

Abstract

Coordination of FeCl3 to the redox-active pyridine–aminophenol ligand NNOH2 in the presence of base and under aerobic conditions generates FeCl2(NNOISQ) (1), featuring high-spin FeIII and an NNOISQ radical ligand. The complex has an overall S = 2 spin state, as deduced from experimental and computational data. The ligand-centered radical couples antiferromagnetically with the Fe center. Readily available, well-defined, and air-stable 1 catalyzes the challenging intramolecular direct C(sp3)–H amination of unactivated organic azides to generate a range of saturated N-heterocycles with the highest turnover number (TON) (1 mol% of 1, 12 h, TON = 62; 0.1 mol% of 1, 7 days, TON = 620) reported to date. The catalyst is easily recycled without noticeable loss of catalytic activity. A detailed kinetic study for C(sp3)–H amination of 1-azido-4-phenylbutane (S1) revealed zero order in the azide substrate and first order in both the catalyst and Boc2O. A cationic iron complex, generated from the neutral precatalyst upon reaction with Boc2O, is proposed as the catalytically active species.
Introduction
The development of efficient methods for the formation of carbon–nitrogen (C–N) bonds is one of the most crucial tasks in chemical synthesis. The installment of C–N bonds by direct functionalization of C(sp3)–H bonds is a powerful and atom-efficient transformation for chemical synthesis. Although the direct installation of nitrogen into a C(sp3)–H bond is extremely challenging due to the thermodynamic and kinetic stability of the C(sp3)–H bond, intra- and intermolecular C(sp3)–H amination has seen much progress in the past decade.1 Particularly, intramolecular C(sp3)–H amination as an atom-economical strategy has found extensive applications for the construction of varieties of important N-heterocycles.2 Four main strategies have been developed for the construction of C(sp3)–H bonds by direct, intramolecular amination of either activated or unactivated C(sp3)–H bonds. A crucial advance in intramolecular C(sp3)–H amination can be traced back to the Hofmann–Löffler–Freytag (HLF) reaction, developed in the early 1880s with the initial discovery by Hofmann.3 The N-halogenated amines are utilized as starting materials in HLF reactions, and the generally accepted mechanism involves a free radical pathway (Scheme 1a).4 Another effective method involves the oxidation of C,N-dianions generated by successive deprotonation of an N–H and a C–H bond, followed by oxidative coupling under strongly basic conditions (Scheme 1b).5 Recently, transition-metal-catalyzed (predominantly palladium) amination has emerged for the activation of aliphatic C–H bonds, which typically requires an electron-withdrawing directing group (Scheme 1c).6 Lastly, nitrene (in situ generated) insertion into a C(sp3)–H bond is an efficient and perhaps the best studied approach for C(sp3)–N bond formation (Scheme 1d). Nitrenes can be generated either from amines by utilizing a combination of PhI(OAc)2 and MgO or from activatived, nonaliphatic organic azides (e.g., sulfonyl azide, aryl azide) or iminoiodinanes in the presence of transition metal catalysts.7
Scheme 1. Intramolecular C(sp3)–H Amination Strategies for the Formation of N-Heterocyles.

Unfortunately, most of the existing C–H amination strategies involve directing groups, preoxidation of substrates, or external chemical oxidants, leading to poor atom economy and waste generation. In contrast, in situ generation of a metal-bound nitrene species from readily available aliphatic organoazides, releasing only molecular nitrogen as the side product, followed by selective insertion into a C(sp3)–H bond would constitute an efficient approach for catalytic C–H amination. Synthesis of N-heterocycles via direct C(sp3)–H amination using aliphatic azide substrates is an appealing strategy, given that N-heterocycles are prevailing building blocks in natural products, pharmaceuticals, and functional materials (Figure 1a).8 Recently, two reports appeared on air-sensitive FeII-catalyzed direct C(sp3)–H amination of linear azides to give saturated Boc-protected N-heterocycles, proposedly proceeding via an FeIII–nitrene radical intermediate (Figure 1b).9,10 Apart from these systems featuring a redox-active metal center (“metalloradical” approach),11 our group recently demonstrated the catalytic PdII-mediated C(sp3)–H amination of aliphatic azide to pyrrolidine, albeit with very modest turnover. This system operates via single electron transfer from an aminophenol-derived redox-active ligand to the substrate to generate a Pd-bound nitrene substrate radical and the one-electron-oxidized iminosemiquinonato (ISQ) ligand radical (Figure 1b).12 Herein, we discuss the synthesis and detailed characterization of a bench-stable iron(III) complex with a redox-active NNO ligand. This air-stable iron species is an effective and recyclable catalyst for direct C(sp3)–H amination of aliphatic azides to N-heterocycles with much improved turnover numbers (TONs) compared to those of the existing catalysts.
Figure 1.

(a) Pyrrolidine-containing natural products and pharmaceuticals and (b) catalysts for direct C(sp3)–H amination of 1-azido-4-phenylbutane as the benchmark substrate.
Results and Discussion
The ligand NNOH2 is readily accessible following a literature procedure.12a Coordination of the neutral ligand NNOH2 to FeCl3 in MeOH at −80 °C followed by the addition of NEt3 in air resulted in the paramagnetic dark green solid 1 in good yield (Scheme 2). UV–vis spectroscopy supports the imino–semiquinonato (ISQ) ligand oxidation state (λmax = 740 nm, ε = 8.37 × 103 M–1 cm–1).13−16 Magnetic susceptibility measurements of 1 at 298 K using Evans’ method revealed an effective magnetic moment (μeff) of 4.86 μB, thus indicating an S = 2 ground state, which is consistent with a high-spin FeIII center (d5) that is strongly antiferromagnetically coupled with a ligand-centered NNOISQ radical. Temperature-dependent solid-state SQUID measurement and zero-field 57Fe Mössbauer spectroscopy confirmed the total S = 2 ground state (χMT = 3.7 cm3 mol–1 K or μeff = 5.44 μB) and an FeIII oxidation state (δ = 0.42 mm/s, ΔEQ = 0.85 mm/s), respectively (Figure 2).17
Scheme 2. Synthesis of 1, with Representation of Three Possible Oxidation States of NNO.

Figure 2.

Solid-state characterization of 1 by (a) variable-temperature SQUID magnetometry and (b) zero-field 57Fe Mössbauer spectroscopy at 80 K.
The formulation of 1 as FeIIICl2(NNOISQ) was further confirmed by single-crystal X-ray structure determination (Figure 3a). The geometry around iron (τ of 0.52) is intermediate between trigonal bipyramidal and square pyramidal. The iron–ligand bond lengths (Fe–O 1.9572(10); Fe–N 2.0136(12) Å) as well as ligand-based interatomic distances (O1–C1 1.2809(17); N1–C6 1.3390(17) Å) are characteristic of the ISQ ligand oxidation state.14,18−20 A metrical oxidation state value of −0.69 (±0.04)21 was determined for the NNOISQ ligand of 1. Density functional theory (DFT) calculations (B3LYP, def2-TZVP) show a broken-symmetry S = 2 spin state (⟨S2⟩ = 6.8) as the ground state (see Supporting Information). The energy difference between the broken-symmetry S = 2 ground state (Figure 3b) and the high-spin S = 3 excited state (Figure 3c) is calculated to be +5.3 kcal mol–1 by DFT. The spin-density plot for S = 2 (Figure 3b) clearly illustrates the observed antiferromagnetic coupling between the NNO radical fragment and the Fe center via the coordinated N and O atoms. A Löwdin population analysis22 (see Supporting Information) shows that the Fe center has a total spin equivalent to four unpaired electrons.
Figure 3.

(a) Displacement ellipsoid plot (50% probability level) of 1; hydrogen atoms and lattice solvent molecules omitted for clarity. Selected bond lengths (Å) and angles (deg): Fe(1)–Cl(1) 2.2512(4); Fe(1)–Cl(2) 2.2366(4); Fe(1)–O(1) 1.9572(10); Fe(1)–N(1) 2.0136(12); Fe(1)–N(2) 2.1024(11); C(1)–O(1) 1.2809(17); C(6)–N(1) 1.3390(17); C(1)–C(6) 1.4634(18); O(1)–Fe(1)–N(1) 78.99(4); N(1)–Fe(1)–N(2) 77.56(4); O(1)–Fe(1)–N(2) 156.18(4); Cl(1)–Fe(1)–Cl(2) 117.32(2). DFT (M06, def2-TZVP) calculated spin density plot of (b) ground state of 1 (S = 2) and (c) high spin state of 1 (S = 3).
Cyclic voltammetry of 1 in CH2Cl2 solution revealed quasi-reversible one-electron oxidation and reduction events at +0.51 V and −0.74 V vs Fc/Fc+, respectively (Figure 4a). Chemical oxidation of 1 using silver salts likely afforded Cl– abstraction, but no clean species was obtained. Chemical reduction of 1 using CoCp2 led to formation of homoleptic FeII(NNOISQ)2 (2). The latter species does not re-form FeIII(NNOISQ) upon reoxidation. The homoleptic complex 2 was characterized by single-crystal X-ray structure determination (Figure 4b and Supporting Information). Two crystallographically independent molecules of 2 were found in the asymmetric unit (P21/n). The geometry around each Fe metal center is distorted octahedral, with meridionally coordinated NNO ligands. The iron–ligand bond lengths, angles, and interatomic distances within the NNO moieties are very similar for both molecules and suggestive of the ISQ ligand oxidation state.14,18−20 The iron–ligand bond lengths in complex 2 (average bond distances: Fe–O 1.928 Å, Fe–Nimino 1.884 Å, Fe–Npyridyl 1.963 Å) are slightly shorter than those in heteroleptic complex 1 (Fe–O 1.957 Å, Fe–Nimino 2.014 Å, Fe–Npyridyl 2.102 Å). On the contrary, the ligand-based interatomic distances in 2 (average bond distances: C–O 1.320 Å, C–Nimino 1.368 Å) are slightly longer than those in complex 1 (C–O 1.281 Å, C–Nimino 1.339 Å). Chemical oxidation of 2 with AgBF4 resulted in the formation of [FeIII(NNOISQ)2]BF4 (3a), which was structurally characterized (see Supporting Information for details). The chloride derivative of this homoleptic FeIII system, [FeIII(NNOISQ)2]Cl (3b), was accessible directly by heating a mixture of FeCl3 and NNOH2 (2 molar equiv) at reflux in the presence of NEt3 under aerobic conditions.
Figure 4.

(a) Cyclic voltammogram of 1 in CH2Cl2 (1 × 10–3 M), scan rate 100 mV s–1 vs Fc/Fc+ on a Pt disk. (b) Displacement ellipsoid plot (50% probability level) of 2 (only one of the two independent molecules is shown); hydrogen atoms and lattice solvent molecules omitted for clarity. Selected bond lengths (Å) and angles (deg): Fe(1)–O(1A) 1.9267(17); Fe(1)–O(1B) 1.9144(17); Fe(1)–N(1A) 1.890(2); Fe(1)–N(1B) 1.877(2); Fe(1)–N(2A) 1.968(2); Fe(1)–N(2B) 1.957(2); C(1A)–O(1A) 1.314(3); C(1B)–O(1B) 1.324(3); C(6A)–N(1A) 1.367(3); C(6B)–N(1B) 1.375(3); C(1A)–C(6A) 1.433(4); C(1B)–C(6B) 1.439(3); O(1A)–Fe(1)–N(1A) 83.32(8); O(1B)–Fe(1)–N(1B) 84.24(8); N(1A)–Fe(1)–N(2A) 81.62(10); N(1B)–Fe(1)–N(2B) 82.31(9); O(1A)–Fe(1)–N(2A) 164.94(9); O(1B)–Fe(1)–N(2B) 166.04(9); N(1A)–Fe(1)–N(1B) 177.21(9).
We set out to investigate the activity of well-defined air-stable 1 for catalytic C(sp3)–H amination, using 1-azido-4-phenylbutane (S1) as standard substrate and di-tert-butyl dicarbonate (Boc2O) as in situ protecting group to avoid catalyst deactivation by pyrrolidine coordination (Table 1). Heating an equimolar mixture of both reagents (100 μmol) at 100 °C in benzene for 24 h in the presence of 10 mol% of 1 as catalyst in a pressure tube resulted in complete conversion of S1 to the desired Boc-protected pyrrolidine P1a (70%) and Boc-protected amine P1b (30%) as the side product (entry 1). Lowering the catalyst loading to 5 mol% led to a slightly different product ratio of 63:37 for P1a/P1b (entry 2).
Table 1. Performance of 1 in Intramolecular C(sp3)–H Amination of Aliphatic Azide S1 to P1a and P1ba.


Conditions: [S1] 20 mM, [Boc2O] 20 mM, 1 (10/5/2/1 mol%), C6H6 (5 mL). 1H NMR yields of P1a and P1b are reported using 1,3,5-trimethoxybenzene as a standard.
Isolated yields.
Recycled catalyst.
[Boc2O] 50 mM.
[Boc2O] 100 mM.
Conditions: [S1] 20 mM, [Boc2O] 20 mM, 1 (0.1 mol%), C6H6 (25 mL).
Conditions: [S1] 20 mM, [Boc2O] 20 mM, 1 (10 and 5 mol%), toluene (5 mL). Key parameters for each entry are indicated in red.
Catalyst 1 was successfully recovered by precipitation (dark green precipitate) from the crude reaction mixture upon addition of pentane, allowing recycling without any loss of catalytic activity (entry 3). Analysis of the recovered dark green solid by UV–vis spectroscopy (λmax: 740 nm) and mass spectrometry (M+: m/z 464.1084) confirmed the structural integrity of complex 1 after catalysis. Based on these observations, we exclude involvement of homoleptic FeII species 2 as the catalytically active species, as this complex cannot regenerate complex 1. Further reduction of the catalyst loading to 2 or 1 mol% gave full conversion with virtually the same ratio of P1a/P1b (entries 4 and 5). Using excess Boc2O at 100 °C did not lead to any change in the product distribution (entries 6 and 7).10 Monitoring the reaction progress with 5 mol% of catalyst loading showed complete conversion after 3 h (entry 9) and also for recovered catalyst (entry 10). Upon reducing the catalyst loading to 2 mol%, we observed approximately 50% conversion of substrate in 3 h (entry 12) and full conversion in 6 h (entry 13). Also in this case, the catalyst was recovered and reused without significant loss of catalytic activity (entry 14). The ratio of P1a/P1b (∼1.6:1) remained constant (entries 12–14). Hence, the catalyst can be recovered and reused without significant loss of catalytic activity, using either 5 mol% (entries 3 and 10) or 2 mol% (entry 14) of catalyst loading. Complete conversion of substrate to products was also obtained with 1 mol% of catalyst loading after 12 h (entry 15). Heating an equimolar mixture of both reagents (500 μmol) at 100 °C in benzene (25 mL) for 12 h in the presence of 1 mol% of 1 (5 μmol, entry 16) also allowed for facile catalyst recovery by precipitating into pentane. In this case, the reaction did not go to completion in 12 h using the recycled catalyst (entry 17). Besides the two products (P1a/P1b ≈ 1.6:1), roughly 16% unreacted azide (S1) was recovered. Hence, a slight loss of catalytic activity was observed with this recycling at 1 mol% of catalyst loading. However, this diminished catalytic activity might be due to partial loss of catalyst during recovery. Thereafter, the catalyst loading was further reduced to 0.1 mol%. We performed the runs with 500 μmol of S1 and Boc2O in 25 mL of solvent, keeping the effective concentration constant (entries 18–20). Lowering the catalyst loading to 0.1 mol% resulted in 17 and 36% conversion of substrate in 24 (entry 18) and 48 h (entry 19), respectively, with a product ratio of ∼1.6:1 (P1a/P1b). The highest TON of 620 was obtained with 0.1 mol% of catalyst loading after a week of heating (entry 20). Changing from benzene to toluene did not have a significant influence on the outcome (entries 21 and 22), and only minor differences in product ratio were observed. A large-scale reaction (500 μmol S1, 500 μmol Boc2O, 5 mol% of catalyst) resulted in an isolated yield for P1a of 62% (see Supporting Information). Therefore, catalyst 1 allows turnover numbers significantly higher than those in previously reported homogeneous Fe-based systems (maximum TON of ∼6)9a,10 for the direct intramolecular C(sp3)–H amination of unactivated organic azide.
We explored several additional substrates for the intramolecular C(sp3)–H amination catalyzed by complex 1 (Table 2 and Supporting Information). Complete conversion of substrates S2 to S10 to the corresponding N-heterocycles and linear amines was observed in 24 h at 100 °C using 5 mol% of catalyst loading.
Table 2. Substrate Screening with 1 for C–H Amination of Aliphatic Azides to N-Heterocycles and Amines.

Conditions: [Sx] 20 mM, [Boc2O] 20 mM, 1 (5 mol%), C6H6 (5 mL), T = 100 °C, 24 h. 1H NMR ratios are reported in brackets, determined using 1,3,5-trimethoxybenzene as an internal standard.
Isolated yields.
Utilizing 1-azido-5-hexene (S2) as substrate, allylic C–H amination occurs cleanly to give five-membered N-heterocycle (P2a, 96% isolated yield), and no linear amine byproduct was detected. Both homoallylic and allylic C–H amination occur using 1-azido-6-heptene (S3), generating five- (P3a, 57%) and six-membered (P3b, 38%) N-heterocycles with traces of undesired amine (P3c). The products P3a and P3b are obtained by homoallylic and allylic C–H bond activation, respectively, with a ratio of 1.5:1.0 (P3a/P3b). This observation can be considered as support for direct nitrene insertion into the C(sp3)–H bond, generating both an N–H and C–N bond simultaneously. Combined with the formation of a favorable five-membered ring, the somewhat stronger homoallylic C–H bond is preferentially activated over the weaker allylic C–H bond. A similar observation (but without explanation) was made by Betley et al. for the C–H amination of 1-azido-5-methylhexane, with five-membered pyrrolidine (from secondary C–H bond activation) being the major and six-membered piperidine (from weaker tertiary C–H bond activation) the minor product in a ratio of 1.5:1.0.9a Substrates S4 and S5, containing secondary C–H bonds adjacent to an electron-withdrawing ester group were transformed to the corresponding pyrrolidines P4a (51%) and P5a (39%). Azide substrate S6, containing an ether linkage in the aliphatic chain, gave the oxazolidine product P6a gave the oxazolidine product P5a in high yield (90% isolated yield). Apart from monocyclic products, bicyclic N-heterocycles also proved accessible via this approach. Starting from 1-azidomethyl-2-ethylbenzene (S7), containing secondary benzylic C–H bonds, or 1-azidomethyl-2-methylbenzene (S8) and 1-(2-azidoethyl)-2-methylbenzene (S9) with primary C–H bonds, yielded the desired N-heterocycles (P7a, 53%; P8a, 46%; P9a, 44%). The scope of the intramolecular C(sp3)–H amination catalyzed by complex 1 was also extended to include 1-azido-1-phenyl-5-hexene (S10) as secondary azide, selectively yielding the desired N-heterocycle P10a (mixture of rotamers and diastereomers) (95% isolated yield). Furthermore, organoazides containing a vinyl functionality (S2, S3, and S10) provide the desired N-heterocyles (P2a, P3a, P3b, and P10a) with very little or no undesired linear amine or aziridine. We could not find a clear correlation between the bond dissociation energy of all types of C–H bonds involved and the outcome of the catalytic reactions.23
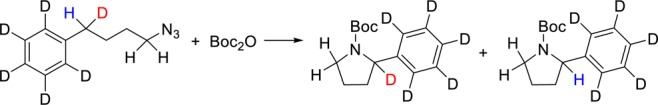
To exclude that the observed catalytic C–H amination activity was due to an impurity originating from the FeCl3 precursor, we tested the FeCl3 used for the synthesis of complex 1 as a catalyst for the conversion of S1. No conversion to N-heterocycle (P1a) or linear amine (P1b) was observed with either 5 or 10 mol% of catalyst loading in the presence of 1 equiv of Boc2O (in benzene or toluene) at 100 °C for 24 h. When using homoleptic FeIII complex 3b, [FeIII(NNOISQ)2]Cl (5 or 10 mol%), prepared directly from FeCl3 and NNOH2, no product formation was detected. Partial poisoning tests using tetramethylthiourea24 (±0.3 molar equiv relative to 1) and elemental mercury gave identical conversion and product ratio compared to the standard reaction without these additives. On the basis of these results, we rule out the active participation of (ligand-stabilized) trace metal impurities present in the FeCl3 precursor.25 Thereafter, we performed kinetic analysis for the intramolecular C(sp3)–H amination of S1 catalyzed by complex 1 (see Tables S4–S6 in the Supporting Information). Monitoring the reaction progress using 2 mol% of catalyst loading (entries 1–5), we observed in a linear decrease in [S1] with time, suggestive of saturation kinetics or zero-order kinetics in S1 (rate constant = 3.5 μM h–1) (Figure 5a). A constant rate of substrate consumption (17.5 μmol h–1) was observed (Figure 5b). The rate of substrate consumption (μmol h–1) against the concentration of 1 (mM) varied between 1 and 7 mol% (entries 6–12) showed first-order kinetics in 1 (Figure 5c). Performing the C(sp3)–H amination of S1 with different concentrations of Boc2O (20–60 mM; entries 13–17) led to a linear increase in the rate of reaction (μmol h–1) (Figure 5d), indicating first-order kinetics in Boc2O. An intramolecular kinetic isotope effect (KIE) of 3.4 was obtained using 1-azido-4-deutero-4-(deuterophenyl)butane as a substrate (Scheme 3). This value is identical to the KIE value (3.4) obtained for the C(sp3)–H amination of 1-azido-4-deutero-4-phenylbutane catalyzed by the Pd catalyst recently published by our group.12a
Figure 5.

Kinetic analysis for C(sp3)–H amination of S1 in the presence of Boc2O catalyzed by 1: (a) rate of substrate consumption vs time, (b) rate of substrate consumption vs substrate concentration, (c) rate of substrate consumption vs concentration of catalyst, and (d) rate of substrate consumption vs concentration of Boc2O.
Scheme 3. Intramolecular Kinetic Isotope Effect in Catalytic C–H Amination of 1-Azido-4-deutero-4-(deuterophenyl)butane as Substrate.
For both Betley’s iron catalyst9a and the palladium catalyst reported by us,12a C(sp3)–H amination of unactivated azides was proposed to occur via rate-limiting azide activation (release of N2) and a subsequent radical pathway involving metal- or ligand-based single-electron transfer, resulting in the formation of a metal–nitrene radical species. Similar “nitrene radical” intermediates have recently been spectroscopically characterized for cobalt porphyrins and other systems.26−28 Formation of the saturated N-heterocycle proceeds via either H atom abstraction followed by radical rebound or direct insertion of the nitrene moiety into the benzylic C–H bond and then forms a pyrrolidine complex. Finally, reaction with Boc2O releases the Boc-protected N-heterocycle and regenerates the catalyst (see the Supporting Information for hypothetical scheme based on 1).
However, this mechanism, as postulated for other systems, is not in agreement with the observed kinetic data when employing catalyst 1. The zero order in substrate (S1) implies that (binding and) activation of the azide substrate is not rate-limiting. Reaction of complex 1 with either a stoichiometric amount or excess (5 and 10 times) S1 in the absence of Boc2O at room temperature (or high temperature) did not yield intermediate A (or B or C; see Supporting Information), and only starting materials were recovered. The first-order kinetics in Boc2O suggests that a reaction with Boc2O is the rate-determining step in the overall C(sp3)–H amination reaction of S1. The active involvement of Boc2O for catalytic turnover is also suggested by the lack of any product (P1a/P1b) formation from S1 in the absence of Boc2O. However, pyrrolidine adduct C proved inaccessible by reaction of 1 with (excess) 2-phenylpyrrolidine, which argues against product inhibition in this case. Therefore, we propose an alternative mechanism to explain the kinetic data (Scheme 4). Initial activation of complex 1 by Boc2O (present in slight excess relative to substrate) at elevated temperature (rate-determining step) leads to the activated FeIII catalyst with a higher affinity for the substrate. Two potential ways for Boc2O to interact with 1 are depicted in Scheme 5: either chloride abstraction or reaction with the phenolate fragment of the redox-active NNO ligand can (pseudo)reversibly generate a cationic FeIII species 1′.29 Thereafter, facile coordination of the azide substrate to the metal center gives adduct A′ and subsequent N2 elimination generates iron(III)-nitrenoid species B′. Either direct nitrene insertion (preferred for (homo)allylic substrates) or H atom abstraction and radical rebound forms the FeIII(pyrrolidine) adduct C′. Finally, reaction with a Boc-containing species (denoted “Boc” in Scheme 4)—either the in situ generated tert-butoxycarbonyl chloride or the carbonate derivative of the NNO ligand; see Scheme 5—releases Boc-protected N-heterocycle (P1a) and tert-butanol with regeneration of complex 1.
Scheme 4. Proposed Cationic Pathway for the Conversion of S1 to P1a with 1 as Catalyst.

Scheme 5. Possible Activation Steps of Catalyst 1 by Boc2O.

It is reasonable to assume that chloride dissociation from complex 1 generates a four-coordinated cationic complex [FeIIICl(NNOISQ)]+ (1′a) or FeIIICl2(NNOISQ-Boc]+ (1′b), which can easily bind an organoazide to generate A′. Release of dinitrogen forms an Fe–NR intermediate B′, which may exist in various spin states. This eventually forms cationic pyrrolidine adduct [FeIIICl(NNOISQ)(2-phenylpyrrolidine)]+ (C′). Upon reaction of 1 with 1 equiv of TlPF6 as redox-inert halide-abstracting agent in the presence of a small excess of S1 (2.5 equiv) in THF, a color change from green to blue-green was observed, concomitant with formation of a white precipitate. Mass spectrometric analysis of the filtrate (m/z 614.3342) is in line with formation of the cationic complex [Fe(NNO)(2-phenylpyrrolidine)(THF)]+.30 This cationic complex was further reacted with Boc2O (1 equiv) to cleanly form Boc-protected pyrrolidine P1a, supporting its possible involvement in the proposed catalytic pathway for the C–H amination of S1.31
Performing the C(sp3)–H amination of S1 in the presence of excess tBuOH, which is generated after coupling of the heterocycle with Boc2O led to the same P1a/P1b ratio of 63:37 as observed under standard reaction conditions, excluding any role of the alcohol in the formation of the side product. No nitrile or imine byproduct was observed under these conditions, which argues against the substrate acting as the H atom donor. When the catalysis was carried out at 10-fold higher absolute concentrations of all components (S1, Boc2O and 1)—to reduce any harmful effects of impurities in the solvent—the P1a/P1b ratio increased significantly (P1a/P1b = 79:21; see Supporting Information for details). Additional distillation of the solvent or switching from C6H6 to C6D6 did not affect the product ratio determined by 1H NMR spectroscopy, which speaks against the solvent acting as a hydrogen source and suggests the involvement of an unknown impurity at low concentration in the side reactions producing the linear Boc-protected amines.
Summary and Conclusions
In summary, straightforward synthesis of complex 1 [FeIIICl2(NNOISQ)] gives access to an air-stable and recoverable Fe catalyst for the efficient direct C(sp3)–H amination of unactivated organic azides to N-heterocycles, providing TONs significantly higher than those previously reported with any homogeneous catalyst for this type of transformation. Experimental and computational data suggest that 1 is best described as an FeIII center that is antiferromagnetically coupled to a ligand-centered NNOISQ radical. In addition to the standard azide substrate S1, the scope of C–H amination was extended to eight other primary azides (S2–S9) and a secondary azide (S10). Based on the experimental evidence, we propose a mechanism for the C–H amination of organoazides involving catalyst activation by Boc2O to form an activated cationic species, followed by a cationic azide activation pathway. In addition to the desired N-heterocycles, unwanted linear amines form in most of the cases. However, organoazides containing a vinyl functionality were converted almost exclusively to the preferred N-heterocycles. The origin of the hydrogen required for the formation of linear amine is unclear to date. The exact mechanism of the C–H amination is currently unknown, and various redox states as well as spin states are possible for the combination of iron, NNO ligand, and a metal-bound nitrene moiety, all of which are potentially redox-active and make for a complex overall system. However, the observed preference for homoallylic versus allylic C–H amination suggests direct nitrene insertion without radical character induced by metal or ligand electron transfer as the most competent pathway. The catalyst integrity as a heteroleptic species, enabling turnover at relatively low catalyst loading as well as catalyst recycling, and the versatile coordination chemistry and the potential proclivity to allow various redox spin states are considered key factors that contribute to the overall performance of this system. Detailed computational investigations are ongoing to unravel the mechanism and to determine the metal, ligand, and substrate redox states of the key intermediates. Additionally, we are exploring the catalytic activity of complex 1 for intermolecular C–H amination and other types of reactions.
Acknowledgments
This research was funded by the European Research Council (ERC) through a Starting Grant (Agreement 279097, EuReCat) to J.I.vdV. V.S. acknowledges the CSER program of NWO-FOM and Shell for funding, and P.F.K. and B.dB. thank NWO (VICI project 016.122.613). We thank Ed Zuidinga for MS analysis, Prof. Joost Reek for interest and support, and Prof. Franc Meyer for generous access to the Mössbauer and SQUID equipment in Göttingen.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b00270.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews, see:; a Collet F.; Dodd R.; Dauban P. Chem. Commun. 2009, 5061–5074. 10.1039/b905820f. [DOI] [PubMed] [Google Scholar]; b Sun C.-L.; Li B.-J.; Shi Z.-J. Chem. Rev. 2011, 111, 1293–1314. 10.1021/cr100198w. [DOI] [PubMed] [Google Scholar]; c Ramirez T.; Zhao B.; Shi Y. Chem. Soc. Rev. 2012, 41, 931–942. 10.1039/C1CS15104E. [DOI] [PubMed] [Google Scholar]; d Gephart R. T.; Warren T. H. Organometallics 2012, 31, 7728–7752. 10.1021/om300840z. [DOI] [Google Scholar]; e Intrieri D.; Zardi P.; Caselli A.; Gallo E. Chem. Commun. 2014, 50, 11440–11453. 10.1039/C4CC03016H. [DOI] [PubMed] [Google Scholar]; f Shin K.; Kim H.; Chang S. Acc. Chem. Res. 2015, 48, 1040–1052. 10.1021/acs.accounts.5b00020. [DOI] [PubMed] [Google Scholar]; g Guo X.-X.; Gu D.-W.; Wu Z.; Zhang W. Chem. Rev. 2015, 115, 1622–1651. 10.1021/cr500410y. [DOI] [PubMed] [Google Scholar]; h Wan J.-P.; Jing Y. Beilstein J. Org. Chem. 2015, 11, 2209–2222. 10.3762/bjoc.11.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey J. L.; Sarpong R. Chem. Sci. 2013, 4, 4092–4106. 10.1039/c3sc51420j. [DOI] [Google Scholar]
- a Hofmann A. W. Ber. Dtsch. Chem. Ges. 1883, 16, 558–560. 10.1002/cber.188301601120. [DOI] [Google Scholar]; b Hofmann A. W. Ber. Dtsch. Chem. Ges. 1885, 18, 5–23. 10.1002/cber.18850180103. [DOI] [Google Scholar]
- a Fan R.; Pu D.; Wen F.; Wu J. J. Org. Chem. 2007, 72, 8994–8997. 10.1021/jo7016982. [DOI] [PubMed] [Google Scholar]; b Chen H.; Sanjaya S.; Wang Y.-F.; Chiba S. Org. Lett. 2013, 15, 212–215. 10.1021/ol303302r. [DOI] [PubMed] [Google Scholar]; c Verma A.; Patel S.; Meenakshi; Kumar A.; Yadav A.; Kumar S.; Jana S.; Sharma S.; Prasad C. D.; Kumar S. Chem. Commun. 2015, 51, 1371–1374. 10.1039/C4CC08717H. [DOI] [PubMed] [Google Scholar]; d Qin Q.; Yu S. Org. Lett. 2015, 17, 1894–1897. 10.1021/acs.orglett.5b00582. [DOI] [PubMed] [Google Scholar]; e Yamamoto C.; Takamatsu K.; Hirano K.; Miura M. J. Org. Chem. 2016, 81, 7675–7684. 10.1021/acs.joc.6b01393. [DOI] [PubMed] [Google Scholar]
- a Bisai A.; West S. P.; Sarpong R. J. Am. Chem. Soc. 2008, 130, 7222–7223. 10.1021/ja8028069. [DOI] [PubMed] [Google Scholar]; b Smith A. C.; Williams R. M. Angew. Chem., Int. Ed. 2008, 47, 1736–1740. 10.1002/anie.200705421. [DOI] [PMC free article] [PubMed] [Google Scholar]; c West S. P.; Bisai A.; Lim A. D.; Narayan R. R.; Sarpong R. J. Am. Chem. Soc. 2009, 131, 11187–11194. 10.1021/ja903868n. [DOI] [PubMed] [Google Scholar]; d Gruver J. M.; West S. P.; Collum D. B.; Sarpong R. J. Am. Chem. Soc. 2010, 132, 13212–13213. 10.1021/ja106852n. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Carpenter R. D.; Verkman A. S. Org. Lett. 2010, 12, 1160–1163. 10.1021/ol902836c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Neumann J. J.; Rakshit S.; Dröge T.; Glorius F. Angew. Chem., Int. Ed. 2009, 48, 6892–6895. 10.1002/anie.200903035. [DOI] [PubMed] [Google Scholar]; b Nadres E. T.; Daugulis O. J. Am. Chem. Soc. 2012, 134, 7–10. 10.1021/ja210959p. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yang M.; Su B.; Wang Y.; Chen K.; Jiang X.; Zhang Y.-F.; Zhang X.-S.; Chen G.; Cheng Y.; Cao Z.; Guo Q.; Wang L.; Shi Z.-J. Nat. Commun. 2014, 5, 4707–4752. 10.1038/ncomms5707. [DOI] [PubMed] [Google Scholar]; d Wu X.; Yang K.; Zhao Y.; Sun H.; Li G.; Ge H. Nat. Commun. 2015, 6, 6462–6471. 10.1038/ncomms7462. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yang M.-Y.; Jiang X.-Y.; Shi Z.-J. Org. Chem. Front. 2015, 2, 51–54. 10.1039/C4QO00282B. [DOI] [Google Scholar]
- a Espino C. G.; DuBois J. Angew. Chem., Int. Ed. 2001, 40, 598–600. . [DOI] [PubMed] [Google Scholar]; b Espino C. G.; Wehn P. M.; Chow J.; Du Bois J. J. Am. Chem. Soc. 2001, 123, 6935–6936. 10.1021/ja011033x. [DOI] [Google Scholar]; c Liang J.-L.; Yuan S.-X.; Huang J.-S.; Yu W.-Y.; Che C.-M. Angew. Chem., Int. Ed. 2002, 41, 3465–3468. . [DOI] [PubMed] [Google Scholar]; d Wehn P. M.; Du Bois J. J. Am. Chem. Soc. 2002, 124, 12950–12951. 10.1021/ja028139s. [DOI] [PubMed] [Google Scholar]; e Hinman A.; Du Bois J. J. Am. Chem. Soc. 2003, 125, 11510–11511. 10.1021/ja0368305. [DOI] [PubMed] [Google Scholar]; f Espino C. G.; Fiori K. W.; Kim M.; Du Bois J. J. Am. Chem. Soc. 2004, 126, 15378–15379. 10.1021/ja0446294. [DOI] [PubMed] [Google Scholar]; g Lebel H.; Huard K.; Lectard S. J. Am. Chem. Soc. 2005, 127, 14198–14199. 10.1021/ja0552850. [DOI] [PubMed] [Google Scholar]; h Kim M.; Mulcahy J. V.; Espino C. G.; Du Bois J. Org. Lett. 2006, 8, 1073–1076. 10.1021/ol052920y. [DOI] [PubMed] [Google Scholar]; i Ruppel J. V.; Kamble R. M.; Zhang X. P. Org. Lett. 2007, 9, 4889–4892. 10.1021/ol702265h. [DOI] [PubMed] [Google Scholar]; j Huard K.; Lebel H. Chem. - Eur. J. 2008, 14, 6222–6230. 10.1002/chem.200702027. [DOI] [PubMed] [Google Scholar]; k Lu H.; Tao J.; Jones J. E.; Wojtas L.; Zhang X. P. Org. Lett. 2010, 12, 1248–1251. 10.1021/ol100110z. [DOI] [PubMed] [Google Scholar]; l Lu H.; Jiang H.; Hu Y.; Wojtas L.; Zhang X. P. Chem. Sci. 2011, 2, 2361–2366. 10.1039/c1sc00366f. [DOI] [Google Scholar]; m Nguyen Q.; Sun K.; Driver T. G. J. Am. Chem. Soc. 2012, 134, 7262–7265. 10.1021/ja301519q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a O’Hagan D. Nat. Prod. Rep. 2000, 17, 435–446. 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]; b Pinder A. R. Nat. Prod. Rep. 1984, 1, 225–230. 10.1039/np9840100225. [DOI] [Google Scholar]
- a Hennessy E. T.; Betley T. A. Science 2013, 340, 591–595. 10.1126/science.1233701. [DOI] [PubMed] [Google Scholar]; b Hennessy E. T.; Liu R. Y.; Iovan D. A.; Duncan R. A.; Betley T. A. Chem. Sci. 2014, 5, 1526–1532. 10.1039/c3sc52533c. [DOI] [Google Scholar]; c Iovan D. A.; Betley T. A. J. Am. Chem. Soc. 2016, 138, 1983–1993. 10.1021/jacs.5b12582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker N. C.; Lin Z.; Zhang T.; Gilhula J. C.; Abney C. W.; Lin W. J. Am. Chem. Soc. 2016, 138, 3501–3509. 10.1021/jacs.5b13394. [DOI] [PubMed] [Google Scholar]
- Recent reviews on metalloradical catalysis:; a Clark A. J. Eur. J. Org. Chem. 2016, 2016, 2231–2243. 10.1002/ejoc.201501571. [DOI] [Google Scholar]; b Xue Z.; He D.; Xie X. Polym. Chem. 2015, 6, 1660–1687. 10.1039/C4PY01457J. [DOI] [Google Scholar]; c Lyaskovskyy V.; de Bruin B. ACS Catal. 2012, 2, 270–279. 10.1021/cs200660v. [DOI] [Google Scholar]; d Jahn U. Top. Curr. Chem. 2011, 320, 191–322. 10.1007/128_2011_285. [DOI] [PubMed] [Google Scholar]
- a Broere D. L. J.; de Bruin B.; Reek J. N. H.; Lutz M.; Dechert S.; van der Vlugt J. I. J. Am. Chem. Soc. 2014, 136, 11574–11577. 10.1021/ja502164f. [DOI] [PubMed] [Google Scholar]; b Broere D. L. J.; van Leest N. P.; de Bruin B.; Siegler M. A.; van der Vlugt J. I. Inorg. Chem. 2016, 55, 8603–8611. 10.1021/acs.inorgchem.6b01192. [DOI] [PubMed] [Google Scholar]
- a Bittner M. M.; Lindeman S. V.; Popescu C. V.; Fiedler A. T. Inorg. Chem. 2014, 53, 4047–4061. 10.1021/ic403126p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bittner M. M.; Lindeman S. V.; Fiedler A. T. J. Am. Chem. Soc. 2012, 134, 5460–5463. 10.1021/ja212163t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Min K. S.; Weyhermüller T.; Wieghardt K. Dalton Trans. 2003, 1126–1132. 10.1039/B211698G. [DOI] [Google Scholar]; b Sun X.; Chun H.; Hildenbrand K.; Bothe E.; Weyhermüller T.; Neese F.; Wieghardt K. Inorg. Chem. 2002, 41, 4295–4303. 10.1021/ic011297k. [DOI] [PubMed] [Google Scholar]
- a Broere D. L. J.; Plessius R.; van der Vlugt J. I. Chem. Soc. Rev. 2015, 44, 6886–6915. 10.1039/C5CS00161G. [DOI] [PubMed] [Google Scholar]; b Broere D. L. J.; Metz L. L.; de Bruin B.; Reek J. N. H.; Siegler M. A.; van der Vlugt J. I. Angew. Chem., Int. Ed. 2015, 54, 1516–1520. 10.1002/anie.201410048. [DOI] [PubMed] [Google Scholar]; c Broere D. L. J.; Demeshko S.; de Bruin B.; Pidko E. A.; Reek J. N. H.; Siegler M. A.; Lutz M.; van der Vlugt J. I. Chem. - Eur. J. 2015, 21, 5879–5886. 10.1002/chem.201500084. [DOI] [PubMed] [Google Scholar]; d Broere D. L. J.; Modder D. K.; Blokker E.; Siegler M. A.; van der Vlugt J. I. Angew. Chem., Int. Ed. 2016, 55, 2406–2410. 10.1002/anie.201509412. [DOI] [PubMed] [Google Scholar]; e Broere D. L. J.; Plessius R.; Tory J.; Demeshko S.; de Bruin B.; Siegler M. A.; Hartl F.; van der Vlugt J. I. Chem. - Eur. J. 2016, 22, 13965–13975. 10.1002/chem.201601900. [DOI] [PubMed] [Google Scholar]; f Bagh B.; Broere D. L. J.; Siegler M. A.; van der Vlugt J. I. Angew. Chem., Int. Ed. 2016, 55, 8381–8385. 10.1002/anie.201603659. [DOI] [PubMed] [Google Scholar]
- Lakshman T. R.; Chatterjee S.; Chakraborty B.; Paine T. K. Dalton Trans. 2016, 45, 8835–8844. 10.1039/C5DT04541J. [DOI] [PubMed] [Google Scholar]
- Gütlich P. Z. Anorg. Allg. Chem. 2012, 638, 15–43. 10.1002/zaac.201100416. [DOI] [Google Scholar]
- a Mukherjee S.; Weyhermüller T.; Bill E.; Wieghardt K.; Chaudhuri P. Inorg. Chem. 2005, 44, 7099–7108. 10.1021/ic050885l. [DOI] [PubMed] [Google Scholar]; b Chun H.; Bill E.; Weyhermüller T.; Wieghardt K. Inorg. Chem. 2003, 42, 5612–5620. 10.1021/ic0301526. [DOI] [PubMed] [Google Scholar]; c Chun H.; Weyhermüller T.; Bill E.; Wieghardt K. Angew. Chem., Int. Ed. 2001, 40, 2489–2492. . [DOI] [PubMed] [Google Scholar]
- a Rajput A.; Sharma A. K.; Barman S. K.; Koley D.; Steinert M.; Mukherjee R. Inorg. Chem. 2014, 53, 36–48. 10.1021/ic401985d. [DOI] [PubMed] [Google Scholar]; b Ali A.; Barman S. K.; Mukherjee R. Inorg. Chem. 2015, 54, 5182–5194. 10.1021/ic503103e. [DOI] [PubMed] [Google Scholar]; c Ali A.; Dhar D.; Barman S. K.; Lloret F.; Mukherjee R. Inorg. Chem. 2016, 55, 5759–5771. 10.1021/acs.inorgchem.5b02688. [DOI] [PubMed] [Google Scholar]
- a Wong J. L.; Sanchez R. F.; Logan J. G.; Zarkesh R. A.; Ziller J. W.; Heyduk A. F. Chem. Sci. 2013, 4, 1906–1910. 10.1039/c3sc22335c. [DOI] [Google Scholar]; b Wong J. L.; Higgins R. F.; Bhowmick I.; Cao D. X.; Szigethy G.; Ziller J. W.; Shores M. P.; Heyduk A. F. Chem. Sci. 2016, 7, 1594–1599. 10.1039/C5SC03006D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Brown S. N. Inorg. Chem. 2012, 51, 1251–1260. 10.1021/ic202764j. [DOI] [PubMed] [Google Scholar]; See also:; b Bhattacharya S.; Gupta P.; Basuli F.; Pierpont C. G. Inorg. Chem. 2002, 41, 5810–5816. 10.1021/ic025766+. [DOI] [PubMed] [Google Scholar]
- Löwdin P. J. Chem. Phys. 1950, 18, 365–375. 10.1063/1.1747632. [DOI] [Google Scholar]
- a Blanksby S. J.; Ellison G. B. Acc. Chem. Res. 2003, 36, 255–263. 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]; b For substrates S7–S9, the substrate with the weaker C–H bond is more easily converted. The bond dissociation energy (BDE) at 298 K (ΔH298) of the benzylic C–H bond in toluene is 89.8 kcal mol–1. The BDE (ΔH298) of benzylic C–H bond of ethylbenzene (77.6 kcal mol–1) is ca. 12 kcal mol–1 lower than that of toluene. Therefore, C–H bond activation of S7 (o-substituted ethylbenzene) is easier than S8 and S9 (o-substituted toluene), and this is reflected in higher yield of P7a (53%) compared to that of P8a (46%) or P9a (44%). Substrates S2 and S10 with similar allylic C–H bonds gave similar yields of products (P2a 96%, P10a 95%). The BDEs (ΔH298) of allylic C–H bonds are very similar to those of benzylic C–H bonds: propene (88.8 kcal mol–1) vs toluene (89.8 kcal mol–1), 1-butene (76.5 kcal mol–1) vs ethylbenzene (77.6 kcal mol–1), 1-pentene (75.4 kcal mol–1) vs n-propylbenzene (76.4 kcal mol–1). However, S2 with an allylic C–H bond gave P2a in high yield (96%), while S1 with a benzylic C–H bond resulted in only 65% of P1a. The BDE (ΔH298) of a benzylic C–H bond adjacent to an alkoxy group is comparable to the BDE (ΔH298) of a benzylic C–H bond adjacent to an alkyl group. However, S6 gave 92% of P6a, whereas S1 gave only 65% of P1a. So, no clear correlation between C–H bond strength of substrates and yields of products can be found for S1, S2, and S6.
- a Korstanje T. K.; van der Vlugt J. I.; Elsevier C. J.; de Bruin B. Science 2015, 350, 298–302. 10.1126/science.aaa8938. [DOI] [PubMed] [Google Scholar]; b Drost R. M.; Rosar V.; Dalla Marta S.; Lutz M.; Demitri N.; Milani B.; de Bruin B.; Elsevier C. J. ChemCatChem 2015, 7, 2095–2107. 10.1002/cctc.201500200. [DOI] [Google Scholar]
- ICP-AAS analysis confirmed <10 ppm levels for Co, Rh, Ir, Ni, Pd, and Cu; see the Supporting Information for details.
- Sharon D. A.; Mallick D.; Wang B.; Shaik S. J. Am. Chem. Soc. 2016, 138, 9597–9610. 10.1021/jacs.6b04636. [DOI] [PubMed] [Google Scholar]
- Review on nitrogen-centered ligand radicals:Olivos Suarez A. I.; Lyaskovskyy V.; Reek J. N. H.; van der Vlugt J. I.; de Bruin B. Angew. Chem., Int. Ed. 2013, 52, 12510–12529. 10.1002/anie.201301487. [DOI] [PubMed] [Google Scholar]
- Experimental detection of cobalt nitrene radical species:; a Goswami M.; Lyaskovskyy V.; Domingos S. R.; Buma W. J.; Woutersen S.; Troeppner O.; Ivanović-Burmazović I.; Lu H.; Cui X.; Zhang X. P.; Reijerse E. J.; DeBeer S.; van Schooneveld M. M.; Pfaff F. F.; Ray K.; de Bruin B. J. Am. Chem. Soc. 2015, 137, 5468–5479. 10.1021/jacs.5b01197. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lyaskovskyy V.; Olivos Suárez A. I.; Lu H.; Jiang H.; Zhang X. P.; de Bruin B. J. Am. Chem. Soc. 2011, 133, 12264–12273. 10.1021/ja204800a. [DOI] [PubMed] [Google Scholar]; For efficient Co-catalyzed intramolecular C(sp3)-H amination of aliphatic azides, see:; c de Bruin B.; Kuijpers P. F.; Tiekink M. J.; Breukelaar W.; Broere D. L. J.; van Leest N. P.; van der Vlugt J. I.; Reek J. N. H. Chem. - Eur. J. 2017, 10.1002/chem.201700358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoichiometric reactions between complex 1 and Boc2O resulted in full recovery of 1. This suggests that the catalyst activation process by Boc2O is most likely an energetically uphill process generating the active form of the catalyst as a short-lived species in rather low concentrations.
- Slow diffusion of pentane into the blue-green solution led to almost quantitative precipitation of a microcrystalline blue solid, but attempts to analyze this species by single-crystal X-ray structure determination were unsuccessful to date.
- The C(sp3)–H amination of S1 in the presence of Boc2O with 1 and TlPF6 (both 5 mol%) led to the formation of P1a and P1b in the same ratio of ∼1.6:1 as observed under the standard conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.