the absence of integration and detectable transcription, as was reported previously (53, 54). These data also lend support to the hypothesis that even in the absence of integration and detectable GFP expression, some HIV-1 DNA may be transcribing at a low level that is undetectable by RNA FISH. Triple labeling confirmed that a significant amount of EdU was pSer175 positive and RNA positive albeit at a lower percentage than in individual stains. We believe that the decreased triple colocalization may be due to the difficulty in labeling three separate components; however, it directly confirmed that RNA and active P-TEFb are associated with HIV-1 DNA. These results, for the first time, offer the opportunity to directly observe actively transcribing HIV-1 DNA in MDM and provide the opportunity to dissect the molecular machinery present in these complexes.
Our newly developed assay using fluorescent click labeling of EdU-incorporated HIV-1 DNA in SAMHD1-depleted MDM allows imaging of HIV-1 independently of genotype and can help uncover the events of both early and late stages of the intracellular HIV-1 life cycle. Our data support a model in which CA uncoating occurs concomitantly with HIV-1 RT during early infection, and at least some CA remains with HIV-1 DNA within the nucleus of infected MDM. Our data demonstrate that later in infection, HIV-1 RT products stably remain in productively infected and nonproductively infected cells at different rates and in differing quantities, and transcription from HIV-1 genomes occurs in a subset of intranuclear HIV-1 genomes. This approach can be used to further our understanding of the architecture of transcribing HIV-1 and ultimately track the entire course of HIV-1 infection in target cells.
MATERIALS AND METHODS
Viruses.
pLAI∂env (55) is an envelope-deleted proviral construct kindly provided by Michael Emerman. Masahiro Yamashita generously provided pLAI∂env-GFP proviral constructs bearing wild-type CA or CA mutants Q63A/67A and N74D, all of which encode enhanced GFP (eGFP) in place of the Nef open reading frame (2). pSIV3+, kindly provided by Andrea Cimarelli, was derived from the SIVmac251 proviral clone and contains multiple mutations that facilitate the production of VLP that are deficient in envelope expression and genome packaging (psi−) (35). Proviral clones containing either inactive vpx or vpx and vpr open reading frames were introduced into pSIV3+ to produce pSIV3+Vpr− and pSIV3+Vpx−Vpr− as previously described (33, 56) and were kindly provided by Jacek Skowronski.
Virus stocks.
Single-round VSV-G-pseudotyped HIV-1 virions were produced by calcium phosphate cotransfection of HEK 293T cells with the appropriate HIV-1 envelope-deleted construct along with a plasmid expressing VSV-G driven by the HIV-1 LTR (pLTR-VSV-G), as described previously (55). Equivalent weights of pLTR-VSV-G and HIV-1 constructs were transfected together for all viruses produced. Transfected cells were washed at 16 h posttransfection, medium was replaced 8 h later, and supernatants containing VSV-G-pseudotyped virus were collected the next morning, at approximately 40 h posttransfection. Supernatants were then centrifuged to clear cellular debris, passed through 0.45-μm filters, aliquoted, and stored in liquid nitrogen prior to infectivity and p24 assays.
SIV-VLP were produced by calcium phosphate cotransfection of HEK 293T cells with pSIV3+, pSIV3+Vpx−Vpr−, or pSIV3+Vpr− in combination with an equivalent weight of pLTR-VSV-G. VLP were collected in the same manner as described above for pseudotyped HIV-1 virions, concentrated by ultracentrifugation, and stored in liquid nitrogen. SIV-VLP were tested for the delivery of Vpx by infection of MDM for 16 h and subsequent imaging for SAMHD1 expression. SIV-VLP stocks were considered effective if they depleted SAMHD1 expression in approximately 90% of the MDM.
The infectivity of HIV-1 stocks was determined by infection of LuSIV (SIV LTR-luciferase) indicator cells as previously described (57). LuSIV cells were infected with serially diluted virus stocks for 40 h and assayed for luciferase activity by using the Bright-Glo luciferase assay system (Promega, Madison, WI). The level of p24 CA was quantified by an enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (XpressBio, Frederick, MD). Titers of viral stocks were determined on MDM, and the viral stocks were used at concentrations just below saturating infectivity.
Cell culture.
LuSIV cells are a CEMx174-derived T cell line with a stably integrated HIV-2 LTR-luciferase construct (57) obtained from the NIH AIDS Reagent Program. LuSIV cells were cultured in RPMI supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA), and 0.3 mg/ml hygromycin B (Sigma-Aldrich, St. Louis, MO). Monocyte-derived macrophages were prepared from peripheral blood mononuclear cells (PBMC) isolated from healthy donors by Ficoll density gradient centrifugation. CD14-positive monocytes were then isolated by positive selection using CD14 magnetic bead separation (Miltenyi Biotech, San Diego, CA). Monocytes were cultured in 24-well plates on glass coverslips (2.5 × 105 cells per well) in Iscove's modified Dulecco's medium (IMDM) without phenol red (Gibco, Waltham, MA) supplemented with 5% human serum (Valley Biomedical, Winchester, VA), 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA), and 1,000 U/ml macrophage colony-stimulating factor (M-CSF) (R&D Systems, Minneapolis, MN). M-CSF was added again at 1,000 U/ml at 2 to 3 days postisolation, and cells were cultured for a total of 6 to 7 days to fully differentiate MDM.
EdU labeling.
The MDM medium was replaced without M-CSF, and SIV-VLP were added where needed. Sixteen to twenty-four hours later, the cells were washed, cultured with 10 μM EdU, and infected with HIV-1 (75 ng p24 CA unless noted otherwise). MDM were washed at 16 to 24 h p.i. to remove cell-free virus and EdU and further cultured. At the indicated time points, cells were rinsed in phosphate-buffered saline (PBS), fixed in 4% electron microscopy (EM)-grade formaldehyde (Polysciences, Warrington, PA) in PBS for 15 min at room temperature, and rinsed in PBS. Cells were then washed twice in 3% bovine serum albumin (BSA) in PBS, permeabilized with 0.5% Triton X-100 in PBS for 15 min, and washed twice with 3% BSA in PBS. For each well in a 24-well plate, cells were labeled for 30 min with the following mixture (added in order): 220 μl of 100 mM Tris-buffered saline (pH 7.5), 10 μl of 68 mM CuSO4, 64 mM Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) (Sigma-Aldrich, St. Louis, MO), 1 μl of 500 μM Alexa Fluor 647 picolyl azide (Life Technologies, Carlsbad, CA), and 25 μl of 1 M sodium ascorbate. When EdU labeling alone was desired, a mixture of 75 mM CuSO4 and 50 mM THPTA was added to increase the EdU signal. Following EdU labeling, MDM were washed once with 3% BSA in PBS and once in PBS alone and either mounted in Prolong Gold Antifade mountant (Life Technologies, Carlsbad, CA) or further processed for immunostaining or FISH labeling.
Antibodies and reagents.
Mouse monoclonal antibodies for HIV-1 p24 CA (clone AG3.0; NIH AIDS Reagent Program, Germantown, MD), lamin A/C (clone 4C11; Sigma-Aldrich, St. Louis, MO), and SAMHD1 (clone 1A1; Origene, Rockville, MD) were used in this study. Donkey anti-mouse antibodies labeled with Cy3, Alexa Fluor 488, or Cy5 were used for immunofluorescence imaging (Jackson Immuno-Research, West Grove, PA). Rabbit polyclonal cyclin T1 antibody was obtained from Santa Cruz Biotechnologies, Dallas, TX. Rabbit polyclonal antiserum to the phosphoserine 175 modification was generated at Covance Research Products as previously described (21) and was a kind gift from Jonathan Karn. Donkey anti-rabbit antibodies labeled with Alexa Fluor 594 or Alexa Fluor 488 were obtained from Life Technologies, Carlsbad, CA. Raltegravir (used at 10 μM) and nevirapine (used at 1 μM) were obtained from the NIH AIDS Reagent Program.
DNA FISH.
DNA FISH was performed by using the FISH Tag DNA Red kit (Invitrogen, Carlsbad, CA). DNA probes were produced by nick translation of plasmid pLAI∂env and subsequent labeling of incorporated amine-labeled nucleotides with the amine-reactive Alexa Fluor 594 fluorophore according to the manufacturer's instructions. Alexa Fluor 647-EdU-labeled MDM were washed twice in PBS at room temperature and serially dehydrated in 70%, 85%, and 100% ethanol at room temperature for 1 min each. MDM were then rehydrated in 70% formamide in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) (UltraPure 20× SSC [Invitrogen, Carlsbad, CA] diluted 1:10 in water) at 72°C for 2 min and serially dehydrated in 70%, 85%, and 100% ethanol at −20°C for 1 min each. Hybridization buffer was then prepared (50% formamide in 2× SSC) with DNA FISH probes at a final concentration of 1 ng/μl. The probe was denatured for 5 min at 72°C and immediately placed on ice before being added to MDM in a humidified chamber for overnight hybridization at 50°C. Following hybridization, cells were washed in 2× SSC–0.1% NP-40 at 37°C for 5 min, washed in 0.4× SSC–0.3% NP-40 at 73°C for 5 min, and finally washed in 2× SSC–0.1% NP-40 at 73°C for 1 min as described in the FISH Tag DNA kit (Invitrogen, Carlsbad, CA). Coverslips were then dried and stained with Hoechst dye (Bis-Benzamide H, 4 μg/ml; Sigma-Aldrich, St. Louis, MO) and mounted in Prolong Gold Antifade mountant (Life Technologies, Carlsbad, CA).
RNA FISH.
Stellaris RNA FISH probes (Biosearch Technologies, Novato, CA) targeting both the 3′ LTR and the 5′ LTR of the HIV-1 provirus were designed from the 551-bp 5′-LTR sequence of the pLAI∂env provirus by using Biosearch Technologies online software. The 18 unique, 20-nucleotide RNA probes were labeled at the 3′ end with a 6-carboxytetramethylrhodamine (TAMRA) fluorophore. In situations where EdU click labeling was followed by RNA FISH, washes were performed with PBS alone, and 5 U/ml RNase inhibitor (RNasin; Roche, Mannheim, Germany) was added to the permeabilization solution. Fixed cells were permeabilized for at least 1 h in 70% ethanol at 4°C. Following permeabilization or EdU click labeling, coverslips were rehydrated in wash buffer (2× SSC, 10% deionized formamide) for 5 min. Coverslips were then hybridized in a humidified chamber at 37°C in hybridization buffer (10% dextran sulfate, 2× SSC, 10% deionized formamide) containing 2.5 μM RNA probes for 4 to 12 h. Following hybridization, cells were washed once in wash buffer at 37°C for 30 min and washed a second time in wash buffer containing 0.5 μg/ml Hoechst dye for 30 min at 37°C. Cells were washed a final time in 2× SSC for 5 min and either immediately immunostained or mounted by using Prolong Gold Antifade mountant (Life Technologies, Carlsbad, CA).
Immunofluorescence.
Fixed, EdU-labeled cells were immunostained by using the indicated primary antibodies for 15 min in staining buffer (SB) (10% normal donkey serum in PBS) at room temperature, washed 3 times with PBS, and stained with an appropriate fluorescent secondary antibody along with Hoechst dye in SB for 15 min at room temperature, followed by 3 more PBS washes. If RNA FISH-labeled MDM were to be immunostained, 5 U/ml RNase inhibitor (Roche, Mannheim, Germany) was added to primary and secondary antibody reagents. Coverslips were mounted onto glass slides by using Prolong Gold Antifade mountant (Life Technologies, Carlsbad, CA).
Cured slides were imaged on a DeltaVision RT epifluorescence microscope (Applied Precision, Inc., Issaquah, WA). Images were captured in z-series by using a charge-coupled-device (CCD) digital camera, and out-of-focus light was digitally removed by deconvolution software (Applied Precision, Inc., Issaquah, WA). Three-dimensionally (3-D) rendered whole-cell volumes were generated by using the DeltaVision SoftWoRx analysis program.
Quantitative image analysis.
To quantify the percentage of GFP-positive cells, low-magnification (20× objective) images were obtained, reconstructed in 3-D, and surface rendered by using Imaris software (Bitplane, South Windsor, CT). The total number of cells was quantified by using surface approximation of Hoechst nuclear staining, and the number of productively infected, GFP+ MDM was quantified by surface approximation of GFP signals, with the lower threshold being set by uninfected control MDM.
EdU puncta were counted from 3-D reconstructions of high-magnification (60× objective) z-stacks taken of the entire volume of the cell with a maximum z-step of 400 nm. Imaris spot modeling classified EdU puncta as intranuclear or cytoplasmic based on complete containment within 3-D-reconstructed lamin or colocalization with Hoechst staining. Data were graphically presented by using GraphPad Prism software. P values were calculated by unpaired t test analysis (two-tailed) or unpaired Mann-Whitney tests with GraphPad Prism software where indicated.
Analysis of the colocalization of EdU with RNA FISH, CycT1, pSer175, or CA p24 was performed by using the Imaris spot colocalization tool, with distances of <200 nm (diffraction resolution limit of the microscope) being considered colocalization. A quality threshold was set when modeling spots based on control infections with treatment with RAL or on unstained samples. For colocalization analysis of triple-colocalized EdU, RNA, and pSer175, colocalized EdU and RNA were modeled as a new spot and analyzed for colocalization with pSer175 by using the Imaris spot colocalization tool.
Quantitative real-time PCR.
Prior to infection of MDM, HIV-1 stocks were treated with 30 U/ml DNase I with 10 mM MgCl2 for 30 min at 37°C to eliminate any plasmid DNA contamination. DNA was harvested from infected MDM by using the DNeasy kit (Qiagen, Germantown, MD). Reaction mixtures contained 20 ng total isolated DNA along with Power SYBR green PCR master mix (Life Technologies, Carlsbad, CA) in a total volume of 25 μl. HIV-1 late RT products were amplified by the MH531 and MH532 primers, and HIV-1 2-LTR circles were amplified by the MH535 and MH536 primers, as previously described (19). The amount of late reverse transcription products was calculated by comparison to a standard curve of total MDM DNA spiked with a serially diluted proviral plasmid. The amount of 2-LTR circles was calculated by comparison to a standard curve of total MDM DNA with a serially diluted plasmid containing a 2-LTR region. The standard curve was created with a chosen number of MDM genomes (using 3 billion base pairs as a measure of genome size) spiked with increasing numbers of copies of the proviral plasmid (around 10,000 bp per copy) or copies of a plasmid containing the 2-LTR region (around 3,000 bp per copy). Thus, by measuring the number of late reverse transcription products from standard samples containing 0, 0.1, 1, 10, and 100 proviral plasmid copies or 2-LTR plasmid copies per genome, we were able to interpolate the number of late reverse transcription products or 2-LTR circles per genome from unknown samples isolated from HIV-infected MDM. Reverse transcription products were finally quantified by using the Bio-Rad MyiQ single-color real-time PCR detection system.
ACKNOWLEDGMENTS
We thank Jennifer Bongorno for technical support and the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, for reagents and antibodies used here. We also thank Jacek Skowronski and Jonathan Karn for helpful discussions and readings of the manuscript. We also thank Jonathan Karn and Uri Mbonye for help and reagents related to the pSer175 and P-TEFb studies described.
This study was supported by the NIAID (AI087511), the NIDCR (DE025464), and the Case/UH Center for AIDS Research (AI036219). R.D.S. was supported by the Case Western Reserve University Medical Scientist Training Program (T32 GM007250).
REFERENCES
- 1.Hulme AE, Perez O, Hope TJ. 2011. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc Natl Acad Sci U S A 108:9975–9980. doi: 10.1073/pnas.1014522108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamashita M, Perez O, Hope TJ, Emerman M. 2007. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog 3:1502–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y, Fricke T, Diaz-Griffero F. 2013. Inhibition of reverse transcriptase activity increases stability of the HIV-1 core. J Virol 87:683–687. doi: 10.1128/JVI.01228-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lukic Z, Dharan A, Fricke T, Diaz-Griffero F, Campbell EM. 2014. HIV-1 uncoating is facilitated by dynein and kinesin 1. J Virol 88:13613–13625. doi: 10.1128/JVI.02219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, Hope TJ. 2002. Visualization of the intracellular behavior of HIV in living cells. J Cell Biol 159:441–452. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu H, Franks T, Gibson G, Huber K, Rahm N, Strambio De Castillia C, Luban J, Aiken C, Watkins S, Sluis-Cremer N, Ambrose Z. 2013. Evidence for biphasic uncoating during HIV-1 infection from a novel imaging assay. Retrovirology 10:70. doi: 10.1186/1742-4690-10-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Francis AC, Marin M, Shi J, Aiken C, Melikyan GB. 2016. Time-resolved imaging of single HIV-1 uncoating in vitro and in living cells. PLoS Pathog 12:e1005709. doi: 10.1371/journal.ppat.1005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arhel NJ, Souquere-Besse S, Munier S, Souque P, Guadagnini S, Rutherford S, Prevost MC, Allen TD, Charneau P. 2007. HIV-1 DNA flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J 26:3025–3037. doi: 10.1038/sj.emboj.7601740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matreyek KA, Engelman A. 2011. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J Virol 85:7818–7827. doi: 10.1128/JVI.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, KewalRamani VN, Chin JW, Towers GJ, James LC. 2012. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog 8:e1002896. doi: 10.1371/journal.ppat.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sowd GA, Serrao E, Wang H, Wang W, Fadel HJ, Poeschla EM, Engelman AN. 2016. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc Natl Acad Sci U S A 113:E1054–E1063. doi: 10.1073/pnas.1524213113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chin CR, Perreira JM, Savidis G, Portmann JM, Aker AM, Feeley EM, Smith MC, Brass AL. 2015. Direct visualization of HIV-1 replication intermediates shows that capsid and CPSF6 modulate HIV-1 intra-nuclear invasion and integration. Cell Rep 13:1717–1731. doi: 10.1016/j.celrep.2015.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hulme AE, Kelley Z, Foley D, Hope TJ. 2015. Complementary assays reveal a low level of CA associated with viral complexes in the nuclei of HIV-1-infected cells. J Virol 89:5350–5361. doi: 10.1128/JVI.00476-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng K, Muranyi W, Glass B, Laketa V, Yant SR, Tsai L, Cihlar T, Muller B, Krausslich HG. 2014. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. eLife 3:e04114. doi: 10.7554/eLife.04114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sloan RD, Wainberg MA. 2011. The role of unintegrated DNA in HIV infection. Retrovirology 8:52. doi: 10.1186/1742-4690-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 18.Bukrinsky MI, Stanwick TL, Dempsey MP, Stevenson M. 1991. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 254:423–427. doi: 10.1126/science.1925601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat Med 7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 20.Karn J, Stoltzfus CM. 2012. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb Perspect Med 2:a006916. doi: 10.1101/cshperspect.a006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mbonye UR, Gokulrangan G, Datt M, Dobrowolski C, Cooper M, Chance MR, Karn J. 2013. Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4(+) T lymphocytes. PLoS Pathog 9:e1003338. doi: 10.1371/journal.ppat.1003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albanese A, Arosio D, Terreni M, Cereseto A. 2008. HIV-1 pre-integration complexes selectively target decondensed chromatin in the nuclear periphery. PLoS One 3:e2413. doi: 10.1371/journal.pone.0002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burdick RC, Hu WS, Pathak VK. 2013. Nuclear import of APOBEC3F-labeled HIV-1 preintegration complexes. Proc Natl Acad Sci U S A 110:E4780–E4789. doi: 10.1073/pnas.1315996110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francis AC, Di Primio C, Quercioli V, Valentini P, Boll A, Girelli G, Demichelis F, Arosio D, Cereseto A. 2014. Second generation imaging of nuclear/cytoplasmic HIV-1 complexes. AIDS Res Hum Retroviruses 30:717–726. doi: 10.1089/aid.2013.0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lelek M, Di Nunzio F, Henriques R, Charneau P, Arhel N, Zimmer C. 2012. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc Natl Acad Sci U S A 109:8564–8569. doi: 10.1073/pnas.1013267109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marini B, Kertesz-Farkas A, Ali H, Lucic B, Lisek K, Manganaro L, Pongor S, Luzzati R, Recchia A, Mavilio F, Giacca M, Lusic M. 2015. Nuclear architecture dictates HIV-1 integration site selection. Nature 521:227–231. doi: 10.1038/nature14226. [DOI] [PubMed] [Google Scholar]
- 27.Di Primio C, Quercioli V, Allouch A, Gijsbers R, Christ F, Debyser Z, Arosio D, Cereseto A. 2013. Single-cell imaging of HIV-1 provirus (SCIP). Proc Natl Acad Sci U S A 110:5636–5641. doi: 10.1073/pnas.1216254110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jambo KC, Banda DH, Kankwatira AM, Sukumar N, Allain TJ, Heyderman RS, Russell DG, Mwandumba HC. 2014. Small alveolar macrophages are infected preferentially by HIV and exhibit impaired phagocytic function. Mucosal Immunol 7:1116–1126. doi: 10.1038/mi.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deleage C, Wietgrefe SW, Del Prete G, Morcock DR, Hao XP, Piatak M Jr, Bess J, Anderson JL, Perkey KE, Reilly C, McCune JM, Haase AT, Lifson JD, Schacker TW, Estes JD. 2016. Defining HIV and SIV reservoirs in lymphoid tissues. Pathog Immun 1:68–106. doi: 10.20411/pai.v1i1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salic A, Mitchison TJ. 2008. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. 1986. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 233:215–219. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 32.Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, Hahn BH, Kappes JC. 2012. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol 86:2715–2728. doi: 10.1128/JVI.06157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Negre D, Mangeot PE, Duisit G, Blanchard S, Vidalain PO, Leissner P, Winter AJ, Rabourdin-Combe C, Mehtali M, Moullier P, Darlix JL, Cosset FL. 2000. Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther 7:1613–1623. doi: 10.1038/sj.gt.3301292. [DOI] [PubMed] [Google Scholar]
- 36.Bregnard C, Benkirane M, Laguette N. 2014. DNA damage repair machinery and HIV escape from innate immune sensing. Front Microbiol 5:176. doi: 10.3389/fmicb.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koyama T, Sun B, Tokunaga K, Tatsumi M, Ishizaka Y. 2013. DNA damage enhances integration of HIV-1 into macrophages by overcoming integrase inhibition. Retrovirology 10:21. doi: 10.1186/1742-4690-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roshal M, Kim B, Zhu Y, Nghiem P, Planelles V. 2003. Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J Biol Chem 278:25879–25886. doi: 10.1074/jbc.M303948200. [DOI] [PubMed] [Google Scholar]
- 39.Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ambrose Z, Lee K, Ndjomou J, Xu H, Oztop I, Matous J, Takemura T, Unutmaz D, Engelman A, Hughes SH, KewalRamani VN. 2012. Human immunodeficiency virus type 1 capsid mutation N74D alters cyclophilin A dependence and impairs macrophage infection. J Virol 86:4708–4714. doi: 10.1128/JVI.05887-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collin M, Gordon S. 1994. The kinetics of human immunodeficiency virus reverse transcription are slower in primary human macrophages than in a lymphoid cell line. Virology 200:114–120. doi: 10.1006/viro.1994.1169. [DOI] [PubMed] [Google Scholar]
- 42.Weinberg JB, Matthews TJ, Cullen BR, Malim MH. 1991. Productive human immunodeficiency virus type 1 (HIV-1) infection of nonproliferating human monocytes. J Exp Med 174:1477–1482. doi: 10.1084/jem.174.6.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. 1998. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92:451–462. doi: 10.1016/S0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 44.Ocwieja KE, Brady TL, Ronen K, Huegel A, Roth SL, Schaller T, James LC, Towers GJ, Young JA, Chanda SK, Konig R, Malani N, Berry CC, Bushman FD. 2011. HIV integration targeting: a pathway involving transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog 7:e1001313. doi: 10.1371/journal.ppat.1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog 7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koh Y, Wu X, Ferris AL, Matreyek KA, Smith SJ, Lee K, KewalRamani VN, Hughes SH, Engelman A. 2013. Differential effects of human immunodeficiency virus type 1 capsid and cellular factors nucleoporin 153 and LEDGF/p75 on the efficiency and specificity of viral DNA integration. J Virol 87:648–658. doi: 10.1128/JVI.01148-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ. 2013. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansen EC, Ransom M, Hesselberth JR, Hosmane NN, Capoferri AA, Bruner KM, Pollack RA, Zhang H, Drummond MB, Siliciano JM, Siliciano R, Stivers JT. 2016. Diverse fates of uracilated HIV-1 DNA during infection of myeloid lineage cells. eLife 5:e18447. doi: 10.7554/eLife.18447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Margolis DM, Garcia JV, Hazuda DJ, Haynes BF. 2016. Latency reversal and viral clearance to cure HIV-1. Science 353:aaf6517. doi: 10.1126/science.aaf6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, Lai J, Strain MC, Lada SM, Hoh R, Ho YC, Richman DD, Deeks SG, Siliciano JD, Siliciano RF. 2016. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med 22:1043–1049. doi: 10.1038/nm.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, Hebbeler AM, Greene WC. 2010. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143:789–801. doi: 10.1016/j.cell.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC. 2014. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poon B, Chen IS. 2003. Human immunodeficiency virus type 1 (HIV-1) Vpr enhances expression from unintegrated HIV-1 DNA. J Virol 77:3962–3972. doi: 10.1128/JVI.77.7.3962-3972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. 1990. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J 9:1551–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bartz SR, Vodicka MA. 1997. Production of high-titer human immunodeficiency virus type 1 pseudotyped with vesicular stomatitis virus glycoprotein. Methods 12:337–342. doi: 10.1006/meth.1997.0487. [DOI] [PubMed] [Google Scholar]
- 56.Srivastava S, Swanson SK, Manel N, Florens L, Washburn MP, Skowronski J. 2008. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog 4:e1000059. doi: 10.1371/journal.ppat.1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roos JW, Maughan MF, Liao Z, Hildreth JE, Clements JE. 2000. LuSIV cells: a reporter cell line for the detection and quantitation of a single cycle of HIV and SIV replication. Virology 273:307–315. doi: 10.1006/viro.2000.0431. [DOI] [PubMed] [Google Scholar]