

Abstract
One of the intriguing questions regarding cell motility concerns the mechanism that makes stationary cells move. Here, we provide the first physical evidence that the onset of breast cancer cell motility in response to insulin-like growth factor I (IGF-I) correlates with lowering of adhesion strength from 2.52 ± 0.20 to 1.52 ± 0.13 μdynes/μm2 in cells attached to fibronectin via α5β1 integrin. The adhesion strength depends on the dose of IGF-I and time of IGF-I treatment. Weakening of cell-matrix adhesion is blocked significantly (p < 0.01) by the catalytically inactive IGF-I receptor (IGF-IR) and the phosphoinositide 3-kinase (PI-3 kinase) inhibitor LY-294002, but it is unaffected by mitogen-activated protein kinase kinase inhibitor UO-126 and Src kinase inhibitor PP2. Sustained blockade of Rho-associated kinase (ROCK) with Y-27632 down-regulates adhesion strength in stationary, but not in IGF-I-treated, cells. Jasplakinolide, a drug that prevents actin filament disassembly, counteracts the effect of IGF-I on integrin-mediated cell adhesion. In the absence of growth factor signaling, ROCK supports a strong adhesion via α5β1 integrin, whereas activation of the IGF-IR kinase reduces cell-matrix adhesion through a PI-3K-dependent, but ROCK-independent, mechanism. We propose that disassembly of the actin filaments via PI-3 kinase pathway contributes to weakening of adhesion strength and induction of cell movement. Understanding how cell adhesion and migration are coordinated has an important application in cancer research, developmental biology, and tissue bioengineering.
INTRODUCTION
Coordinated movement of epithelial cells drives blastocyst implantation and tissue morphogenesis at early stages of embryonic development (Wang and Armant, 2002). Throughout adult life, movement of epithelial cells continues to play a key role in tissue reparative and regenerative processes and evidently contributes to the neoplastic disease termed carcinoma (Polyak, 2001; Casanova, 2002). Behavior of epithelial cells is believed to be tightly controlled by tissue microenvironment consisting of secreted hormones, growth factors, cytokines, and filamentous components of the extracellular matrix (ECM) (Birchmeier et al., 1995; Boudreau and Bissell, 1998). Although many aspects of this important regulatory mechanism remain unknown, normal cellular responses to external signals find a parallel in pathological changes that accompany malignant transformation. For instance, in the developing mammary gland, epithelial cells display many of the properties associated with tumor progression, including rapid cell growth and movement into surrounding stromal tissue (Wiseman and Werb, 2002). Also, many factors necessary for the development of the mammary gland have been implicated in breast cancer. IGF-I, a physiological peptide with endocrine, paracrine, and autocrine roles, is one of the factors that has been postulated to mediate directly the cross-talk between the stroma and epithelium during mammary gland branching morphogenesis and carcinoma progression (Kleinberg et al., 2000; Sachdev and Yee, 2001). At the molecular level, signaling effects of IGF-I are mediated through the type I receptor for IGF-I (IGF-IR), a cell surface protein with intrinsic tyrosine kinase activity (Adams et al., 2000). While acting through the IGF-IR, IGF-I is capable of promoting migration in a wide range of normal and tumor cell types, including carcinomas of the lung, the pancreas, and the breast (Leventhal and Feldman, 1997). The interruption of IGF-IR expression by antisense mRNA (Long et al., 1998), inhibition of IGF-I interaction with its receptor (Dunn et al., 1998), or introduction of the inactive form of the IGF-IR (Guvakova et al., 2002; Sachdev et al., 2004) restricts cell motile behavior, suggesting that targeting IGF-IR function may inhibit the metastatic potential of the cell.
Studies in different cells have indicated that IGF-I may have an impact on cell motility through regulation of cell adhesion to the ECM (Jones et al., 1995; Doerr and Jones, 1996). Among the transmembrane receptors mediating cell attachment to the matrix, the best recognized are integrins (Geiger et al., 2001). These receptors bind both ECM proteins and intracellular cytoskeleton-associated proteins and provide means of cell anchorage needed for tissue organization and traction during migration. It is believed that during migration cell-matrix adhesion is dynamically regulated by cycles of reversible interactions between integrins and their extra- and intracellular molecular partners (Ridley et al., 2003). Although growth factors are known to induce cell migration, whether and how these peptides affect adhesive function of integrins is unclear. It has been predicted mathematically that cell migration depends on the physical strength of cell-substratum attachment (DiMilla et al., 1991). It also has been envisioned that weak cell-substratum interactions would not provide a sufficient traction force for cell spreading and hence movement, whereas strong adhesion to the substratum would limit generation of traction force and prevent cell locomotion. Only when the strength of cell-matrix interaction reaches a certain intermediate, or so-called “optimal” level, can cell movement be permitted (DiMilla et al., 1993). Although the existence of an optimal strength of cell-substratum adhesive interactions that favors cell migration has been suggested, direct evidence demonstrating biological mechanisms that down-regulate the adhesive strength has been lacking. Determination of dynamic transition in adhesion strength requires application of adhesion assays in which the strength of attachment is measured quantitatively. Previous studies of cell adhesion have focused on either assessing the fraction of cells that resist detachment of a low force in a conventional washing assay or measurements of initial and therefore relatively low attachment strength to characterize the short-term cell-matrix interactions (Palecek et al., 1997). Those studies demonstrated that variations in either integrin cell surface expression levels (Keely et al., 1995), the adsorbed concentration of substratum protein, a ligand for integrins (DiMilla et al., 1993), or integrin-ligand affinity (Huttenlocher et al., 1996) could affect the adhesion strength and a speed of cell movement.
To our knowledge, there are no reports to date on mechanism by which growth factors control adhesion strength of integrins. In this study, we applied original biophysical approach and investigated two new aspects of cell-matrix interactions: 1) dynamics of the physical strength of long-term adhesion in stationary and moving breast carcinoma cells and 2) the nature of intracellular mechanisms that control the ligand binding function of integrins and contribute to the onset of carcinoma cell movement.
MATERIALS AND METHODS
Cells and Reagents
HT-1080 human fibrosarcoma and T47D and MDA-MB-231 human breast cancer cell lines were purchased from the American Type Culture Collection (Manassas, VA) and cultured according to its protocols. Generation of clonal derivatives of MCF-7 human breast cancer cells overexpressing ∼18- to 20-fold increase in either the wild-type (WT) or dead kinase mutant (DK) of the human IGF-IR have been reported previously (Guvakova and Surmacz, 1997; Guvakova et al., 2002). These clones were grown in Dulbecco's modified Eagle's medium/F-12 (1:1) containing 5% calf serum (CS). For overnight serum starvation, cells were incubated in phenol red and serum-free DMEM/F-12 containing 0.5 mg/ml bovine serum albumin (BSA). Culture media and CS were purchased from Invitrogen (Carlsbad, CA). Recombinant human IGF-I was purchased from Bachem California (Torrance, CA), and it was used at the concentration of 100 ng/ml unless indicated otherwise. Pharmacological inhibitors LY-294002, Y-27632, jasplakinolide, PP2, and PP3 were from Calbiochem (San Diego, CA). UO-126 was purchased from Promega (Madison, WI). BSA fraction V was from Sigma-Aldrich (St. Louis, MO), human fibronectin was from Invitrogen, bovine collagen was from Vitrogen (Cohesive Technologies, Palo Alto, CA), mouse laminin from Invitrogen, ethidium homodimer was from Molecular Probes (Eugene, OR), and 25-mm-diameter glass cover slips were from Bellco Glass (Vineland, NJ).
Spinning Disk Detachment Assay
Cell adhesion was measured using a spinning disk device that exposes a cell population to a linear hydrodynamic shear gradient. The shear stress required to detach 50% of the cells, called adhesion strength τ50, has been shown to be directly proportional to the number of integrin-ligand bonds (Garcia et al., 1998; Shi and Boettiger, 2003). Coverslips were coated in 10 μg/ml human fibronectin in phosphate-buffered saline (PBS) for 30 min and blocked in 1% heat-inactivated BSA in PBS at room temperature. Then, 105 cells were plated on a coverslip, incubated overnight in serum-free medium, and treated with IGF-I at different concentrations or for different times in the absence or presence of the pharmacological inhibitors. The coverslips were then placed on a spinning disk device, consisting of a rotating shaft bathed in buffer (24 mM HEPES, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, and 1 mM glucose, pH 7.4) within a baffled chamber. The flow of buffer applies a shear force, τ, which is greatest at the coverslip's edges and negligible at its center. After a 5-min spin, cells were fixed in 95% ethanol, permeabilized with 1% Triton X-100, and cell nuclei were visualized with 0.5 μg/ml ethidium homodimer. In each experiment, 61 fields (0.5 mm2 each) of stained nuclei were counted using liquid-cooled charge-coupled device (CCD) camera and image analysis software (Phase 3 Imaging, Glenn Mills, PA). A range of τ applied resulted in a cell detachment profile from which τ50, required to detach 50% cells was calculated. For this, cell counts were plotted relative to radial position on the coverslip and normalized to cell count in the center, where the shear force is zero. The fraction of adherent cells was fitted to a sigmoid curve, where the inflection point represents τ50. Cells were analyzed in a series of experiments over a period of 17 mo with at least three replicates in each experiment. In total, 8-10 × 103 cells were analyzed for each data point.
Flow Cytometry Analysis
Suspended cells were incubated with human α5 integrin (BIIG2) rat monoclonal antibody (mAb) (hybridoma was a gift from C. Damsky, University California San Francisco) in PBS for 1 h at 4°C, followed by three washings in ice-cold PBS and incubation with anti-rat fluorescein isothiocyanate (FITC)-conjugated secondary IgG (Oncogene Science, Cambridge, MA) for 30 min at 4°C. To detect β1 integrin, cells were incubated with human β1 integrin (TS2/16) mouse hybridoma (American Type Culture Collection) and then with anti-mouse FITC-conjugated secondary IgG (Vector Laboratories, Burlingame, CA). The primary antibodies were omitted in the control experiments. Cell surface immunofluorescence of each integrin was analyzed in a FACScan (BD Biosciences, San Jose, CA) in two independent experiments.
Short-Term Adhesion Assay
Cells were fluorescently labeled with 4 μg/ml calcein AM (Molecular Probes) for 1 h at 37°C. Suspended labeled cells were incubated with function blocking monoclonal antibodies to β1 integrin (AIIB2) (gift from C. Damsky), α5 integrin (BIIG2), or noninhibitory β1 integrin antibody (TS2/16). In the control samples, antibody was omitted. After 30 min, cells were seeded into 96-well plates coated with a range of concentrations of fibronectin (0.0-10.0 μg/ml) and incubated for 1 h at 37°C. After nonadherent cells were removed by three washings, adhesion of fluorescent dye-labeled cells to fibronectin-coated 96-well plates was measured using MicroFLUOR reader (Dynatech Labs, Chantilly, VA) with fluorescein optical filter (488/520 nm).
Immunoprecipitation (IP) and Western Blot (WB) Analysis
Cells were lysed in buffer containing 50 mM HEPES pH 7.5, 150 mM NaCl, 1.5 mM MgCl, 1 mM EGTA, 10% glycerol, 20 μg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride, 2 mM Na orthovanadate, and 1% Triton X-100. For WBs, 50 μg of total protein was resolved by SDS-PAGE and transferred to Hybond ECL nitrocellulose (Amersham Biosciences, Piscataway, NJ). Proteins were precipitated from 500 μg of total protein extracts in 500 μl of HNTG buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% Triton X-100, and 10% glycerol) overnight at 4°C. For IP of β1 integrin, extracts were incubated with 4 μg of β1 integrin monoclonal IgG1, clone P4C10 (Invitrogen) and 50 μl of anti-mouse IgG-agarose (Sigma-Aldrich). For IP of IGF-IR, extracts were incubated with 5 μg/ml IGF-IR mAb (Calbiochem) and 50 μl of anti-mouse IgG-agarose. The immunoprecipitated proteins were resolved by SDS-PAGE. The blots were probed with antibodies against IGF-IR β subunit (polyclonal, C-20; Santa Cruz Biotechnology, Santa Cruz, CA), β1 integrin (polyclonal to cytoplasmic domain); phoshorylated c-Src (pY 418 Src; BioSource International, Camarillo, CA) and total c-Src (monoclonal IgG1, clone GD11; Upstate Biotechnology, Lake Placid, NY), phospho-mitogen-activated protein kinase (MAPK) (clone E10; New England Biolabs, Beverly, MA), extracellular signal-regulated kinase (ERK)1/2 (polyclonal k23; Santa Cruz Biotechnology); phosphor-Akt Ser 473 and Akt (polyclonal; Cell Signaling Tehcnology, Beverly, MA); and anti-phosphotyrosine (monoclonal PY-20; BD Transduction Laboratories, Lexington, KY). To visualize primary antibody-bound protein, the secondary antibodies conjugated to horseradish peroxidase (1:5000 dilution; Oncogene Research) and ECL detection solutions (Amersham Biosciences UK, Little Chalfont, Buckinghamshire, United Kingdom) were applied. The chemiluminescent intensity of bands was digitized using the Image Analyser LAS-1000 Plus system and the Image Reader LAS-1000 Lite version 1.0 software (Fuji, Tokyo, Japan). When several proteins were to be detected on the same membrane, nitrocellulose was incubated in stripping buffer (Pierce Chemical, Rockford, IL), washed in buffer (100 mM Tris, pH 7.5, 1 M NaCl, and 1% Tween 20), and reprobed. The relative activity of c-Src kinase was quantified as the ratio of phosphorylated protein at autophosphorylation sites (tyrosine 418 for human c-Src) to the amount of total protein detected on the same blot.
Rho-GTPase Pull-Down Assay
The active GTP-bound form of RhoA (RhoA-GTP) was purified by affinity precipitation by using a glutathione S-transferase (GST) fusion protein of Rho-binding domain (RBD) of the Rho effector protein Rhotekin as described previously. Cells were serum starved overnight, treated with 100 ng/ml IGF-I, and then lysed in 1% Triton X-100 buffer. Clarified cell lysates were immediately incubated with 30 μg of GST fusion protein of RBD Rhotekin bound to glutathione-conjugated beads (Upstate Biotechnology) for 45 min at 4°C. Complexes bound to the beads were washed three times in ice-cold HNTG buffer and then removed from the beads in reducing Laemmli buffer. To detect activated GTP-bound RhoA, the samples were analyzed by 12% SDS-PAGE and Western blotting by using polyclonal antibody recognizing RhoA (SC). To determine the total quantities of RhoA GTPase in the samples, 50 μg of total cellular protein was analyzed in parallel. The relative activity of RhoA is a ratio of GTP-bound RhoA protein to the amount of total protein detected on the same sample.
Microscopy
Phase-contrast microscopy was used with a 20× objective lens on a Nikon (Diaphot) inverted microscope linked to CCD camera; images were captured using MetaMorph software version 4.6 (Universal Imaging, West Chester, PA). To visualize filamentous actin (F-actin), cells were fixed with 3.7% formaldehyde in PBS for 15 min, permeabilized with 0.05% Triton X-100 in PBS for 5 min, and stained with 1 μg/ml tetramethylrhodamine B isothiocyanate-phalloidin (Sigma-Aldrich) for 30 min. In some experiments, actin was visualized with mAb to actin (Clone AC-40; Sigma-Aldrich) in cells fixed in methanol. The samples were examined under a Nikon Eclipse E600 MRC 1024 confocal laser-scanning microscope (Bio-Rad, Hercules, CA) with a Plan Apo 60×/1.4 oil objective lens (Nikon, Melville, NY). Z-series images were acquired using the Bio-Rad LaserSharp 2000 software run under the OS/2 Warp ConnectTM operating system.
Migration Assay
Cell layers were grown to 70-90% confluence on 10 μg/ml fibronectin-coated coverslips. Control, serum-starved cells, and cells pretreated with drugs for 1 h were scratched with a tip of micropipette and morphology of the wounded monolayer was recorded under a Nikon Diaphot microscope with a Plan Fluor 10×/0.30 objective lens (Nikon). Then, samples were exposed to IGF-I (100 ng/ml) at 37°C, and morphology of the wounded monolayer was recorded in the same microscopic field after 30, 60 and 120 min. Images were quantified to determining the mean distance between the center of the scratch and cells at the edge of the wound by taking 42 measurements before and after addition of IGF-I in each sample in two independent experiments.
Statistical Analysis
The statistical evaluation of results was performed using single-factor analysis of variance (ANOVA). The significance level was taken as p ≤ 0.05.
RESULTS
IGF-I-induced Breast Cancer Cell Motile Behavior Involves β1 Integrins
We have previously established that the treatment of MCF-7 human breast cancer cells with IGF-I results in the development of asymmetric cell shape, reorganization of the actin cytoskeleton, and the quick onset of cell motility. The signaling mediated by the IGF-IR and motile behavior was enhanced in MCF-7 cells overexpressing the wild-type (WT) IGF-IR and blocked in the cells expressing a catalytically inactive DK mutant of this receptor (Figure 1A; Guvakova et al., 2002). These events were coupled with structural and biochemical changes in focal adhesions, strongly suggesting that the induction of cell migration is linked with the ability of IGF-I to modulate cell-matrix interactions (Guvakova and Surmacz, 1999). The most prominent integrins found in MCF-7 cells are heterodimers of α subunits with β1 integrin (Doerr and Jones, 1996). To test whether IGF-IR signaling controls function of β1 integrins, we examined the effects of IGF-I on adhesive behavior mediated by α2β1, α3β1, and α5β1 integrins in the cells adherent to collagen, laminin, and fibronectin, respectively. Clones of MCF-7 cells overexpressing either the WT or DK mutant of the IGF-IR were plated on matrix-coated dishes, serum-starved, and then stimulated with IGF-I for 1 h. Although under serum-free conditions both WT and DK cells were able to attach and spread, only the WT cells developed the motile phenotype in response to IGF-I (Figure 1, B and C). These findings revealed the regulatory role of IGF-I in motile behavior of breast carcinoma cells via several β1 class integrins. To better understand the mechanisms that drive migration of breast cancer cells, we focused our analysis on the adhesive function of α5β1 integrin, the fibronectin receptor.
Figure 1.

IGF-I induces breast cancer cell motility on various biological substrata. (A) IGF-IR expression levels and signaling in MCF-7-derived cells. Left, cells were stimulated with 100 ng/ml IGF-I for 15 min. Expression of the IGF-IR was compared in MCF-7 cells (parental), MCF-7 cells transfected with pcDNA3 vector (mock), pcDNA3 plasmids encoding the full-length human IGF-IR (WT), and its DK form. Total proteins (50 μg) were resolved by 6% PAGE, blotted with an antibody recognizing the C terminus of the IGF-IR β subunit, stripped, and reprobed with a PY-20 antibody. Total (IGF-IR) and phosphorylated (pIGF-IR) levels of the receptor are shown. Right, mock, WT, and DK cells were stimulated with 100 ng/ml IGF-I for indicated times. Total (ERK1/2 and Akt) and phosphorylated (pERK1/2 and pAkt) levels of ERK1/2 and Akt were assessed in the whole cellular extracts resolved by 12% PAGE; IGF-IR levels were assessed in the immunoprecipitates resolved by 6% PAGE and probed with IGF-IR (IGF-IR) and PY-20 (pIGF-IR) antibodies (see Materials and Methods). Molecular weight markers are shown in kilodaltons. Note, IGF-I-induced phosphorylation of IGF-IR, and its downstream targets Akt and ERK1/2 MAP kinases, was enhanced in WT cells compared with mock MCF-7 cells; in DK cells, IGF-I-induced signaling events were completely blocked. (B and C) Phase-contrast micrographs of WT (B) and DK (C) cells plated on glass coverslips coated with collagen (100 μg/ml), laminin (10 μg/ml), and fibronectin (10 μg/ml). Left, cells incubated in serum-free medium overnight. Right, serum-starved cells treated with 100 ng/ml IGF-I for 1 h. 100% of the WT cells developed motile phenotype in response to IGF-I. Arrows in B point at the leading edge of lamellipodia in moving cells. Bar, 10 μm.
Induction of Cell Motility by IGF-I Is Not Related to the Changes in α5 and β1 Integrin Expression Levels
In mammary epithelial cells, motility and adhesive interactions with the matrix may be mediated by the level of surface integrin expression (Keely et al., 1995). To test the possibility that forced overexpression of the IGF-IR in MCF-7 transfectants affected α5β1 integrin levels, possibly interfering with our measurements, we first examined the cell surface expression levels of α5 and β1 integrins. Flow cytometry analysis with antibodies recognizing human integrins clearly indicated that the levels of α5 and β1 integrins were comparable in the WT and DK cells (Figure 2A).
Figure 2.

α5 and β1 integrin expression in MCF-7-derived cells. (A) Flow cytometry for MCF-7 parental cell line, WT cells overexpressing wild-type human IGF-IR, and DK cells expressing kinase inactive IGF-IR. Cells were treated with primary antibodies for β1 (mouse anti-human TS2/16) and α5 (mouse anti-human BIIG2) and anti-mouse secondary antibody conjugated to FITC. Dotted line, control (no primary antibody); solid line, cells labeled with primary and secondary antibodies. (B) Short-term adhesion of WT cells to tissue culture plates coated with 0.32, 0.63, 1.25, 2.5, 5.0, and 10.0 μg/ml fibronectin by using standard protocols for coating cover slips and adhesion assay (see Materials and Methods). Adhesion was detected in the presence of AIIB2 (□), a function blocking antibody for β1 integrin; BIIG2 (▵), a function blocking antibody for α5 integrin; and TS2/16 (○), a noninhibitory antibody for b1 integrin. Control (⋄), no antibody. Results are means of triplicates in two independent repeats. Error bar, SEM. (C) β1 integrin expression in WT and DK cells treated with 100 ng/ml IGF-I for 0, 5, 15, 30, and 60 min. The mAb P4C10 was used for IP, and a polyclonal antibody to the C terminus of β1 integrin (cyto) was used for the WB.
Expression of the fibronectin receptor α5β1 integrin, rather than αvβ1 integrin, has been reported in MCF-7 cells (Doerr and Jones, 1996). Here, we used α5 and β1 integrin function blocking antibodies to confirm that attachment of MCF-7-derived cells to fibronectin is indeed mediated by the α5β1 integrin (Figure 2B). In addition, we evaluated the protein expression levels of α5 and β1 integrins in the immunoprecipitates of the WT and DK cells treated with IGF-I. Due to low levels of α5 integrin expression in MCF-7-derived cells, as evident from fluorescence-activated cell sorting analysis (Figure 2A), the levels of α5 were below the detection limits in the Western blot. The immunoprecipitated β1 integrin was readily detectable in the extracts of both WT and DK cells. In serum-starved cells and cells exposed to IGF-I for 5, 15, 30, and 60 min, β1 integrin levels were similar (Figure 2C). Thus, neither overexpression nor ligand-induced activation of the IGF-IR altered the levels of α5 and β1 integrins in MCF-7 cells. This implied a role for the IGF-IR kinase in the regulation of adhesive function of α5β1 integrins during initiation of breast cancer cell migration.
IGF-IR Signaling Down-Regulates the Adhesion Strength in Cells Attached to Fibronectin
Current concept that only the intermediate levels of cell-matrix adhesion are fully permissive for cell migration comes from works in which the short-term cell adhesion was measured 20-30 min after cell plating on substratum, whereas cell migration parameters were assessed 12-24 h later (DiMilla et al., 1993; Palecek et al., 1997). A disaccord in a time scale of two assays makes it difficult to directly compare the adhesive and motile responses and to identify the mechanisms coordinating these processes. In MCF-7 cells, IGF-I increases motile behavior within first hour of treatment (Guvakova et al., 2002). Hence, to investigate mechanisms by which IGF-I contributes to the initiation of carcinoma cell motility, we decided to use a spinning disk detachment assay and to measure adhesion strength at the time when cell motility is increased.
We hypothesized that activation of the IGF-IR triggers the intercellular signaling that relieves the stress mediated by integrins on the ECM and thereby reduces cell-matrix adhesion and facilitates cell movements. We began to test this hypothesis by the measuring adhesion strength τ50 in the cells exposed to doses of IGF-I ranging from 0.1 to 100.0 ng/ml. The increase in IGF-I correlated with a decrease in τ50 from 251 to152 dynes/cm2 in the WT cells (Figure 3, A-F). In the WT, but not in DK, cells exposed to 1.0 ng/ml IGF-I, which is the minimum dose sufficient to induce IGF-IR autophosphorylation in these cells, the adhesion strength decreased significantly (p < 0.05) compared with the untreated control cells (Figure 3F).
Figure 3.

Adherent fraction profiles for MCF-7 cells treated with IGF-I. Cells were serum-starved overnight on fibronectin-coated coverslips and treated with IGF-I for 1 h before exposure to shear stress on a spinning disk device. τ50 is the shear stress at which 50% of the cells are removed from the coverslip. The adherent fraction is the number of cells remaining, normalized to the number at the center, where shear stress is negligible. Experiment (•); sigmoid fit (□); r2, regression coefficient, indicates how accurate the experimental data fit into a sigmoid model. (A) WT cells serum-starved. (B) IGF-I, 0.1 ng/ml. (C) IGF-I, 1.0 ng/ml. (D) IGF-I, 10 ng/ml. (E) IGF-I, 100 ng/ml. (F) τ50 for each treatment plotted as a function of IGF-I concentration. WT cells (•), DK cells (▪). p values < 0.05 by ANOVA are marked with an asterisk for any τ50 significantly different from concentration of IGF-I of 0.0 ng/ml. Results are presented as means of τ50± SEM.
To determine directly whether the enzymatic activity of the IGF-IR kinase plays a role in down-regulating integrin-mediated adhesion, τ50 was compared in cells expressing functional WT or catalytically inactive DK mutant of the IGF-IR. Under serum-free conditions, τ50 was the same in WT and DK cells (∼250 and 240 dynes/cm2, respectively). After a 60-min treatment with IGF-I, τ50 in WT cells declined linearly to ∼150 dynes/cm2 and remained at this level for another hour. In sharp contrast, IGF-I had no significant effect on adhesiveness in the DK cells exposed to this peptide for 2 h (Figure 4A).
Figure 4.
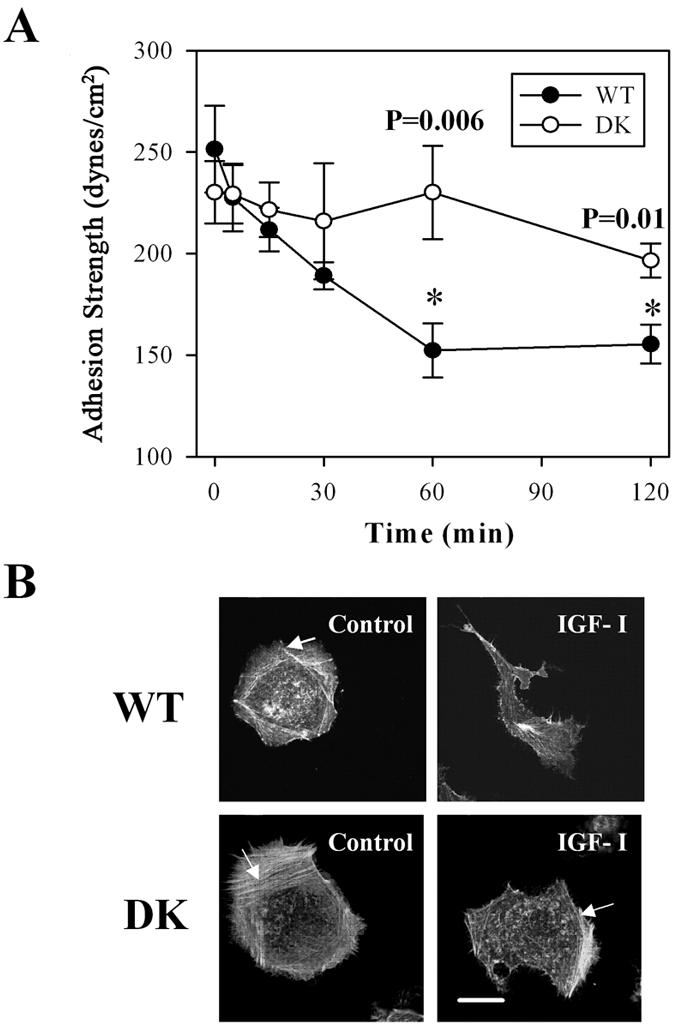
Adhesion strength and F-actin reorganization is dependent on the function of the IGF-IR kinase. (A) Adhesion strength as a function of time for the WT and DK MCF-7 cells treated with 100 ng/ml IGF-I over 2 h. Adhesion strength is measured as the shear stress at which 50% of cells on a spinning disk device remain. p values < 0.05 are marked with asterisks for any τ50 in WT cells that are significantly different from that in DK cells. Results are presented as means of τ50± SEM. (B) F-actin in cells starved in serum-free medium (SFM); starved and then treated with 100 ng/ml IGF-I for 1 h (IGF-I). Arrows point at peripheral stress fibers.
We next examined whether changes in cell-matrix adhesion could be linked to the reorganization of the actin cytoskeleton. Staining with phalloidin revealed the alterations in the distribution of F-actin associated with motile phenotype. The peripheral actin fibers normally present in the serum-starved WT and DK cells (arrows in Figure 4B) disappeared in the WT cells treated with IGF-I. These changes do not occur in DK cells that remained mainly stationary in the presence of IGF-I. Together, these results provide first and direct evidence that the initiation of breast cancer cell migration is associated with down-regulation of a steadystate cell adhesion via integrins as a result of IGF-IR tyrosine kinase activation. The adhesive changes coincided with the altered assembly of F-actin in response to IGF-I.
Inhibition of c-Src Autophosphorylation Does Not Affect the Adhesion Strength
The growth factor-induced signaling regulating integrin-mediated adhesion strength has not been documented. The only signaling molecule reported so far to directly control the strength of integrin adhesion and only during initial cell attachment to the substratum is Src (Felsenfeld et al., 1999; Datta et al., 2002). There are several ways in which Src can be activated, including the intramolecular interactions of Src involving enhanced phosphotyrosine residues 418 in the kinase domain (Frame et al., 2002). Phosphorylation of Src also may be controlled by the receptor type tyrosine kinases. We therefore addressed whether autophosphorylation of c-Src is regulated by IGF-IR signaling in breast cancer cells exposed to IGF-I. We made use of PP2, a compound known to block c-Src autophosphorylation at the tyrosine residue 418, and PP3, a negative control compound for PP2 inhibitor. Using phospho-specific c-Src antibody, we found that in the immobile, serum-starved cells c-Src was phosphorylated at the tyrosine residues 418. IGF-I had minor stimulatory effect on basal phoshorylation at the residues 418 (Figure 5A, first two left lanes). We also found that PP2, unlike PP3, selectively and by 63% on average inhibited phosphorylation at tyrosine 418 in the catalytic domain of Src (Figure 5, A and B).
Figure 5.

Effects of the PP2 Src kinase inhibitor on c-Src autophosphorylation, adhesion strength, and F-actin. (A) c-Src autophosphorylation at tyrosine 418 measured in the absence or presence of 100 ng/ml IGF-I. Serum-starved cells were pretreated with 1.0 μM PP2 or 1.0 μM PP3 for 1 h and then stimulated with IGF-I for 30 min. Drugs were omitted in the control experiments. Whole cell extracts were resolved by 8% PAGE. Blot was probed with antibody recognizing Src phosphorylated at tyrosine 418 (pY418 Src) and then with pan Src antibody. (B). Summary of three independent experiments. Bar graph shows mean value of relative phosphorylation of c-Src (phospho-Src/total Src). Error bar, SEM. (C) τ50 was measured in WT cells the absence (-IGF-I) or presence (+IGF-I) of IGF-I at 100 ng/ml. Serum-starved cells were treated with 1 μM PP2 or 1 μM PP3 alone for 2 h or pretreated with drug for 1 h and then exposed to IGF-I and drug for another hour. In the control experiments, drugs were omitted. Summary of four independent experiments. Error bar, SEM. Asterisks, significance of reduction in τ50 caused by IGF-I. (D) F-actin in cells treated with the drug alone for 2 h (PP2 and PP3) or pretreated with drug for 1 h and then exposed to IGF-I and drug for 1 h (PP2 + IGF-I, PP3 + IGF-I). Without IGF-I, cells assembled prominent stress fibers at periphery (arrows); cell shape is nearly circular. Exposure to IGF-I results in a loss of long actin filaments and development of an asymmetric cell shape. Bar, 10 μm.
To test whether inhibition of the catalytic activity of Src with PP2 affects the adhesion strength, τ50 was measured in cells exposed to PP2, PP3 and in cells stimulated with IGF-I after pretreatment with these inhibitors. We found no significant effect of these drugs on either basal adhesion to fibronectin or IGF-I-stimulated weakening of cell-matrix adhesion, because τ50 significantly decreased in the presence of IGF-I in spite of pretreatment with PP2 and PP3 (Figure 5C). In parallel experiments, we assessed F-actin redistribution in the WT cells stimulated with IGF-I in the presence or absence of the drugs and found that, in response to IGF-I, cells were losing peripheral stress fibers and adopting an asymmetric morphology regardless of the type of drug pretreatment (Figure 5D). These findings confirm that PP2 selectively inhibited Src kinase activity; nevertheless, PP2 did not prevent weakening of cell-matrix adhesion, remodeling of F-actin and initiation of cell motility in response to IGF-I.
Activation of the PI-3K Significantly Decreases the Adhesion Strength
The disappearance of the long actin filaments is a PI-3K-dependent effect of IGF-I that precedes the development of the motile cell protrusions in MCF-7 cells (Guvakova and Surmacz, 1999). Also, the pharmacological inhibition of mitogen-activated protein kinase kinase (MEK)1/2 impairs actin reorganization by IGF-I (Guvakova et al., 2002). To test quantitatively the possibility that weakening of cell-matrix adhesion is coupled with reorganization of the actin cytoskeleton, we examined the effects of IGF-I, PI-3K, and MEK1/2 inhibitors on the measurements of τ50 and related that to F-actin. We found that weakening α5β1 integrin-mediated adhesion in response to IGF-I was blocked significantly (p = 0.001) by the PI-3K inhibitor LY-294002 and to a lesser extent by MEK1/2 inhibitor UO-126 (p > 0.05); both used at 25 μM (Figure 6A). This concentration of drugs was sufficient to block a sixfold increase in the activity of MEK1/2 and a ninefold increase in the activity of the PI-3K toward their substrates ERK1/2 and Akt induced by IGF-I (Figure 6B; our unpublished data). Inhibition of the PI-3K prevented the loss of peripheral stress fibers in response to IGF-I, whereas the inhibition of MEK1/2 did not (Figure 6C). Thus, the transition of the adhesion strength from strong to intermediate level, which is optimal for cell migration, requires chiefly PI-3K activity downstream of the IGF-IR.
Figure 6.

Effects of the PI-3K and ERK1/2 MAPK inhibitors on adhesion strength and F-actin. (A) τ50 was measured in WT cells the absence (-IGF-I) or presence (+IGF-I) of IGF-I at 100 ng/ml. Summary of three independent experiments in serum-starved cells (control) and in the cells pretreated with the PI-3K inhibitor or MEK1/2 inhibitor at 25 μM for 1 h (LY-294002 and UO-126, respectively) and then exposed or not to IGF-I and drug for 1 h. Error bar, SEM. Asterisk, significance of prevention in a decrease of τ50 caused by IGF-I. (B) Serum-starved cells (0.0), cells treated with 100 ng/ml IGF-I for 15 min after exposure to indicated concentrations of UO-126 for 1 h. Top, ERK1/2 phosphorylation on threonine 202 and tyrosine 204 detected with specific phospo-MAPK antibody (pERK1/2); bottom, the same blot stripped and reprobed with antibody recognizing total ERK1/2. (C) F-actin in serum-starved cells treated with LY-294002 and UO-126 at 25 μM for 2 h. One-half of the samples was pretreated with the same inhibitor for 1 h and then exposed to IGF-I and inhibitor for 1 h (LY-294002 + IGF-I, UO-126+ IGF-I). Cells treated with LY-294002 had a circular shape and a similar pattern of F-actin whether exposed or not to IGF-I. Bar, 10 μm.
ROCK Supports Strong Adhesion via α5β1 Integrins in Stationary Cancer Cells
The integrity of the cytoskeleton depends on actomyosin tension that may be controlled by Rho-associated kinase (ROCK) activated by the small GTPase Rho (Amano et al., 1997). Blocking ROCK with Y-27632 is known to decrease cell-generated tension. We, therefore, investigated whether ROCK plays a role in the control of adhesion strength in the stationary and motile carcinoma cells. Strikingly, ROCK inhibitor at 10 μM caused a marked (p = 0.003) reduction of τ50 in serum-starved, but not in IGF-I-treated, cells (Figure 7A). The inhibition of ROCK was associated with a nearly total disappearance of the prominent actin filaments and retraction of cellular membrane in majority of the cells. Although some cells looked more spread than control cells (Figure 7B), they did not show polarized morphology with a leading edge lamellipodia (compare Figure 7B with WT cells in Figure 4B).
Figure 7.

Effects of the ROCK inhibitor on adhesion strength and F-actin. (A) τ50 was measured in WT cells in the absence (-IGF-I) or presence (+IGF-I) of IGF-I at 100 ng/ml. Summary of three independent experiments in serum-starved (control) and in serum-starved cells pretreated with ROCK inhibitor at 10 μM (Y-27632) for 1 h and then exposed to IGF-I for an additional hour. Error bar, SEM. Asterisk, significance of reduction in τ50 caused by Y-27632. (B) F-actin in serum-starved treated with 10 μM Y-27632 for 2 h. Cells also were pretreated with 10 μM Y-27632 and 1 h later stimulated with IGF-I and inhibitor for another hour (Y-27632 + IGF-I). Cells exposed to Y-27632 lost noticeable actin filaments and retracted Individual cells remain spread, left image. Bar, 10 μm. (C) Effect of ROCK inhibitor on adhesion strength of MCF-7 DK, HT-1080, MDA-MB-231, and T47D cells. τ50 was measured in serum-starved cells (control) and in cells treated with 10 μM Y-27632 for 2 h. Summary of three independent experiments is shown in a bar graph. Error bar, SEM. Asterisk, significance of reduction in τ50 caused by Y-27632. (D) Effects of varying concentrations of Y-27632 on adhesion strength and cell morphology. τ50 was measured in WT cells in the absence (-IGF-I) or presence (+IGF-I) of 100 ng/ml IGF-I. Cells were treated with Y-27632 at concentrations of 0.3, 3.0, and 30.0 μM for 2 h, or pretreated with inhibitor for 1 h and then exposed to IGF-I and inhibitor for 1 h. Error bar, SEM. Asterisk, significance of reduction in τ50 caused by IGF-I. (E) Cells were prepared and treated as in D. Phase-contrast images were taken before the spinning assay. Thin black arrows, retracted and rounded cells; bold black arrows, lamellipodia with peripheral ruffles; white arrowheads, abnormally elongated protrusions. (F) Stimulatory effect of IGF-I on RhoA-GTP loading. Serum-starved WT and DK cells were treated with 100 ng/ml IGF-I for indicated times and then processed for RhoA activation. Top, representative blots of active GTP-bound RhoA; bottom, loading control, or total RhoA protein in the cellular extracts. (G) Summary of three to four independent RhoA pull-down assays in WT and DK cells. Error bar, SEM. * and ** denote p = 0.01 and p = 0.03 relative to control, respectively.
To test whether ROCK is needed for adhesion in other stationary cancer cells expressing α5β1 integrin, we compared τ50 in MCF-7 DK, MDA-MB-231, and T47D breast carcinoma and HT-1080 fibrosarcoma cells plated on fibronectin and then exposed to ROCK inhibitor. We found that Y-27632 also reduced adhesion strength in the DK cells, MDA-MB-231, HT-1080 cells, but did not affect T47D cells (Figure 7C). Based on our findings, we conclude that in the immobile cancer cells, ROCK plays a role in the maintenance of strong adhesion mediated by α5β1 integrins, perhaps through generation of the cytoskeleton tension.
Weakening of α5β1 Integrin-mediated Adhesion Is ROCK Independent
Striking down-regulation of adhesion strength caused by ROCK inhibitor led us to suggest that reducing ROCK activity and decreasing cytoskeleton tension might be a meaningful biological mechanism by which IGF-IR signaling could contribute to weakening of breast carcinoma adhesion to the ECM and to the increased motility. To test this idea, we compared τ50 in the cells stimulated with IGF-I in the absence or presence of increasing concentrations of Y-27632 (0-30 μM) (Figure 7D). The cell morphology was recorded before the spinning assay by phase-contrast microscopy that allows visualization of ruffles at the leading edge of lamellipodia in motile cells (Figure 7E). We found that Y-27632 at 3.0 and 30 μM had a similar effect on τ50 compared with that caused by IGF-I alone (Figure 7D); however, changes in cell morphology associated with the decreased adhesion were markedly different. The cells exposed to 3.0 and 30 μM of Y-27632 retracted and rounded up, whereas the cells stimulated with IGF-I polarized and developed fan-like classic lamellipodia with intensive ruffling at the leading edge (bold arrows, Figure 7E). Notably, the cells pretreated with 3.0 and 30 μM and 1 h later stimulated with IGF-I also polarized, but instead of lamellipodia exhibited abnormally elongated protrusions (Figure 7E, arrowheads). The finding that inhibition of ROCK activity affected lamellipodia identified the ROCK involvement in carcinoma cell motility by IGF-I. Thus, it seemed unlikely that IGF-IR signaling inhibited activity of ROCK.
To examine the effects of IGF-I on ROCK activation, we measured the relative activity of the small GTPase RhoA, which is a well recognized activator of ROCK (Amano et al., 2000). Using cellular extracts in a RhoA pull-down assay, we found that in response to IGF-I the relative RhoA activity increased in WT but not in DK cells (Figure 7, F and G). Our findings suggest that activation of the IGF-IR kinase induces up-regulation of ROCK by increasing the intercellular levels of active GTP-bound RhoA. Therefore, we conclude that the effect of the activated IGF-IR on α5β1 integrin-mediated adhesion is independent of its effect on the Rho-ROCK pathway.
Blocking Disassembly of Actin Filaments Prevents Decrease of Adhesion Strength
The correlation between a decrease in adhesion strength and reorganization of F-actin suggested that actin dynamics control the adhesion strength mediated by integrins. Because disassembly of F-actin is the early effect of IGF-IR activation, we sought to have an experimental means to prevent actin disassembly, while preserving other IGF-IR signaling mechanisms. We made use of jasplakinolide, a macrocyclic peptide known to compete with phalloidin for actin binding, inhibit actin filament disassembly, and induce actin polymerization (Cramer, 1999). Jasplakinolide is a cell permeable compound and can be used as an F-actin stabilizer while measuring adhesion strength in live cells. Initial titration experiments showed that 0.01-0.05 μM jasplakinolide had no effect on the morphology of WT cells within 30-40 min of treatment, whereas higher concentrations or longer exposures to the drug resulted in a retraction of cell edges, indicating the toxic effect of the drug (our unpublished data). A time course in serum-starved WT cells revealed that after a 30-min treatment with IGF-I, a reduction of adhesion strength became significant (p = 0.04), whereas in the presence of 0.05 μM jasplakinolide, IGF-I was no longer able to reduce adhesion strength (Figure 8A). Staining with antibody to actin further confirmed the stabilizing effects of jasplakinolide on F-actin (Figure 8B). Due to drug toxicity, however, exposure of cells to 0.05 μM jasplakinolide for 1 h, whether in the absence or presence of IGF-I, caused a marked decrease of adhesion strength below the corresponding controls (Figure 8A). Despite the limitation of manipulation with jasplakinolide, our findings provide first evidence that blocking disassembly of the actin filaments may counteract the regulatory effects of IGF-I on adhesion strength.
Figure 8.
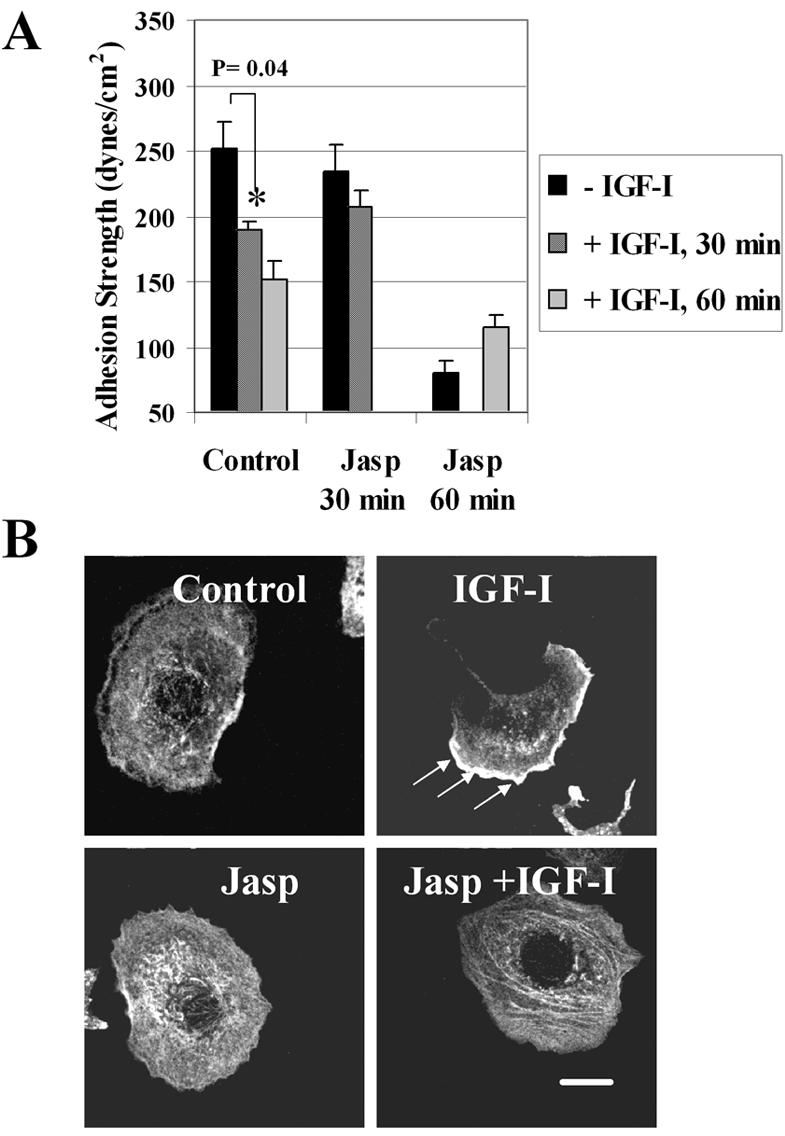
Effects of jasplakinolide on adhesion strength and actin. (A) τ50 was measured in WT cells the absence (-IGF-I) or presence of IGF-I (100 ng/ml) for 30 and 60 min (+IGF-I, 30 min; +IGF-I, 60 min). Serum-starved cells were exposed to jasplakinolide alone (-IGF-I, Jasp 30 min; -IGF-I, Jasp 60 min) or mixed with IGF-I (+IGF-I, Jasp 30 min; +IGF-I, Jasp 60 min). In the control experiments, the drug was omitted. Summary of two independent experiments with triplicate samples. Error bar, SEM. Asterisks, significance of reduction in τ50 caused by IGF-I. (B) Actin in cells serum-starved (control) or treated with 100 ng/ml IGF-I (IGF-I), 0.05 μM jasplakinolide, and a mix of these peptides (Jasp + IGF-I) for 30 min. Exposure to IGF-I results in development of an asymmetric cell shape and concentration of actin at the leading edge of motile lamellipodium (arrows); presence of the drug blocks these effects. Bar, 10 μm. Note, because jasplakinolide competes with phalloidin, actin was visualized by staining with a monoclonal anti-actin antibody that recognizes an epitope located on the C-terminal end in all actin isoforms (Materials and Methods).
Effects of IGF-I on Adhesion Strength and Cell Migration Are Linked
When added to a monolayer of serum-starved MCF-7 cells, IGF-I induced sequential motile responses (Guvakova et al., 2002). The distinct features of the responses were rapid loss of cell-cell contacts and development of motile lamellipodia, followed by movement of the cell body over substratum. To relate effects of IGF-I on cell adhesive and motile behavior, we compared migration of WT and DK cells in a modified “wound closure” assay, in which cell movement toward the center of the scratched areas was modulated by IGF-I and pharmacological inhibitors. The rates of wound closure were calculated based on image analysis and summarized in the bar graph (Figure 9). In the control experiments, IGF-I markedly stimulated migration of WT, but not of DK, cells. In the WT cells, motile responses in the presence of MEK inhibitor, UO-126, were comparable with that induced by IGF-I alone. PP2 and PP3 moderately reduced WT cell migration, but because their effect was similar, it seems to be Src kinase independent. Jasplakinolide blocked cell migration, but its inhibitory effect was likely due to drug toxicity in this 2-h assay. The PI-3K (LY-294002) and ROCK (Y-27632) inhibitors both reduced WT cell wound closure by ∼40%. However, inhibition of the PI-3K blocked lamellipodia and cell-cell separation, whereas inhibition of ROCK allowed lamellipodium formation but prevented cell body translocation (Figures 7E and 9). Together, our data show that in motile cells signaling through the IGF-IR and PI-3K is a key pathway controlling both the reduction of adhesion strength and initiation of cell motile responses, whereas ROCK is required for movement of the cell body.
Figure 9.

Migration of WT and DK cells in response to IGF-I. Images were acquired after scratching in serum-starved WT and DK cells (control) and WT cells pretreated with 25 μM UO-126 (UO-126), 1 μM PP2 and PP3 (shown for PP2), 25 μM LY-294002 (LY-2954002), 10 μM Y-27632 (Y-27632), 0.05 μM jasplakinolide (Jasp) before (-IGF-I) and after (+IGF-I) addition of 100 ng/ml IGF-I for 2 h. The mean distance from the center of the scratch and to the edge of the wound was determined by taking a similar number of measurements before and after exposure to IGF-I (example is shown in UO-126). The relative closure of the wound was calculated as D0 - D2h/D0 × 100%, where D is a distance from the center of the scratch to the edge of the wound before (D0) and after (D2h) exposure to IGF-I. Bar graph, summary of two independent experiments. Error bar, SEM.
DISCUSSION
Despite growing evidence on the motility promoting role of growth factors in normal and pathological processes, very little is known about the function of integrins in growth factor-induced cell motility. Previous studies on the molecular mechanisms that couple growth factor receptors and integrins revealed the physical interactions of surface receptors and cross talk between their signaling pathways (Miyamoto et al., 1996; Schneller et al., 1997). In this study, we established that growth factor-driven carcinoma cell motility involves translation of biochemical signals generated by growth factor receptor tyrosine kinase into down-regulation of adhesion strength to permit cell movement. These results provide physical evidence in support of previously proposed key mechanisms of cell migration and identify novel principle by which IGF-I initiates movement of stationary cells.
Motion versus Immobility: Is There a Relationship between Adhesion Strength and Cell Motility?
Early studies addressing the relationship between cell adhesion and motility implied that the limited attachment of cells to the ECM is essential for rendering cells motile. Through an antisense approach to reduce (Keely et al., 1995) or overexpression techniques to amplify (Palecek et al., 1997) levels of integrins, it has been shown that the speed of cell migration may depend on integrin expression levels. Others exposed cells to the increasing concentrations of the ECM and found that migratory behavior of cells varied with absorbed concentration of matrix proteins, exhibiting biphasic dependence with cell speed reaching a maximum at intermediate concentrations of the matrix (DiMilla et al., 1993). Experimental manipulation of binding affinities of integrins to their ligands also affected cell motile behavior (Huttenlocher et al., 1996). Thus, it has been hypothesized that variations in either integrin cell surface expression levels, or ligand surface density, or integrin-ligand affinity could affect the adhesion strength and a speed of cell movement.
The experimental paradigm of early works, however, includes the untested assumption that the short-term adhesion measurements accurately reflect adhesive behavior of cells hours later when the motility measurements were made. Because the methods used to measure cell adhesion could not be applied at the time of cell migration, it was difficult to determine whether cells do regulate adhesion strength to control cell motility. The spinning disk detachment assay overcomes limitations of other methods because it can be used several hours after cell plating and a linear dependence of adhesion strength on fibronectin density is maintained (Shi and Boettiger, 2003). The application of the spinning disk detachment method to our well characterized model of IGF-I-stimulated migration of MCF-7 cells demonstrated a down-regulation of α5β1 integrin-mediated adhesion at the time when cell motility increased.
Because adhesion strength (τ50) is directly proportional to the number of bonds formed by intergrins and extracellular matrix proteins (Garcia et al., 1998), our data suggest the existence of IGF-IR-mediated mechanism that reduces the number of α5β1 intergrins coupled to the fibronectin molecules. Little is known about the mechanisms of adhesion bond disengagement. In theory, decreases in ECM ligand concentration, integrin cell surface expression, and integrin affinity could allow the cells to form fewer bonds with the substrate, resulting in the decreased adhesion. Cell adhesion might be weakened during postligand binding events through the processes of loss of integrin clustering and lateral mobility, so-called avidity modulation.
In our studies, cells were exposed to a uniform concentration of the adsorbed fibronectin. Because the adhesion strength had fallen rapidly within a 30-min exposure of WT cells to IGF-I, it is unlikely that IGF-IR signaling affects either deposition or degradation of the extracellular fibronectin. The production of fibronectin by mammary stroma rather than epithelia (Yamazaki and Eyden, 1998) makes it even less probable that IGF-IR signaling alters the composition of the ECM and weakens adhesion strength in MCF-7 cells.
There are examples of growth factors selectively modulating integrin expression levels. Overexpression of c-erbB2 in mammary epithelial cells is coupled with the decreased expression of α2 integrin (D'Souza et al., 1993). Furthermore, lower levels of α2β1 integrin expression were related to a lower degree of tumor cell differentiation, suggesting that a loss of α2 integrin might increase cell invasiveness. On the other hand in endothelial cells, the normally low expression of αVβ3 integrin is up-regulated by basic fibroblast growth factor and vascular endothelial growth factor, whereas transforming growth factor β increases expression of β1 integrins (Sepp et al., 1994; Senger et al., 1996; Smyth and Patterson, 2002). The observed changes in the cell surface expression levels of integrins occurred after a 24-h stimulation with growth factors. Thus, it is unlikely that a drastic difference in motile behavior of WT and DK cells exposed to IGF-I for 60 min is due to the altered total number of integrins.
Although it is tempting to speculate that IGF-IR signaling impairs affinity of α5β1 integrins for fibronectin, it is unclear whether reduced adhesion is due to effects on affinity of integrin-matrix interactions or perhaps due to a decrease in integrin clustering and avidity modulation. This view is supported by our unpublished data showing that average τ50 in WT cells measured 30 min after seeding cells on fibronectin-coated surface is 163.0 ± 19.7 dynes/cm2. Overnight, adhesion strength increased to its maximal level of 251.5 ± 20.2 dynes/cm2, suggesting that post integrin ligand-binding strengthening of adhesion has taken place. Conceivably, the maturation of the actin filamentous network might contribute to increase of adhesion strength, whereas disruption of F-actin integrity by IGF-IR signaling reduces cell adhesiveness.
The impairment of adhesion strength only in the WT cells with the functional IGF-IR but not in the DK cells with catalytically inactive receptor identifies the key regulatory role of IGF-IR signaling in the adhesive properties of breast carcinoma cells. The enzymatic activity of the IGF-IR is tightly controlled by the presence of its ligands in the tissue microenvironment. Unlike some other receptor tyrosine kinases, the IGF-IR cannot be activated as a result of overexpression alone and therefore availability of IGFs is imperative for activation of the receptor (Sachdev and Yee, 2001). Our findings here emphasize the importance of relatively small fluctuations in the extracellular IGF-I that may change drastically the motile behavior of carcinoma cells.
What Biochemical Signals Control the Adhesion Strength?
In this work, we investigated the nature of signaling mechanisms that regulate function of integrins in a steady-state adhesion and during initiation of cell movement. Previous studies examined only the initial cell attachment to the substrate and found that the physical strength of αVβ3 integrincytoskeleton bonds was modulated by the tyrosine kinase activity of Src (Felsenfeld et al., 1999; Datta et al., 2002). These data, together with the evidence that the linked activities of Src and focal adhesion kinase (FAK) may control adhesion turnover and migration (Fincham and Frame, 1998; Parsons et al., 2000), led us to investigate the possibility that the kinase activity of Src could regulate adhesion strength in human breast carcinoma cells. Selective decrease the autophosphorylation of Src affected neither the general appearance of F-actin nor adhesion strength in MCF-7 cells. Surprisingly, we found similar inhibitory effects of PP2 and PP3 on the rate of wound closure, pointing to Src phosphorylation-independent effect of these compounds on cell movement. On the bases of these findings, we conclude that in breast carcinoma cells modulation of c-Src autophosphorylation does not influence the adhesion strength mediated by α5β1 integrin. This, of course, does not rule out possible scaffolding role of the Src molecule in the regulation of integrins in focal adhesions.
With respect to cell migration, activity of the PI-3K has been related to the increased actin polymerization at the leading edge of lamellipodia (Ridley et al., 2003). We, and others, have previously shown that the PI-3K inhibitors blocked a loss of stress fibers and changes in focal adhesions in response to IGF-I (Guvakova and Surmacz, 1999; Cheng et al., 2000). The findings in this work are consistent with the previous results, but they suggest the additional involvement of a PI-3K-dependent mechanism in the control of the adhesion strength mediated by α5β1 integrins. Because the effect of the PI-3K inhibitor on F-actin, integrins, and cell migration is coupled, we reason that PI-3K signaling is necessary for destabilizing the integrin-cytoskeleton interface. This idea was further corroborated by data obtained with an actin stabilizer, jasplakinolide, which prevented weakening of adhesion strength by IGF-I. Also, activation of the PI-3K by IGF-I causes redistribution of α-actinin, a key actin binding protein, connecting β1 integrins to the actin network (Guvakova et al., 2002). Furthermore, in MCF-7 cells changes in the actin cytoskeleton and cell motility correlate with intercellular relocalization and protein tyrosine phosphatase-mediated dephosphorylation of focal adhesion proteins FAK, p130 Cas, and paxillin (Guvakova and Surmacz, 1999; Manes et al., 1999). In light of more recent findings in other cell types, down-regulation of FAK phosphorylation seems to be general mechanism of growth factor-induced motility in epithelial cells (Miao et al., 2000; Lu et al., 2001). Our preliminary data also show that adhesion strength of the WT cells attached to collagen is down-regulated by IGF-I, implicating modulation of integrin-cytoskeleton link as a general mechanism controlling β1 integrins. Therefore, we now propose that PI-3K signaling affects the β1 integrin-cytoskeleton linkage via effects on the assembly of actin filaments and dynamic modulation of multiprotein focal adhesion complexes associated with the intracellular tail of integrins. This may result in a reduction of number of integrins capable of binding to the extracellular matrix and contribute to the initiation of cell movement.
It is thought that the small GTPase RhoA plays a role in epithelial cell motility, at least in part through its downstream effector ROCK (Oxford and Theodorescu, 2003). From data obtained in different cell types, it has been suggested that ROCK affects cell motility by regulating assembly of stress fibers, dynamics of focal adhesions, or cell contraction via phosphorylation of myosin light chain. Although ROCK has been implicated in carcinoma migration (Somlyo et al., 2000; Jo et al., 2002; Sahai and Marshall, 2003), the functions that are regulated by this kinase in carcinoma cells have not been specified. In this study, we provide direct evidence that in stationary carcinoma cells ROCK activity is required to support adhesion strength mediated by α5β1 integrins. We found that similar control exists for HT-1080 human fibrosarcoma cells. We also found that sensitivity to the ROCK inhibitor varied among carcinoma cells. In T47D cells, in contrast to other tested breast cancer cells, the strength of α5β1 integrin adhesion has been slightly increased by Y-27632, although the change in τ50 did not reach statistical significance. The negative feedback effect from ROCK to Rho has been proposed for these cells attached to collagen gel via the α2β1 integrins (Wozniak et al., 2003). Hence, it would be interesting to test in the future whether such a regulatory loop exists in T47D cells attached to fibronectin.
Signaling events downstream of ERK MAPK may contribute to motility of colon carcinoma cells by inactivating β1 integrin and keeping RhoA and ROCK activities low (Vial et al., 2003). Our measurements showed that block of activities of ERK1/2 MAPKs had no significant effect on weakening of adhesion strength, and RhoA activity was not decreased in motile MCF-7 cells. Inhibitors of MEK and ROCK were unable to prevent the initial loss of cell-cell contacts. However, inhibition of ROCK, but not MEK, altered the ability of cells to move forward, suggesting that activity of ROCK is required for motile responses to IGF-I after cell-cell separation. Our finding that ROCK inhibitor causes extensive retraction of cell body contrasts with a previous study in which 10 μM of Y-27632 increased lamellipodia in the tail region of smooth muscle cells (Katsumi et al., 2002), implying distinct roles of ROCK in different cell types. Most importantly, we found that the effect of ROCK on α5β1 integrin-mediated adhesion depends on the context of the intercellular activity of other molecules. In a model, which we propose in Figure 10, ROCK supports strong adhesion only in the absence of IGF-IR signaling in stationary cells. When the activated IGF-IR kinase triggers the PI-3K, ROCK is no longer able to support strong adhesion via integrins, but its activity is needed for movement of the cell body.
Figure 10.

Model for regulation of α5β1 integrin adhesion strength in carcinoma cells. Black arrows indicate effects and white arrows indicate increased activity of signaling molecules. Bold arrows demonstrate prevailing pathways. Plus and minus symbols represent positive and negative effects, respectively. Left, in the absence of growth factor receptor signaling, basal activities of RhoA and ROCK keep actin cytoskeleton tension in a balance with a strong adhesion force mediated by integrins. Carcinoma cells strongly attach to the ECM and do not move. Right, growth factors, such as IGFs, trigger cell motility through the activation of the receptor tyrosine kinase, and subsequent increase in the activities of the PI-3K, Rho A, and ROCK. Apart from actin polymerization, activation of the PI3-K causes disassembly of F-actin and weakens the integrin-cytoskeleton linkage, leading to reduced adhesion strength via integrins. Once the cytoskeleton is reorganized by a PI-3K-dependent mechanism, ROCK is no longer able to support strong adhesion. ROCK generated cytoskeleton tension is involved in forwarding cell body.
With respect to cancer treatment, targeting RhoA and ROCK has been suggested for the development of agents that aimed at reducing the invasion of tumor cells (Itoh et al., 1999; Sahai and Marshall, 2003). In light of our findings, it seems that strategies directed at blocking ROCK need to be discreetly evaluated, and it would be necessary to investigate whether ROCK supports strong adhesion and integrity in normal tissues. The major finding in this work that in adherent cells signaling from growth factor receptor reduces cell attachment to the ECM via β1 integrin and renders cells motile may have direct applications not only in cancer research but also in developmental biology and tissue engineering. Indeed, the IGF-IR is expressed in a variety of normal and tumor cells (Khandwala et al., 2000), and β1 integrin is the most common integrin detected on adherent cells (Juliano, 1993), implying that cells of disparate phenotype could share mechanisms of cell migration. For instance, it would be interesting to investigate the link between the IGF-IR and integrins in blastocyst implantation, a key process in the formation of placenta. Current investigations suggest that β1 and β3 class integrins mediate blastocyst binding to fibronectin, whereas mesenchymally derived IGF-I acting via the IGF-IR provides a paracrine stimulus for migration of extravillous trophoblast cells (Lacey et al., 2002; Kabir-Salmani et al., 2004). Undoubtedly, knowledge of mechanisms modulating cell adhesive properties provides indispensable information for tissue engineers who design synthetic materials, surrogate ECM, for the purpose of reparative medicine aimed to restore or improve function of tissues and organs (Vogel and Baneyx, 2003).
Acknowledgments
We thank C. H. Damsky for AIIB2 and BIIG2 antibodies, and D. R. Armant and A. Hall for helpful discussions. This study was supported by grants to M.A.G. from the American Cancer Society IRG# 78-002-26 and the University Research Foundation, and in part by National Institutes of Health CA-16502 and GM-57388 grants to D. B.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E04-05-0399. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-05-0399.
References
- Adams, T. E., Epa, V. C., Garrett, T. P., and Ward, C. W. (2000). Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol. Life Sci. 57, 1050-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano, M., Chihara, K., Kimura, K., Fukata, Y., Nakamura, N., Matsuura, Y., and Kaibuchi, K. (1997). Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science 275, 1308-1311. [DOI] [PubMed] [Google Scholar]
- Amano, M., Fukata, Y., and Kaibuchi, K. (2000). Regulation and functions of Rho-associated kinase. Exp. Cell Res. 261, 44-51. [DOI] [PubMed] [Google Scholar]
- Birchmeier, C., Meyer, D., and Riethmacher, D. (1995). Factors controlling growth, motility, and morphogenesis of normal and malignant epithelial cells. Int. Rev. Cytol. 160, 221-266. [DOI] [PubMed] [Google Scholar]
- Boudreau, N., and Bissell, M. J. (1998). Extracellular matrix signaling: integration of form and function in normal and malignant cells. Curr. Opin. Cell Biol. 10, 640-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova, J. E. (2002). Epithelial cell cytoskeleton and intracellular trafficking V. Confluence of membrane trafficking and motility in epithelial cell models. Am. J. Physiol. 283, G1015-G1019. [DOI] [PubMed] [Google Scholar]
- Cheng, H. L., Steinway, M. L., Russell, J. W., and Feldman, E. L. (2000). GTPases and phosphatidylinositol 3-kinase are critical for insulin-like growth factor-I-mediated Schwann cell motility. J. Biol. Chem. 275, 27197-27204. [DOI] [PubMed] [Google Scholar]
- Cramer, L. P. (1999). Role of actin-filament disassembly in lamellipodium protrusion in motile cells revealed using the drug jasplakinolide. Curr. Biol. 9, 1095-1105. [DOI] [PubMed] [Google Scholar]
- D'Souza, B., Berdichevsky, F., Kyprianou, N., and Taylor-Papadimitriou, J. (1993). Collagen-induced morphogenesis and expression of the alpha 2-integrin subunit is inhibited in c-erbB2-transfected human mammary epithelial cells. Oncogene 8, 1797-1806. [PubMed] [Google Scholar]
- Datta, A., Huber, F., and Boettiger, D. (2002). Phosphorylation of beta3 integrin controls ligand binding strength. J. Biol. Chem. 277, 3943-3949. [DOI] [PubMed] [Google Scholar]
- DiMilla, P. A., Barbee, K., and Lauffenburger, D. A. (1991). Mathematical model for the effects of adhesion and mechanics on cell migration speed. Biophys. J. 60, 15-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMilla, P. A., Stone, J. A., Quinn, J. A., Albelda, S. M., and Lauffenburger, D. A. (1993). Maximal migration of human smooth muscle cells on fibronectin and type IV collagen occurs at an intermediate attachment strength. J. Cell Biol. 122, 729-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr, M. E., and Jones, J. I. (1996). The roles of integrins and extracellular matrix proteins in the insulin-like growth factor I-stimulated chemotaxis of human breast cancer cells. J. Biol. Chem. 271, 2443-2447. [DOI] [PubMed] [Google Scholar]
- Dunn, S. E., Ehrlich, M., Sharp, N. J., Reiss, K., Solomon, G., Hawkins, R., Baserga, R., and Barrett, J. C. (1998). A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res. 58, 3353-3361. [PubMed] [Google Scholar]
- Felsenfeld, D. P., Schwartzberg, P. L., Venegas, A., Tse, R., and Sheetz, M. P. (1999). Selective regulation of integrincytoskeleton interactions by the tyrosine kinase Src. Nat. Cell Biol. 1, 200-206. [DOI] [PubMed] [Google Scholar]
- Fincham, V. J., and Frame, M. C. (1998). The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J. 17, 81-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame, M. C., Fincham, V. J., Carragher, N. O., and Wyke, J. A. (2002). v-Src's hold over actin and cell adhesions. Nat. Rev. Mol. Cell Biol. 3, 233-245. [DOI] [PubMed] [Google Scholar]
- Garcia, A. J., Huber, F., and Boettiger, D. (1998). Force required to break alpha5beta1 integrin-fibronectin bonds in intact adherent cells is sensitive to integrin activation state. J. Biol. Chem. 273, 10988-10993. [DOI] [PubMed] [Google Scholar]
- Geiger, B., Bershadsky, A., Pankov, R., and Yamada, K. M. (2001). Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2, 793-805. [DOI] [PubMed] [Google Scholar]
- Guvakova, M. A., Adams, J. C., and Boettiger, D. (2002). Functional role of alpha-actinin, PI 3-kinase and MEK1/2 in insulin-like growth factor I receptor kinase regulated motility of human breast carcinoma cells. J. Cell Sci. 115, 4149-4165. [DOI] [PubMed] [Google Scholar]
- Guvakova, M. A., and Surmacz, E. (1997). Overexpressed IGF-I receptors reduce estrogen growth requirements, enhance survival, and promote E-cadherin-mediated cell-cell adhesion in human breast cancer cells. Exp. Cell Res. 231, 149-162. [DOI] [PubMed] [Google Scholar]
- Guvakova, M. A., and Surmacz, E. (1999). The activated insulin-like growth factor I receptor induces depolarization in breast epithelial cells characterized by actin filament disassembly and tyrosine dephosphorylation of FAK, Cas, and paxillin. Exp. Cell Res. 251, 244-255. [DOI] [PubMed] [Google Scholar]
- Huttenlocher, A., Ginsberg, M. H., and Horwitz, A. F. (1996). Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J. CELL BIOL. 134, 1551-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, K., Yoshioka, K., Akedo, H., Uehata, M., Ishizaki, T., and Narumiya, S. (1999). An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat. Med. 5, 221-225. [DOI] [PubMed] [Google Scholar]
- Jo, M., Thomas, K. S., Somlyo, A. V., Somlyo, A. P., and Gonias, S. L. (2002). Cooperativity between the Ras-ERK and Rho-Rho kinase pathways in urokinase-type plasminogen activator-stimulated cell migration. J. Biol. Chem. 277, 12479-12485. [DOI] [PubMed] [Google Scholar]
- Jones, J. I., Doerr, M. E., and Clemmons, D. R. (1995). Cell migration: interactions among integrins, IGFs and IGFBPs. Prog. Growth Factor Res. 6, 319-327. [DOI] [PubMed] [Google Scholar]
- Juliano, R. L. (1993). The role of beta 1 integrins in tumors. Semin. Cancer Biol. 4, 277-283. [PubMed] [Google Scholar]
- Kabir-Salmani, M., Shiokawa, S., Akimoto, Y., Sakai, K., and Iwashita, M. (2004). The role of alpha(5)beta(1)-integrin in the IGF-I-induced migration of extravillous trophoblast cells during the process of implantation. Mol. Hum. Reprod. 10, 91-97. [DOI] [PubMed] [Google Scholar]
- Katsumi, A., Milanini, J., Kiosses, W. B., del Pozo, M. A., Kaunas, R., Chien, S., Hahn, K. M., and Schwartz, M. A. (2002). Effects of cell tension on the small GTPase Rac. J. Cell Biol. 158, 153-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely, P. J., Fong, A. M., Zutter, M. M., and Santoro, S. A. (1995). Alteration of collagen-dependent adhesion, motility, and morphogenesis by the expression of antisense alpha 2 integrin mRNA in mammary cells. J. Cell Sci. 108, 595-607. [DOI] [PubMed] [Google Scholar]
- Khandwala, H. M., McCutcheon, I. E., Flyvbjerg, A., and Friend, K. E. (2000). The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr. Rev. 21, 215-244. [DOI] [PubMed] [Google Scholar]
- Kleinberg, D. L., Feldman, M., and Ruan, W. (2000). IGF-I: an essential factor in terminal end bud formation and ductal morphogenesis. J. Mammary Gland Biol. Neoplasia 5, 7-17. [DOI] [PubMed] [Google Scholar]
- Lacey, H., Haigh, T., Westwood, M., and Aplin, J. D. (2002). Mesenchymally-derived insulin-like growth factor 1 provides a paracrine stimulus for trophoblast migration. BMC Dev. Biol. 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventhal, P. S., and Feldman, E. L. (1997). Insulin-like growth factors as regulators of cell motility signaling mechanisms. Trends Endocrinol. Metab. 8, 1-6. [DOI] [PubMed] [Google Scholar]
- Long, L., Navab, R., and Brodt, P. (1998). Regulation of the Mr 72,000 type IV collagenase by the type I insulin-like growth factor receptor. Cancer Res. 58, 3243-3247. [PubMed] [Google Scholar]
- Lu, Z., Jiang, G., Blume-Jensen, P., and Hunter, T. (2001). Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol. Cell Biol. 21, 4016-4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes, S., Mira, E., Gomez-Mouton, C., Zhao, Z. J., Lacalle, R. A., and Martinez, A. (1999). Concerted activity of tyrosine phosphatase SHP-2 and focal adhesion kinase in regulation of cell motility. Mol. Cell Biol. 19, 3125-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, H., Burnett, E., Kinch, M., Simon, E., and Wang, B. (2000). Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 2, 62-69. [DOI] [PubMed] [Google Scholar]
- Miyamoto, S., Teramoto, H., Gutkind, J. S., and Yamada, K. M. (1996). Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J. Cell Biol. 135, 1633-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxford, G., and Theodorescu, D. (2003). Ras superfamily monomeric G proteins in carcinoma cell motility. Cancer Lett. 189, 117-128. [DOI] [PubMed] [Google Scholar]
- Palecek, S. P., Loftus, J. C., Ginsberg, M. H., Lauffenburger, D. A., and Horwitz, A. F. (1997). Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 385, 537-540. [DOI] [PubMed] [Google Scholar]
- Parsons, J. T., Martin, K. H., Slack, J. K., Taylor, J. M., and Weed, S. A. (2000). Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene 19, 5606-5613. [DOI] [PubMed] [Google Scholar]
- Polyak, K. (2001). On the birth of breast cancer. Biochim. Biophys. Acta 1552, 1-13. [DOI] [PubMed] [Google Scholar]
- Ridley, A. J., Schwartz, M. A., Burridge, K., Firtel, R. A., Ginsberg, M. H., Borisy, G., Parsons, J. T., and Horwitz, A. R. (2003). Cell migration: integrating signals from front to back. Science 302, 1704-1709. [DOI] [PubMed] [Google Scholar]
- Sachdev, D., Hartell, J. S., Lee, A. V., Zhang, X., and Yee, D. (2004). A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells. J. Biol. Chem. 279, 5017-5024. [DOI] [PubMed] [Google Scholar]
- Sachdev, D., and Yee, D. (2001). The IGF system and breast cancer. Endocr. Relat. Cancer 8, 197-209. [DOI] [PubMed] [Google Scholar]
- Sahai, E., and Marshall, C. J. (2003). Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 5, 711-719. [DOI] [PubMed] [Google Scholar]
- Schneller, M., Vuori, K., and Ruoslahti, E. (1997). Alphavbeta3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. EMBO J. 16, 5600-5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger, D. R., Ledbetter, S. R., Claffey, K. P., Papadopoulos-Sergiou, A., Peruzzi, C. A., and Detmar, M. (1996). Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the alphavbeta3 integrin, osteopontin, and thrombin. Am. J. Pathol. 149, 293-305. [PMC free article] [PubMed] [Google Scholar]
- Sepp, N. T., Li, L. J., Lee, K. H., Brown, E. J., Caughman, S. W., Lawley, T. J., and Swerlick, R. A. (1994). Basic fibroblast growth factor increases expression of the alpha v beta 3 integrin complex on human microvascular endothelial cells. J. Investig. Dermatol. 103, 295-299. [DOI] [PubMed] [Google Scholar]
- Shi, Q., and Boettiger, D. (2003). A novel mode for integrin-mediated signaling: tethering is required for phosphorylation of FAK Y397. Mol. Biol. Cell 14, 4306-4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth, S. S., and Patterson, C. (2002). Tiny dancers: the integrin-growth factor nexus in angiogenic signaling. J. Cell Biol. 158, 17-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo, A. V., Bradshaw, D., Ramos, S., Murphy, C., Myers, C. E., and Somlyo, A. P. (2000). Rho-kinase inhibitor retards migration and in vivo dissemination of human prostate cancer cells. Biochem. Biophys. Res. Commun. 269, 652-659. [DOI] [PubMed] [Google Scholar]
- Vial, E., Sahai, E., and Marshall, C. J. (2003). ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell 4, 67-79. [DOI] [PubMed] [Google Scholar]
- Vogel, V., and Baneyx, G. (2003). The tissue engineering puzzle: a molecular perspective. Annu. Rev. Biomed. Eng. 5, 441-463. [DOI] [PubMed] [Google Scholar]
- Wang, J., and Armant, D. R. (2002). Integrin-mediated adhesion and signaling during blastocyst implantation. Cells Tissues. Organs 172, 190-201. [DOI] [PubMed] [Google Scholar]
- Wiseman, B. S., and Werb, Z. (2002). Stromal effects on mammary gland development and breast cancer. Science 296, 1046-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak, M. A., Desai, R., Solski, P. A., Der, C. J., and Keely, P. J. (2003). ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J. Cell Biol. 163, 583-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki, K., and Eyden, B. P. (1998). Characterisation of breast stromal fibroblasts: cell surface distribution of collagen type IV, laminin and fibronectin. J. Submicrosc. Cytol. Pathol. 30, 217-226. [PubMed] [Google Scholar]